Multi-Omic Analysis Reveals Different Effects of Sulforaphane on the Microbiome and Metabolome in Old Compared to Young Mice

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design and Sample Collection
2.2. Sample Preparation for Microbiome Analysis
2.3. Sample Preparation for Metabolomics Analysis
2.4. Bioinformatic Processing with 16S rRNA Amplicon Sequencing Data
2.5. Computation Frameworks to Integrate Microbiome and Metabolome for the Identification of Potential Mechanistic Links
3. Results
3.1. Data Annotation and Overview of Samples
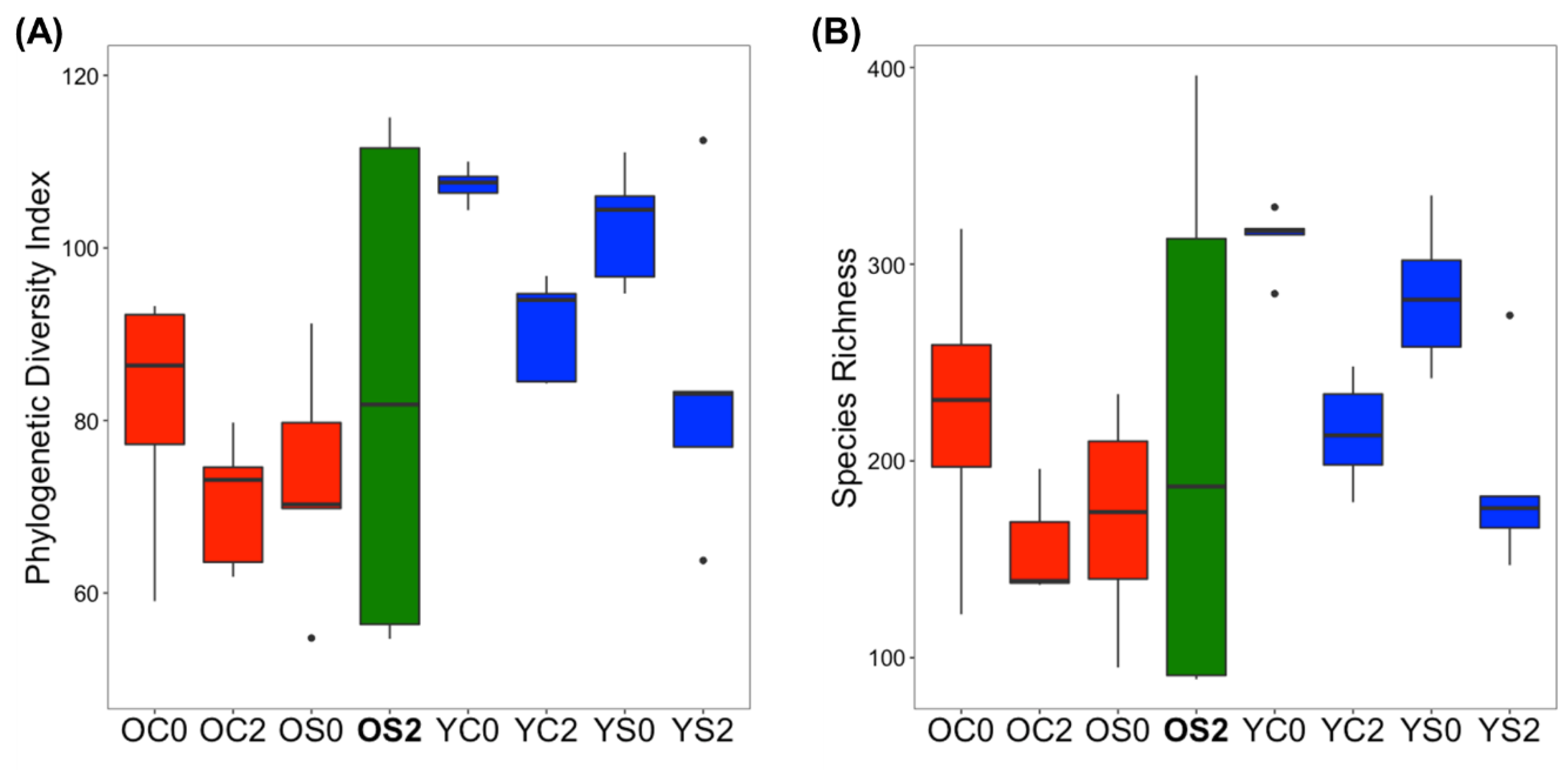
3.2. Gut Microbiome Alpha Diversity Analysis
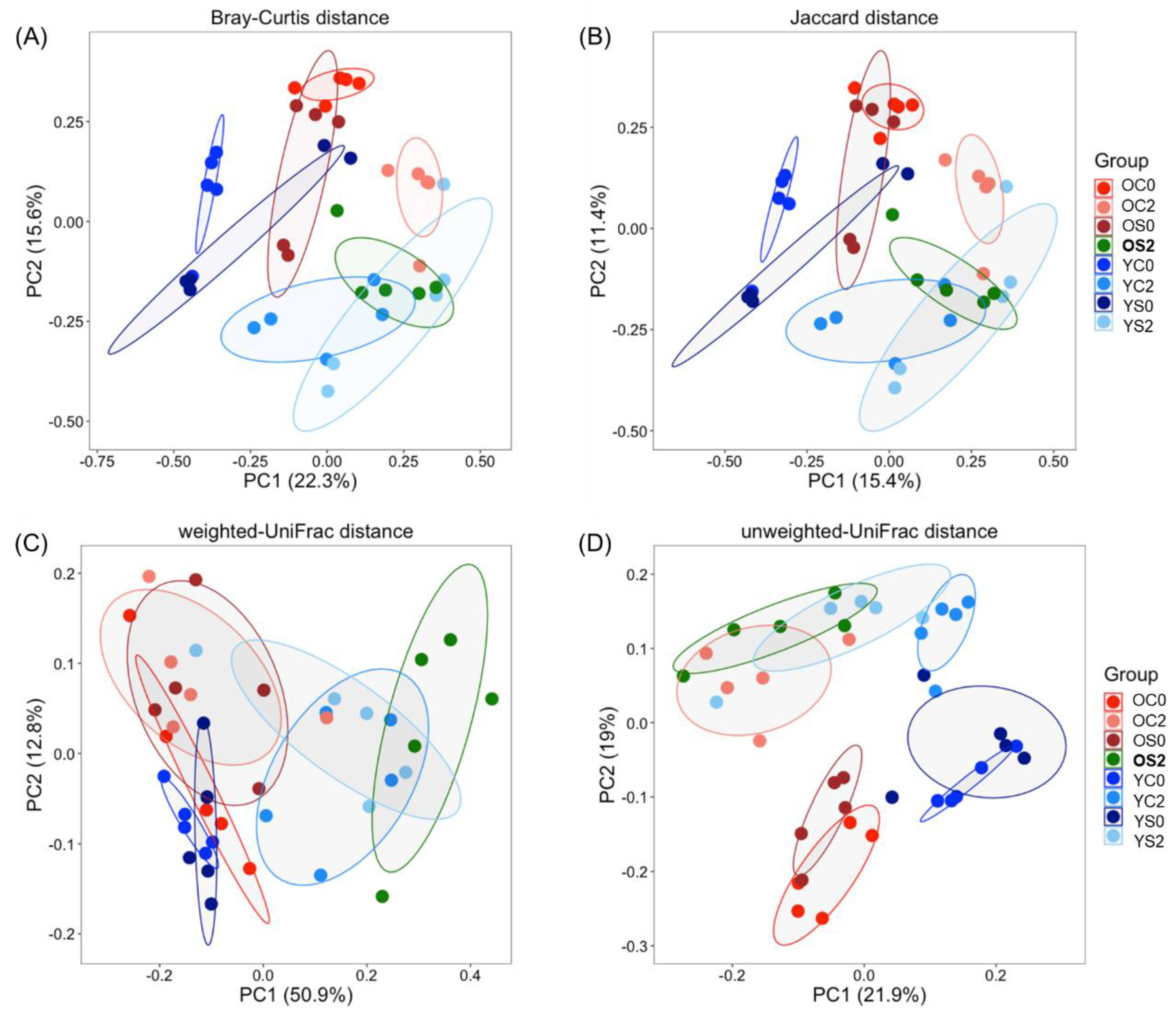
3.3. Gut Microbiome Beta Diversity Analysis
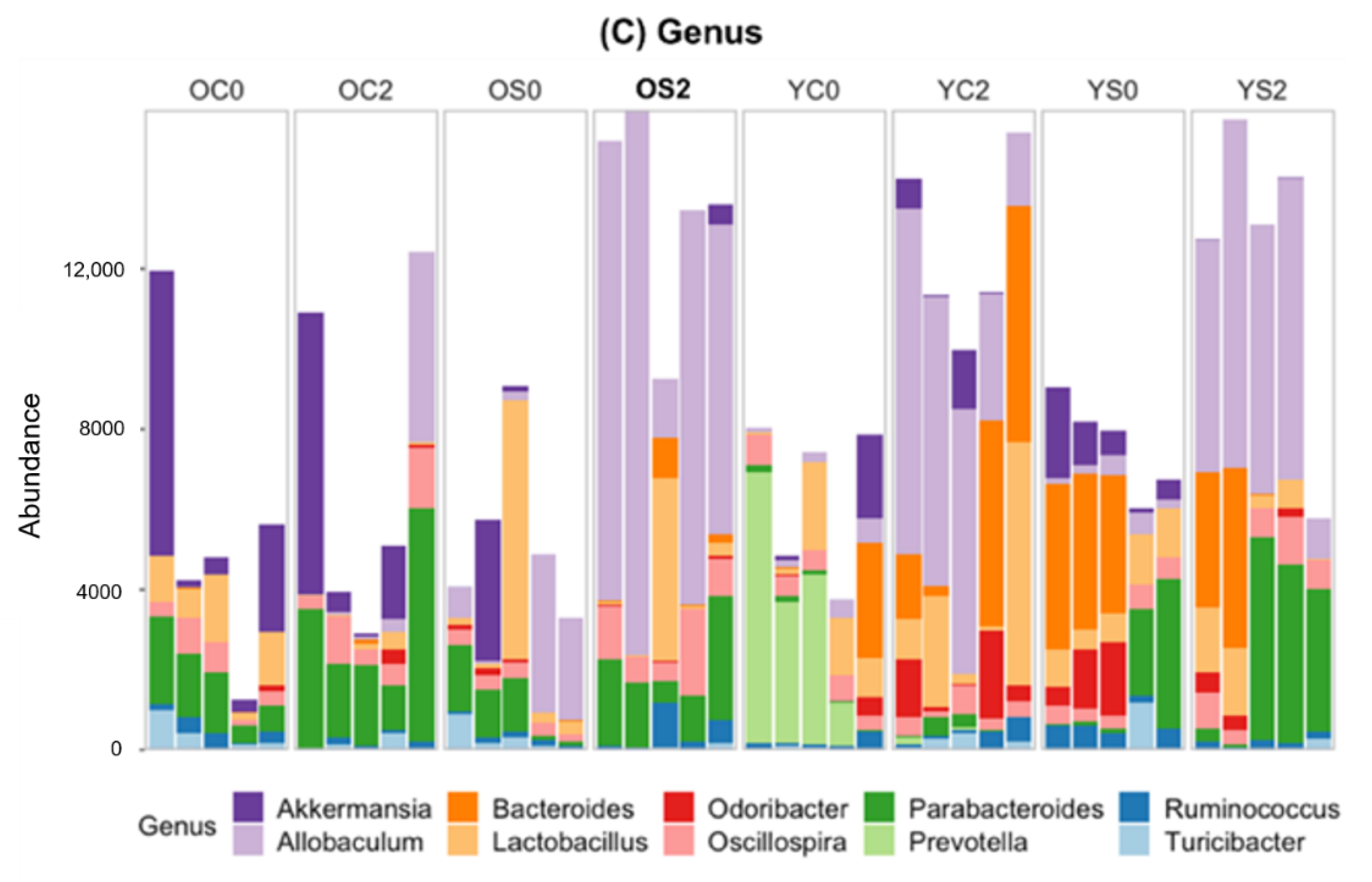
3.4. Gut microbiome Taxonomic Profiling
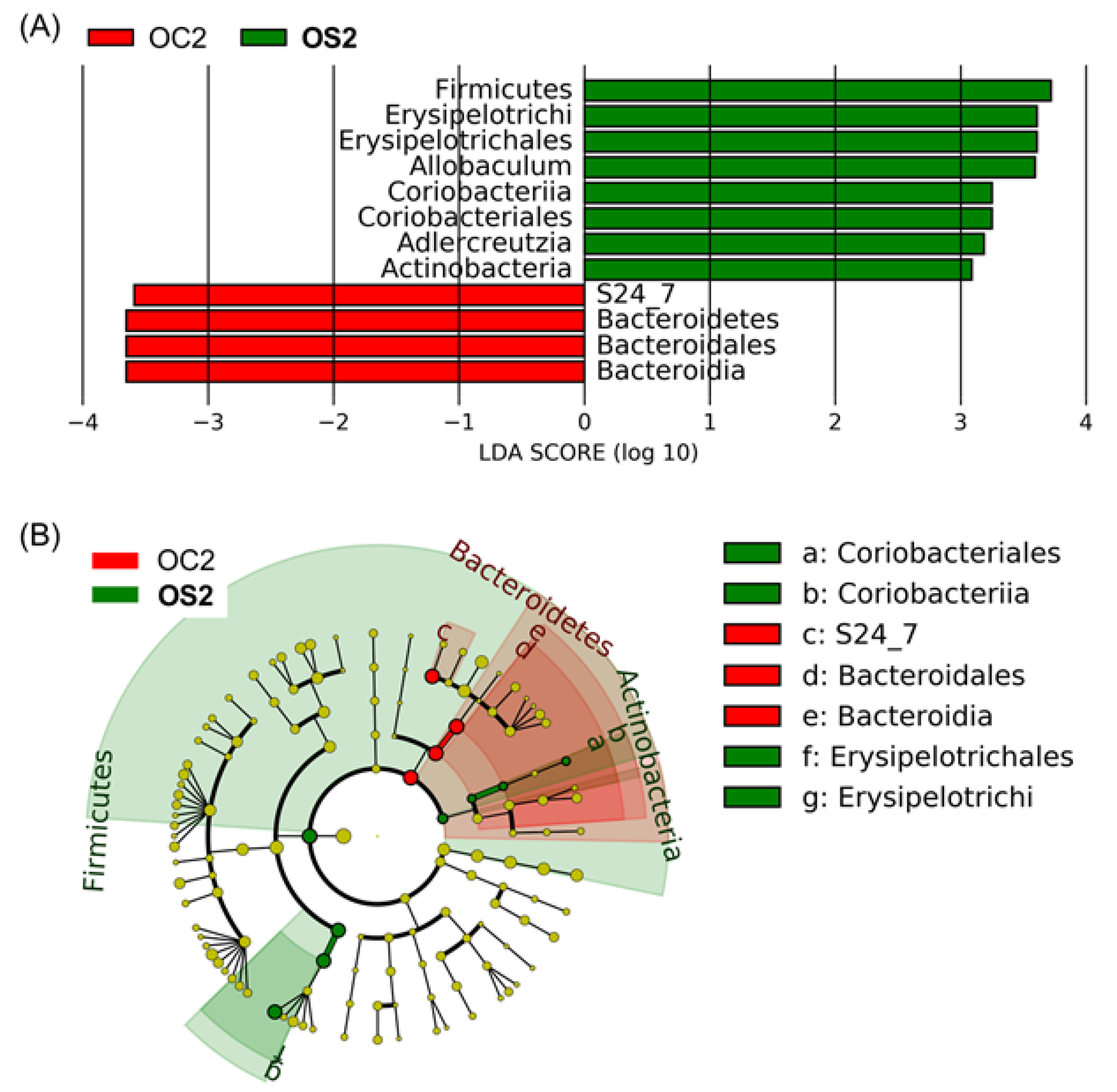
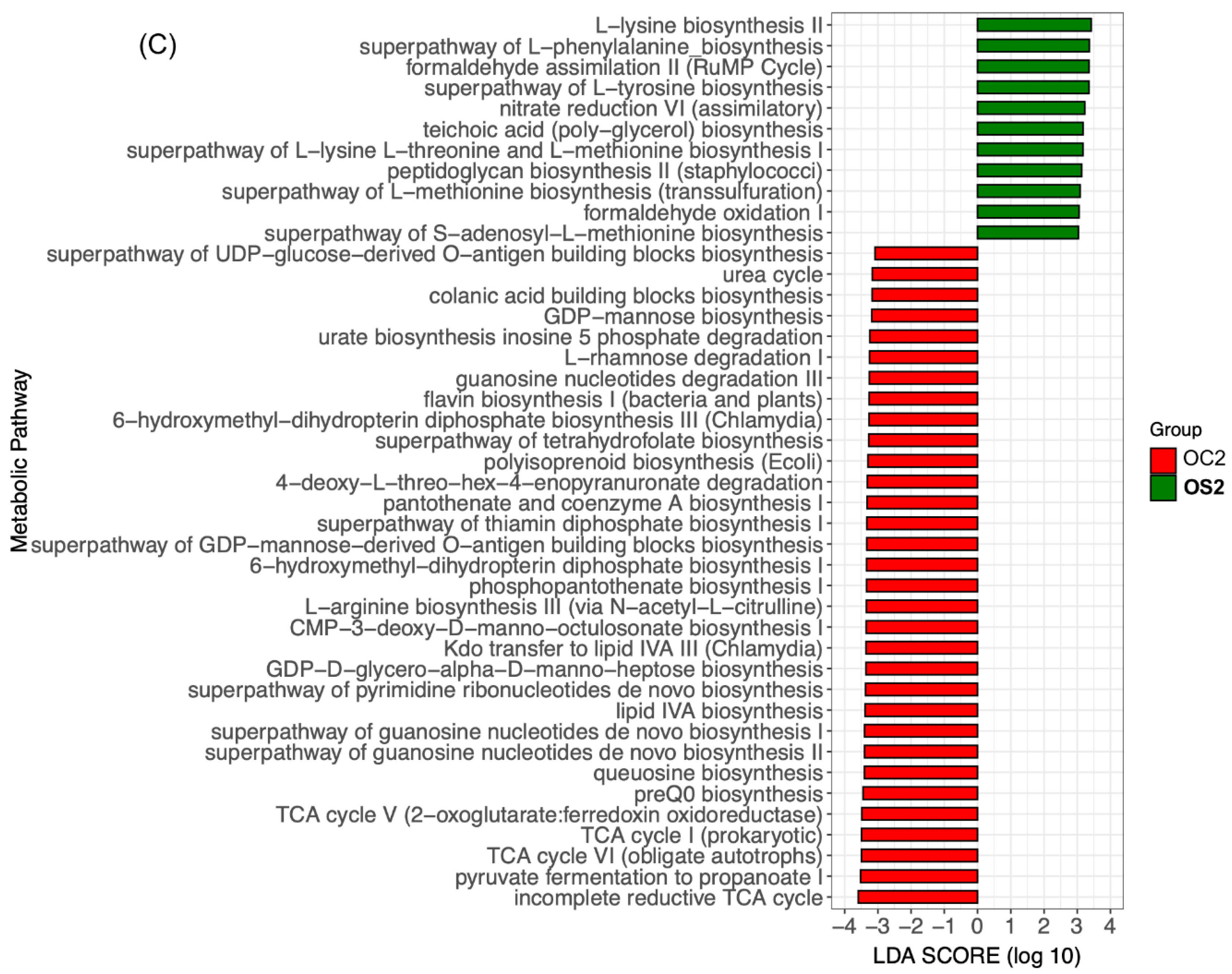
3.5. Linear Discriminant Effect Size Analysis (LEfSe) of Gut Microbiota
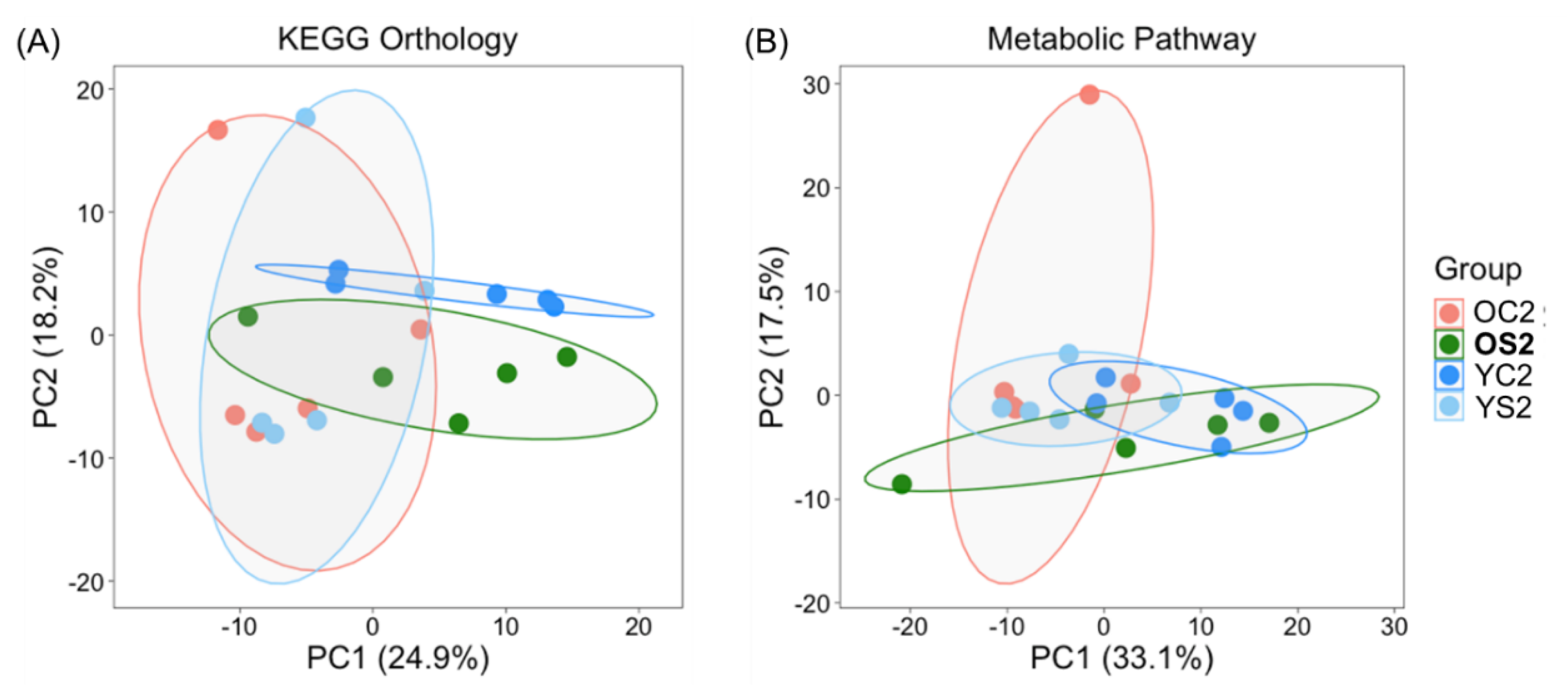
3.6. Potential Functional Annotations of Gut Microbiota in Two Age Groups with and without SFN Diet
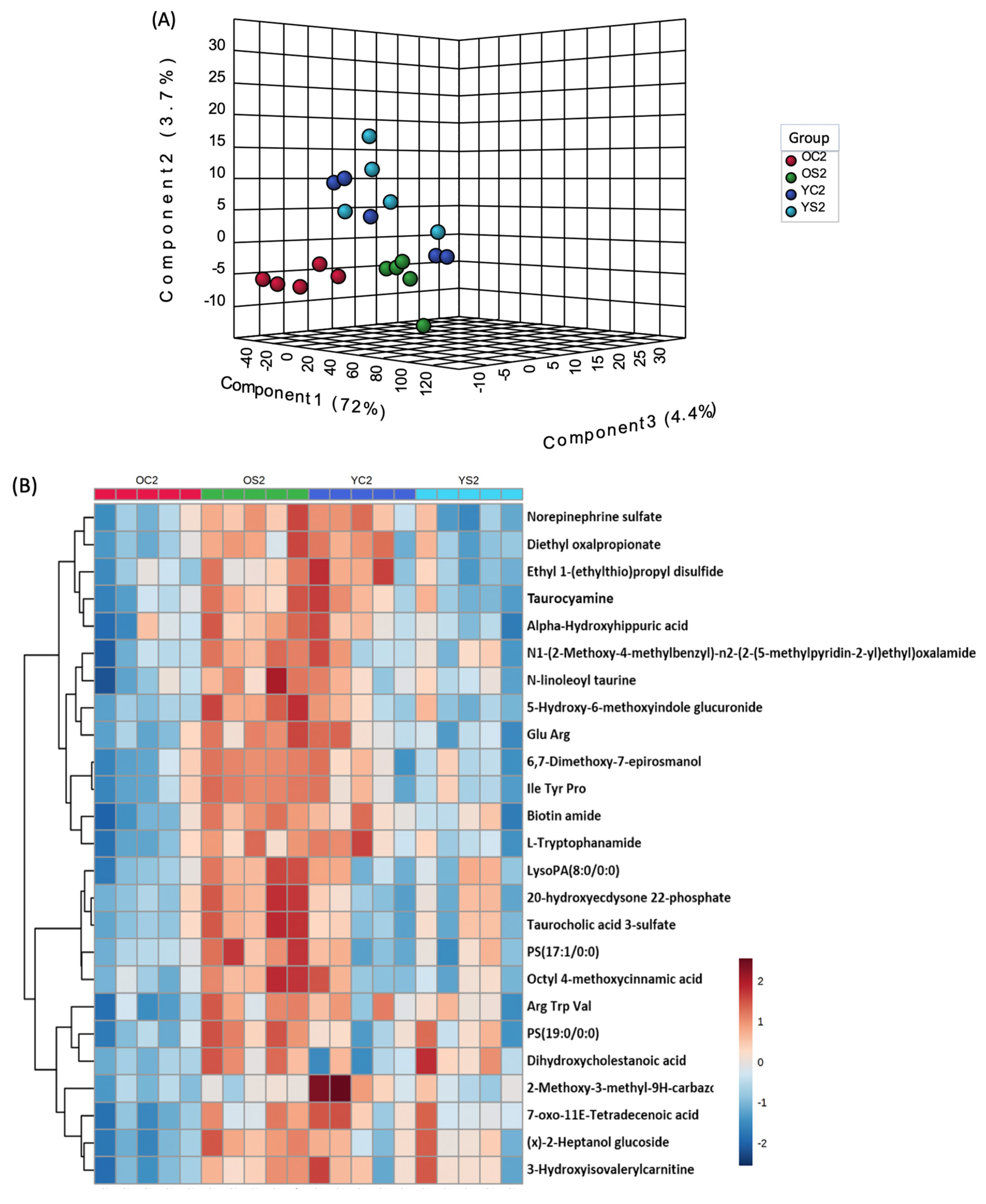
3.7. Gut Metabolome of Two AGE Groups with and without SFN Diet
3.8. Microbiome and Metabolome Data Integration Analysis Reveals Microbiome-Dependent Metabolic Changes
4. Discussion
4.1. Age-Dependent Microbial Signatures of the Mouse Gut Microbiome
4.2. SFN Diet-Dependent Microbial Signatures in the Mouse Gut Microbiome
4.3. SFN Diet-Dependent Microbial Functional Signatures in the Mouse Gut Microbiome
4.4. SFN Diet-Dependent Microbiome-Dependent Metabolites
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Zhang, Z.; Tang, H.; Chen, P.; Xie, H.; Tao, Y. Demystifying the manipulation of host immunity, metabolism, and extraintestinal tumors by the gut microbiome. Signal. Transduct. Target. Ther. 2019, 4, 41. [Google Scholar] [CrossRef]
- Jandhyala, S.M.; Talukdar, R.; Subramanyam, C.; Vuyyuru, H.; Sasikala, M.; Reddy, D.N. Role of the normal gut microbiota. World J. Gastroenterol. 2015, 21, 8787–8803. [Google Scholar] [CrossRef]
- Zheng, D.; Liwinski, T.; Elinav, E. Interaction between microbiota and immunity in health and disease. Cell Res. 2020, 30, 492–506. [Google Scholar] [CrossRef]
- Visconti, A.; Le Roy, C.I.; Rosa, F.; Rossi, N.; Martin, T.C.; Mohney, R.P.; Li, W.; De Rinaldis, E.; Bell, J.T.; Venter, J.C.; et al. Interplay between the human gut microbiome and host metabolism. Nat. Commun. 2019, 10, 4505. [Google Scholar] [CrossRef] [Green Version]
- Levy, M.; Blacher, E.; Elinav, E. Microbiome, metabolites and host immunity. Curr. Opin. Microbiol. 2017, 35, 8–15. [Google Scholar] [CrossRef]
- Claesson, M.J.; Jeffery, I.B.; Conde, S.; Power, S.E.; O’Connor, E.M.; Cusack, S.; Harris, H.M.B.; Coakley, M.; Lakshminarayanan, B.; O’Sullivan, O.; et al. Gut microbiota composition correlates with diet and health in the elderly. Nature 2012, 488, 178–184. [Google Scholar] [CrossRef]
- Cotillard, A.; ANR MicroObes Consortium; Kennedy, S.; Kong, L.C.; Prifti, E.; Pons, N.; Le Chatelier, E.; Almeida, M.; Quinquis, B.; Levenez, F.; et al. Dietary intervention impact on gut microbial gene richness. Nature 2013, 500, 585–588. [Google Scholar] [CrossRef]
- Nagpal, R.; Mainali, R.; Ahmadi, S.; Wang, S.; Singh, R.; Kavanagh, K.; Kitzman, D.W.; Kushugulova, A.; Marotta, F.; Yadav, H. Gut microbiome and aging: Physiological and mechanistic insights. Nutr. Health Aging 2018, 4, 267–285. [Google Scholar] [CrossRef] [Green Version]
- Bose, C.; Alves, I.; Singh, P.; Palade, P.T.; Carvalho, E.; Børsheim, E.; Jun, S.-R.; Cheema, A.; Boerma, M.; Awasthi, S.; et al. Sulforaphane prevents age-associated cardiac and muscular dysfunction through Nrf2 signaling. Aging Cell 2020, in press. [Google Scholar]
- Mathers, J.C. Nutrigenomics in the modern era. Proc. Nutr. Soc. 2017, 76, 265–275. [Google Scholar] [CrossRef]
- Kim, J.K.; Park, S.U. Current potential health benefits of sulforaphane. EXCLI J. 2016, 15, 571–577. [Google Scholar] [CrossRef] [PubMed]
- Santin-Marquez, R.; Alarcon-Aguilar, A.; Lopez-Diazguerrero, N.E.; Chondrogianni, N.; Konigsberg, M. Sulforaphane—Role in aging and neurodegeneration. GeroScience 2019, 41, 655–670. [Google Scholar] [CrossRef] [PubMed]
- Casero, D.; Gill, K.; Sridharan, V.; Koturbash, I.; Nelson, G.; Hauer-Jensen, M.; Boerma, M.; Braun, J.M.; Cheema, A.K. Space-type radiation induces multimodal responses in the mouse gut microbiome and metabolome. Microbiome 2017, 5, 105. [Google Scholar] [CrossRef] [PubMed]
- Sinha, R.; Ahn, J.; Sampson, J.N.; Shi, J.; Yu, G.; Xiong, X.; Hayes, R.B.; Goedert, J. Fecal Microbiota, Fecal Metabolome, and Colorectal Cancer Interrelations. PLoS ONE 2016, 11, e0152126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, P.; Rizk, N.M.; Ibrahim, S.; Luo, Y.; Younes, N.; Perry, B.; Dennis, K.; Zirie, M.; Luta, G.; Cheema, A.K. Quantitative metabolomic and lipidomic profiling reveals aberrant amino acid metabolism in type 2 diabetes. Mol. BioSyst. 2013, 9, 307–317. [Google Scholar] [CrossRef]
- Tian, F.; Gu, L.; Si, A.; Yao, Q.; Zhang, X.; Zhao, J.; Hu, D. Metabolomic study on the faecal extracts of atherosclerosis mice and its application in a Traditional Chinese Medicine. J. Chromatogr. B 2015, 1007, 140–148. [Google Scholar] [CrossRef]
- Varghese, R.S.; Cheema, A.K.; Cheema, P.; Bourbeau, M.; Tuli, L.; Zhou, B.; Jung, M.; Dritschilo, A.; Ressom, H.W. Analysis of LC−MS Data for Characterizing the Metabolic Changes in Response to Radiation. J. Proteome Res. 2010, 9, 2786–2793. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adpater sequences from high-throughput sequencing reads. Embnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; A Johnson, A.J.; Holmes, S. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Bokulich, N.A.; Kaehler, B.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.; Caporaso, J.G. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 2018, 6, 90. [Google Scholar] [CrossRef] [PubMed]
- McDonald, D.; Price, M.N.; Goodrich, J.; Nawrocki, E.P.; DeSantis, T.Z.; Probst, A.J.; Andersen, G.L.; Knight, R.; Hugenholtz, P. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 2011, 6, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Bokulich, N.A.; Subramanian, S.; Faith, J.J.; Gevers, D.; Gordon, J.I.; Knight, R.; Mills, D.A.; Caporaso, J.G. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods 2013, 10, 57–59. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Faith, D.P. Conservation evaluation and phylogenetic diversity. Biol. Conserv. 1992, 61, 1–10. [Google Scholar] [CrossRef]
- Lozupone, C.A.; Hamady, M.; Kelley, S.T.; Knight, R. Quantitative and Qualitative beta Diversity Measures Lead to Different Insights into Factors That Structure Microbial Communities. Appl. Environ. Microbiol. 2007, 73, 1576–1585. [Google Scholar] [CrossRef] [Green Version]
- Lozupone, C.; Knight, R. UniFrac: A New Phylogenetic Method for Comparing Microbial Communities. Appl. Environ. Microbiol. 2005, 71, 8228–8235. [Google Scholar] [CrossRef] [Green Version]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahe, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2: An improved and customizable approach for metagenome inference. BioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caspi, R.; Billington, R.; Fulcher, C.A.; Keseler, I.M.; Kothari, A.; Krummenacker, M.; Latendresse, M.; Midford, P.E.; Ong, Q.; Ong, W.K.; et al. The MetaCyc database of metabolic pathways and enzymes. Nucleic Acids Res. 2018, 46, D633–D639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [Green Version]
- Noecker, C.; Eng, A.; Srinivasan, S.; Theriot, C.M.; Young, V.B.; Jansson, J.K.; Fredricks, D.N.; Borenstein, E. Metabolic Model-Based Integration of Microbiome Taxonomic and Metabolomic Profiles Elucidates Mechanistic Links between Ecological and Metabolic Variation. mSystems 2016, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mallick, H.; Franzosa, E.A.; Mclver, L.J.; Banerjee, S.; Sirota-Madi, A.; Kostic, A.D.; Clish, C.B.; Vlamakis, H.; Xavier, R.J.; Huttenhower, C. Predictive metabolomic profiling of microbial communities using amplicon or metagenomic sequences. Nat. Commun. 2019, 10, 3136. [Google Scholar] [CrossRef] [Green Version]
- Larsen, P.E.; Collart, F.R.; Field, D.; Meyer, F.; Keegan, K.P.; Henry, C.S.; McGrath, J.; Quinn, J.; Gilbert, J.A. Predicted Relative Metabolomic Turnover (PRMT): Determining metabolic turnover from a coastal marine metagenomic dataset. Microb. Inf. Exp. 2011, 1, 4. [Google Scholar] [CrossRef]
- Zou, H.; Hastie, T. Regularization and variable selection via the elastic net. J. R. Stat. Soc. B 2005, 67, 301–320. [Google Scholar] [CrossRef] [Green Version]
- Fox, J.G. The Mouse in Biomedical Researchs, 2nd ed.; Elsevier, AP: Amsterdam, The Netherlands; Boston, MA, USA, 2006; Volume 3, pp. 1–816. [Google Scholar]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef]
- E Ley, R. Prevotella in the gut: Choose carefully. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 69–70. [Google Scholar] [CrossRef]
- Kovatcheva-Datchary, P.; Nilsson, A.; Akrami, R.; Lee, Y.S.; De Vadder, F.; Arora, T.; Hallen, A.; Martens, E.; Björck, I.; Bäckhed, F. Dietary Fiber-Induced Improvement in Glucose Metabolism Is Associated with Increased Abundance of Prevotella. Cell Metab. 2015, 22, 971–982. [Google Scholar] [CrossRef] [Green Version]
- Dillon, S.M.; Lee, E.J.; Kotter, C.V.; Austin, G.L.; Gianella, S.; Siewe, B.; Smith, D.; Landay, A.L.; McManus, M.C.; E Robertson, C.; et al. Gut dendritic cell activation links an altered colonic microbiome to mucosal and systemic T-cell activation in untreated HIV-1 infection. Mucosal Immunol. 2015, 9, 24–37. [Google Scholar] [CrossRef] [PubMed]
- Hjorth, M.F.; Blaedel, T.; Bendtsen, L.Q.; Lorenzen, J.K.; Holm, J.B.; Kiilerich, P.; Roager, H.M.; Kristiansen, K.; Larsen, L.H.; Astrup, A. Prevotella-to-Bacteroides ratio predicts body weight and fat loss success on 24-week diets varying in macronutrient composition and dietary fiber: Results from a post-hoc analysis. Int. J. Obes. 2019, 43, 149–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, F.; Li, Y.; Zhao, J. The gut microbiome of healthy long-living people. Aging 2019, 11, 289–290. [Google Scholar] [CrossRef] [PubMed]
- Zapata, H.J.; Quagliarello, V.J. The microbiota and microbiome in aging: Potential implications in health and age-related diseases. J. Am. Geriatr. Soc. 2015, 63, 776–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez, G.P.; Bauerl, C.; Collado, M.C. Understanding gut microbiota in elderly’s health will enable intervention through probiotics. Benef. Microbes 2014, 5, 235–246. [Google Scholar] [CrossRef]
- Rondanelli, M.; Giacosa, A.; Faliva, M.A.; Perna, S.; Allieri, F.; Castellazzi, A.M. Review on microbiota and effectiveness of probiotics use in older. World J. Clin. Cases 2015, 3, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Li, S.; Yang, L.; Huang, P.; Li, W.; Wang, S.; Zhao, G.; Zhang, M.; Pang, X.; Yan, Z.; et al. Structural modulation of gut microbiota in life-long calorie-restricted mice. Nat. Commun. 2013, 4, 2163. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Zhang, M.; Wang, S.; Han, R.; Cao, Y.; Hua, W.; Mao, Y.; Zhang, X.; Pang, X.; Wei, C.; et al. Interactions between gut microbiota, host genetics and diet relevant to development of metabolic syndromes in mice. ISME J. 2010, 4, 232–241. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Zhao, Y.; Zhang, M.; Pang, X.; Xu, J.; Kang, C.; Li, M.; Zhang, C.; Zhang, Z.; Zhang, Y.; et al. Structural Changes of Gut Microbiota during Berberine-Mediated Prevention of Obesity and Insulin Resistance in High-Fat Diet-Fed Rats. PLoS ONE 2012, 7, e42529. [Google Scholar] [CrossRef] [Green Version]
- Ravussin, Y.; Koren, O.; Spor, A.; LeDuc, C.; Gutman, R.; Stombaugh, J.; Knight, R.; Ley, R.E.; Leibel, R.L. Responses of Gut Microbiota to Diet Composition and Weight Loss in Lean and Obese Mice. Obesity 2012, 20, 738–747. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; De Vos, W.M. Next-Generation Beneficial Microbes: The Case of Akkermansia muciniphila. Front. Microbiol. 2017, 8, 1765. [Google Scholar] [CrossRef] [PubMed]
- Vital, M.; Howe, A.; Tiedje, J.M. Revealing the Bacterial Butyrate Synthesis Pathways by Analyzing (Meta)genomic Data. mBio 2014, 5, e00889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, X.; Gao, X.; Peng, Y.; Wu, Q.; Zhu, J.; Tan, C.; Xia, G.; You, C.; Xu, R.; Pan, S.; et al. Higher Risk of Stroke Is Correlated With Increased Opportunistic Pathogen Load and Reduced Levels of Butyrate-Producing Bacteria in the Gut. Front. Cell. Infect. Microbiol. 2019, 9, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zoetendal, E.G.; Plugge, C.M.; Akkermans, A.D.L.; De Vos, W.M. Victivallis vadensis gen. nov., sp. nov., a sugar-fermenting anaerobe from human faeces. Int. J. Syst. Evol. Microbiol. 2003, 53, 211–215. [Google Scholar] [CrossRef] [Green Version]
- Biragyn, A.; Ferrucci, L. Gut dysbiosis: A potential link between increased cancer risk in ageing and inflammaging. Lancet Oncol. 2018, 19, e295–e304. [Google Scholar] [CrossRef]
- Derrien, M.; Vaughan, E.E.; Plugge, C.M.; De Vos, W.M. Akkermansia muciniphila gen. nov., sp. nov., a human intestinal mucin-degrading bacterium. Int. J. Syst. Evol. Microbiol. 2004, 54, 1469–1476. [Google Scholar] [CrossRef] [Green Version]
- Louis, P.; Flint, H.J. Formation of propionate and butyrate by the human colonic microbiota. Environ. Microbiol. 2017, 19, 29–41. [Google Scholar] [CrossRef] [Green Version]
- Wong, A.C.; Levy, M. New Approaches to Microbiome-Based Therapies. mSystems 2019, 4. [Google Scholar] [CrossRef] [Green Version]
- Mariat, D.; Firmesse, O.; Levenez, F.; Guimarães, V.; Sokol, H.; Dore, J.; Corthier, G.; Furet, J.-P. The Firmicutes/Bacteroidetes ratio of the human microbiota changes with age. BMC Microbiol. 2009, 9, 123. [Google Scholar] [CrossRef]
- E Jakobsson, H.; Rodríguez-Piñeiro, A.M.; Schutte, A.; Ermund, A.; Boysen, P.; Bemark, M.; Sommer, F.; Backhed, F.; Hansson, G.C.; Johansson, M.E. The composition of the gut microbiota shapes the colon mucus barrier. EMBO Rep. 2014, 16, 164–177. [Google Scholar] [CrossRef]
- Bai, J.; Hu, Y.; Bruner, D.W. Composition of gut microbiota and its association with body mass index and lifestyle factors in a cohort of 7–18 years old children from the American Gut Project. Pediatr. Obes. 2019, 14, e12480. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, E.; Young, W.; Rosendale, D.; Reichert-Grimm, V.; Riedel, C.U.; Conrad, R.; Egert, M. RNA-Based Stable Isotope Probing Suggests Allobaculum spp. as Particularly Active Glucose Assimilators in a Complex Murine Microbiota Cultured In Vitro. BioMed Res. Int. 2017, 2017, 1829685. [Google Scholar] [CrossRef] [Green Version]
- Everard, A.; Lazarevic, V.; Gaia, N.; Johansson, M.; Stahlman, M.; Backhed, F.; Delzenne, N.M.; Schrenzel, J.; Francois, P.; Cani, P.D. Microbiome of prebiotic-treated mice reveals novel targets involved in host response during obesity. ISME J. 2014, 8, 2116–2130. [Google Scholar] [CrossRef]
- Houghton, D.; Stewart, C.J.; Stamp, C.; Nelson, A.; Ami, N.J.A.; Petrosino, J.F.; Wipat, A.; Trenell, M.I.; Turnbull, D.; Greaves, L.C. Impact of Age-Related Mitochondrial Dysfunction and Exercise on Intestinal Microbiota Composition. J. Gerontol. Ser. A Boil. Sci. Med. Sci. 2017, 73, 571–578. [Google Scholar] [CrossRef] [Green Version]
- Man, A.; Gicheva, N.; Nicoletti, C. The impact of ageing on the intestinal epithelial barrier and immune system. Cell. Immunol. 2014, 289, 112–118. [Google Scholar] [CrossRef]
- Gophna, U.; Konikoff, T.; Nielsen, H.B. Oscillospiraand related bacteria—From metagenomic species to metabolic features. Environ. Microbiol. 2017, 19, 835–841. [Google Scholar] [CrossRef] [Green Version]
- Escobar, J.S.; Klotz, B.; E Valdes, B.; Agudelo, G.M. The gut microbiota of Colombians differs from that of Americans, Europeans and Asians. BMC Microbiol. 2014, 14, 311. [Google Scholar] [CrossRef] [Green Version]
- Goodrich, J.K.; Waters, J.L.; Poole, A.C.; Sutter, J.L.; Koren, O.; Blekhman, R.; Beaumont, M.; Van Treuren, W.; Knight, R.; Bell, J.T.; et al. Human Genetics Shape the Gut Microbiome. Cell 2014, 159, 789–799. [Google Scholar] [CrossRef] [Green Version]
- Tims, S.; Derom, C.; Jonkers, D.M.; Vlietinck, R.; Saris, W.H.; Kleerebezem, M.; De Vos, W.M.; Zoetendal, E.G. Microbiota conservation and BMI signatures in adult monozygotic twins. ISME J. 2013, 7, 707–717. [Google Scholar] [CrossRef] [Green Version]
- Verdam, F.J.; Fuentes, S.; De Jonge, C.; Zoetendal, E.G.; Erbil, R.; Greve, J.W.; Buurman, W.A.; De Vos, W.M.; Rensen, S.S. Human intestinal microbiota composition is associated with local and systemic inflammation in obesity. Obesity 2013, 21, E607–E615. [Google Scholar] [CrossRef]
- Martínez-Reyes, I.; Diebold, L.P.; Kong, H.; Schieber, M.; Huang, H.; Hensley, C.T.; Mehta, M.M.; Wang, T.; Santos, J.H.; Woychik, R.; et al. TCA Cycle and Mitochondrial Membrane Potential Are Necessary for Diverse Biological Functions. Mol. Cell 2016, 61, 199–209. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Zeng, T.; Lu, W.; Rubino, S.; Kelvin, D.J.; Carru, C. A Cross-Sectional Study of Compositional and Functional Profiles of Gut Microbiota in Sardinian Centenarians. mSystems 2019, 4. [Google Scholar] [CrossRef] [Green Version]
- Jiao, N.; Baker, S.S.; Nugent, C.A.; Tsompana, M.; Cai, L.; Wang, Y.; Buck, M.J.; Genco, R.J.; Baker, R.D.; Zhu, R.; et al. Gut microbiome may contribute to insulin resistance and systemic inflammation in obese rodents: A meta-analysis. Physiol. Genom. 2018, 50, 244–254. [Google Scholar] [CrossRef] [Green Version]
- Ma, S.; Upneja, A.; Galecki, A.; Tsai, Y.-M.; Burant, C.F.; Raskind, S.; Zhang, Q.; Zhang, Z.D.; Seluanov, A.; Gorbunova, V.; et al. Cell culture-based profiling across mammals reveals DNA repair and metabolism as determinants of species longevity. eLife 2016, 5. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.-H.; Kim, B.-K.; Park, S.; Park, S.-K. Phosphatidylcholine Extends Lifespan via DAF-16 and Reduces Amyloid-Beta-Induced Toxicity in Caenorhabditis elegans. Oxidative Med. Cell. Longev. 2019, 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taofiq, O.; Gonzalez-Paramas, A.M.; Barreiro, M.F.; Ferreira, I.C. Hydroxycinnamic Acids and Their Derivatives: Cosmeceutical Significance, Challenges and Future Perspectives, a Review. Molecules 2017, 22, 281. [Google Scholar] [CrossRef]
- Caperelli, C.A.; Giroux, E.L. The Human Glycinamide Ribonucleotide Transformylase Domain: Purification, Characterization, and Kinetic Mechanism. Arch. Biochem. Biophys. 1997, 341, 98–103. [Google Scholar] [CrossRef]
- Zarse, K.; Bossecker, A.; Muller-Kuhrt, L.; Siems, K.; Hernandez, M.A.; Berendsohn, W.G.; Birringer, M.; Ristow, M. The Phytochemical Glaucarubinone Promotes Mitochondrial Metabolism, Reduces Body Fat, and Extends Lifespan of Caenorhabditis elegans. Horm. Metab. Res. 2011, 43, 241–243. [Google Scholar] [CrossRef]
- Sun, Y.; Yang, T.; Mao, L.; Zhang, F. Sulforaphane Protects against Brain Diseases: Roles of Cytoprotective Enzymes. Austin J. Cereb. Dis. Stroke 2017, 4. [Google Scholar] [CrossRef] [Green Version]
- Hida, K.; Suzuki, N.; Kwee, I.L.; Nakada, T. pH-lactate dissociation in neonatal anoxia: Proton and31P NMR spectroscopic studies in rat pups. Magn. Reson. Med. 1991, 22, 128–132. [Google Scholar] [CrossRef]
- Sergeeva, O.A.; Chepkova, A.N.; Haas, H.L. Guanidinoethyl sulphonate is a glycine receptor antagonist in striatum. Br. J. Pharmacol. 2002, 137, 855–860. [Google Scholar] [CrossRef] [PubMed]
- Suez, J.; Zmora, N.; Zilberman-Schapira, G.; Mor, U.; Dori-Bachash, M.; Bashiardes, S.; Zur, M.; Regev-Lehavi, D.; Brik, R.B.-Z.; Federici, S.; et al. Post-Antibiotic Gut Mucosal Microbiome Reconstitution Is Impaired by Probiotics and Improved by Autologous FMT. Cell 2018, 174, 1406–1423.e16. [Google Scholar] [CrossRef] [Green Version]
- Zmora, N.; Zilberman-Schapira, G.; Suez, J.; Mor, U.; Dori-Bachash, M.; Bashiardes, S.; Kotler, E.; Zur, M.; Regev-Lehavi, D.; Brik, R.B.-Z.; et al. Personalized Gut Mucosal Colonization Resistance to Empiric Probiotics Is Associated with Unique Host and Microbiome Features. Cell 2018, 174, 1388–1405.e21. [Google Scholar] [CrossRef] [Green Version]
- Suez, J.; Zmora, N.; Segal, E.; Elinav, E. The pros, cons, and many unknowns of probiotics. Nat. Med. 2019, 25, 716–729. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Age | Diet | Time (Month) | Number of Subjects |
|---|---|---|---|---|
| YC0 | Young | Control | 0 | 5 |
| YC2 | Young | Control | 2 | 5 |
| YS0 | Young | SFN | 0 | 5 |
| YS2 | Young | SFN | 2 | 5 |
| OC0 | Old | Control | 0 | 5 |
| OC2 | Old | Control | 2 | 5 |
| OS0 | Old | SFN | 0 | 5 |
| OS2 | Old | SFN | 2 | 5 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jun, S.-R.; Cheema, A.; Bose, C.; Boerma, M.; Palade, P.T.; Carvalho, E.; Awasthi, S.; Singh, S.P. Multi-Omic Analysis Reveals Different Effects of Sulforaphane on the Microbiome and Metabolome in Old Compared to Young Mice. Microorganisms 2020, 8, 1500. https://doi.org/10.3390/microorganisms8101500
Jun S-R, Cheema A, Bose C, Boerma M, Palade PT, Carvalho E, Awasthi S, Singh SP. Multi-Omic Analysis Reveals Different Effects of Sulforaphane on the Microbiome and Metabolome in Old Compared to Young Mice. Microorganisms. 2020; 8(10):1500. https://doi.org/10.3390/microorganisms8101500
Chicago/Turabian StyleJun, Se-Ran, Amrita Cheema, Chhanda Bose, Marjan Boerma, Philip T. Palade, Eugenia Carvalho, Sanjay Awasthi, and Sharda P. Singh. 2020. "Multi-Omic Analysis Reveals Different Effects of Sulforaphane on the Microbiome and Metabolome in Old Compared to Young Mice" Microorganisms 8, no. 10: 1500. https://doi.org/10.3390/microorganisms8101500