Methane-Oxidizing Communities in Lichen-Dominated Forested Tundra Are Composed Exclusively of High-Affinity USCα Methanotrophs

Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Site
2.2. Methane Flux Measurements
2.3. Determination of CH4 Oxidation Activity of Soil Samples
2.4. Chemical Analysis of the Soil Samples
2.5. DNA Extraction, PCR Amplification, and Illumina Sequencing
2.6. Bioinformatic Analysis of Amplicon Sequences
2.7. Molecular Identification of Methanotrophic Bacteria
2.8. qPCR-based Quantification of Methanotrophs
2.9. Sequence Accession Numbers
3. Results and Discussion
3.1. Measurements of CH4 Fluxes
3.2. Distribution of Methane Oxidation Activity over the Soil Profile
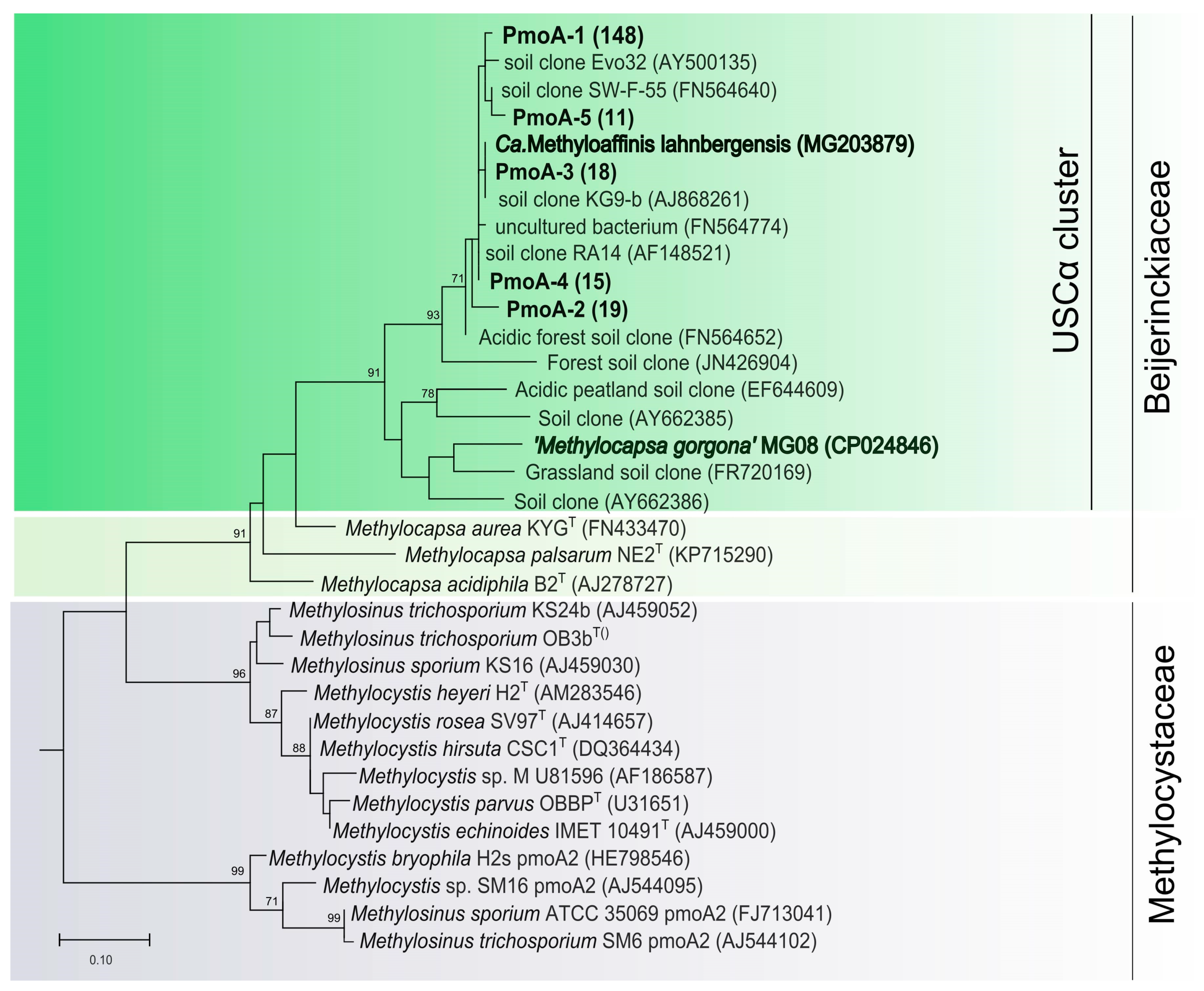
3.3. Identification and Quantification of Methanotrophs Using pmoA-Based Analysis
3.4. Molecular Analysis of Bacterial Diversity in Tundra Soil
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dlugokencky, E.J.; Nisbet, E.G.; Fisher, R.; Lowry, D. Global atmospheric methane: Budget, changes and dangers. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2011, 369, 2058–2072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirschke, S.; Bousquet, P.; Ciais, P.; Saunois, M.; Canadell, J.G.; Dlugokencky, E.J.; Bergamaschi, P.; Bergmann, D.; Blake, D.R.; Bruhwiler, L.; et al. Three decades of global methane sources and sinks. Nat. Geosci. 2013, 6, 813–823. [Google Scholar] [CrossRef]
- Ciais, P.; Sabine, C.; Bala, G.; Bopp, L.; Brovkin, V.; Canadell, J.; Chhabra, A.; DeFries, R.; Galloway, J.; Heimann, M.; et al. Carbon and other biogeochemical cycles. In Climate Change 2013 the Physical Science Basis: Working Group I Contribution to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change; Stocker, T.F., Qin, D., Plattner, G.-K., Tignor, M., Allen, S.K., Boschung, J., Nauels, A., Xia, Y., Bex, V., Midgley, P.M., Eds.; Cambridge University Press: Cambridge, UK, 2013; pp. 465–570. [Google Scholar]
- Prather, M.J.; Holmes, C.D. Overexplaining or underexplaining methane’s role in climate change. Proc. Natl. Acad. Sci. USA 2017, 114, 5324–5326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunfield, P.F. The soil methane sink. In Greenhouse Gas Sinks; Reay, D.S., Hewitt, C.N., Smith, K.A., Grace, J., Eds.; CABI: Wallingford, UK, 2007; pp. 152–170. [Google Scholar]
- Shallcross, D.E.; Butenhoff, C. The atmospheric methane sink. In Greenhouse Gas Sinks; Reay, D.S., Hewitt, C.N., Smith, K.A., Grace, J., Eds.; CABI: Wallingford, UK, 2007; pp. 171–184. [Google Scholar]
- Kolb, S. The quest for atmospheric methane oxidizers in forest soils. Environ. Microbiol. Rep. 2009, 1, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Hanson, R.S.; Hanson, T.E. Methanotrophic bacteria. Microbiol. Rev. 1996, 60, 439–471. [Google Scholar] [CrossRef] [PubMed]
- Trotsenko, Y.A.; Murrell, J.C. Metabolic aspects of aerobic obligate methanotrophy. Adv. Appl. Microbiol. 2008, 63, 183–229. [Google Scholar]
- Chistoserdova, L.; Lidstrom, M.E. Aerobic methylotrophic prokaryotes. In The Prokaryotes: Prokaryotic Physiology and Biochemistry; Rosenberg, E., Delong, E., Lory, S., Stackebrandt, E., Thompson, F., Eds.; Springer-Verlag: Berlin/Heidelberg, Germany, 2013; pp. 267–285. [Google Scholar]
- Khmelenina, V.N.; Colin Murrell, J.; Smith, T.J.; Trotsenko, Y.A. Physiology and biochemistry of the aerobic methanotrophs. In Aerobic Utilization of Hydrocarbons, Oils, and Lipids; Rojo, F., Ed.; Springer: Cham, Switzerland, 2019; pp. 1–25. [Google Scholar]
- Dedysh, S.N.; Knief, C. Diversity and phylogeny of described aerobic methanotrophs. In Methane Biocatalysis: Paving the Way to Sustainability; Kalyuzhnaya, M.G., Xing, X.-H., Eds.; Springer: Cham, Switzerland, 2018; pp. 17–42. [Google Scholar]
- Knief, C. Diversity and habitat preferences of cultivated and uncultivated aerobic methanotrophic bacteria evaluated based on pmoA as molecular marker. Front. Microbiol. 2015, 6, 1346. [Google Scholar] [CrossRef] [Green Version]
- Bender, M.; Conrad, R. Kinetics of CH4 oxidation in oxic soils exposed to ambient air or high CH4 mixing ratios. FEMS Microbiol. Lett. 1992, 101, 261–270. [Google Scholar] [CrossRef]
- Holmes, A.J.; Roslev, P.; McDonald, I.R.; Iversen, N.; Henriksen, K.; Murrell, J.C. Characterization of methanotrophic bacterial populations in soils showing atmospheric methane uptake. Appl. Environ. Microbiol. 1999, 65, 3312–3318. [Google Scholar] [CrossRef] [Green Version]
- Dedysh, S.N.; Horz, H.P.; Dunfield, P.F.; Liesack, W. A novel pmoA lineage represented by the acidophilic methanotrophic bacterium Methylocapsa acidiphila [correction of acidophila] B2. Arch. Microbiol. 2001, 177, 117–121. [Google Scholar] [CrossRef]
- Dedysh, S.N.; Khmelenina, V.N.; Suzina, N.E.; Trotsenko, Y.A.; Semrau, J.D.; Liesack, W.; Tiedje, J.M. Methylocapsa acidiphila gen. nov., sp. nov., a novel methane-oxidizing and dinitrogen-fixing acidophilic bacterium from Sphagnum bog. Int. J. Syst. Evol. Microbiol. 2002, 52, 251–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knief, C.; Lipski, A.; Dunfield, P.F. Diversity and activity of methanotrophic bacteria in different upland soils. Appl. Environ. Microbiol. 2003, 69, 6703–6714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henckel, T.; Jäckel, U.; Schnell, S.; Conrad, R. Molecular analyses of novel methanotrophic communities in forest soil that oxidize atmospheric methane. Appl. Environ. Microbiol. 2000, 66, 1801–1808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen, S.; Holmes, A.J.; Olsen, R.A.; Murrell, J.C. Detection of methane oxidizing bacteria in forest soil by monooxygenase PCR amplification. Microb. Ecol. 2000, 39, 282–289. [Google Scholar] [PubMed]
- Degelmann, D.M.; Borken, W.; Drake, H.L.; Kolb, S. Different atmospheric methane-oxidizing communities in european beech and norway spruce soils. Appl. Environ. Microbiol. 2010, 10, 3228–3235. [Google Scholar] [CrossRef] [Green Version]
- Dörr, N.; Glaser, B.; Kolb, S. Methanotrophic communities in brazilian ferralsols from naturally forested, afforested, and agricultural sites. Appl. Environ. Microbiol. 2010, 76, 1307–1310. [Google Scholar] [CrossRef] [Green Version]
- Martineau, C.; Pan, Y.; Bodrossy, L.; Yergeau, E.; Whyte, L.G.; Greer, C.W. Atmospheric methane oxidizers are present and active in Canadian high Arctic soils. FEMS Microbiol. Ecol. 2014, 89, 257–269. [Google Scholar] [CrossRef] [Green Version]
- Lau, M.C.Y.; Stackhouse, B.T.; Layton, A.C.; Chauhan, A.; Vishnivetskaya, T.A.; Chourey, K.; Ronholm, J.; Mykytczuk, N.C.S.; Bennett, P.C.; Lamarche-Gagnon, G.; et al. An active atmospheric methane sink in high Arctic mineral cryosols. ISME J. 2015, 9, 1880–1891. [Google Scholar] [CrossRef] [Green Version]
- Ricke, P.; Kolb, S.; Braker, G. Application of a newly developed ARB software-integrated tool for in silico terminal restriction fragment length polymorphism analysis reveals the dominance of a novel pmoA cluster in a forest soil. Appl. Environ. Microbiol. 2005, 71, 1671–1673. [Google Scholar] [CrossRef] [Green Version]
- Singleton, C.M.; McCalley, C.K.; Woodcroft, B.J.; Boyd, J.A.; Evans, P.N.; Hodgkins, S.B.; Chanton, J.P.; Frolking, S.; Crill, P.M.; Saleska, S.R.; et al. Methanotrophy across a natural permafrost thaw environment. ISME J. 2018, 12, 2544–2558. [Google Scholar] [CrossRef] [Green Version]
- Pratscher, J.; Vollmers, J.; Wiegand, S.; Dumont, M.G.; Kaster, A.K. Unravelling the identity, metabolic potential and global biogeography of the atmospheric methane-oxidizing upland soil cluster α. Environ. Microbiol. 2018, 20, 1016–1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tveit, A.T.; Hestnes, A.G.; Robinson, S.L.; Schintlmeister, A.; Dedysh, S.N.; Jehmlich, N.; von Bergen, M.; Herbold, C.; Wagner, M.; Richter, A.; et al. Widespread soil bacterium that oxidizes atmospheric methane. Proc. Natl. Acad. Sci. USA 2019, 10, 589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glagolev, M.; Kleptsova, I.; Filippov, I.; Maksyutov, S.; MacHida, T. Regional methane emission from West Siberia mire landscapes. Environ. Res. Lett. 2011, 6, 045214. [Google Scholar] [CrossRef]
- Glagolev, M.V.; Sabrekov, A.F.; Kleptsova, I.E.; Filippov, I.V.; Lapshina, E.D.; Machida, T.; Maksyutov, S.S. Methane emission from bogs in the subtaiga of Western Siberia: The development of standard model. Eurasian Soil Sci. 2012, 45, 947–957. [Google Scholar] [CrossRef]
- Danilova, O.V.; Belova, S.E.; Gagarinova, I.V.; Dedysh, S.N. Microbial community composition and methanotroph diversity of a subarctic wetland in Russia. Microbiology 2016, 85, 583–591. [Google Scholar] [CrossRef]
- Ivanova, A.A.; Kulichevskaya, I.S.; Merkel, A.Y.; Toshchakov, S.V.; Dedysh, S.N. High diversity of Planctomycetes in soils of two lichen-dominated sub-arctic ecosystems of northwestern Siberia. Front. Microbiol. 2016, 7, 2065. [Google Scholar] [CrossRef] [Green Version]
- Caporaso, J.; Kuczynski, J.; Stombaugh, J. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Glöckner, F.O.; Yilmaz, P.; Quast, C.; Gerken, J.; Beccati, A.; Ciuprina, A.; Bruns, G.; Yarza, P.; Peplies, J.; Westram, R.; et al. 25 years of serving the community with ribosomal RNA gene reference databases and tools. J. Biotechnol. 2017, 261, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Holmes, A.J.; Costello, A.; Lidstrom, M.E.; Murrell, J.C. Evidence that participate methane monooxygenase and ammonia monooxygenase may be evolutionarily related. FEMS Microbiol. Lett. 1995, 132, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, W.; Strunk, O.; Westram, R.; Richter, L.; Meier, H.; Yadhukumar, A.; Buchner, A.; Lai, T.; Steppi, S.; Jacob, G.; et al. ARB: A software environment for sequence data. Nucleic Acids Res. 2004, 32, 1363–1371. [Google Scholar] [CrossRef] [Green Version]
- Kubista, M.; Andrade, J.M.; Bengtsson, M.; Forootan, A.; Jonák, J.; Lind, K.; Sindelka, R.; Sjöback, R.; Sjögreen, B.; Strömbom, L.; et al. The real-time polymerase chain reaction. Mol. Aspects Med. 2006, 2–3, 95–125. [Google Scholar] [CrossRef] [PubMed]
- Ashelford, K.E.; Weightman, A.J.; Fry, J.C. Primrose: A computer program for generating and estimating the phylogenetic range of 16S rRNA oligonucleotide probes and primers in conjunction with the RDP-II database. Nucleic Acids Res. 2002, 30, 3481–3489. [Google Scholar] [CrossRef] [Green Version]
- Emmerton, C.A.; St. Louis, V.L.; Lehnherr, I.; Humphreys, E.R.; Rydz, E.; Kosolofski, H.R. The net exchange of methane with high Arctic landscapes during the summer growing season. Biogeosciences 2014, 11, 3095–3106. [Google Scholar] [CrossRef] [Green Version]
- Luo, G.J.; Kiese, R.; Wolf, B.; Butterbach-Bahl, K. Effects of soil temperature and moisture on methane uptake and nitrous oxide emissions across three different ecosystem types. Biogeosciences 2013, 10, 3205–3219. [Google Scholar] [CrossRef] [Green Version]
- Kolb, S.; Knief, C.; Dunfield, P.F.; Conrad, R. Abundance and activity of uncultured methanotrophic bacteria involved in the consumption of atmospheric methane in two forest soils. Environ. Microbiol. 2005, 7, 1150–1161. [Google Scholar] [CrossRef]
- Pratscher, J.; Dumont, M.G.; Conrad, R. Assimilation of acetate by the putative atmospheric methane oxidizers belonging to the USCα clade. Environ. Microbiol. 2011, 13, 2692–2701. [Google Scholar] [CrossRef]
- Dunfield, P.F.; Yimga, M. Isolation of a Methylocystis strain containing a novel pmoA-like gene. FEMS Microbiol. Ecol. 2002, 41, 17–26. [Google Scholar] [CrossRef]
- Knief, C.; Dunfield, P.F. Response and adaptation of different methanotrophic bacteria to low methane mixing ratios. Environ. Microbiol. 2005, 7, 1307–1317. [Google Scholar] [CrossRef] [PubMed]
- Baani, M.; Liesack, W. Two isozymes of particulate methane monooxygenase with different methane oxidation kinetics are found in Methylocystis sp. strain SC2. Proc. Natl. Acad. Sci. USA 2008, 105, 10203–10208. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Organic Layer | Gray Sand | |
|---|---|---|---|
| C org | % | 1.51 ± 0.30 | 0.11 ± 0.02 |
| N total | 0.06 ± 0.01 | 0.01 ± 0.002 | |
| N-NO3 | mg/kg | 7.30 ± 0.55 | 7.00 ± 0.53 |
| N-NH4 | 14.00 ± 1.05 | 9.00 ± 0.68 | |
| P total | 165.0 ± 33.0 | 39.0 ± 7.8 | |
| SO42- | 19.40 ± 4.85 | 1.50 ± 0.38 | |
| Ca | 13600 ± 2176 | 440.0 ± 70.4 | |
| Mg | 450.0 ± 72.0 | 26.0 ± 4.16 | |
| Fe | 1821.0 ± 291.0 | 1621.0 ± 259.4 | |
| pH | 4.1 ± 0.1 | 6.1 ± 0.1 | |
| Phylotype | Number of Sequences | GenBank Accession Numbers | Identity to Cand. M. lahnbergensis (%) | Identity to M. gorgona (%) |
|---|---|---|---|---|
| pmoA-1 * | 148 | KX534008, KX534009, KX534010, KX534011, KX534012, KX534013, KX534014, KX534015, KX534016, KX534017, KX534018, KX534020, KX534021, KX534022, KX534023, KX534025, KX534026, KX534027, KX534028, KX534029, KX534031, KX534035, KX534037, KX534038, KX534039, KX534040, KX534041, KX534044, KX534046, KX534048, KX534051, KX534052, KX534055, KX534056 | 97.5 | 84.2 |
| pmoA-2 * | 19 | KX534053, KX534045, KX534030, KX534047, KX534036, KX534033 | 96.4 | 83.1 |
| pmoA-3 * | 18 | KX534034, KX534049, KX534019 | 99.2 | 83.5 |
| pmoA-4 * | 15 | KX534032, KX534042 | 95.8 | 84.0 |
| pmoA-5 * | 11 | KX534043 | 95.0 | 80.8 |
| pmoA-6 | 8 | KX534007, KX534054 | 96.4 | 83.1 |
| pmoA-7 | 3 | KX534050 | 97.5 | 83.8 |
| OTU ID | Relative Abundance (%) | Taxonomy | Closest Silva Match (Similarity, %) | Reported Habitat | Similarity MG203879 (%) | Similarity M. gorgona (%) |
|---|---|---|---|---|---|---|
| 16S-1 | 2.358 | Beijerinckiaceae uncultured | AY913480 (100) | bulk soil | 99 | 98.6 |
| 16S-2 | 1.902 | Beijerinckiaceae uncultured | AY913598 (100) | limestone cave | 100 | 97.6 |
| 16S-3 | 0.167 | Beijerinckiaceae | KX509291 (100) | rainwater | 98.6 | 98.1 |
| 16S-4 | 0.021 | Beijerinckiaceae | KT182565 (100) | bioreactor | 96.6 | 96.2 |
| 16S-5 | 0.019 | Beijerinckiaceae | JQ905994 (99) | acid mine | 96.6 | 97.1 |
| 16S-6 | 0.016 | Beijerinckiaceae | FPLS01031837 (100) | unknown | 98.6 | 98.1 |
| 16S-7 | 0.01 | Beijerinckiaceae | KF100807 (99) | human skin | 96.6 | 95.7 |
| 16S-8 | 0.004 | Beijerinckiaceae uncultured | HG528987 (100) | peatland | 98.1 | 98.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Belova, S.E.; Danilova, O.V.; Ivanova, A.A.; Merkel, A.Y.; Dedysh, S.N. Methane-Oxidizing Communities in Lichen-Dominated Forested Tundra Are Composed Exclusively of High-Affinity USCα Methanotrophs. Microorganisms 2020, 8, 2047. https://doi.org/10.3390/microorganisms8122047
Belova SE, Danilova OV, Ivanova AA, Merkel AY, Dedysh SN. Methane-Oxidizing Communities in Lichen-Dominated Forested Tundra Are Composed Exclusively of High-Affinity USCα Methanotrophs. Microorganisms. 2020; 8(12):2047. https://doi.org/10.3390/microorganisms8122047
Chicago/Turabian StyleBelova, Svetlana E., Olga V. Danilova, Anastasia A. Ivanova, Alexander Y. Merkel, and Svetlana N. Dedysh. 2020. "Methane-Oxidizing Communities in Lichen-Dominated Forested Tundra Are Composed Exclusively of High-Affinity USCα Methanotrophs" Microorganisms 8, no. 12: 2047. https://doi.org/10.3390/microorganisms8122047