Uncovering Bifidobacteria via Targeted Sequencing of the Mammalian Gut Microbiota


Abstract
:1. Introduction
2. Materials and Methods
2.1. DNA Extraction
2.2. Identification of Bifidobacteria by 16S rRNA Gene Sequencing
2.3. Bifidobacterial ITS Sequencing
2.4. Experimental Design of myBaits for The Targeted WMS
2.5. Bifidobacterial DNA Targeted Enrichment
2.6. Metagenomic Analyses
2.7. Comparative Genomics
2.8. Data Availability
3. Results and Discussion
3.1. Identification of Bifidobacteria in the Gut of Mammals
3.2. Targeted Sequencing of the Bifidobacterium genus
3.3. Bifidobacterial Genome Reconstruction
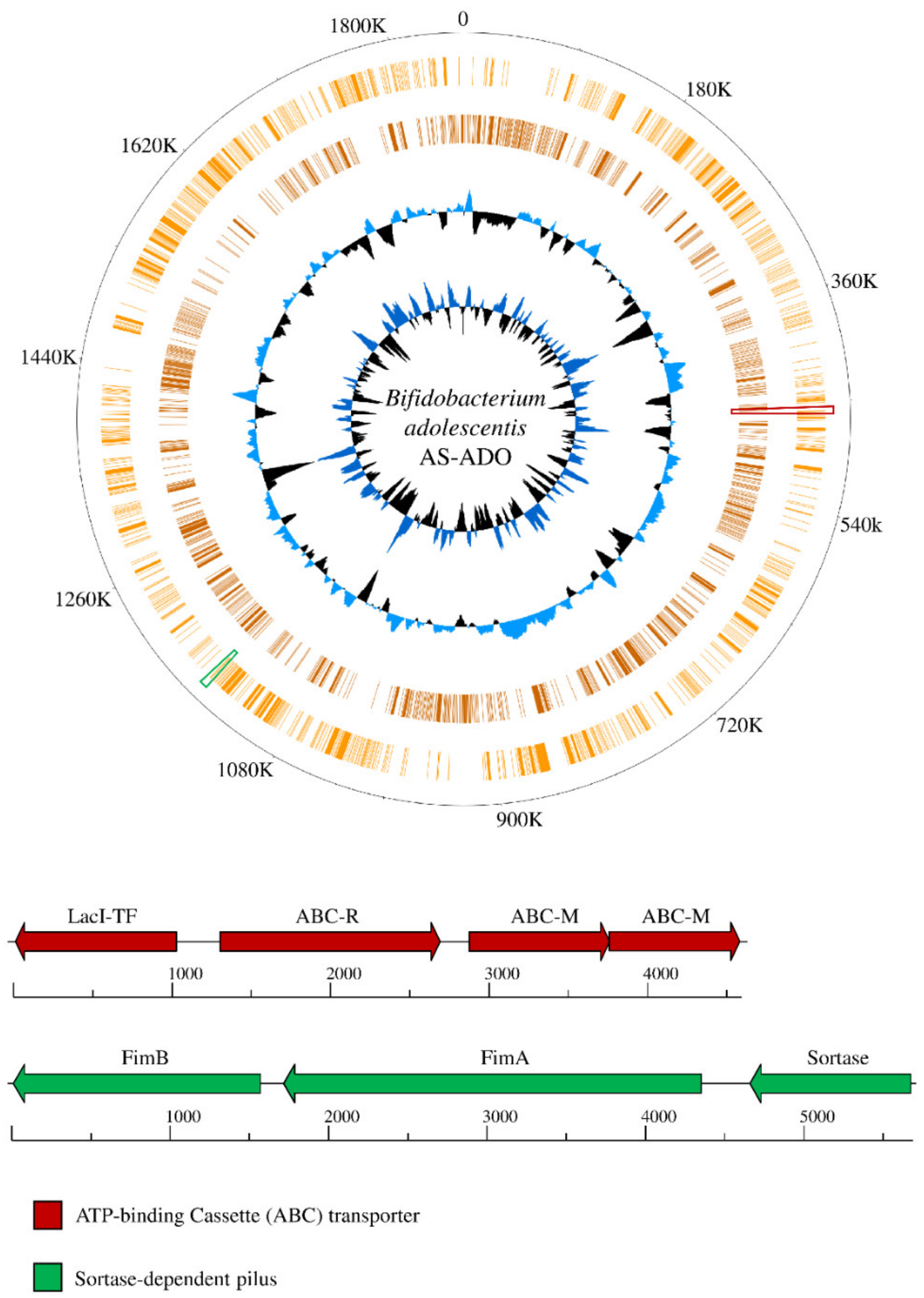
3.4. Insights into The Genetics of The Reconstructed Bifidobacterial Strains
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fischbach, M.A.; Segre, J.A. Signaling in host-associated microbial communities. Cell 2016, 164, 1288–1300. [Google Scholar] [CrossRef]
- Yatsunenko, T.; Rey, F.E.; Manary, M.J.; Trehan, I.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Baldassano, R.N.; Anokhin, A.P.; et al. Human gut microbiome viewed across age and geography. Nature 2012, 486, 222–227. [Google Scholar] [CrossRef]
- Lozupone, C.A.; Stombaugh, J.I.; Gordon, J.I.; Jansson, J.K.; Knight, R. Diversity, stability and resilience of the human gut microbiota. Nature 2012, 489, 220–230. [Google Scholar] [CrossRef] [Green Version]
- Milani, C.; Duranti, S.; Bottacini, F.; Casey, E.; Turroni, F.; Mahony, J.; Belzer, C.; Delgado Palacio, S.; Arboleya Montes, S.; Mancabelli, L.; et al. The first microbial colonizers of the human gut: Composition, activities, and health implications of the infant gut microbiota. Microbiol. Mol. Biol. Rev. MMBR 2017, 81, e00036-17. [Google Scholar] [CrossRef] [PubMed]
- Morgan, X.C.; Huttenhower, C. Meta’omic analytic techniques for studying the intestinal microbiome. Gastroenterology 2014, 146, 1437–1448. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Ley, R.E.; Hamady, M.; Fraser-Liggett, C.M.; Knight, R.; Gordon, J.I. The human microbiome project. Nature 2007, 449, 804–810. [Google Scholar] [CrossRef] [PubMed]
- Turroni, F.; Milani, C.; Duranti, S.; Ferrario, C.; Lugli, G.A.; Mancabelli, L.; van Sinderen, D.; Ventura, M. Bifidobacteria and the infant gut: An example of co-evolution and natural selection. Cell. Mol. Life Sci. 2018, 75, 103–118. [Google Scholar] [CrossRef]
- Hidalgo-Cantabrana, C.; Delgado, S.; Ruiz, L.; Ruas-Madiedo, P.; Sanchez, B.; Margolles, A. Bifidobacteria and their health-promoting effects. Microbiol. Spectr. 2017, 5, 3. [Google Scholar]
- Turroni, F.; Taverniti, V.; Ruas-Madiedo, P.; Duranti, S.; Guglielmetti, S.; Lugli, G.A.; Gioiosa, L.; Palanza, P.; Margolles, A.; van Sinderen, D.; et al. Bifidobacterium bifidum prl2010 modulates the host innate immune response. Appl. Environ. Microbiol. 2014, 80, 730–740. [Google Scholar] [CrossRef]
- Egan, M.; Motherway, M.O.; Kilcoyne, M.; Kane, M.; Joshi, L.; Ventura, M.; van Sinderen, D. Cross-feeding by bifidobacterium breve ucc2003 during co-cultivation with bifidobacterium bifidum prl2010 in a mucin-based medium. BMC Microbiol. 2014, 14, 282. [Google Scholar] [CrossRef]
- Lugli, G.A.; Milani, C.; Duranti, S.; Mancabelli, L.; Mangifesta, M.; Turroni, F.; Viappiani, A.; van Sinderen, D.; Ventura, M. Tracking the taxonomy of the genus bifidobacterium based on a phylogenomic approach. Appl. Environ. Microbiol. 2018, 84, e02249-17. [Google Scholar] [PubMed]
- Odamaki, T.; Kato, K.; Sugahara, H.; Hashikura, N.; Takahashi, S.; Xiao, J.Z.; Abe, F.; Osawa, R. Age-related changes in gut microbiota composition from newborn to centenarian: A cross-sectional study. BMC Microbiol. 2016, 16, 90. [Google Scholar] [CrossRef] [PubMed]
- Koboldt, D.C.; Steinberg, K.M.; Larson, D.E.; Wilson, R.K.; Mardis, E.R. The next-generation sequencing revolution and its impact on genomics. Cell 2013, 155, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Paez-Espino, D.; Eloe-Fadrosh, E.A.; Pavlopoulos, G.A.; Thomas, A.D.; Huntemann, M.; Mikhailova, N.; Rubin, E.; Ivanova, N.N.; Kyrpides, N.C. Uncovering earth’s virome. Nature 2016, 536, 425–430. [Google Scholar] [CrossRef]
- Klemm, E.; Dougan, G. Advances in understanding bacterial pathogenesis gained from whole-genome sequencing and phylogenetics. Cell Host Microbe 2016, 19, 599–610. [Google Scholar] [CrossRef]
- Clark, S.A.; Doyle, R.; Lucidarme, J.; Borrow, R.; Breuer, J. Targeted DNA enrichment and whole genome sequencing of neisseria meningitidis directly from clinical specimens. Int. J. Med. Microbiol. 2018, 308, 256–262. [Google Scholar] [CrossRef]
- Vezzulli, L.; Grande, C.; Tassistro, G.; Brettar, I.; Hofle, M.G.; Pereira, R.P.; Mushi, D.; Pallavicini, A.; Vassallo, P.; Pruzzo, C. Whole-genome enrichment provides deep insights into vibrio cholerae metagenome from an african river. Microb. Ecol. 2017, 73, 734–738. [Google Scholar] [CrossRef]
- Maixner, F.; Krause-Kyora, B.; Turaev, D.; Herbig, A.; Hoopmann, M.R.; Hallows, J.L.; Kusebauch, U.; Vigl, E.E.; Malfertheiner, P.; Megraud, F.; et al. The 5300-year-old helicobacter pylori genome of the iceman. Science 2016, 351, 162–165. [Google Scholar] [CrossRef]
- Lugli, G.A.; Milani, C.; Duranti, S.; Alessandri, G.; Turroni, F.; Mancabelli, L.; Tatoni, D.; Ossiprandi, M.C.; van Sinderen, D.; Ventura, M. Isolation of novel gut bifidobacteria using a combination of metagenomic and cultivation approaches. Genome Biol. 2019, 20, 96. [Google Scholar] [CrossRef]
- Milani, C.; Mangifesta, M.; Mancabelli, L.; Lugli, G.A.; James, K.; Duranti, S.; Turroni, F.; Ferrario, C.; Ossiprandi, M.C.; van Sinderen, D.; et al. Unveiling bifidobacterial biogeography across the mammalian branch of the tree of life. ISME J. 2017, 11, 2834–2847. [Google Scholar] [CrossRef] [Green Version]
- Duranti, S.; Lugli, G.A.; Mancabelli, L.; Armanini, F.; Turroni, F.; James, K.; Ferretti, P.; Gorfer, V.; Ferrario, C.; Milani, C.; et al. Maternal inheritance of bifidobacterial communities and bifidophages in infants through vertical transmission. Microbiome 2017, 5, 66. [Google Scholar] [CrossRef] [PubMed]
- Milani, C.; Hevia, A.; Foroni, E.; Duranti, S.; Turroni, F.; Lugli, G.A.; Sanchez, B.; Martin, R.; Gueimonde, M.; van Sinderen, D.; et al. Assessing the fecal microbiota: An optimized ion torrent 16s rrna gene-based analysis protocol. PLoS ONE 2013, 8, e68739. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. Qiime allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. Dada2: High-resolution sample inference from illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glockner, F.O. The silva ribosomal rna gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Milani, C.; Lugli, G.A.; Turroni, F.; Mancabelli, L.; Duranti, S.; Viappiani, A.; Mangifesta, M.; Segata, N.; van Sinderen, D.; Ventura, M. Evaluation of bifidobacterial community composition in the human gut by means of a targeted amplicon sequencing (its) protocol. FEMS Microbiol. Ecol. 2014, 90, 493–503. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and clustering orders of magnitude faster than blast. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. Spades: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. A J. Comput. Mol. Cell Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Zhao, Y.; Tang, H.; Ye, Y. Rapsearch2: A fast and memory-efficient protein similarity search tool for next-generation sequencing data. Bioinformatics 2012, 28, 125–126. [Google Scholar] [CrossRef]
- Milani, C.; Casey, E.; Lugli, G.A.; Moore, R.; Kaczorowska, J.; Feehily, C.; Mangifesta, M.; Mancabelli, L.; Duranti, S.; Turroni, F.; et al. Tracing mother-infant transmission of bacteriophages by means of a novel analytical tool for shotgun metagenomic datasets: Metannotatorx. Microbiome 2018, 6, 145. [Google Scholar] [CrossRef] [PubMed]
- Hyatt, D.; Chen, G.L.; Locascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef] [PubMed]
- Lugli, G.A.; Milani, C.; Mancabelli, L.; van Sinderen, D.; Ventura, M. Megannotator: A user-friendly pipeline for microbial genomes assembly and annotation. FEMS Microbiol. Lett. 2016, 363, 7. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wu, J.; Yang, J.; Sun, S.; Xiao, J.; Yu, J. Pgap: Pan-genomes analysis pipeline. Bioinformatics 2012, 28, 416–418. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Enright, A.J.; Van Dongen, S.; Ouzounis, C.A. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002, 30, 1575–1584. [Google Scholar] [CrossRef]
- Richter, M.; Rossello-Mora, R.; Oliver Glockner, F.; Peplies, J. Jspeciesws: A web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics 2016, 32, 929–931. [Google Scholar] [CrossRef]
- Cohrs, R.J.; Lee, K.S.; Beach, A.; Sanford, B.; Baird, N.L.; Como, C.; Graybill, C.; Jones, D.; Tekeste, E.; Ballard, M.; et al. Targeted genome sequencing reveals varicella-zoster virus open reading frame 12 deletion. J. Virol. 2017, 9, e01141-17. [Google Scholar] [CrossRef]
- Lugli, G.A.; Milani, C.; Mancabelli, L.; Turroni, F.; Ferrario, C.; Duranti, S.; van Sinderen, D.; Ventura, M. Ancient bacteria of the otzi’s microbiome: A genomic tale from the copper age. Microbiome 2017, 5, 5. [Google Scholar] [CrossRef]
- Lugli, G.A.; Milani, C.; Turroni, F.; Duranti, S.; Ferrario, C.; Viappiani, A.; Mancabelli, L.; Mangifesta, M.; Taminiau, B.; Delcenserie, V.; et al. Investigation of the evolutionary development of the genus bifidobacterium by comparative genomics. Appl. Environ. Microbiol. 2014, 80, 6383–6394. [Google Scholar] [CrossRef]
- Lugli, G.A.; Milani, C.; Turroni, F.; Duranti, S.; Mancabelli, L.; Mangifesta, M.; Ferrario, C.; Modesto, M.; Mattarelli, P.; Jiri, K.; et al. Comparative genomic and phylogenomic analyses of the bifidobacteriaceae family. BMC Genom. 2017, 18, 568. [Google Scholar] [CrossRef] [PubMed]
- Milani, C.; Mangifesta, M.; Mancabelli, L.; Lugli, G.A.; Mancino, W.; Viappiani, A.; Faccini, A.; van Sinderen, D.; Ventura, M.; Turroni, F. The sortase-dependent fimbriome of the genus bifidobacterium: Extracellular structures with potential to modulate microbe-host dialogue. Appl. Environ. Microbiol. 2017, 83, e01295-17. [Google Scholar] [CrossRef] [PubMed]
- Arboleya, S.; Bottacini, F.; O’Connell-Motherway, M.; Ryan, C.A.; Ross, R.P.; van Sinderen, D.; Stanton, C. Gene-trait matching across the bifidobacterium longum pan-genome reveals considerable diversity in carbohydrate catabolism among human infant strains. BMC Genom. 2018, 19, 33. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Features | B. adolescentis AS-ADO | B. longum subsp. longum IS-LON |
|---|---|---|
| Biological origin | Hippopotamus amphibius | Homo sapiens |
| Average Coverage | 1057 | 1855 |
| Contigs | 89 | 43 |
| Genome length | 1,857,949 | 2,366,427 |
| Average GC percentage | 59.44 | 59.85 |
| Predicted ORFs | 1549 | 2,024 |
| ANI value (%, species) | 97.9, B. adolescentis ATCC 15703 | 97.1, B. longum subsp. longum DSM 20088 |
| TUGs | 33 | 19 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lugli, G.A.; Duranti, S.; Milani, C.; Mancabelli, L.; Turroni, F.; Sinderen, D.v.; Ventura, M. Uncovering Bifidobacteria via Targeted Sequencing of the Mammalian Gut Microbiota. Microorganisms 2019, 7, 535. https://doi.org/10.3390/microorganisms7110535
Lugli GA, Duranti S, Milani C, Mancabelli L, Turroni F, Sinderen Dv, Ventura M. Uncovering Bifidobacteria via Targeted Sequencing of the Mammalian Gut Microbiota. Microorganisms. 2019; 7(11):535. https://doi.org/10.3390/microorganisms7110535
Chicago/Turabian StyleLugli, Gabriele Andrea, Sabrina Duranti, Christian Milani, Leonardo Mancabelli, Francesca Turroni, Douwe van Sinderen, and Marco Ventura. 2019. "Uncovering Bifidobacteria via Targeted Sequencing of the Mammalian Gut Microbiota" Microorganisms 7, no. 11: 535. https://doi.org/10.3390/microorganisms7110535