Type IA Topoisomerases as Targets for Infectious Disease Treatments


1
Department of Chemistry and Biochemistry, Florida International University, Miami, FL 33199, USA
2
Biomolecular Sciences Institute, Florida International University, Miami, FL 33199, USA
*
Author to whom correspondence should be addressed.
Microorganisms 2021, 9(1), 86; https://doi.org/10.3390/microorganisms9010086
Submission received: 15 November 2020
/
Revised: 13 December 2020
/
Accepted: 17 December 2020
/
Published: 1 January 2021
(This article belongs to the Section Antimicrobial Agents and Resistance)
Abstract
:Infectious diseases are one of the main causes of death all over the world, with antimicrobial resistance presenting a great challenge. New antibiotics need to be developed to provide therapeutic treatment options, requiring novel drug targets to be identified and pursued. DNA topoisomerases control the topology of DNA via DNA cleavage–rejoining coupled to DNA strand passage. The change in DNA topological features must be controlled in vital processes including DNA replication, transcription, and DNA repair. Type IIA topoisomerases are well established targets for antibiotics. In this review, type IA topoisomerases in bacteria are discussed as potential targets for new antibiotics. In certain bacterial pathogens, topoisomerase I is the only type IA topoisomerase present, which makes it a valuable antibiotic target. This review will summarize recent attempts that have been made to identify inhibitors of bacterial topoisomerase I as potential leads for antibiotics and use of these inhibitors as molecular probes in cellular studies. Crystal structures of inhibitor–enzyme complexes and more in-depth knowledge of their mechanisms of actions will help to establish the structure–activity relationship of potential drug leads and develop potent and selective therapeutics that can aid in combating the drug resistant bacterial infections that threaten public health.
1. Introduction
Infectious diseases are diseases where certain microorganisms grow and replicate inside a host, leading to damage or injury to body tissues of the host. Even normal flora can cause diseases to the host if the host is immunocompromised, for instance in cancer and acquired immunodeficiency syndrome (AIDS). Many effective antimicrobial agents have been discovered and marketed so far. However, there has been a constant issue that counteracts the effects of well-established antimicrobial agents, i.e., antimicrobial drug resistance [1,2,3].
Antimicrobial resistance has been a major global health concern in recent years. Mechanisms of resistance vary [4,5], such as degrading or changing the drug molecule to an inactive form inside the microorganism [6], modifying or protecting the target for the antimicrobial agent by the microorganism [7,8], or pumping out the drug molecules from the microorganism cell, known as efflux [9,10]. Resistance to antimicrobial agents is usually genetically encoded by the microorganism, either on the bacterial chromosome, or on a plasmid that can be spread among different strains of microorganisms, resulting in the emergence of new resistant strains [11,12].
The number of deaths attributable to antimicrobial resistance is expected to continue growing, hitting around 10 million cases in 2050 [13]. Worldwide, tuberculosis (TB) is one of the top 10 causes of death. According to the WHO, TB is the leading cause of death from a single infectious agent. TB caused an estimated 1.4 million deaths worldwide in 2019, with an estimated 10.0 million new cases of TB, equivalent to 130 cases per 100,000 population [14]. Globally, there were an estimated 558,000 new cases of rifampicin-resistant TB (RR-TB). Among RR-TB cases, an estimated 82% were multidrug-resistant TB (MDR-TB). Furthermore, 3.5% of new TB cases and 18% of previously treated cases were MDR/RR-TB. Drug resistant TB is very difficult to treat [15]. Other examples of drug resistant bacteria include the Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa and Enterobacter spp. (ESKAPE) pathogens [16,17].
Following the discovery of daptomycin in late 1980s [18], the process of novel antibiotic discovery stagnated and could not keep up with the emergence of antibiotic resistant pathogens [19,20]. Hence, the discovery of novel antimicrobial agents is an urgent need. DNA topoisomerases are well known targets for antibacterial drug discovery [7,21,22,23,24,25]. Topoisomerases are responsible for controlling the topology of the genome, including local and global DNA supercoiling [26,27]. DNA topology and topoisomerase functions play important roles in replication, transcription, and genome stability [21,27,28,29,30,31,32].
In general, all DNA topoisomerases work by binding to the target DNA molecule, cleaving one or both strands of the double helix, passing another single or double strand through the break, and resealing the DNA [21,30]. A covalent complex is transiently formed between an active tyrosine residue of the topoisomerase enzyme and one end of the broken strand during the cleavage process. [33]. The covalent complex with cleaved DNA is a vulnerable intermediate that can lead to cell death if trapped.
The interests in studying the structures, functions, mechanisms of actions, and inhibitors of DNA topoisomerases are mainly due to two reasons: (1) the pivotal role of DNA topoisomerases in the management and control of DNA topology [21,30,32] and (2) development of topoisomerase inhibitors into widely used clinical therapies for cancer and bacterial infections [21,24,25,34,35,36]. Many efforts have been exerted so far to either synthesize new molecules, or discover new bioactive molecules, which could potentially inhibit specific members of the DNA topoisomerase family, to provide new treatment for cancer and infectious diseases.
2. Type IA Topoisomerases
DNA topoisomerases are divided into two main categories: type I topoisomerases and type II topoisomerases. Differences in DNA strand passage activity and catalysis are the basis of this categorization, and each main category is subdivided into subcategories according to the mode of DNA binding and relaxation. Type I topoisomerases break and rejoin a single DNA strand during catalysis. On the other hand, type II topoisomerases break and rejoin the two strands of a double helix in order to pass another intact double helix through the break, promoting catenation/decatenation, unknotting/knotting, and relaxation of positively and negatively supercoiled DNA [37]. Type II topoisomerases are subcategorized into two subfamilies: topoisomerases IIA and topoisomerases IIB. All type II topoisomerases covalently attach to the 5′ end of the DNA through a phosphotyrosine bond, requiring divalent magnesium cation and ATP hydrolysis. Archaeal and bacterial type IIA topoisomerases, such as Escherichia coli DNA gyrase, are A2B2 heterotetramers. In contrast, eukaryotic type IIA topoisomerases are all homodimeric enzymes. The discovery of topoisomerase VI, an A2B2 heterotetrameric type II topoisomerase found in all archaea, added a new type IIB subfamily to the type II topoisomerases. The relative arrangement of elements required for type II topoisomerase’s mechanism as well as the overall structural organization are dissimilar between the type IIA and type IIB topoisomerases [30]. Type IIA topoisomerases cleave the opposing strands of duplex DNA with a four-base stagger, while two-base staggered cuts are made by type IIB topoisomerases [37].
Type I topoisomerases are subcategorized into three subfamilies: topoisomerases IA, topoisomerases IB, and topoisomerases IC [38]. Members of the topoisomerase IA subfamily share common mechanistic features, such as being monomeric proteins and covalent attachment to the cleaved DNA end through a 5’ phosphotyrosine bond. Divalent cation is needed as a cofactor to complete the catalytic process. Finally, the members of this subfamily can relax negative supercoils because they only require single stranded regions of the target DNA molecule for binding [39]. In the protein-mediated DNA gate model for strand passage by type IA topoisomerases [40,41], a segment of single-stranded DNA (ssDNA) binds through domains I and IV, and the active tyrosine residue in domain III performs the nucleophilic attack on this G-strand ssDNA to create a break. Movement of domains I and III away from each other results in gate opening (Figure 1) to allow passage of another ssDNA strand (T-strand). Gate-opening dynamics of E. coli topoisomerase I and III were observed recently in single-molecule studies [42]. The presence of Mg2+ divalent ions is necessary for the process of DNA rejoining by type IA topoisomerases [43,44,45]. The divalent ion is hypothesized to help position the 3′-hydroxyl group of the cleaved DNA in proximity to the phosphotyrosine linkage, where the 3′-hydroxyl group performs a nucleophilic attack on the phosphotyrosine linkage leading to rejoining of the DNA backbone [46]. The requirement for divalent ions and formation of 5-phosphotyrosine bond are mechanistic similarities between type IA and type IIA topoisomerase. In contrast, type IB and type IC topoisomerases do not require divalent ions for catalysis and are linked to the 3′-phosphate in the covalent intermediate. There is no sequence or structural homology between type IA, type IB and type IC topoisomerases [47]. Type IA topoisomerases are the only family of topoisomerases that can catalyze topological changes on both DNA and RNA molecules [48]. The activity of type IA topoisomerases is needed for overcoming DNA or RNA topological barriers that require strand passage through the break mediated by the topoisomerase IA on a single strand of nucleic acid [32,49]. At least one type IA topoisomerase is present in all free-living organisms [50] found in the three kingdoms of life. Drugs targeting bacterial or parasitic type IA topoisomerases for treatment of infectious diseases must be selective in not inhibiting the human type IA topoisomerases TOP3A and TOP3B.
2.1. Rationale for Type IA Topoisomerases as Drug Targets
Since topoisomerases play key roles in managing the topology of DNA, which, in turn, is a major factor impacting cell viability, they have been widely targeted for clinical treatment of cancer and infectious diseases [24,25,51]. Topoisomerase inhibitors can be classified based on the mechanism of action as poison inhibitors or catalytic inhibitors [52,53]. Poison inhibitors are agents that work by stabilizing the topoisomerase covalent complex that is formed as an intermediate during catalysis, while catalytic inhibitors are agents that work by interfering with the catalytic cycle of the enzyme in any other step, such as inhibitors that prevent initial substrate binding or formation of the cleavage complex. Topoisomerase targeting drugs in current clinical use are poison inhibitors against type IB and type IIA topoisomerase [52,53,54]. Topoisomerase poison inhibitors cause loss of cellular viability through accumulation of breaks in the chromosome, triggering apoptosis in cancer cells [55] and oxidative damage cell death pathway in bacteria [56]. This mechanism of cell death is highly efficient since it does not require the activity of most of the target topoisomerase molecules to be blocked and the topoisomerase activity does not need to be essential. Even though there are currently no approved drugs that utilize type IA topoisomerases as cellular targets, it can be argued that since all topoisomerases utilize an active site tyrosine to form the covalent intermediate, it should also be possible for a poison inhibitor to trap the covalent complex formed by type IA topoisomerases.
2.2. Validation for Bacterial Topoisomerase IA as a Novel Antibiotics Target
Topoisomerase I and topoisomerase III are type IA topoisomerases that are present in bacteria, with reverse gyrase being found in thermophilic bacteria. E. coli has both topoisomerase I and topoisomerase III present. Bacterial topoisomerase I is much more efficient for relaxation of negatively supercoiled DNA during transcription while topoisomerase III has a stronger decatenation activity for resolution of replication and recombination intermediates [57,58,59,60]. Null mutation of topA in E. coli can be compensated by mutations that reduce the activity of gyrase that has the opposite effect on DNA supercoiling [61,62,63]. Nevertheless, endogenous inhibitors of topoisomerase I activity including overexpressed Tn5 transposase [64,65], T4 gp55.2 [66] and toxin YjhX [67] can inhibit cell growth and result in a loss of viability. Furthermore, even though the essentiality of topoisomerase I in some bacteria is uncertain, poison inhibitors against the type IA topoisomerase present in every bacterium should generate bactericidal DNA lesions. The bactericidal consequence of topoisomerase I covalent complex accumulation in E. coli was demonstrated through identification of bacterial topoisomerase I mutants deficient in DNA rejoining [44,45,68,69,70]. The mutations involved include mutations in the conserved D111, D113 and E115 (DDE) triad that bind Mg2+ required for DNA religation [69]. These dominant lethal topoisomerase I mutations mimic the expected action of type IA topoisomerase poison inhibitors. It should also be noted that YjhX overexpression resulted in E. coli cell death, even though YjhX has been shown not to be a poison inhibitor of E. coli topoisomerase I [67]. Therefore, inhibitors of bacterial topoisomerase I do not necessarily have to act as topoisomerase poisons to be bactericidal.
Studies have shown that topoisomerase I is essential for cellular viability of a number of important bacterial pathogens, including Mycobacterium tuberculosis [71,72], Streptococcus pneumoniae [73], P. aeruginosa [74] and Helicobacter pylori [75]. In bacteria that only have one type IA topoisomerase, the catalytic activity of the enzyme should be essential for overcoming topological barriers that require passage of one strand of DNA through another single strand [32], and catalytic inhibitors of topoisomerase I would have antibacterial efficacy regardless of the mechanism of topoisomerase inhibition. Hence, bacterial topoisomerase IA represents a valid target for novel antibiotics aimed at overcoming antimicrobial resistance.
2.3. Eukaryotic Type IA Topoisomerases as Potential Target for Infectious Disease Treatment
Fungi have only one type IA topoisomerase, TOP3, that has high degree of similarity to bacterial topoisomerase III. The TOP3 mutant is not viable in Saccharomyces pombe [76,77] and has a slow growth phenotype in Saccharomyces cerevisiae [78]. In higher eukaryotes, the TOP3 gene has been duplicated into TOP3A and TOP3B isoforms [50]. TOP3A is essential for mice embryo development [79]. TOP3B knockout mice developed to maturity but had a shortened life span [80]. Human topoisomerase III beta (TOP3B) has been shown to have both DNA and RNA topoisomerase activities [81,82]. It relaxes hyper-negatively supercoiled DNA in transcription and prevents R-loop accumulation [83,84]. Interaction between TOP3B and mRNAs [85] is required for neurodevelopment and synapse formation [81,82]. Human TOP3B is stabilized by Tudor domain containing 3 protein (TDRD3) [84,86,87], which was previously identified as a host protein needed for efficient replication of flaviviruses, such as dengue and yellow fever virus [88]. However, subsequent follow-up studies on the role of TDRD3 in viral replication have revealed that TOP3B is the actual host factor required for the efficient replication of all positive-sense single-stranded RNA viruses, with TDRD3 playing a role in the stabilization of TOP3B [49]. Specific inhibitors for TOP3B could potentially be useful as broad-spectrum antiviral treatments against flaviviruses and coronaviruses including SARS COV-2 [49].
Topoisomerases in kinetoplastid parasites are potential targets for the development of improved therapeutic options against deadly parasitic diseases [89,90]. Three type IA topoisomerases are present in the kinetoplastids. In addition to functionally active TOP3A [91] and TOP3B [92], a prokaryotic-like mitochondrial TOP1A is also present. The mitochondrial TOP1A in Trypanosoma brucei is essential for late theta replication intermediates structure resolution [93]. Since the mitochondrial TOP1A has no close homologs in humans, it could be a target for selectively toxic new antiprotozoal treatments [93].
3. Screening Approaches
3.1. In Silico Screening
Several strategies and techniques can be used to identify novel topoisomerase inhibitors for treatment of infectious diseases. A commonly used strategy is screening chemical libraries composed of a large number of molecules for the desired activity. This strategy has been widely used for decades in drug discovery. The chemical libraries used could comprise of synthetic compounds, natural products, or both. In silico virtual screening employing docking and molecular dynamics (MD) simulations can be used first to identify potential inhibitors from compound libraries, followed by biochemical assays on a small number of in silico hits with the highest predicted binding affinity to confirm the interaction and assess the topoisomerase inhibition and antimicrobial activity. This approach has been applied to identify inhibitors for E. coli and M. tuberculosis topoisomerase I using either crystal structures or target structures generated by computational modeling [94,95,96,97,98].
3.2. Biochemical Screening Assays
Various biochemical assays can be used to assess the ability of a compound to inhibit the activity of a specific topoisomerase, and to determine the mechanism of inhibition so that the hit can be classified either as a catalytic inhibitor or as a poison inhibitor. Examples include, but are not limited to, gel based-assays, fluorescence-based assays, and detection of DNA-enzyme covalent complex via the rapid approach to DNA adduct recovery (RADAR) assay.
Agarose gel electrophoresis can be used to determine the inhibition of the bacterial topoisomerase I, since this enzyme relaxes supercoiled plasmid DNA. A supercoiled plasmid DNA, representing inhibited enzyme or control reaction without enzyme, will migrate the fastest, while the fully relaxed plasmid DNA will migrate the slowest in an agarose gel electrophoresis experiment [99]. DNA can be visualized using ethidium bromide staining. The presence of an effective inhibitor will prevent the enzyme from relaxing the plasmid DNA, and it will instead remain supercoiled until the concentration of the inhibitor is sufficiently low for the enzyme to regain its activity. To further determine the mode of inhibition, single-stranded DNA or oligonucleotide substrate labeled with 32P at the 5′-end can be used to visualize the shortened cleavage product that results from topoisomerase I cleaving the substrate following electrophoresis of the reaction products in denaturing acrylamide gel [100]. A topoisomerase poison inhibitor is expected to result in the increase in accumulated cleavage product.
Since gel electrophoresis is time consuming and low in throughput capacity, several fluorescence-based assays have been developed to monitor topoisomerase activities and detect potential inhibitors with high throughput capacity. One such assay involved the use of an immobilized triplex-forming oligonucleotide capable of selectively capturing supercoiled plasmids that can be quantified by SYBR Green dye [101]. Another group of researchers have developed a homogenous assay that measures the fluorescence anisotropy of the triplex-forming oligonucleotide and can be applied to assay the activity of both E. coli gyrase and topoisomerase I [102]. A different approach is to insert into a plasmid an inverted repeat containing a fluorophore-quencher pair that can emit fluorescence depending on whether cruciform formation is facilitated by negative supercoiling of the plasmid [103,104]. An assay based on supercoiling-dependent fluorescence quenching has been used to study the kinetics of relaxation of negatively supercoiled DNA by E. coli and Mycobacterium smegmatis topoisomerase I [105]. However, these fluorescence-based assays designed for the measurement of supercoiling/relaxation of plasmid DNA have not yet been applied to screen for bacterial topoisomerase I inhibitors from a large compound library. An oligonucleotide substrate with fluorophore and quencher at the 5′ and 3′ ends of the stem-loop structure was designed to detect the inhibition of DNA religation by topoisomerase I poison inhibitors [106]. This assay identified organomercury compounds that act as poison inhibitors against E. coli and Y. pestis topoisomerase I that bind Zn(II) with cysteines [100].
The rapid approach to DNA adduct recovery (“RADAR”) assay was first developed to purify and measure the level of topoisomerase–DNA complex trapped by anticancer drugs that act as topoisomerase poison inhibitors in mammalian cells [107]. A modified RADAR/ELISA assay can also measure the level of gyrase covalent complex trapped by fluoroquinolones in bacterial cells [108]. Recently, it has been shown that this RADAR/ELISA assay can detect in E. coli and M. smegmatis increased accumulation of covalent complex formed by bacterial topoisomerase I with mutations that inhibit DNA rejoining, providing a potential assay for the discovery and optimization of drugs that act as bacterial topoisomerase I poison inhibitors [109].
4. Recent Attempts to Discover Novel Bacterial Topoisomerase I Inhibitors
During the past decade, various attempts have been made to find inhibitors of bacterial topoisomerase I to provide potential leads for the development of novel antibiotics, aiming to combat antimicrobial resistance. These inhibitors are discussed below and the half maximal inhibitory concentrations (IC50s) for inhibition of E. coli or M. tuberculosis topoisomerase I activity are shown in Table 1.
4.1. Bis-Benzimidazoles
Bis-benzimidazole derivatives have gained much attention as potential therapeutics since the development of Hoechst bis-benzimidazole 33,258 and 33,342 as DNA minor groove binders that inhibit human topoisomerase I and show activity against certain types of cancers [117,118], but the drug development efforts did not progress to clinical trials due to high cytotoxicity [119]. A series of bis-benzimidazoles with closely related structures were synthesized by two different groups and found to provide selective inhibition of type IA E. coli topoisomerase I over inhibition of type IB human topoisomerase I [110,120]. The bis-benzimidazoles specific for E. coli topoisomerase I exhibited low minimum inhibitory concentrations (MICs) of 8 µg/mL against bacterial pathogens that included methicillin-resistant S. aureus (MRSA), vancomycin-resistant Enterococcus faecalis, Staphylococcus epidermidis, A. baumannii, Shigella flexneri and clinical isolates of E. coli [110,111,121]. However, most of these derivatives still had cytotoxicity concerns, due to their DNA-binding activity [110,121]. DPA 154, shown in Figure 2, was also reported to elicit changes in the cellular ultrastructure including induction of spheroplasts and membrane lysis, suggesting a dual mechanism of antibacterial action that includes compromising cell membrane integrity [122]. In vivo efficacy against E. coli was demonstrated in the mouse systemic infection model as well as the mouse neutropenic thigh model [111] for one of the bis-benzimidazoles, 2′-(4-ethoxyphenyl)-5-(4-propylpiperazin-1-yl)-1H,1′H-2,5′-bibenzo[d]imidazole (PPEF), shown in Figure 2. It was proposed that PPEF shifts the cleavage–religation equilibrium of E. coli topoisomerase I by binding to the strictly conserved acidic triad D111, D113 and E115 (DDE) based on results from a site-directed mutagenesis study [123] where the aspartate and glutamate residues in the acidic triad were replaced with alanine residues. Circular dichroism studies and in silico docking have shown reduced binding of PPEF to these mutants, especially for the double mutants where two out of the three DDE residues were replaced by alanine [123]. This acidic triad of bacterial topoisomerase I is known to be involved in interactions with Mg2+ ions [124,125]. Substitution of the first aspartate to asparagine has been shown to affect metal binding and result in deficiency of DNA religation [69].
4.2. Tricyclic Antidepressants
In silico docking was conducted to screen a library of FDA-approved drugs for their potential activity against a homolog model of M. tuberculosis topoisomerase I with Mg2+ and a DNA fragment bound in the open state [96]. Clinically prescribed tricyclic antidepressant imipramine and norclomipramine, which is the active metabolite of another tricyclic antidepressant (clomipramine), were then selected based on the docking results for biochemical and whole cell assays. Imipramine and norclomipramine (Figure 3) were found to inhibit M. tuberculosis topoisomerase I relaxation activity at <0.1 µM concentration following 15 min preincubation with the enzyme [96]. However, inhibition of E. coli topoisomerase I was not detected at up to 25 µM compound concentration. In silico docking with the M. tuberculosis topoisomerase I homology model has placed imipramine and norclomipramine near the strictly conserved DDE acidic triad. Alanine substitution at the glutamate in the third position abolished inhibition by the compounds while alanine substitution at the first aspartate had no effect on the inhibition. Both compounds showed a slight increase in cleavage product from a 32P-labeled double stranded oligonucleotide substrate and enhanced cytotoxicity in M. smegmatis overexpressing recombinant mycobacterial topoisomerase I, consistent with the compounds acting with a poison inhibitor mechanism [96]. However, the MICs required to stop the growth of M. tuberculosis were relatively high (60 µM for norclomipramine and 250 µM for imipramine). The tricyclic antidepressants have hard-to-tolerate side effects and several drug–drug interactions, which have recently led to the clinical preference of other classes of antidepressants [126]. Therefore, the tricyclic antidepressants are probably not a practical clinical option for targeting bacterial topoisomerase I.
4.3. Inhibitors Based on Polyamine Scaffold
The collection of small molecules at Torrey Pines Institute (TPI) now consists of more than 80 million small molecules organized into a small number of scaffold-ranking libraries so that promising molecular scaffolds can be identified quickly from a positive result in a biological assay. Structural analogs in the selected scaffold library mixtures can be studied with position scanning to identify the most favorable substitutions in the specific molecular scaffold [127,128,129]. This approach has been applied previously for the discovery of bacterial tyrosine recombinase and site-specific recombinase inhibitors [130]. In efforts to discover small molecules targeting bacterial topoisomerase I, inhibition of E. coli topoisomerase I relaxation activity was assayed first to identify the most promising scaffold and then the most inhibitory substitutions at three different R positions of the selected polyamine scaffold (Figure 4A). Fourteen related compounds that were synthesized as part of the 2471 series based on the position scanning data showed IC50 values < 20 µM against E. coli and M. tuberculosis topoisomerase I, and MIC values < 50 µM against M. smegmatis [112]. Among these 14 compounds, four polyamine analogues (Figure 4B) were confirmed to be also active against M. tuberculosis with selectivity index (50% cytotoxic concentration (CC50) against J774 macrophage cell line/IC50 against M. tuberculosis) values of up to 15. The observed inhibition of bacterial type IA topoisomerases by the 2471 compounds is unlikely due to non-specific inhibition of the type IA topoisomerase activity by the positively charged polyamine structure, or non-specific binding of the polyamine inhibitors to DNA, as the IC50 values for inhibition of human topoisomerase IB and topoisomerase II activities were >10 fold higher. Viable colony counts obtained following treatment of M. smegmatis and M. tuberculosis showed that both MICs and cell killing efficacies of these four polyamine analogs are sensitive to the level of topoisomerase IA activity. The bacteria became less sensitive when recombinant M. tuberculosis topoisomerase I was overexpressed, indicating catalytic inhibition of the topoisomerase IA activity as part of the antibacterial mechanism of action. Further improvement of potency and selectivity is desirable for this class of type IA topoisomerase inhibitors.
4.4. Gold(III) Complexes
Metal-containing complexes have been used frequently as therapeutics throughout history, such as the well-known anti-cancer drug cisplatin [131] and the gold-containing auranofin used to treat rheumatoid arthritis [132]. Gold(III) tetra-aryl porphyrins with anticancer activity were found to bind DNA and inhibit human topoisomerase I [133]. That led to an increased interest in using gold compounds to inhibit topoisomerases. A number of pyrrole-based gold(III) macrocycles were synthesized and tested for anticancer activity and inhibition of human topoisomerase I [134]. The gold(III) macrocycle 10 illustrated in Figure 5A was found to be a catalytic inhibitor of human topoisomerase I with selectivity over human topoisomerase IIA. Gold(III) macrocycle 10 also caused lethality to many of the NCI’s panel of 60 human cancer cell lines [134]. In a follow up study investigating potential antibacterial application, some of the gold(III) macrocycles and additional bis(pyrrolide-imine) gold(III) chelates were found to inhibit the growth of both M. tuberculosis and Mycobacterium abscessus. The gold(III) chelate 14 (Figure 5B) showed a MIC of 1 µM against M. tuberculosis and 10 µM against M. abscessus, with bactericidal activity against mycobacteria as well. Gold(III) macrocycle 10 had even lower MICs against the mycobacteria but was not bactericidal, so these two compounds may have differences in their mechanism of action. Gold(III) chelate 14 showed no activity against Gram-negative species and only low level inhibition against other Gram-positives, suggesting that it has a specific activity against mycobacteria, including fluoroquinolone-resistant strains [113]. Both compounds shown in Figure 5 inhibit the relaxation activity of M. tuberculosis and E. coli topoisomerase I (Table 1), without any effect on bacterial DNA gyrase activity, suggesting the specificity these two compounds have for bacterial type IA topoisomerases inhibition [113]. Further studies are required to characterize the mechanism of antimycobacterial action of these gold(III) compounds and provide more insights in their structure–activity relationship (SAR) to limit the cytotoxicity.
4.5. Fluoroquinophenoxazine Derivatives
A fluoroquinophenoxazine NSC648059 was initially identified in enzyme-based screening of E. coli topoisomerase I inhibitors [114]. A series of fluoroquinophenoxazine derivatives were synthesized to evaluate the SAR and to probe the structural elements of the fluoroquinophenoxazine core towards type IA topoisomerase target recognition. These derivatives were assayed for selectivity in inhibition of type IA topoisomerases, and antibacterial activity. Comparative molecular field analysis (CoMFA) of the three-dimensional quantitative structure–activity relationship (3D-QSAR) of these fluoroquinophenoxazine derivatives resulted in a model with reasonable statistics (q2 = 0.688 and r2 = 0.806) and predictive power (predictive correlation coefficient r2pred = 0.767). While the structures of these fluoroquinophenoxazines have similarities to fluoroquinolones that target bacterial type IIA topoisomerases, this series of fluoroquinophenoxazines tested for selectivity exhibited much greater potency for inhibition of the type IA bacterial topoisomerase than inhibition of E. coli gyrase, human type topoisomerase I and IIα [114]. Derivative 11a with 6-methylpiperazinyl and 9-amino groups (Figure 6, IC50 = 0.48 µM) showed broad spectrum antibacterial activity (MICs = 0.78–7.6 µM) against bacteria that include Gram-negative E. coli and M. tuberculosis. Derivative 11g with the 6-bipiperidinyl lipophilic substitution showed the most promising antitubercular activity (MIC = 2.5 µM, selectivity index (SI) = 9.8) and was also active (MIC = 50 µM) against a clinical isolate of Mycobacterium abscessus [114,115]. The mechanism of resistance in mycobacteria was investigated by stepwise isolation of resistant mutants in M. smegmatis followed by comparison of the whole-genome sequence of the original strain and mutant isolates. Mutations in genes that affect compound entry and retention were identified instead of mutations in the topoisomerase I gene that might confirm the enzyme as the antibacterial target [115]. Biophysical analysis showed that DNA binding by fluoroquinophenoxazine 11g may contribute to the antibacterial mechanism, but could not account entirely for the direct binding and potent inhibition of the mycobacterial topoisomerase I [115].
4.6. Vichem’s Benzo(g)quinoxaline Compound
Compounds with antitubercular activity were identified with whole-cell assays from approximately 17,000 molecules in Vichem’s Nested Chemical Library (NCL) [135]. The 639 compounds with MIC values < 6 μM were assayed for inhibition of M. tuberculosis topoisomerase I activity, and the results on the 108 compounds found to be active at 100 μM were used for building machine learning models for M. tuberculosis topoisomerase I inhibitors [97]. A selected set of seven Vichem NCL molecules with benzo(g)-quinoxaline, quinoxaline, or styryl-benzo(g)-quinazoline scaffolds was further characterized [98]. Cytotoxicity was measured against three human cell lines. In silico docking predicted strong binding affinity to specific sites on M. tuberculosis topoisomerase I through interactions with R167 and R114 [97,98]. The interaction of the selected hits with the human ATP-binding cassette (ABC) multi-drug transporters was assessed for potential drug resistance. The human ABCB (MDR/TAP) transporter family plays a major role in multiple drug resistance (MDR) against a variety of tuberculosis therapies, such as fluoroquinolones and aminoglycosides [98]. Most of the hits demonstrated considerable interaction with the transporters, except for VCC891909 (Figure 7). This compound also showed no cytotoxicity in all the human cells lines tested in the cytotoxicity assays with efficient inhibition of M. tuberculosis growth (MIC90 = 10.4 µM against H37Rv) and topoisomerase I activity (complete inhibition at 7.5–10 µM), suggesting that VCC891909 might be a good candidate for further antimycobacterial drug development [98].
4.7. Alkaloids SCN and N-SCN
Two natural products and boldine-derivative alkaloids, seconeolitsine (SCN) and N-methyl-seconeolitsine (N-SCN) (Figure 8), were previously found to inhibit both topoisomerase I activity and cell growth of Streptococcus pneumoniae at approximately 17 µM [136]. The targeting of topoisomerase I by these compounds was supported by evidence of hypernegative supercoiling of DNA in treated cells along with protection from overexpression of topoisomerase I [136]. SCN has been used to demonstrate the homeostatic regulation of DNA supercoiling in S. pneumoniae by the topoisomerase I gene and the transcriptomic response to changes in local and global supercoiling [137,138]. These two alkaloids were tested more recently as potential inhibitors of M. tuberculosis topoisomerase I [116]. SCN and N-SCN exhibited MIC values below 16 µM against multiple isolates of M. tuberculosis and low IC50 values against the M. tuberculosis topoisomerase I (Table 1). SCN was shown to inhibit the cleavage of a 5′-biotin-labeled 32-mer oligonucleotide by the enzyme. The impact of N-SCN on DNA relaxation by topoisomerase I was confirmed by results from the analysis of topoisomer distribution of the mycobacterial plasmid by two-dimensional agarose gel, showing that the compound induced a 20% increase in plasmid supercoiling [116]. The action of both SCN and N-SCN as topoisomerase I catalytic inhibitors instead of poison inhibitors I was further confirmed by the decreasing MIC values of both compounds against M. smegmatis strain that expresses decreased levels of topoisomerase I [116]. Partial inhibition of human topoisomerase I could be observed at 50 µM concentration of SCN and N-SCN, with a small but significant effect on the viability of neutrophils at 100 µM concentration [136].
4.8. Additional Small Molecule Inhibitors Identified from Virtual Screening
Virtual screening was conducted sequentially to identify in silico hits against M. tuberculosis topoisomerase I for assays of enzyme inhibition. The Asinex elite library of 104,000 compounds was first docked against the crystal structure (Protein Data Bank (PDB) 5D5H) of the truncated apo-enzyme of M. tuberculosis topoisomerase I, consisting of N-terminal domains D1–D4 plus C-terminal domain D5. The top 1000 hits were then docked against 1000 structures of 5D5H generated by molecular-dynamics to allow deeper binding of the molecules in the DNA-binding pockets. Six of the 82 in silico hits that were found to show inhibition of M. tuberculosis topoisomerase I relaxation activity share a common piperidine amide structural motif in the center that appears to interact with conserved R167 and E115 residues. Docking was then conducted for 200 compounds from the ChemBridge library that have this piperidine amide motif. Among the 96 top-scoring ChemBridge compounds purchased for testing, 18 compounds exhibited IC50 < 125 µM. Compound seven (Figure 9) has the lowest IC50 (2 µM), and >250-fold selectivity over inhibition of human topoisomerase I and E. coli gyrase. Antibacterial activity of the ChemBridge hit compounds against M. smegmatis measured in the presence of efflux pump inhibitor thioridazine showed higher MICs when M. tuberculosis topoisomerase I was overexpressed, consistent with these compounds acting as catalytic inhibitors of topoisomerase I. Further utilization of piperidine amide compounds for antimycobacterial activity would require modifications that improve penetrance and retention of the compounds in mycobacteria.
To identify candidates for bacterial topoisomerase I poison inhibitors, stabilization of the covalent intermediate formed after DNA cleavage was targeted for in silico screening [94]. The C-terminal domains of E. coli topoisomerase I (from full length structure PDB 4RUL) were connected to the 67 kDa N-terminal domains in covalent complex with cleaved DNA (PDB 3PX7) based on alignments of the two structures. CGenFF force field parameters were applied to create the 5′-phospotyrosine linkage between the enzyme and cleaved DNA, followed by the energy minimization of the structure. To demonstrate the utility of this minimized E. coli topoisomerase I covalent complex model for in silico screening, docking was conducted on 2263 molecules selected from the NCI DTP chemical library. Direct interaction between the enzyme and one of the top predicted hits (NSC76027, Figure 9) was confirmed with surface plasmon resonance (dissociation constant KD = ~80 nM). The IC50 value of NSC76027 against E. coli topoisomerase I was estimated to be 2.2 µM [94], with a similar IC50 value of ~4 µM for inhibition of M. tuberculosis topoisomerase I relaxation activity (to be published), NSC76027 has no detectable antibacterial activity against E. coli MG1655 but was found to inhibit growth of M. tuberculosis (MIC = 35 µM) [94]. Further experiments are needed to determine the mechanism of inhibition and selectivity of NSC76027.
4.9. DNA Molecules as Bacterial Topoisomerase I Inhibitors
A distinct approach to target bacterial topoisomerase I is by using DNA molecules as specific inhibitors. The first step in the catalytic cycle of the enzyme is the recognition of the single-stranded regions in duplex DNA [139]. A series of 84 bp double-stranded oligonucleotides with variations in the number of bulge bases in a single-stranded loop were synthesized and tested for inhibition of the relaxation activity of E. coli topoisomerase I [140]. Inhibition efficiency improves as the size of the single-stranded loop increases from 1 to 10 base in length with IC50 reaching 63.1 nM. Inhibition of eukaryotic calf thymus topoisomerase I relaxation activity was not observed. It was hypothesized that because the single-stranded loops in the bulge oligonucleotides are not base-paired, the bulge oligonucleotides may be acting as irreversible competitive inhibitors [140].
In a second study, small circular DNA molecules with curvature that can be recognized by bacterial topoisomerase I were also investigated as competitive catalytic inhibitors [141]. Several duplex small circular oligodeoxyribonucleotides (cODNs) of varying sizes were designed, synthesized, and evaluated for inhibition of E. coli topoisomerase I relaxation activity. The sizes of the DNA circles synthesized mattered because the degree of bending and torsional stress in the DNA molecule increases with decreasing ring size, and the bending and torsional stress can be recognized by the enzyme. The biochemical evaluation showed inhibition of DNA relaxation activity with a IC50 value of 36.4 nM for the 66 bp cODN. Small single-stranded DNA circles or cODNs with seven base single-stranded mismatches have even lower IC50s (~10 nM) [141]. The cODNs have greater thermal stability than the bulge oligonucleotides.
5. Future Perspectives
Type IA topoisomerases are novel targets for infectious diseases for which no clinically approved drug has been developed so far. Effect of overexpression of the target topoisomerase should be informative on the mechanism of inhibition, overexpression of the target topoisomerase would increase the lethality of poison inhibitors. Conversely, the increased level of the target topoisomerase would help to overcome the effect of the catalytic inhibitors. The homeostatic regulation of bacterial topoisomerase I promoter activity by DNA supercoiling could increase the level of topoisomerase I expression in response to a catalytic inhibitor. The goal of finding and developing a successful drug targeting this class of topoisomerases would be greatly facilitated by the availability of the three-dimensional structure of topoisomerase–inhibitor complex, or ternary complex with DNA also present. Identification of inhibitors with high binding affinity from specific interactions with the topoisomerase protein residues would enable such structural studies. The structural information on the molecular interactions between enzyme and inhibitor would further validate the targeting of the type IA topoisomerase and allow structure-based design/optimization of type IA topoisomerase inhibitors for the development of a new therapy in the fight against the antimicrobial resistance. Determining the structure of a topoisomerase–RNA complex may also be useful for utilizing human TOP3B as a host target for providing options of antiviral therapy during flaviviruses and the coronavirus pandemics [49].
Author Contributions
Literature survey and the writing of the first draft were completed by A.S., T.A. and Y.-C.T.-D. contributed to the final draft. All authors have read and agreed to the published version of the manuscript.
Funding
The authors’ research was funded by NIH grants R01-AI069313 and R01-GM054226.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviation
| WHO | World Health Organization. |
| PDB | Protein Data Bank. |
| ELISA | enzyme-linked immunosorbent assay. |
| MIC | minimum inhibitory concentration: The lowest concentration of an agent that stops microbial growth. |
| CC50 | the 50% cytotoxic concentration: The concentration that reduces the cellular viability by 50%. |
| IC50 | the half maximal inhibitory concentration: The concentration that reduces enzymatic activity by 50%. |
| FDA | Food and Drug Administration |
| NCI | National Cancer Institute |
| SI | selectivity index: A ratio of cytotoxicity to antimicrobial activity. |
| SAR | structure–activity relationship: The relationship between the chemical structure of a molecule and its biological activities. |
| 3D-QSAR | three-dimensional quantitative structure–activity relationship: A quantification of SAR via mathematical relationships between physicochemical properties of a molecule and its biological activities. |
| CoMFA | comparative molecular field analysis: One of the QSAR methods, where mathematical relationships between steric and electrostatic properties of a ligand and the resulting receptor–ligand interactions are determined. |
| CGenFF | CHARMM General Force Field |
| DTP | Developmental Therapeutics Program |
References
- Marston, H.D.; Dixon, D.M.; Knisely, J.M.; Palmore, T.N.; Fauci, A.S. Antimicrobial Resistance. JAMA 2016, 316, 1193–1204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boucher, H.W. Bad Bugs, No Drugs 2002–2020: Progress, Challenges, and Call to Action. Trans. Am. Clin. Climatol. Assoc. 2020, 131, 65–71. [Google Scholar] [PubMed]
- Schrader, S.M.; Vaubourgeix, J.; Nathan, C. Biology of antimicrobial resistance and approaches to combat it. Sci. Transl. Med. 2020, 12, eaaz6992. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.; Davies, D. Origins and Evolution of Antibiotic Resistance. Microbiol. Mol. Biol. Rev. 2010, 74, 417–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bin Zaman, S.; Hussain, M.A.; Nye, R.; Mehta, V.; Mamun, K.T.; Hossain, N. A Review on Antibiotic Resistance: Alarm Bells are Ringing. Cureus 2017, 9, e1403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bush, K.; Jacoby, G.A. Updated Functional Classification of beta-Lactamases. Antimicrob. Agents Chemother. 2010, 54, 969–976. [Google Scholar] [CrossRef] [Green Version]
- Aldred, K.J.; Kerns, R.J.; Osheroff, N. Mechanism of Quinolone Action and Resistance. Biochemistry 2014, 53, 1565–1574. [Google Scholar] [CrossRef]
- Wilson, D.N.; Hauryliuk, V.; Atkinson, G.C.; O’Neill, A.J. Target protection as a key antibiotic resistance mechanism. Nat. Rev. Microbiol. 2020, 637–648. [Google Scholar] [CrossRef]
- Li, X.-Z.; Plésiat, P.; Nikaido, H. The Challenge of Efflux-Mediated Antibiotic Resistance in Gram-Negative Bacteria. Clin. Microbiol. Rev. 2015, 28, 337–418. [Google Scholar] [CrossRef] [Green Version]
- Blair, J.M.A.; Richmond, G.E.; Piddock, L.J.V. Multidrug efflux pumps in Gram-negative bacteria and their role in antibiotic resistance. Future Microbiol. 2014, 9, 1165–1177. [Google Scholar] [CrossRef]
- Andersson, D.I.; Hughes, D. Persistence of antibiotic resistance in bacterial populations. FEMS Microbiol. Rev. 2011, 35, 901–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunn, S.J.; Connor, C.; McNally, A. The evolution and transmission of multi-drug resistant Escherichia coli and Klebsiella pneumoniae: The complexity of clones and plasmids. Curr. Opin. Microbiol. 2019, 51, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Sugden, R.; Kelly, R.; Davies, S. Combatting antimicrobial resistance globally. Nat. Microbiol. 2016, 1, 16187. [Google Scholar] [CrossRef] [PubMed]
- Harding, E. WHO global progress report on tuberculosis elimination. Lancet Respir. Med. 2020, 8, 19. [Google Scholar] [CrossRef]
- WHO. Consolidated Guidelines on Tuberculosis: Module 4: Treatment—Drug-Resistant Tuberculosis Treatment; WHO Guidelines Approved by the Guidelines Review Committee; WHO: Geneva, Switzerland, 2020. [Google Scholar]
- Pendleton, J.N.; Gorman, S.P.; Gilmore, B.F. Clinical relevance of the ESKAPE pathogens. Expert Rev. Anti-Infect. Ther. 2013, 11, 297–308. [Google Scholar] [CrossRef]
- De Oliveira, D.M.P.; Forde, B.M.; Kidd, T.J.; Harris, P.N.A.; Schembri, M.A.; Beatson, S.A.; Paterson, D.L.; Walker, M.J. Antimicrobial Resistance in ESKAPE Pathogens. Clin. Microbiol. Rev. 2020, 33. [Google Scholar] [CrossRef]
- Beiras-Fernandez, A.; Vogt, F.; Sodian, R.; Weis, F. Daptomycin: A novel lipopeptide antibiotic against Gram-positive pathogens. Infect. Drug Resist. 2010, 3, 95–101. [Google Scholar] [CrossRef] [Green Version]
- Boucher, H.W.; Talbot, G.H.; Bradley, J.S.; Edwards, J.E.; Gilbert, D.; Rice, L.B.; Scheld, M.; Spellberg, B.; Bartlett, J. Bad Bugs, No Drugs: No ESKAPE! An Update from the Infectious Diseases Society of America. Clin. Infect. Dis. 2009, 48, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Hutchings, M.I.; Truman, A.W.; Wilkinson, B. Antibiotics: Past, present and future. Curr. Opin. Microbiol. 2019, 51, 72–80. [Google Scholar] [CrossRef]
- Bush, N.G.; Evans-Roberts, K.; Maxwell, A. DNA Topoisomerases. EcoSal Plus 2015, 6. [Google Scholar] [CrossRef]
- Nagaraja, V.; Godbole, A.A.; Henderson, S.R.; Maxwell, A. DNA topoisomerase I and DNA gyrase as targets for TB therapy. Drug Discov. Today 2017, 22, 510–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tse-Dinh, Y.-C. Exploring DNA Topoisomerases as Targets of Novel Therapeutic Agents in the Treatment of Infectious Diseases. Infect. Disord. Drug Targets 2007, 7, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Hiasa, H. DNA Topoisomerases as Targets for Antibacterial Agents. Methods Mol. Biol. 2018, 1703, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y. Drugging Topoisomerases: Lessons and Challenges. ACS Chem. Biol. 2013, 8, 82–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zechiedrich, E.L.; Khodursky, A.B.; Bachellier, S.; Schneider, R.; Chen, D.; Lilley, D.M.J.; Cozzarelli, N.R. Roles of Topoisomerases in Maintaining Steady-state DNA Supercoiling in Escherichia coli. J. Biol. Chem. 2000, 275, 8103–8113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brochu, J.; Breton, É.-V.; Drolet, M. Supercoiling, R-Loops, Replication and the Functions of Bacterial Type 1A Topoisomerases. Genes 2020, 11, 249. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.F.; Wang, J.C. Supercoiling of the DNA template during transcription. Proc. Natl. Acad. Sci. USA 1987, 84, 7024–7027. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Wang, M.D. DNA supercoiling during transcription. Biophys. Rev. 2016, 8 (Suppl. S1), 75–87. [Google Scholar] [CrossRef]
- Vos, S.M.; Tretter, E.M.; Schmidt, B.H.; Berger, J.M. All tangled up: How cells direct, manage and exploit topoisomerase function. Nat. Rev. Mol. Cell Biol. 2011, 12, 827–841. [Google Scholar] [CrossRef] [Green Version]
- Pommier, Y.; Sun, Y.; Huang, S.-Y.N.; Nitiss, J.L. Roles of eukaryotic topoisomerases in transcription, replication and genomic stability. Nat. Rev. Mol. Cell Biol. 2016, 17, 703–721. [Google Scholar] [CrossRef]
- Wang, J.C. Cellular roles of DNA topoisomerases: A molecular perspective. Nat. Rev. Mol. Cell Biol. 2002, 3, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Tse, Y.C.; Kirkegaard, K.; Wang, J.C. Covalent bonds between protein and DNA. Formation of phosphotyrosine linkage between certain DNA topoisomerases and DNA. J. Biol. Chem. 1980, 255, 5560–5565. [Google Scholar] [PubMed]
- Delgado, J.L.; Hsieh, C.-M.; Chan, N.-L.; Hiasa, H. Topoisomerases as anticancer targets. Biochem. J. 2018, 475, 373–398. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.; Pommier, Y. Targeting Topoisomerase I in the Era of Precision Medicine. Clin. Cancer Res. 2019, 25, 6581–6589. [Google Scholar] [CrossRef] [PubMed]
- Nitiss, J.L. Targeting DNA topoisomerase II in cancer chemotherapy. Nat. Rev. Cancer 2009, 9, 338–350. [Google Scholar] [CrossRef] [Green Version]
- Champoux, J.J. DNA Topoisomerases: Structure, Function, and Mechanism. Annu. Rev. Biochem. 2001, 70, 369–413. [Google Scholar] [CrossRef] [Green Version]
- Baker, N.M.; Rajan, R.; Mondragón, A. Structural studies of type I topoisomerases. Nucleic Acids Res. 2009, 37, 693–701. [Google Scholar] [CrossRef] [Green Version]
- Kirkegaard, K.; Wang, J.C. Bacterial DNA topoisomerase I can relax positively supercoiled DNA containing a single-stranded loop. J. Mol. Biol. 1985, 185, 625–637. [Google Scholar] [CrossRef]
- Lima, C.D.; Wang, J.C.; Mondragón, A. Three-dimensional structure of the 67K N-terminal fragment of E. coli DNA topoisomerase I. Nature 1994, 367, 138–146. [Google Scholar] [CrossRef]
- Changela, A.; DiGate, R.J.; Mondragón, A. Crystal structure of a complex of a type IA DNA topoisomerase with a single-stranded DNA molecule. Nature 2001, 411, 1077–1081. [Google Scholar] [CrossRef]
- Mills, M.; Tse-Dinh, Y.-C.; Neuman, K.C. Direct observation of topoisomerase IA gate dynamics. Nat. Struct. Mol. Biol. 2018, 25, 1111–1118. [Google Scholar] [CrossRef] [PubMed]
- Tse-Dinh, Y.C. Uncoupling of the DNA breaking and rejoining steps of Escherichia coli type I DNA topoisomerase. Demonstration of an active covalent protein-DNA complex. J. Biol. Chem. 1986, 261, 10931–10935. [Google Scholar] [PubMed]
- Sorokin, E.P.; Cheng, B.; Rathi, S.; Aedo, S.J.; Abrenica, M.V.; Tse-Dinh, Y.-C. Inhibition of Mg2+ binding and DNA religation by bacterial topoisomerase I via introduction of an additional positive charge into the active site region. Nucleic Acids Res. 2008, 36, 4788–4796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhat, A.G.; Leelaram, M.N.; Hegde, S.; Nagaraja, V. Deciphering the Distinct Role for the Metal Coordination Motif in the Catalytic Activity of Mycobacterium smegmatis Topoisomerase I. J. Mol. Biol. 2009, 393, 788–802. [Google Scholar] [CrossRef] [PubMed]
- Cao, N.; Tan, K.; Annamalai, T.; Joachimiak, A.; Tse-Dinh, Y.-C. Investigating mycobacterial topoisomerase I mechanism from the analysis of metal and DNA substrate interactions at the active site. Nucleic Acids Res. 2018, 46, 7296–7308. [Google Scholar] [CrossRef] [PubMed]
- Corbett, K.D.; Berger, J.M. Structure, Molecular Mechanisms, and Evolutionary Relationships in DNA Topoisomerases. Annu. Rev. Biophys. Biomol. Struct. 2004, 33, 95–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, M.; Xue, Y.; Lee, S.K.; Martindale, J.L.; Shen, W.; Li, W.; Zou, S.; Ciaramella, M.; Debat, H.; Nadal, M.; et al. RNA topoisomerase is prevalent in all domains of life and associates with polyribosomes in animals. Nucleic Acids Res. 2016, 44, 6335–6349. [Google Scholar] [CrossRef] [Green Version]
- Prasanth, K.R.; Hirano, M.; Fagg, W.S.; McAnarney, E.T.; Shan, C.; Xie, X.; Hage, A.; Pietzsch, C.A.; Bukreyev, A.; Rajsbaum, R.; et al. Topoisomerase III-β is required for efficient replication of positive-sense RNA viruses. Antivir. Res. 2020, 182, 104874. [Google Scholar] [CrossRef]
- Forterre, P.; Gadelle, D. Phylogenomics of DNA topoisomerases: Their origin and putative roles in the emergence of modern organisms. Nucleic Acids Res. 2009, 37, 679–692. [Google Scholar] [CrossRef] [Green Version]
- Bjornsti, M.-A.; Kaufmann, S.H. Topoisomerases and cancer chemotherapy: Recent advances and unanswered questions. F1000Research 2019, 8, 1704. [Google Scholar] [CrossRef]
- Pommier, Y.; Leo, E.; Zhang, H.; Marchand, C. DNA Topoisomerases and Their Poisoning by Anticancer and Antibacterial Drugs. Chem. Biol. 2010, 17, 421–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.F. DNA Topoisomerase Poisons as Antitumor Drugs. Annu. Rev. Biochem. 1989, 58, 351–375. [Google Scholar] [CrossRef] [PubMed]
- Pham, T.D.M.; Ziora, Z.; Blaskovich, M.A.T. Quinolone antibiotics. MedChemComm 2019, 10, 1719–1739. [Google Scholar] [CrossRef] [PubMed]
- Solary, E.; Bertrand, R.; Pommier, Y. Apoptosis Induced by DNA Topoisomerase I and II Inhibitors in Human Leukemic HL-60 Cells. Leuk. Lymphoma 1994, 15, 21–32. [Google Scholar] [CrossRef]
- Dwyer, D.J.; Kohanski, M.A.; Hayete, B.; Collins, J.J. Gyrase inhibitors induce an oxidative damage cellular death pathway in Escherichia coli. Mol. Syst. Biol. 2007, 3, 91. [Google Scholar] [CrossRef]
- Terekhova, K.; Gunn, K.H.; Marko, J.F.; Mondragón, A. Bacterial topoisomerase I and topoisomerase III relax supercoiled DNA via distinct pathways. Nucleic Acids Res. 2012, 40, 10432–10440. [Google Scholar] [CrossRef] [Green Version]
- Terekhova, K.; Marko, J.F.; Mondragón, A. Single-molecule analysis uncovers the difference between the kinetics of DNA decatenation by bacterial topoisomerases I and III. Nucleic Acids Res. 2014, 42, 11657–11667. [Google Scholar] [CrossRef] [Green Version]
- Massé, E.; Drolet, M. Escherichia coliDNA Topoisomerase I Inhibits R-loop Formation by Relaxing Transcription-induced Negative Supercoiling. J. Biol. Chem. 1999, 274, 16659–16664. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.M.; Wang, G.; Pertsinidis, A.; Marians, K.J. Topoisomerase III Acts at the Replication Fork To Remove Precatenanes. J. Bacteriol. 2019, 201. [Google Scholar] [CrossRef] [Green Version]
- Raji, A.; Zabel, D.J.; Laufer, C.S.; Depew, R.E. Genetic analysis of mutations that compensate for loss of Escherichia coli DNA topoisomerase I. J. Bacteriol. 1985, 162, 1173–1179. [Google Scholar] [CrossRef] [Green Version]
- Dinardo, S.; Voelkel, K.A.; Sternglanz, R.; Reynolds, A.E.; Wright, A. Escherichia coli DNA topoisomerase I mutants have compensatory mutations in DNA gyrase genes. Cell 1982, 31, 43–51. [Google Scholar] [CrossRef]
- Pruss, G.J.; Manes, S.H.; Drlica, K. Escherichia coli DNA topoisomerase I mutants: Increased supercoiling is corrected by mutations near gyrase genes. Cell 1982, 31, 35–42. [Google Scholar] [CrossRef]
- Yigit, H.; Reznikoff, W.S. Escherichia coli DNA Topoisomerase I and Suppression of Killing by Tn5 Transposase Overproduction: Topoisomerase I Modulates Tn5 Transposition. J. Bacteriol. 1998, 180, 5866–5874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yigit, H.; Reznikoff, W.S. Escherichia coli DNA Topoisomerase I Copurifies with Tn5 Transposase, and Tn5 Transposase Inhibits Topoisomerase I. J. Bacteriol. 1999, 181, 3185–3192. [Google Scholar] [CrossRef] [Green Version]
- Mattenberger, Y.; Silva, F.; Belin, D. 55.2, a Phage T4 ORFan Gene, Encodes an Inhibitor of Escherichia coli Topoisomerase I and Increases Phage Fitness. PLoS ONE 2015, 10, e0124309. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, Y.; Inouye, M. An endogenous protein inhibitor, YjhX (TopAI), for topoisomerase I from Escherichia coli. Nucleic Acids Res. 2015, 43, 10387–10396. [Google Scholar] [CrossRef] [Green Version]
- Cheng, B.; Shukla, S.; Vasunilashorn, S.; Mukhopadhyay, S.; Tse-Dinh, Y.-C. Bacterial Cell Killing Mediated by Topoisomerase I DNA Cleavage Activity. J. Biol. Chem. 2005, 280, 38489–38495. [Google Scholar] [CrossRef] [Green Version]
- Cheng, B.; Annamalai, T.; Sorokin, E.; Abrenica, M.; Aedo, S.; Tse-Dinh, Y.-C. Asp-to-Asn Substitution at the First Position of the DxD TOPRIM Motif of Recombinant Bacterial Topoisomerase I Is Extremely Lethal to E. coli. J. Mol. Biol. 2009, 385, 558–567. [Google Scholar] [CrossRef] [Green Version]
- Narula, G.; Annamalai, T.; Aedo, S.; Cheng, B.; Sorokin, E.; Wong, A.; Tse-Dinh, Y.-C. The Strictly Conserved Arg-321 Residue in the Active Site of Escherichia coli Topoisomerase I Plays a Critical Role in DNA Rejoining. J. Biol. Chem. 2011, 286, 18673–18680. [Google Scholar] [CrossRef] [Green Version]
- Ravishankar, S.; Ambady, A.; Ramachandran, V.; Eyermann, C.J.; Reck, F.; Rudrapatna, S.; Sambandamurthy, V.K.; Sharma, U.K.; Awasthy, D.; Mudugal, N.V.; et al. Genetic and chemical validation identifies Mycobacterium tuberculosis topoisomerase I as an attractive anti-tubercular target. Tuberculosis 2015, 95, 589–598. [Google Scholar] [CrossRef]
- Ahmed, W.; Menon, S.; Godbole, A.A.; Karthik, P.V.; Nagaraja, V. Conditional silencing of topoisomerase I gene of Mycobacterium tuberculosis validates its essentiality for cell survival. FEMS Microbiol. Lett. 2014, 353, 116–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Gallay, C.; Kjos, M.; Domenech, A.; Slager, J.; Van Kessel, S.P.; Knoops, K.; Sorg, R.A.; Zhang, J.-R.; Veening, J.-W. High-throughput CRISPRi phenotyping identifies new essential genes in Streptococcus pneumoniae. Mol. Syst. Biol. 2017, 13, 931. [Google Scholar] [CrossRef] [PubMed]
- Yan, R.; Hu, S.; Ma, N.; Song, P.; Liang, Q.; Zhang, H.; Li, Y.; Shen, L.; Duan, K.; Chen, L. Regulatory Effect of DNA Topoisomerase I on T3SS Activity, Antibiotic Susceptibility and Quorum- Sensing-Independent Pyocyanin Synthesis in Pseudomonas aeruginosa. Int. J. Mol. Sci. 2019, 20, 1116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suerbaum, S.; Brauer-Steppkes, T.; Labigne, A.; Cameron, B.; Drlica, K. Topoisomerase I of Helicobacter pylori: Juxtaposition with a flagellin gene (flaB) and functional requirement of a fourth zinc finger motif. Gene 1998, 210, 151–161. [Google Scholar] [CrossRef]
- Goodwin, A.; Wang, S.-W.; Toda, T.; Norbury, C.; Hickso, I.D. Topoisomerase III is essential for accurate nuclear division in Schizosaccharomyces pombe. Nucleic Acids Res. 1999, 27, 4050–4058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maftahi, M.; Han, C.S.; Langston, L.D.; Hope, J.C.; Zigouras, N.; Freyer, G.A. The top3+ gene is essential in Schizosaccharomyces pombe and the lethality associated with its loss is caused by Rad12 helicase activity. Nucleic Acids Res. 1999, 27, 4715–4724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallis, J.W.; Chrebet, G.; Brodsky, G.; Rolfe, M.; Rothstein, R. A hyper-recombination mutation in S. cerevisiae identifies a novel eukaryotic topoisomerase. Cell 1989, 58, 409–419. [Google Scholar] [CrossRef]
- Li, W.; Wang, J.C. Mammalian DNA topoisomerase IIIα is essential in early embryogenesis. Proc. Natl. Acad. Sci. USA 1998, 95, 1010–1013. [Google Scholar] [CrossRef] [Green Version]
- Kwan, K.Y.; Wang, J.C. Mice lacking DNA topoisomerase IIIβ develop to maturity but show a reduced mean lifespan. Proc. Natl. Acad. Sci. USA 2001, 98, 5717–5721. [Google Scholar] [CrossRef] [Green Version]
- Stoll, G.; Pietiläinen, O.P.H.; Linder, B.; Suvisaari, J.; Brosi, C.; Hennah, W.; Leppa, V.; Torniainen, M.; Ripatti, S.; Ala-Mello, S.; et al. Deletion of TOP3β, a component of FMRP-containing mRNPs, contributes to neurodevelopmental disorders. Nat. Neurosci. 2013, 16, 1228–1237. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.; Shen, W.; Guo, R.; Xue, Y.; Peng, W.; Sima, J.; Yang, J.; Sharov, A.; Srikantan, S.; Yang, J.; et al. Top3β is an RNA topoisomerase that works with fragile X syndrome protein to promote synapse formation. Nat. Neurosci. 2013, 16, 1238–1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, T.; Wallis, M.; Petrovic, V.; Challis, J.; Kalitsis, P.; Hudson, D.F. Loss of TOP3B leads to increased R-loop formation and genome instability. Open Biol. 2019, 9, 190222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; McBride, K.M.; Hensley, S.; Lu, Y.; Chedin, F.; Bedford, M.T. Arginine Methylation Facilitates the Recruitment of TOP3B to Chromatin to Prevent R Loop Accumulation. Mol. Cell 2014, 53, 484–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, M.; Shen, W.; Li, W.; Xue, Y.; Zou, S.; Xu, D.; Wang, W. Topoisomerase 3β is the major topoisomerase for mRNAs and linked to neurodevelopment and mental dysfunction. Nucleic Acids Res. 2016, 45, 2704–2713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siaw, G.E.-L.; Liu, I.-F.; Lin, P.-Y.; Been, M.D.; Hsieh, T.-S. DNA and RNA topoisomerase activities of Top3β are promoted by mediator protein Tudor domain-containing protein 3. Proc. Natl. Acad. Sci. USA 2016, 113, E5544–E5551. [Google Scholar] [CrossRef] [Green Version]
- Goto-Ito, S.; Yamagata, A.; Takahashi, T.S.; Sato, Y.; Fukai, S. Structural basis of the interaction between Topoisomerase IIIβ and the TDRD3 auxiliary factor. Sci. Rep. 2017, 7, 42123. [Google Scholar] [CrossRef] [Green Version]
- Barrows, N.J.; Anglero-Rodriguez, Y.; Kim, B.; Jamison, S.F.; Le Sommer, C.; McGee, C.E.; Pearson, J.L.; Dimopoulos, G.; Ascano, M.; Bradrick, S.S.; et al. Dual roles for the ER membrane protein complex in flavivirus infection: Viral entry and protein biogenesis. Sci. Rep. 2019, 9, 9711. [Google Scholar] [CrossRef] [Green Version]
- Saha, S.; Chowdhury, S.R.; Majumder, H.K. DNA Topoisomerases of Kinetoplastid Parasites: Brief Overview and Recent Perspectives. Curr. Issues Mol. Biol. 2019, 31, 45–62. [Google Scholar] [CrossRef]
- Balaña-Fouce, R.; Alvarez-Velilla, R.; Fernandez-Prada, C.; García-Estrada, C.; Reguera, R.M. Trypanosomatids topoisomerase re-visited. New structural findings and role in drug discovery. Int. J. Parasitol. Drugs Drug Resist. 2014, 4, 326–337. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.-S.; Cross, G.A.M. TOPO3α Influences Antigenic Variation by Monitoring Expression-Site-Associated VSG Switching in Trypanosoma brucei. PLoS Pathog. 2010, 6, e1000992. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, B.; Sen, N.; Majumder, H.K. Identification of a Functional Type IA Topoisomerase, LdTopIIIβ, from Kinetoplastid Parasite Leishmania donovani. Enzym. Res. 2011, 2011, 230542. [Google Scholar] [CrossRef] [Green Version]
- Scocca, J.R.; Shapiro, T.A. A mitochondrial topoisomerase IA essential for late theta structure resolution in African trypanosomes. Mol. Microbiol. 2008, 67, 820–829. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, P.B.; Chapagain, P.P.; Seddek, A.; Annamalai, T.; Üren, A.; Tse-Dinh, Y. Covalent Complex of DNA and Bacterial Topoisomerase: Implications in Antibacterial Drug Development. ChemMedChem 2020, 15, 623–631. [Google Scholar] [CrossRef] [PubMed]
- Sandhaus, S.; Chapagain, P.P.; Tse-Dinh, Y. Discovery of novel bacterial topoisomerase I inhibitors by use of in silico docking and in vitro assays. Sci. Rep. 2018, 8, 1437. [Google Scholar] [CrossRef]
- Godbole, A.A.; Ahmed, W.; Bhat, R.S.; Bradley, E.K.; Ekins, S.; Nagaraja, V. Targeting Mycobacterium tuberculosis Topoisomerase I by Small-Molecule Inhibitors. Antimicrob. Agents Chemother. 2015, 59, 1549–1557. [Google Scholar] [CrossRef] [Green Version]
- Ekins, S.; Godbole, A.A.; Kéri, G.; Őrfi, L.; Pato, J.; Bhat, R.S.; Verma, R.; Bradley, E.K.; Nagaraja, V. Machine learning and docking models for Mycobacterium tuberculosis topoisomerase I. Tuberculosis 2017, 103, 52–60. [Google Scholar] [CrossRef]
- Temesszentandrási-Ambrus, C.; Tóth, S.; Szakács, G.; Őrfi, L.; Nagaraja, V.; Ekins, S.; Telbisz, Á.; Verma, R.; Bánhegyi, P.; Szabadkai, I.; et al. Characterization of new, efficient Mycobacterium tuberculosis topoisomerase-I inhibitors and their interaction with human ABC multidrug transporters. PLoS ONE 2018, 13, e0202749. [Google Scholar] [CrossRef]
- Levene, S.D. Analysis of DNA Topoisomers, Knots, and Catenanes by Agarose Gel Electrophoresis. Methods Mol. Biol. 2009, 582, 11–25. [Google Scholar] [CrossRef]
- Cheng, B.; Annamalai, T.; Sandhaus, S.; Bansod, P.; Tse-Dinh, Y.-C. Inhibition of Zn(II) Binding Type IA Topoisomerases by Organomercury Compounds and Hg(II). PLoS ONE 2015, 10, e0120022. [Google Scholar] [CrossRef] [Green Version]
- Maxwell, A.; Burton, N.P.; O’Hagan, N. High-throughput assays for DNA gyrase and other topoisomerases. Nucleic Acids Res. 2006, 34, e104. [Google Scholar] [CrossRef] [Green Version]
- Shapiro, A.B.; Jahic, H.; Prasad, S.; Ehmann, D.; Thresher, J.; Gao, N.; Hajec, L. A Homogeneous, High-Throughput Fluorescence Anisotropy-Based DNA Supercoiling Assay. J. Biomol. Screen. 2010, 15, 1088–1098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jude, K.M.; Hartland, A.; Berger, J.M. Real-time detection of DNA topological changes with a fluorescently labeled cruciform. Nucleic Acids Res. 2013, 41, e133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, M.; Berrido, A.; Gonzalez, W.G.; Miksovska, J.; Chambers, J.W.; Leng, F. Fluorescently labeled circular DNA molecules for DNA topology and topoisomerases. Sci. Rep. 2016, 6, 36006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Rakela, S.; Chambers, J.W.; Hua, Z.-C.; Muller, M.T.; Nitiss, J.L.; Tse-Dinh, Y.-C.; Leng, F. Kinetic Study of DNA Topoisomerases by Supercoiling-Dependent Fluorescence Quenching. ACS Omega 2019, 4, 18413–18422. [Google Scholar] [CrossRef]
- Annamalai, T.; Cheng, B.; Keswani, N.; Tse-Dinh, Y.-C. A Fluorescence-Based Assay for Identification of Bacterial Topoisomerase I Poisons. Methods Mol. Biol. 2017, 1703, 259–268. [Google Scholar] [CrossRef]
- Kiianitsa, K.; Maizels, N. A rapid and sensitive assay for DNA–protein covalent complexes in living cells. Nucleic Acids Res. 2013, 41, e104. [Google Scholar] [CrossRef] [Green Version]
- Kiianitsa, K.; Maizels, N. Ultrasensitive isolation, identification and quantification of DNA–protein adducts by ELISA-based RADAR assay. Nucleic Acids Res. 2014, 42, e108. [Google Scholar] [CrossRef] [Green Version]
- Sinha, D.; Kiianitsa, K.; Sherman, D.R.; Maizels, N. Rapid, direct detection of bacterial topoisomerase 1-DNA adducts by RADAR/ELISA. Anal. Biochem. 2020, 608, 113827. [Google Scholar] [CrossRef]
- Ranjan, N.; Fulcrand, G.; King, A.; Brown, J.; Jiang, X.; Leng, F.; Arya, D.P. Selective inhibition of bacterial topoisomerase I by alkynyl-bisbenzimidazoles. MedChemComm 2014, 5, 816–825. [Google Scholar] [CrossRef] [Green Version]
- Nimesh, H.; Sur, S.; Sinha, D.; Yadav, P.; Anand, P.; Bajaj, P.; Virdi, J.S.; Tandon, V. Synthesis and Biological Evaluation of Novel Bisbenzimidazoles as Escherichia coli Topoisomerase IA Inhibitors and Potential Antibacterial Agents. J. Med. Chem. 2014, 57, 5238–5257. [Google Scholar] [CrossRef]
- Sandhaus, S.; Annamalai, T.; Welmaker, G.; Houghten, R.A.; Paz, C.; Garcia, P.K.; Andres, A.; Narula, G.; Felix, C.R.; Geden, S.; et al. Small-Molecule Inhibitors Targeting Topoisomerase I as Novel Antituberculosis Agents. Antimicrob. Agents Chemother. 2016, 60, 4028–4036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, R.; Felix, C.R.; Akerman, M.P.; Akerman, K.J.; Slabber, C.A.; Wang, W.; Adams, J.; Shaw, L.N.; Tse-Dinh, Y.-C.; Munro, O.Q.; et al. Evidence for Inhibition of Topoisomerase 1A by Gold(III) Macrocycles and Chelates Targeting Mycobacterium tuberculosis and Mycobacterium abscessus. Antimicrob. Agents Chemother. 2018, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuk-Ching, T.-D.; Zhang, M.; Annamalai, T.; Bansod, P.; Narula, G.; Tse-Dinh, Y.-C.; Sun, D. Synthesis, evaluation, and CoMFA study of fluoroquinophenoxazine derivatives as bacterial topoisomerase IA inhibitors. Eur. J. Med. Chem. 2017, 125, 515–527. [Google Scholar] [CrossRef] [Green Version]
- Garcia, P.K.; Annamalai, T.; Wang, W.; Bell, R.S.; Le, D.; Pancorbo, P.M.; Sikandar, S.; Seddek, A.; Yu, X.; Sun, D.; et al. Mechanism and resistance for antimycobacterial activity of a fluoroquinophenoxazine compound. PLoS ONE 2019, 14, e0207733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García, M.T.; Carreño, D.; Tirado-Vélez, J.M.; Ferrándiz, M.J.; Rodrigues, L.; Gracia, B.; Amblar, M.; Aínsa, J.A.; De La Campa, A.G. Boldine-Derived Alkaloids Inhibit the Activity of DNA Topoisomerase I and Growth of Mycobacterium tuberculosis. Front. Microbiol. 2018, 9, 1659. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.Y.; Yu, C.; Gatto, B.; Liu, L.F. DNA minor groove-binding ligands: A different class of mammalian DNA topoisomerase I inhibitors. Proc. Natl. Acad. Sci. USA 1993, 90, 8131–8135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, A.Y.; Yu, C.; Bodley, A.; Peng, L.F.; Liu, L.F. A new mammalian DNA topoisomerase I poison Hoechst 33342: Cytotoxicity and drug resistance in human cell cultures. Cancer Res. 1993, 53, 1332–1337. [Google Scholar]
- Baraldi, P.G.; Bovero, A.; Fruttarolo, F.; Preti, D.; Tabrizi, M.A.; Pavani, M.G.; Romagnoli, R. DNA minor groove binders as potential antitumor and antimicrobial agents. Med. Res. Rev. 2004, 24, 475–528. [Google Scholar] [CrossRef]
- Bansal, S.; Sinha, D.; Singh, M.; Cheng, B.; Tse-Dinh, Y.-C.; Tandon, V. 3,4-Dimethoxyphenyl bis-benzimidazole, a novel DNA topoisomerase inhibitor that preferentially targets Escherichia coli topoisomerase I. J. Antimicrob. Chemother. 2012, 67, 2882–2891. [Google Scholar] [CrossRef] [Green Version]
- Ranjan, N.; Story, S.; Fulcrand, G.; Leng, F.; Ahmad, M.; King, A.; Sur, S.; Wang, W.; Tse-Dinh, Y.; Arya, D.P. Selective Inhibition of Escherichia coli RNA and DNA Topoisomerase I by Hoechst 33258 Derived Mono- and Bisbenzimidazoles. J. Med. Chem. 2017, 60, 4904–4922. [Google Scholar] [CrossRef]
- Chamberlin, J.; Story, S.; Ranjan, N.; Chesser, G.; Arya, D.P. Gram-negative synergy and mechanism of action of alkynyl bisbenzimidazoles. Sci. Rep. 2019, 9, 14171. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Pandey, S.; Sur, S.; Tandon, V. PPEF: A bisbenzimdazole potent antimicrobial agent interacts at acidic triad of catalytic domain of E. coli topoisomerase IA. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 1524–1535. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.-X.; Tse-Dinh, Y.-C. The Acidic Triad Conserved in Type IA DNA Topoisomerases Is Required for Binding of Mg(II) and Subsequent Conformational Change. J. Biol. Chem. 2000, 275, 5318–5322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Cheng, B.; Tse-Dinh, Y.-C. Crystal structure of a covalent intermediate in DNA cleavage and rejoining by Escherichia coli DNA topoisomerase I. Proc. Natl. Acad. Sci. USA 2011, 108, 6939–6944. [Google Scholar] [CrossRef] [Green Version]
- Gillman, P.K. Tricyclic antidepressant pharmacology and therapeutic drug interactions updated. Br. J. Pharmacol. 2007, 151, 737–748. [Google Scholar] [CrossRef] [Green Version]
- Houghten, R.A.; Pinilla, C.; Giulianotti, M.A.; Appel, J.R.; Dooley, C.T.; Nefzi, A.; Ostresh, J.M.; Yu, Y.; Maggiora, G.; Medina-Franco, J.L.; et al. Strategies for the Use of Mixture-Based Synthetic Combinatorial Libraries: Scaffold Ranking, Direct Testing In Vivo, and Enhanced Deconvolution by Computational Methods. J. Comb. Chem. 2008, 10, 3–19. [Google Scholar] [CrossRef]
- Wu, J.; Zhang, Y.; Maida, L.E.; Santos, R.G.; Welmaker, G.S.; LaVoi, T.M.; Nefzi, A.; Yu, Y.; Houghten, R.A.; Toll, L.; et al. Scaffold Ranking and Positional Scanning Utilized in the Discovery of nAChR-Selective Compounds Suitable for Optimization Studies. J. Med. Chem. 2013, 56, 10103–10117. [Google Scholar] [CrossRef] [Green Version]
- Al-Ali, H.; Debevec, G.; Santos, R.G.; Houghten, R.A.; Davis, J.C.; Nefzi, A.; Lemmon, V.P.; Bixby, J.L.; Giulianotti, M.A. Scaffold Ranking and Positional Scanning Identify Novel Neurite Outgrowth Promoters with Nanomolar Potency. ACS Med. Chem. Lett. 2018, 9, 1057–1062. [Google Scholar] [CrossRef]
- Rideout, M.C.; Boldt, J.L.; Vahi-Ferguson, G.; Salamon, P.; Nefzi, A.; Ostresh, J.M.; Giulianotti, M.; Pinilla, C.; Segall, A.M. Potent antimicrobial small molecules screened as inhibitors of tyrosine recombinases and Holliday junction-resolving enzymes. Mol. Divers. 2011, 15, 989–1005. [Google Scholar] [CrossRef]
- Boulikas, T.; Vougiouka, M. Cisplatin and platinum drugs at the molecular level. (Review). Oncol. Rep. 2003, 10, 1663–1682. [Google Scholar] [CrossRef]
- Chaffman, M.; Brogden, R.N.; Heel, R.C.; Speight, T.M.; Avery, G.S. Auranofin. A preliminary review of its pharmacological properties and therapeutic use in rheumatoid arthritis. Drugs 1984, 27, 378–424. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.W.-Y.; Li, C.K.-L.; Ma, D.-L.; Yan, J.J.; Lok, C.-N.; Leung, C.-H.; Zhu, N.; Che, C.-M. Stable Anticancer Gold(III)-Porphyrin Complexes: Effects of Porphyrin Structure. Chemistry 2010, 16, 3097–3113. [Google Scholar] [CrossRef] [PubMed]
- Akerman, K.J.; Fagenson, A.M.; Cyril, V.; Taylor, M.; Muller, M.T.; Akerman, M.P.; Munro, O.Q. Gold(III) Macrocycles: Nucleotide-Specific Unconventional Catalytic Inhibitors of Human Topoisomerase I. J. Am. Chem. Soc. 2014, 136, 5670–5682. [Google Scholar] [CrossRef] [PubMed]
- Sipos, A.; Pató, J.; Nagaraja, V.; Godbole, A.A.; Bush, N.; Collin, F.; Maxwell, A.; Cole, S.T.; Kéri, G.; Székely, R.; et al. Lead selection and characterization of antitubercular compounds using the Nested Chemical Library. Tuberculosis 2015, 95 (Suppl. S1), S200–S206. [Google Scholar] [CrossRef]
- García, M.T.; Blázquez, M.A.; Ferrándiz, M.-J.; Sanz, M.-J.; Silva-Martín, N.; Hermoso, J.A.; De La Campa, A.G. New Alkaloid Antibiotics That Target the DNA Topoisomerase I of Streptococcus pneumoniae. J. Biol. Chem. 2011, 286, 6402–6413. [Google Scholar] [CrossRef] [Green Version]
- De La Campa, A.G.; Ferrándiz, M.J.; Martín-Galiano, A.J.; García, M.T.; Tirado-Vélez, J.M. The Transcriptome of Streptococcus pneumoniae Induced by Local and Global Changes in Supercoiling. Front. Microbiol. 2017, 8, 1447. [Google Scholar] [CrossRef] [Green Version]
- Ferrándiz, M.-J.; Martín-Galiano, A.J.; Arnanz, C.; Camacho-Soguero, I.; Tirado-Vélez, J.-M.; De La Campa, A.G. An increase in negative supercoiling in bacteria reveals topology-reacting gene clusters and a homeostatic response mediated by the DNA topoisomerase I gene. Nucleic Acids Res. 2016, 44, 7292–7303. [Google Scholar] [CrossRef] [Green Version]
- Dasgupta, T.; Ferdous, S.; Tse-Dinh, Y.-C. Mechanism of Type IA Topoisomerases. Molecules 2020, 25, 4769. [Google Scholar] [CrossRef]
- Yang, Z.; Jiang, T.; Zhong, H.; Kang, Y. Bulge oligonucleotide as an inhibitory agent of bacterial topoisomerase I. J. Enzym. Inhib. Med. Chem. 2018, 33, 319–323. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Wang, Q.; Zhou, B.; Zhuge, Q.; Lv, B. Small DNA circles as bacterial topoisomerase I inhibitors. RSC Adv. 2019, 9, 18415–18419. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
DNA-gate formed by cleavage of G-strand DNA by type IA topoisomerase (A) Crystal structure of covalent complex between single-stranded DNA (ssDNA) and E. coli topoisomerase I N-terminal domains I to IV (Protein Data Bank (PDB) 3PX7). The active tyrosine residue Y319 is illustrated with the red color, and ssDNA is represented by the blue color. (B) Gate opening and closing dynamics of bacterial topoisomerase IA. The enzyme is represented by the green shape and the ssDNA is represented by the blue wavy line. The letter Y stands for the active site tyrosine residue Y319.
Figure 1.
DNA-gate formed by cleavage of G-strand DNA by type IA topoisomerase (A) Crystal structure of covalent complex between single-stranded DNA (ssDNA) and E. coli topoisomerase I N-terminal domains I to IV (Protein Data Bank (PDB) 3PX7). The active tyrosine residue Y319 is illustrated with the red color, and ssDNA is represented by the blue color. (B) Gate opening and closing dynamics of bacterial topoisomerase IA. The enzyme is represented by the green shape and the ssDNA is represented by the blue wavy line. The letter Y stands for the active site tyrosine residue Y319.

Figure 2.
Structures of bis-benzimidazoles Hoechst 33258, Hoechst 33342, 2′-(4-ethoxyphenyl)-5-(4-propylpiperazin-1-yl)-1H,1′H-2,5′-bibenzo[d]imidazole (PPEF) and DPA 154.
Figure 2.
Structures of bis-benzimidazoles Hoechst 33258, Hoechst 33342, 2′-(4-ethoxyphenyl)-5-(4-propylpiperazin-1-yl)-1H,1′H-2,5′-bibenzo[d]imidazole (PPEF) and DPA 154.

Figure 3.
Structures of tricyclic antidepressant imipramine and metabolite norclomipramine.

Figure 4.
(A) Polyamine scaffold and (B) individual polyamine inhibitors of bacterial topoisomerase I with antimycobacterial activity [112].
Figure 4.
(A) Polyamine scaffold and (B) individual polyamine inhibitors of bacterial topoisomerase I with antimycobacterial activity [112].

Figure 5.
Gold(III) inhibitors of bacterial topoisomerase I [113]. (A) Gold(III) macrocycle 10 and (B) gold(III) chelate 14.
Figure 5.
Gold(III) inhibitors of bacterial topoisomerase I [113]. (A) Gold(III) macrocycle 10 and (B) gold(III) chelate 14.

Figure 6.
Structures of fluoroquinophenoxazine 11a and 11g [114].
Figure 6.
Structures of fluoroquinophenoxazine 11a and 11g [114].

Figure 7.
VCC891909 from Vichem’s Nested Chemical Library (NCL) [98].
Figure 7.
VCC891909 from Vichem’s Nested Chemical Library (NCL) [98].


Figure 9.
Bacterial topoisomerase I inhibitors identified by in silico screening—piperidine amide compound 7 [95] and NSC76027 [94].


{kind=link}
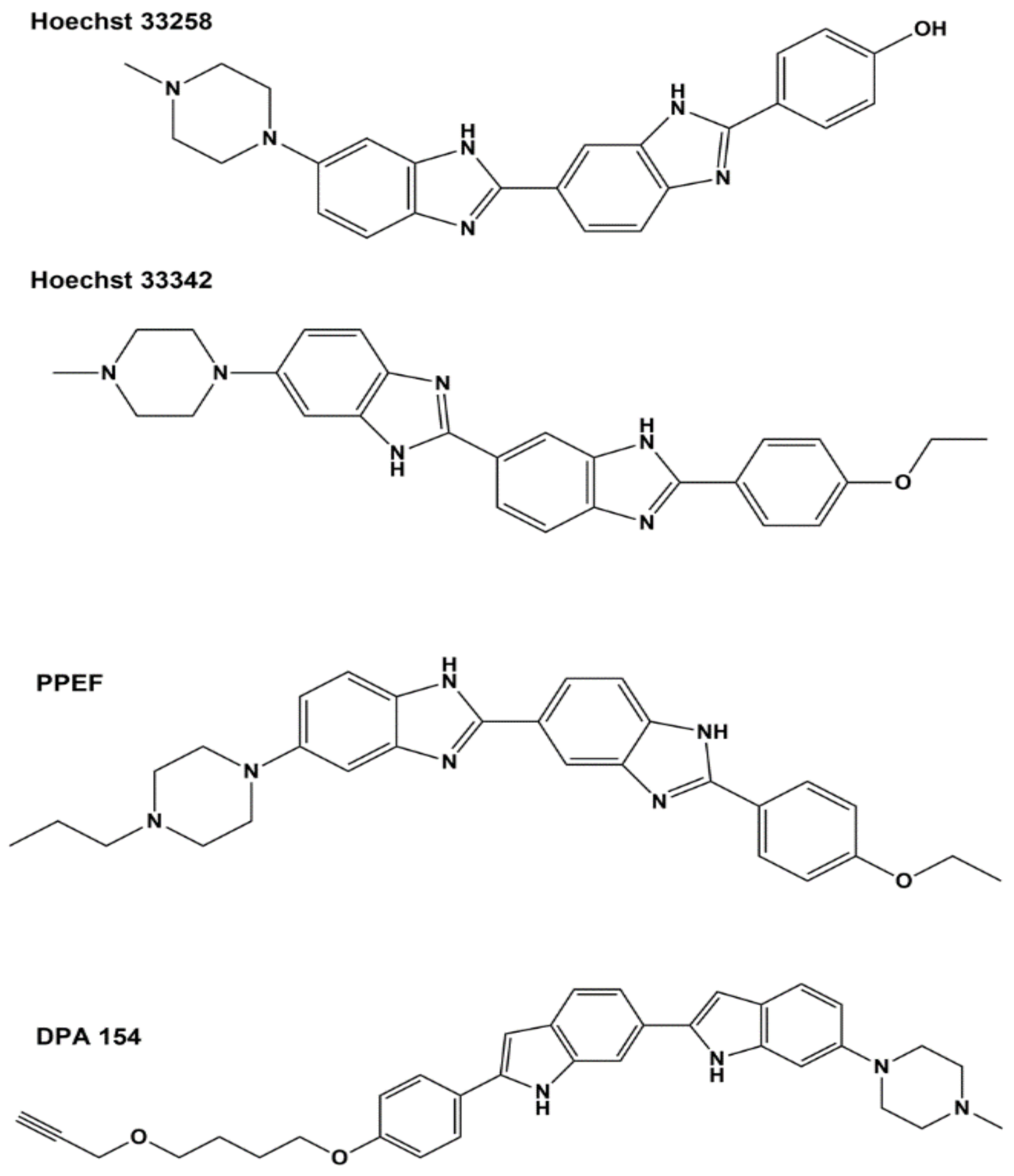{kind=link}
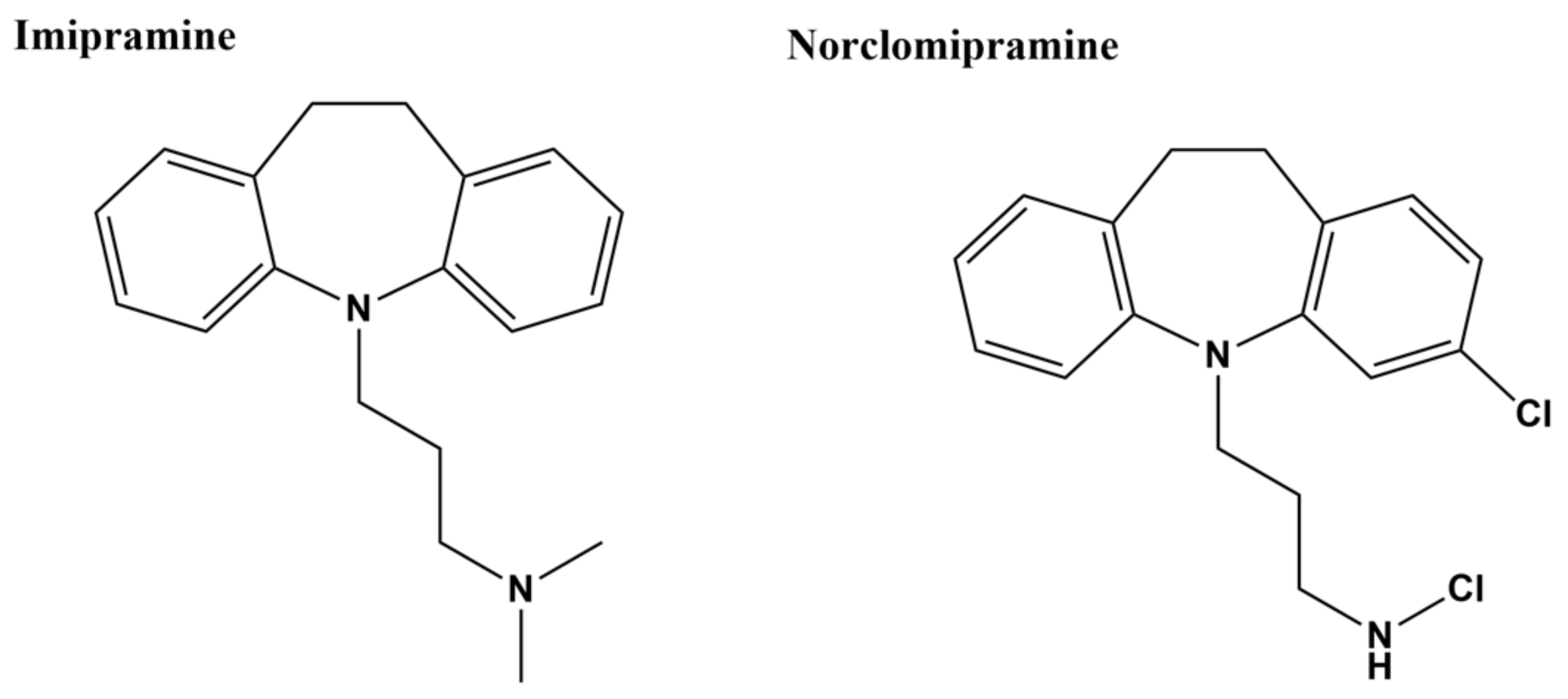{kind=link}
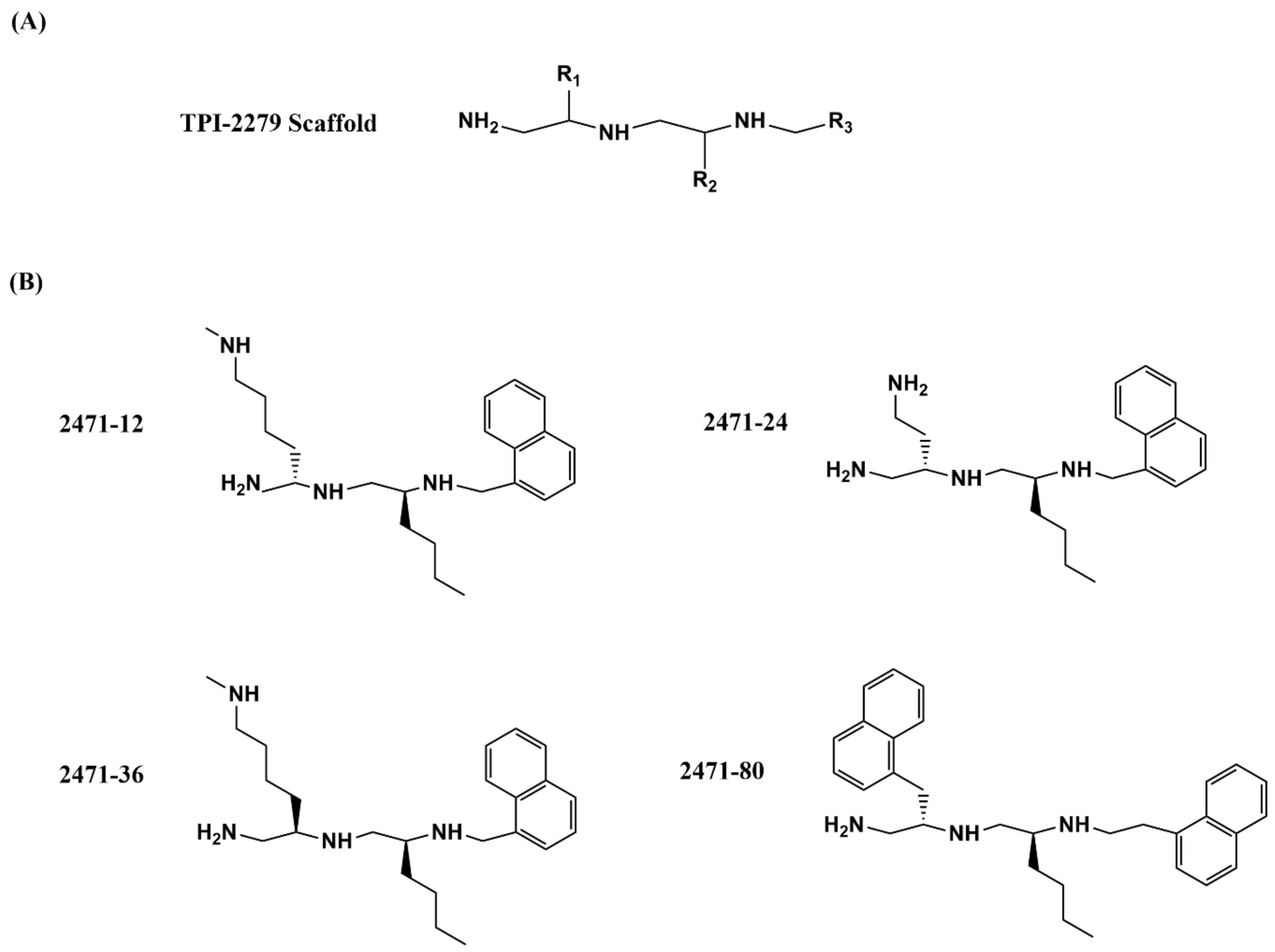{kind=link}
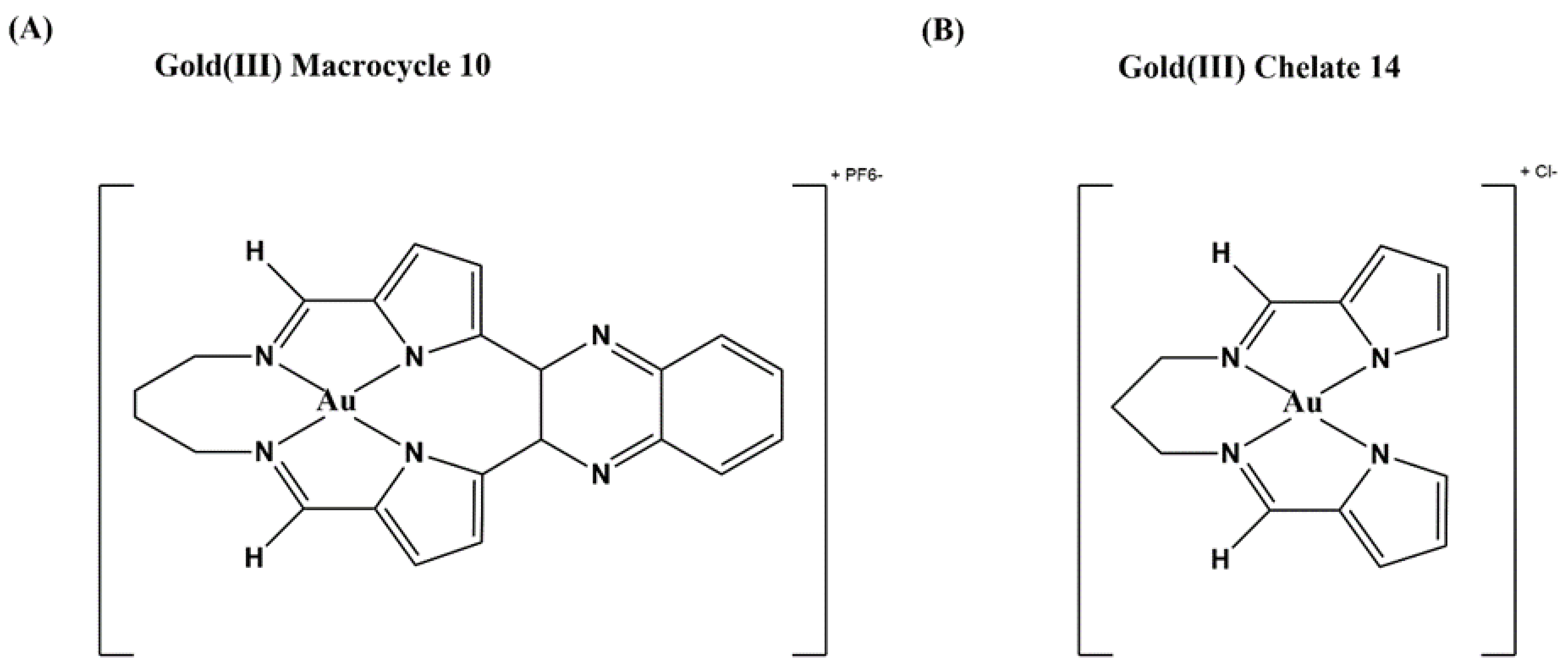{kind=link}
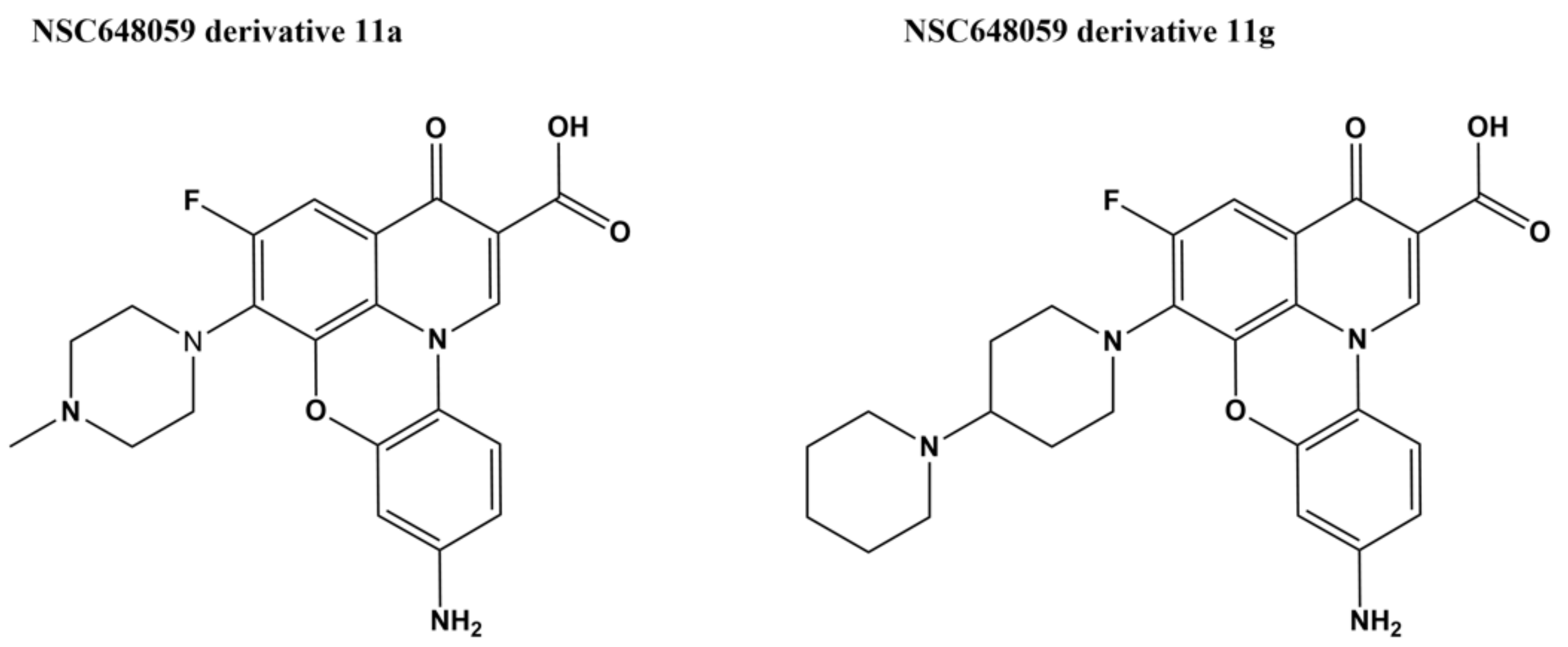{kind=link}
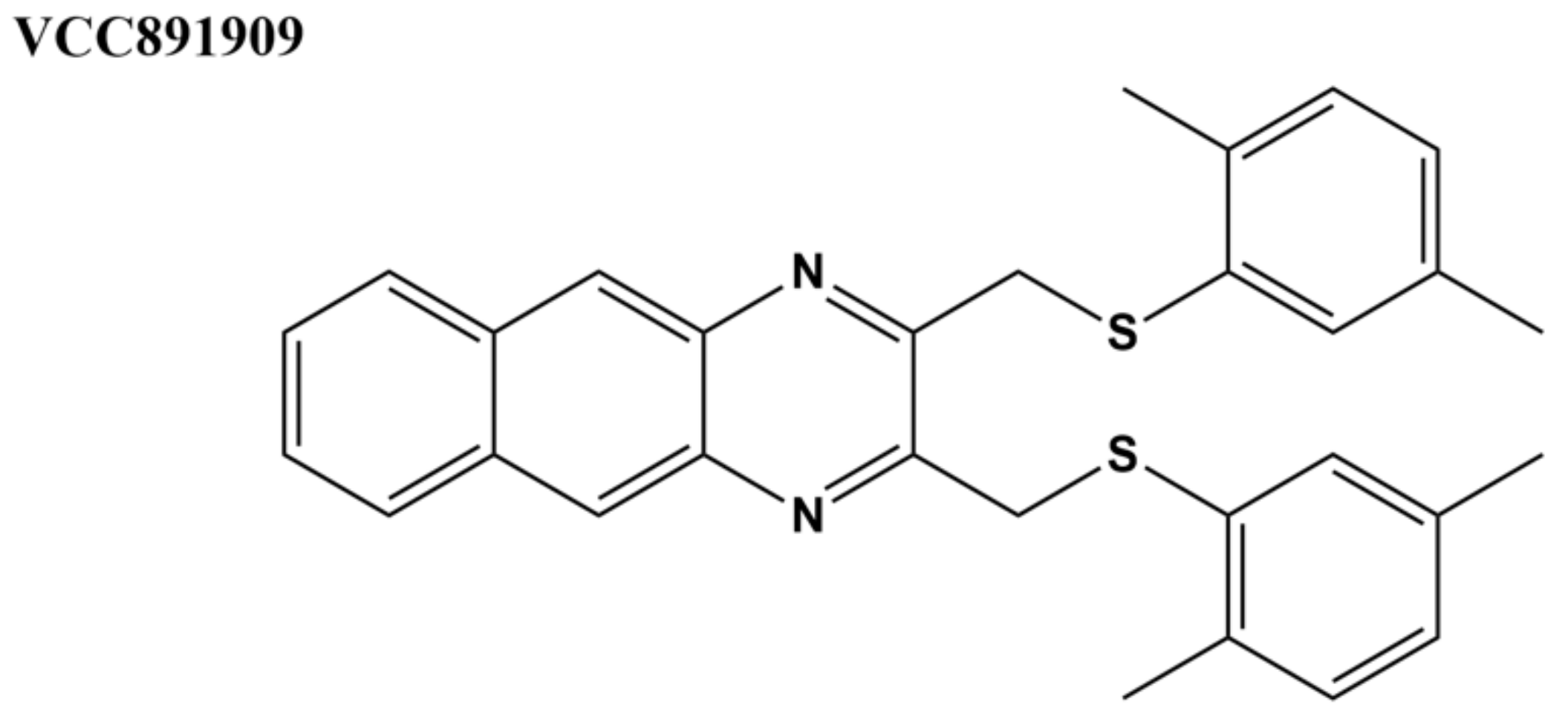{kind=link}
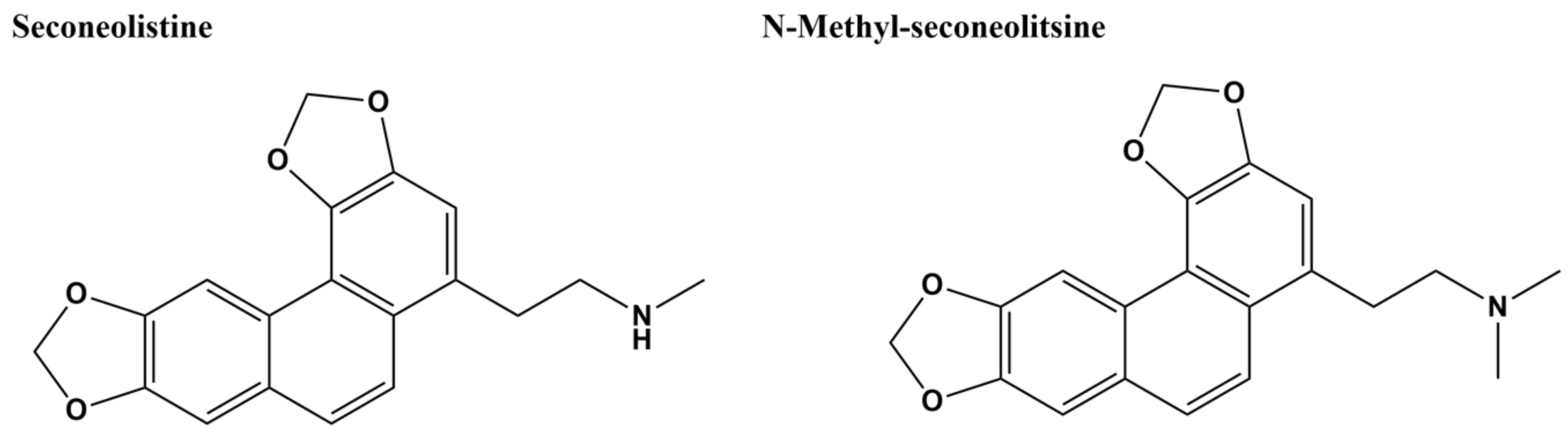{kind=link}
{kind=link}
Table 1.
Recently described small molecule inhibitors of bacterial topoisomerase I activity.
| Inhibitor | IC50 E. coli Topo I | IC50 M. tuberculosis Topo I |
|---|---|---|
| Bis-benzimidazole DPA 154 [110] | 6.6 µM | n.d. 1 |
| Bis-benzimidazole PPEF [111] | 9.4 µM | n.d. |
| Imipramine [96] | No inhibition at 25 µM | <0.1 µM |
| Norclomipramine [96] | No inhibition at 25 µM | <0.1 µM |
| Polyamine 2471-12 [112] | 7.5 µM | 7.5 µM |
| Polyamine 2471-24 [112] | 10 µM | 7.5 µM |
| Gold(III) macrocycle 10 [113] | 5 µM | 10 µM |
| Gold(III) chelate 14 [113] | 1.3 µM | 1.3 µM |
| Fluoroquinophenoxazine 11a [114] | 0.48 µM | 0.98 µM |
| Fluoroquinophenoxazine 11g [114,115] | 0.48 µM | 0.24 µM |
| VCC891909 [98] | n.d. | <7.5 µM |
| Seconeolitsine (SCN) [116] | n.d. | 5.6 µM |
| N-methyl-seconeolitsine (N-SCN) [116] | n.d. | 8.4 µM |
| Piperidine amide 7 [95] | 15.6–31.3 µM | 2 µM |
| NSC76027 [94] | 2.2 µM | 4 µM |
1 n.d.: Not determined.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Seddek, A.; Annamalai, T.; Tse-Dinh, Y.-C. Type IA Topoisomerases as Targets for Infectious Disease Treatments. Microorganisms 2021, 9, 86. https://doi.org/10.3390/microorganisms9010086
AMA Style
Seddek A, Annamalai T, Tse-Dinh Y-C. Type IA Topoisomerases as Targets for Infectious Disease Treatments. Microorganisms. 2021; 9(1):86. https://doi.org/10.3390/microorganisms9010086
Chicago/Turabian StyleSeddek, Ahmed, Thirunavukkarasu Annamalai, and Yuk-Ching Tse-Dinh. 2021. "Type IA Topoisomerases as Targets for Infectious Disease Treatments" Microorganisms 9, no. 1: 86. https://doi.org/10.3390/microorganisms9010086
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.