Genomic Characterization and Environmental Distribution of a Thermophilic Anaerobe Dissulfurirhabdus thermomarina SH388T Involved in Disproportionation of Sulfur Compounds in Shallow Sea Hydrothermal Vents

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Genome Sequencing and Assembly
2.2. Genome Annotation
2.3. Clustered Regularly Interspaced Short Palindromic Repeats (CRISPRs) and Genomic Islands
2.4. Geographical Distribution
2.5. Taxonomical Analyses and Comparative Genomics
3. Results and Discussions
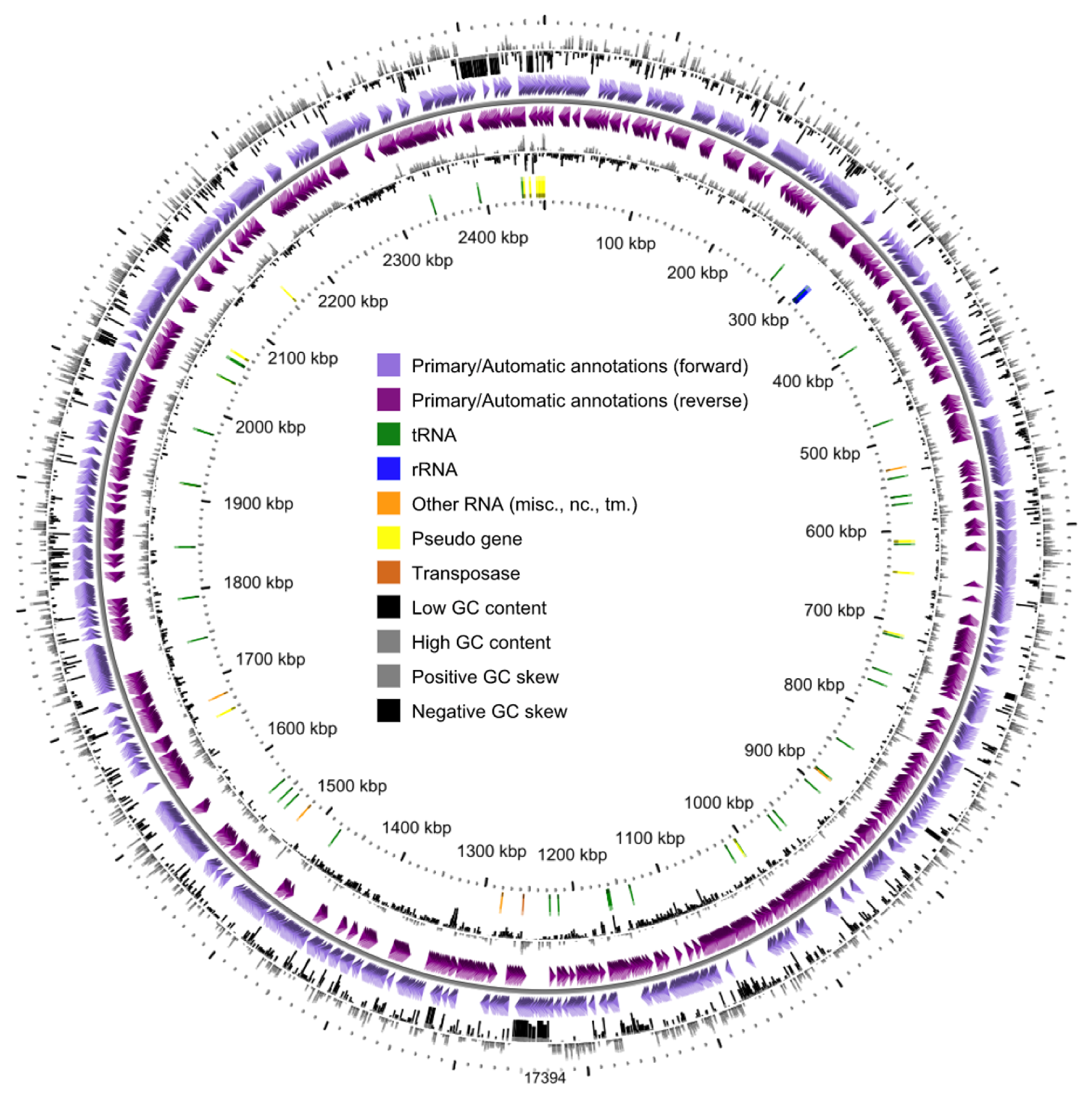
3.1. General Genome Properties and Genomic Islands
3.2. Central Carbon Metabolism
3.3. Hydrogen Metabolism
3.4. Nitrogen Metabolism
3.5. Sulfur Metabolism
3.6. Comparative Genomics
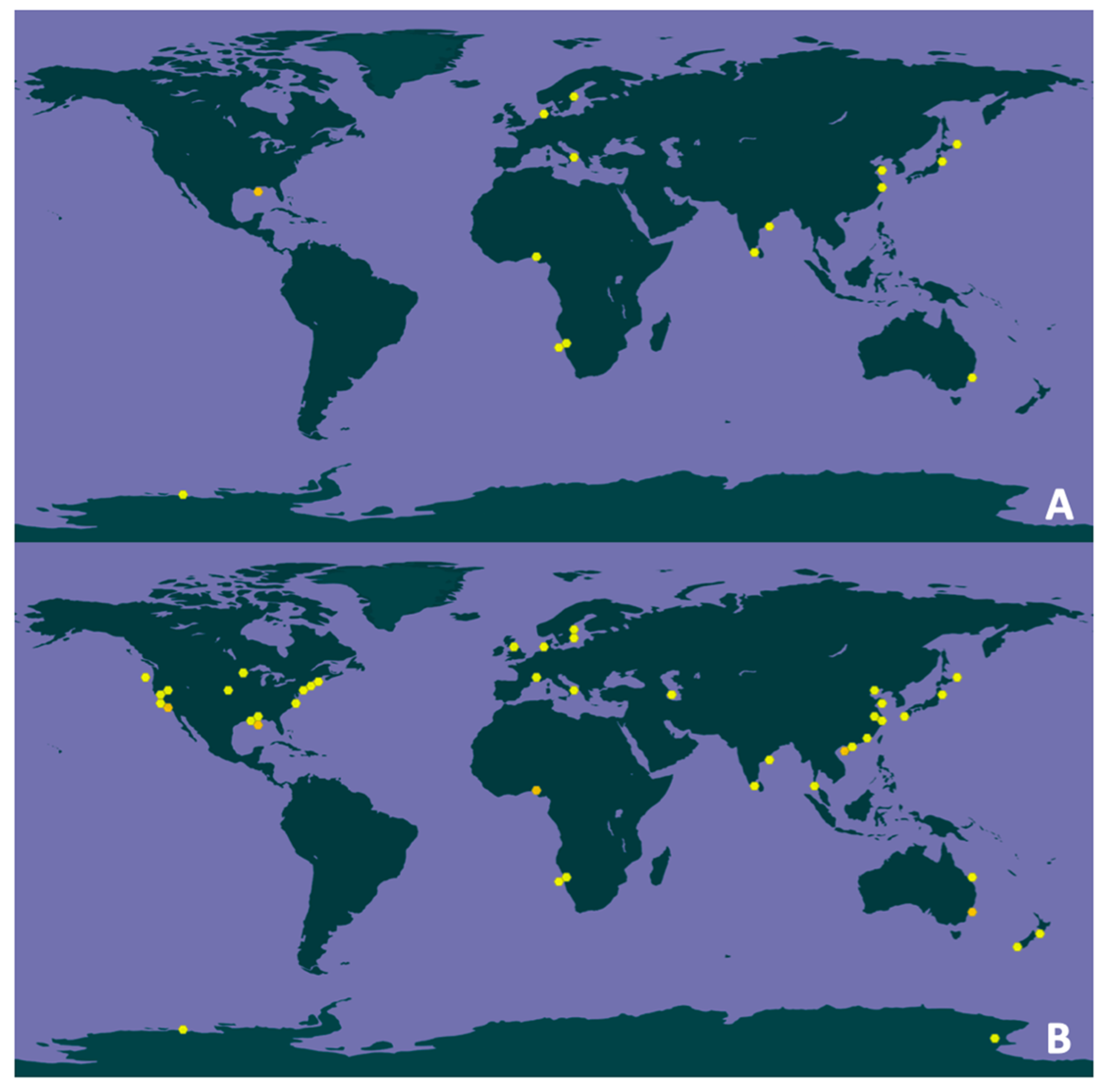
3.7. Geographical and Environmental Distribution
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ANI | Average nucleotide identity |
| CDS | Coding DNA sequences |
| COGs | Clusters of orthologous groups |
| CRISPR | Clustered regularly interspaced short palindromic repeats |
| DNRA | Dissimilatory nitrate reduction to ammonium |
| ENA | European Nucleotide Archive |
| G6P | Glucose-6-phosphate |
| INSDC | International Nucleotide Sequence Database Collaboration |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| MIGS | Minimum information about a genome sequence |
| NCBI | National Center for Biotechnology Information |
| PAHs | Polycyclic aromatic hydrocarbons |
| PGAP | Prokaryotic genome annotation pipeline |
| bp | Base pair |
| SOR | Sulfur oxygenase reductase |
| TCA | Tricarboxylic acid cycle |
References
- Tarasov, V.G.; Gebruk, A.V.; Mironov, A.N.; Moskalev, L.I. Deep-sea and shallow-water hydrothermal vent communities: Two different phenomena? Chem. Geol. 2005, 224, 5–39. [Google Scholar] [CrossRef]
- Dick, G.J. The microbiomes of deep-sea hydrothermal vents: Distributed globally, shaped locally. Nat. Rev. Microbiol. 2019, 17, 271–283. [Google Scholar] [CrossRef] [PubMed]
- Godfroy, A.; François, D.; Hartunians, J.; Moalic, Y.; Alain, K. Physiology, metabolism and ecology of thermophiles from deep-sea vents. In The Microbiology of Deep-Sea; Vetriani, C., Giovannelli, D., Eds.; Springer International: New York, NY, USA, in press.
- Slobodkin, A.I.; Reysenbach, A.L.; Slobodkina, G.B.; Baslerov, R.V.; Kostrikina, N.A.; Wagner, I.D.; Bonch-Osmolovskaya, E.A. Thermosulfurimonas dismutans gen. nov.; sp. nov.; an extremely thermophilic sulfur-disproportionating bacterium from a deep-sea hydrothermal vent. Int. J. Syst. Evol. Microbiol. 2012, 62, 2565–2571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slobodkin, A.I.; Reysenbach, A.L.; Slobodkina, G.B.; Kolganova, T.V.; Kostrikina, N.A.; Bonch-Osmolovskaya, E.A. Dissulfuribacter thermophilus gen. nov., sp. nov.; a thermophilic, autotrophic, sulfur disproportionating, deeply branching deltaproteobacterium from a deep-sea hydrothermal vent. Int. J. Syst. Evol. Microbiol. 2013, 63, 1967–1971. [Google Scholar] [CrossRef]
- Slobodkina, G.B.; Reysenbach, A.L.; Kolganova, T.V.; Novikov, A.A.; Bonch-Osmolovskaya, E.A.; Slobodkin, A.I. Thermosulfuriphilus ammonigenes gen. nov.; sp. nov.; a thermophilic, chemolithoautotrophic bacterium capable of respiratory ammonification of nitrate with elemental sulfur. Int. J. Syst. Evol. Microbiol. 2017, 67, 3474–3479. [Google Scholar] [CrossRef]
- Slobodkina, G.B.; Kolganova, T.V.; Kopitsyn, D.S.; Viryasov, M.B.; Bonch-Osmolovskaya, E.A.; Slobodkin, A.I. Dissulfurirhabdus thermomarina gen. nov.; sp. nov.; a thermophilic, autotrophic, sulfite-reducing and disproportionating deltaproteobacterium isolated from a shallow-sea hydrothermal vent. Int. J. Syst. Evol. Microbiol. 2016, 66, 2515–2519. [Google Scholar] [CrossRef]
- Frolova, A.A.; Slobodkina, G.B.; Baslerov, R.V.; Novikov, A.A.; Slobodkin, A.I. Thermosulfurimonas marina sp. nov., an Autotrophic Sulfur- Disproportionating and Nitrate-Reducing Bacterium Isolated from a Shallow-Sea Hydrothermal Vent. Microbiology 2018, 87, 502–507. [Google Scholar] [CrossRef]
- Bak, F.; Pfennig, N. Chemolithotrophic growth of Desulfovibrio sulfodismutans sp. nov. by disproportionation of inorganic sulfur compounds. Arch. Microbiol. 1987, 147, 184–189. [Google Scholar] [CrossRef]
- Bak, F.; Cypionka, H. A novel type of energy metabolism involving fermentation of inorganic sulphur compounds. Nature 1987, 326, 891–892. [Google Scholar] [CrossRef]
- Finster, K. Microbiological disproportionation of inorganic sulfur compounds. J. Sulfur Chem. 2008, 29, 281–292. [Google Scholar] [CrossRef]
- Slobodkin, A.I.; Slobodkina, G.B. Diversity of Sulfur-Disproportionating Microorganisms. Microbiology 2019, 88, 509–522. [Google Scholar] [CrossRef]
- Philippot, P.; Van Zuilen, M.; Lepot, K.; Thomazo, C.; Farquhar, J.; Van Kranendonk, M.J. Early Archaean microorganisms preferred elemental sulfur, not sulfate. Science 2007, 317, 1534–1537. [Google Scholar] [CrossRef] [PubMed]
- Wacey, D.; Kilburn, M.R.; Saunders, M.; Cliff, J.; Brasier, M.D. Microfossils of sulphur-metabolizing cells in 3.4-billion-year-old rocks of Western Australia. Nat. Geosci. 2011, 4, 698–702. [Google Scholar] [CrossRef]
- Ollivier, B.; Zeyen, N.; Gales, G.; Hickman-Lewis, K.; Gaboyer, F.; Benzerara, K.; Westall, F. Importance of Prokaryotes in the Functioning and Evolution of the Present and Past Geosphere and Biosphere. In Prokaryotes and Evolution; Bertrand, J.C., Normand, P., Ollivier, B., Sime-Ngando, T., Eds.; Springer: Cham, Switzerland, 2018. [Google Scholar] [CrossRef]
- Wasmund, K.; Mußmann, M.; Loy, A. The life sulfuric: Microbial ecology of sulfur cycling in marine sediments. Environ. Microbiol. Rep. 2017, 9, 323–344. [Google Scholar] [CrossRef]
- Jørgensen, B.B.; Findlay, A.J.; Pellerin, A. The Biogeochemical Sulfur Cycle of Marine Sediments. Front. Microbiol. 2019, 10, 849. [Google Scholar] [CrossRef]
- Mardanov, A.V.; Beletsky, A.V.; Kadnikov, V.V.; Slobodkin, A.I.; Ravin, N.V. Genome analysis of Thermosulfurimonas dismutans, the first thermophilic sulfur-disproportionating bacterium of the phylum Thermodesulfobacteria. Front. Microbiol. 2016, 7, 950. [Google Scholar] [CrossRef]
- Thorup, C.; Schramm, A.; Findlay, A.J.; Finster, K.W.; Schreiber, L. Disguised as a sulfate reducer: Growth of the deltaproteobacterium Desulfurivibrio alkaliphilus by sulfide oxidation with nitrate. mBio 2017, 8, e00671-17. [Google Scholar] [CrossRef] [Green Version]
- Florentino, A.P.; Pereira, I.A.C.; Boeren, S.; van den Born, M.; Stams, A.J.M.; Sánchez-Andrea, I. Insight into the sulfur metabolism of Desulfurella amilsii by differential proteomics. Environ. Microbiol. 2019, 21, 209–225. [Google Scholar] [CrossRef] [Green Version]
- Ward, L.M.; Bertran, E.; Johnston, D.T. Genomic sequence analysis of Dissulfurirhabdus thermomarina SH388 and proposed reassignment to Dissulfurirhabdaceae fam. nov. Microb. Genom. 2020. [Google Scholar] [CrossRef]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef] [Green Version]
- Wick, R.R.; Schultz, M.B.; Zobel, J.; Holt, K.E. Bandage: Interactive visualization of de novo genome assemblies. Bioinformatics 2015, 31, 3350–3352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antipov, D.; Raiko, M.; Lapidus, A.; Pevzner, P.A. Plasmid detection and assembly in genomic and metagenomic data sets. Genome Res. 2019, 29, 961–968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Brettin, T.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Olsen, G.J.; Olson, R.; Overbeek, R.; Parrello, B.; Pusch, G.D.; et al. RASTtk: A modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci. Rep. 2015, 5, 8365. [Google Scholar] [CrossRef] [Green Version]
- Tatusova, T.; DiCuccio, M.; Badretdin, A.; Chetvernin, V.; Nawrocki, E.P.; Zaslavsky, L.; Lomsadze, A.; Pruitt, K.D.; Borodovsky, M.; Ostell, J. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016, 44, 6614–6624. [Google Scholar] [CrossRef]
- Vallenet, D.; Calteau, A.; Cruveiller, S.; Gachet, M.; Lajus, A.; Josso, A.; Médigue, C. MicroScope in 2017: An expanding and evolving integrated resource for community expertise of microbial genomes. Nucleic Acids Res. 2017, 45, D517–D528. [Google Scholar] [CrossRef]
- Tanizawa, Y.; Fujisawa, T.; Nakamura, Y. DFAST: A flexible prokaryotic genome annotation pipeline for faster genome publication. Bioinformatics 2018, 34, 1037–1039. [Google Scholar] [CrossRef] [Green Version]
- Søndergaard, D.; Pedersen, C.N.S.; Greening, C. HydDB: A web tool for hydrogenase classification and analysis. Sci. Rep. 2016, 6, 1–8. [Google Scholar] [CrossRef]
- Couvin, D.; Bernheim, A.; Toffano-Nioche, C.; Touchon, M.; Michalik, J.; Néron, B.; Rocha, E.; Vergnaud, G.; Gautheret, D.; Pourcel, C. CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Res. 2018, 46, W246–W251. [Google Scholar] [CrossRef] [Green Version]
- Bertelli, C.; Laird, M.R.; Williams, K.P.; Simon Fraser University Research Computing Group; Lau, B.Y.; Hoad, G.; Winsor, G.L.; Brinkman, F. IslandViewer 4: Expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 2017, 45, W30–W35. [Google Scholar] [CrossRef]
- Moussard, H.; L’Haridon, S.; Tindall, B.J.; Banta, A.; Schumann, P.; Stackebrandt, E.; Reysenbach, A.L.; Jeanthon, C. Thermodesulfatator indicus gen. nov.; sp. nov.; a novel thermophilic chemolithoautotrophic sulfate-reducing bacterium isolated from the Central Indian Ridge. Int. J. Syst. Evol. Microbiol. 2004, 54, 227–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alain, K.; Postec, A.; Grinsard, E.; Lesongeur, F.; Prieur, D.; Godfroy, A. Thermodesulfatator atlanticus sp. nov., a thermophilic, chemolithoautotrophic, sulfate-reducing bacterium isolated from a Mid-Atlantic Ridge hydrothermal vent. Int. J. Syst. Evol. Microbiol. 2010, 60, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Lai, Q.; Cao, J.; Dupont, S.; Shao, Z.; Jebbar, M.; Alain, K. Thermodesulfatator autotrophicus sp. nov.; a thermophilic sulfate-reducing bacterium from the Indian Ocean. Int. J. Syst. Evol. Microbiol. 2016, 66, 3978–3982. [Google Scholar] [CrossRef]
- Slobodkina, G.B.; Mardanov, A.V.; Ravin, N.V.; Frolova, A.A.; Chernyh, N.A.; Bonch-Osmolovskaya, E.A.; Slobodkin, A.I. Respiratory Ammonification of Nitrate Coupled to Anaerobic Oxidation of Elemental Sulfur in Deep-Sea Autotrophic Thermophilic Bacteria. Front. Microbiol. 2017, 8, 87. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, N.; Leroy, G.; Guiral, M.; Giudici-Orticoni, M.T.; Aubert, C. First characterization of the active oligomer form of sulfur oxygenase reductase from the bacterium Aquifex aeolicus. Extremophiles 2008, 12, 205–215. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Item | Description |
|---|---|
| Investigation | |
| Strain | Dissulfurirhabdus thermomarina strain SH388T |
| Submitted to INSDC | GenBank |
| Investigation type | Bacteria |
| Project name | JAATWC000000000 |
| Geographic location (latitude and longitude) | 44°29.469′ N, 146°06.247′ E |
| Geographic location (country and/or sea, region) | Sea of Okhotsk, 250 m from the Kunashir Island shore (Sakhalin oblast, Russia) |
| Collection date | June 2013 |
| Environment (biome) | marine hydrothermal vent biome ENVO:01000030 |
| Environment (feature) | marine hydrothermal vent ENVO:01000122 |
| Environment (material) | marine hydrothermal vent chimney ENVO:01000129 |
| Depth | −12 m |
| General features | |
| Classification | Domain Bacteria |
| Phylum Proteobacteria | |
| Class Deltaproteobacteria | |
| Not assigned to an Order | |
| Not assigned to a Family | |
| Genus Dissulfurirhabdus | |
| Species Dissulfurirhabdus thermomarina | |
| Gram stain | Negative |
| Cell shape | short rods |
| Motility | Motile |
| Growth temperature | Thermophilic, optimum at 50 °C |
| Relationship to oxygen | Anaerobic |
| Trophic level | Chemolithoautotrophic |
| Biotic relationship | free-living |
| Isolation and growth conditions | DOI 10.1099/ijsem.0.001083 |
| Sequencing | |
| Sequencing technology | Illumina MiSeq 2 × 150 bp |
| Sequencing platform | Molecular Research, MrDNA (Shallowater, TX, USA) |
| Assembler | Unicycler (version: 0.4.8-beta) |
| Contig number | 36 |
| N50 | 240,491 |
| Genome coverage | 1384.362× |
| Genome assembly NCBI | ASM1297923v1 |
| Assembly level | Contigs |
| Genomic features: | |
| Genome size (bp) | 2,461,642 |
| GC content (mol%) | 71.1 |
| Protein coding genes | 2267 |
| Number of RNAs | 50 |
| tRNAs | 47 |
| 16S-23S-5S rRNAs | 1-1-1 |
| Genus | Class | Occurrence | Georeferenced Records | ||
|---|---|---|---|---|---|
| Genus | Species | Genus | Species | ||
| Dissulfurirhabdus | Deltaproteobacteria | 200 | 35 a | 114 | 25 a |
| Dissulfuribacter | Deltaproteobacteria | 230 | 27 b | 130 | 14 b |
| Caldimicrobium | Thermodesulfobacteria | 85 | 2 c | 39 | 1 c |
| Thermosulfurimonas | Thermodesulfobacteria | 27 | 17 d | 5 | 3 d |
| Thermosulfuriphilus | Thermodesulfobacteria | 0 | NA | 0 | NA |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Allioux, M.; Yvenou, S.; Slobodkina, G.; Slobodkin, A.; Shao, Z.; Jebbar, M.; Alain, K. Genomic Characterization and Environmental Distribution of a Thermophilic Anaerobe Dissulfurirhabdus thermomarina SH388T Involved in Disproportionation of Sulfur Compounds in Shallow Sea Hydrothermal Vents. Microorganisms 2020, 8, 1132. https://doi.org/10.3390/microorganisms8081132
Allioux M, Yvenou S, Slobodkina G, Slobodkin A, Shao Z, Jebbar M, Alain K. Genomic Characterization and Environmental Distribution of a Thermophilic Anaerobe Dissulfurirhabdus thermomarina SH388T Involved in Disproportionation of Sulfur Compounds in Shallow Sea Hydrothermal Vents. Microorganisms. 2020; 8(8):1132. https://doi.org/10.3390/microorganisms8081132
Chicago/Turabian StyleAllioux, Maxime, Stéven Yvenou, Galina Slobodkina, Alexander Slobodkin, Zongze Shao, Mohamed Jebbar, and Karine Alain. 2020. "Genomic Characterization and Environmental Distribution of a Thermophilic Anaerobe Dissulfurirhabdus thermomarina SH388T Involved in Disproportionation of Sulfur Compounds in Shallow Sea Hydrothermal Vents" Microorganisms 8, no. 8: 1132. https://doi.org/10.3390/microorganisms8081132