
Rhizocarpon geographicum Lichen Discloses a Highly Diversified Microbiota Carrying Antibiotic Resistance and Persistent Organic Pollutant Tolerance
 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
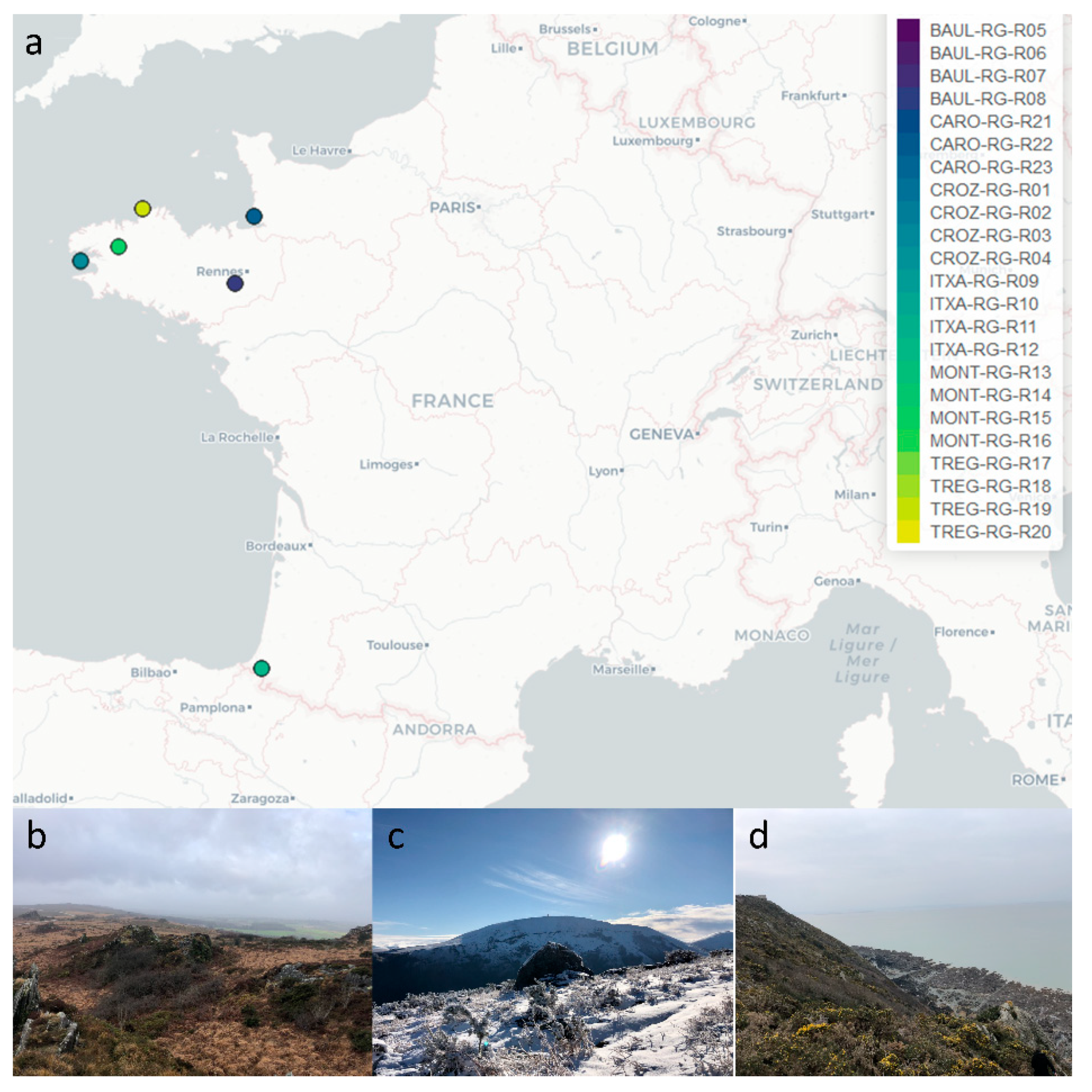
2.1. Collection of Rhizocarpon geographicum Populations
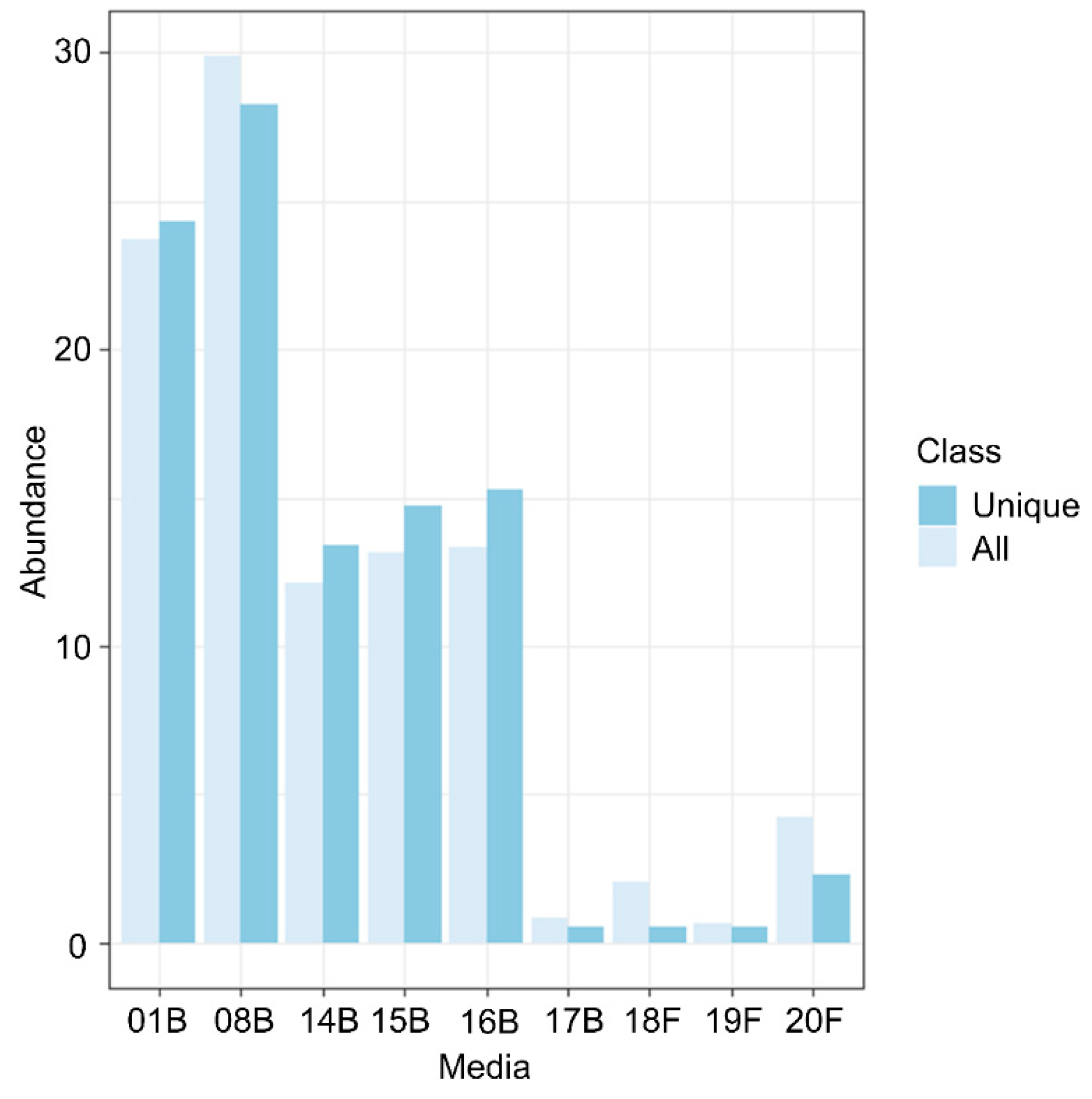
2.2. Culturomics
2.3. Bacterial Isolation and Characterization
2.4. Characterization of Antibiotic-Resistant Strains
2.5. Tolerance to Persistent Organic Pollutants
3. Results
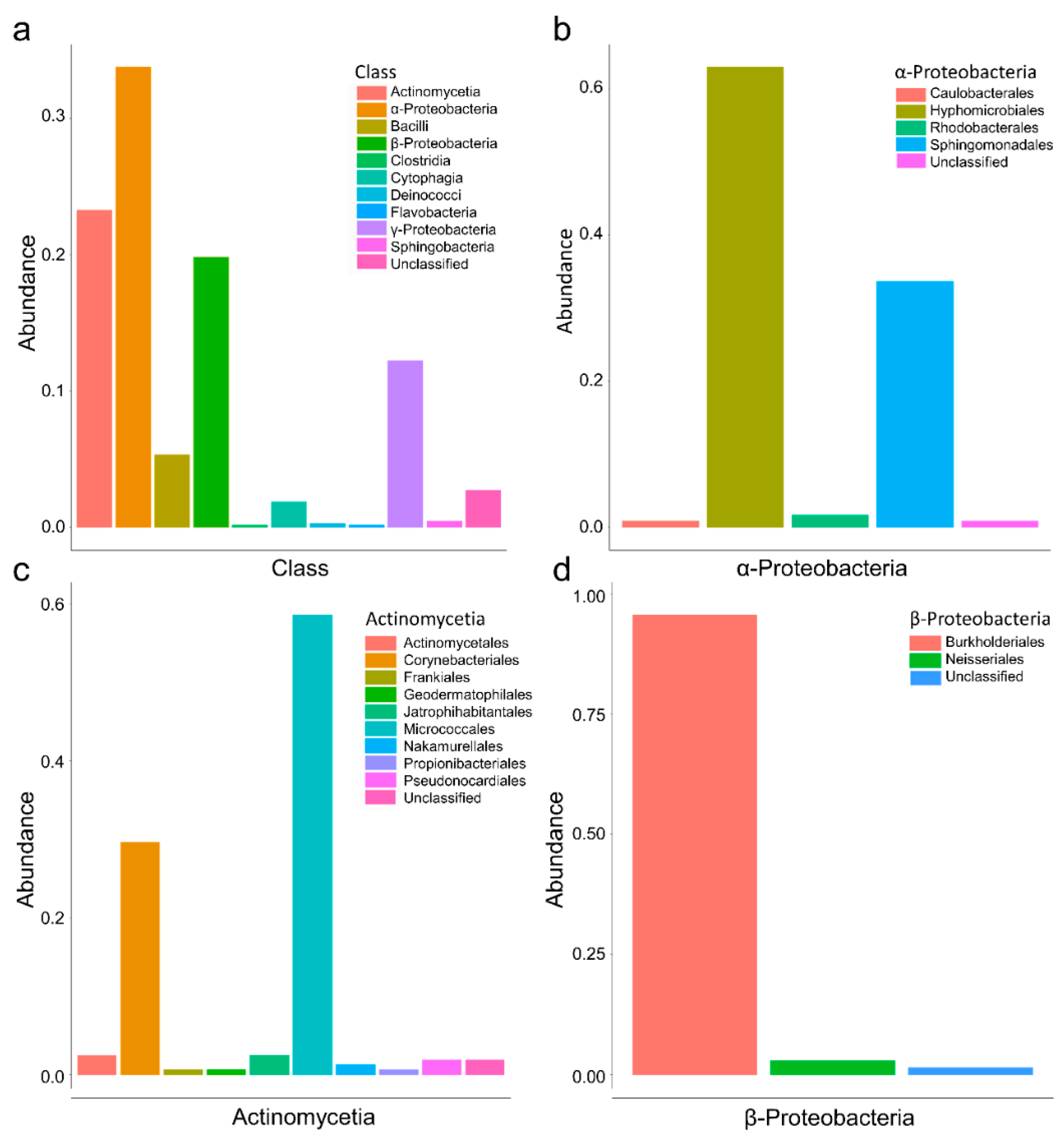
3.1. Culturomics Revealed A Rhizocarpon geographicum Highly Diversified Microbiota
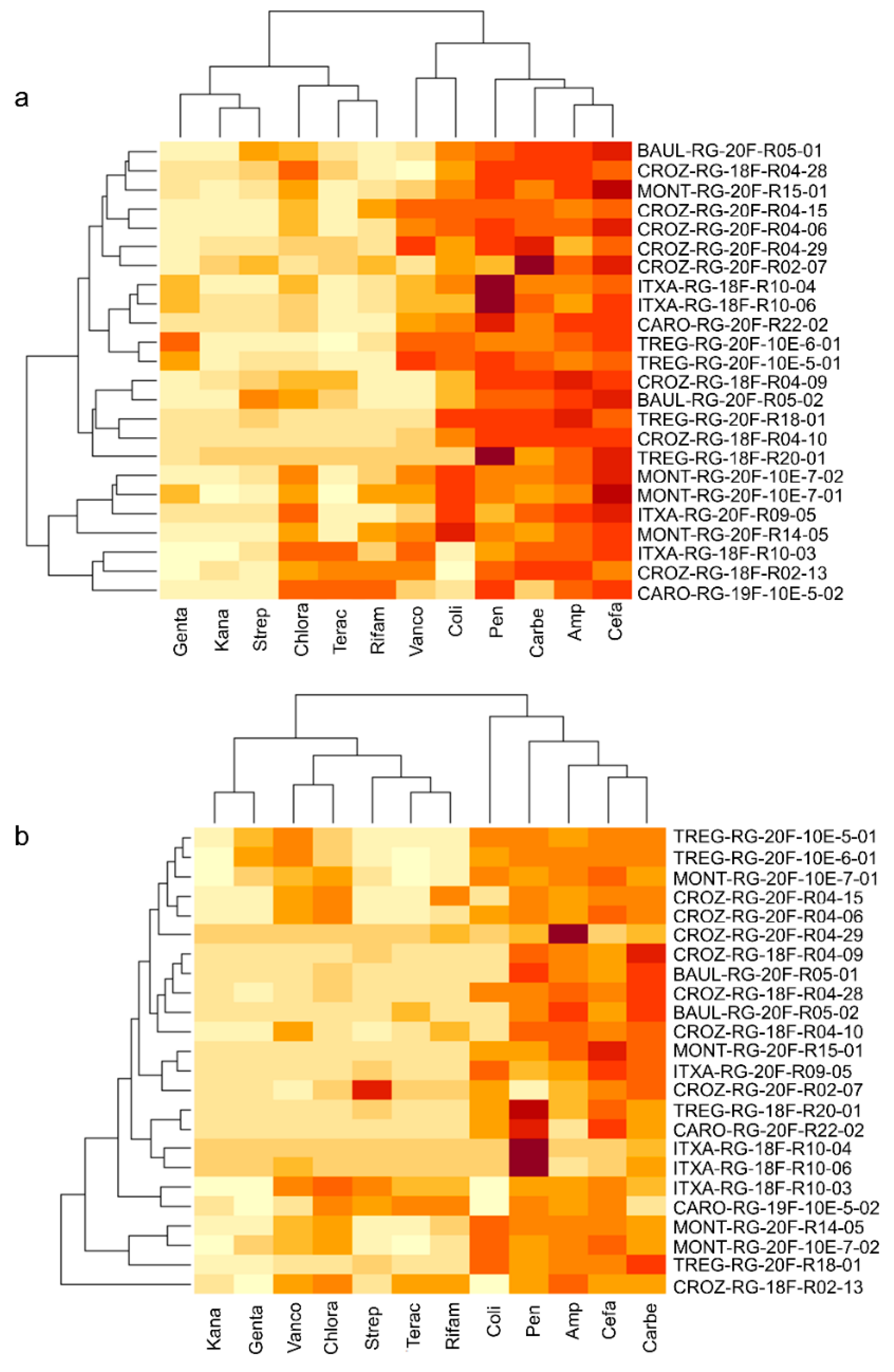
3.2. Culturomics Allows Isolating Taxonomically Diversified Antibiotic-Resistant Bacterial Species
3.3. Whole-Genome Sequencing of Antibiotic-Resistant Bacteria Suggests Potential Novel Species Harboring Antimicrobial Resistance Genes
3.4. Lichen Bacterial Strains Harbor Tolerance to Persistent Organic Pollutants That Is Not Correlated with the Antimicrobial Activity
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hawksworth, D.L.; Grube, M. Lichens redefined as complex ecosystems. New Phytol. 2020, 227, 1281–1283. [Google Scholar] [CrossRef] [PubMed]
- Grimm, M.; Grube, M.; Schiefelbein, U.; Zühlke, D.; Bernhardt, J.; Riedel, K. The Lichens’ Microbiota, Still a Mystery? Front. Microbiol. 2021, 12, 623839. [Google Scholar] [CrossRef]
- Keller, J.; Puginier, C.; Libourel, C.; Otte, J.; Skaloud, P.; Delaux, P.-M.; Keller, J.; Delauz, P.-M.; Berrin, J.-G.; Peterson, M.; et al. Phylogenomics reveals the evolutionary origin of lichenization in chlorophyte algae. bioRxiv 2022. [Google Scholar] [CrossRef]
- Spribille, T.; Tuovinen, V.; Resl, P.; Vanderpool, D.; Wolinski, H.; Aime, M.C.; McCutcheon, J.P.; Johannesson, H.; Thor, G.; Stabentheiner, E.; et al. Basidiomycete yeasts in the cortex of ascomycete macrolichens. Science 2016, 353, 488–492. [Google Scholar] [CrossRef]
- Grube, M.; Cardinale, M.; de Castro, J.V.; Müller, H.; Berg, G. Species-specific structural and functional diversity of bacterial communities in lichen symbioses. ISME J. 2009, 3, 1105–1115. [Google Scholar] [CrossRef] [PubMed]
- Grube, M.; Spribille, T. Exploring symbiont management in lichens. Mol. Ecol. 2012, 21, 3098–3099. [Google Scholar] [CrossRef]
- Simon, J.-C.; Marchesi, J.R.; Mougel, C.; Selosse, M.-A. Host-microbiota interactions, from holobiont theory to analysis. Microbiome 2019, 7, 5. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, S.F.; Sapp, J.; Tauber, A.I. A Symbiotic view of life, we have never been individuals. Quar. Rev. Biol. 2012, 87, 325–341. [Google Scholar] [CrossRef]
- Rosenberg, E.; Zilber-Rosenberg, I. The hologenome concept of evolution after 10 years. Microbiome 2018, 6, 78. [Google Scholar] [CrossRef]
- Berg, G.; Rybakova, D.; Fischer, D.; Cernava, T.; Vergès, M.C.C.; Charles, T.; Chen, X.; Cocolin, L.; Eversole, K.; Corral, G.H.; et al. Microbiome definition re-visited, old concepts and new challenges. Microbiome 2020, 8, 103. [Google Scholar] [CrossRef]
- Liba, C.; Ferrara, F.; Manfio, G.; Fantinatti-Garboggini, F.; Albuquerque, R.; Pavan, C.; Ramos, P.L.; Moreira-Filho, C.; Barbosa, H. Deciphering functional diversification within the lichen microbiota by meta-omics. Microbiome 2017, 5, 82. [Google Scholar]
- Liba, C.; Ferrara, F.; Manfio, G.; Fantinatti-Garboggini, F.; Albuquerque, R.; Pavan, C.; Ramos, P.L.; Moreira-Filho, C.; Barbosa, H. Nitrogen-fixing chemo-organotrophic bacteria isolated from cyanobacteria-deprived lichens and their ability to solubilize phosphate and to release amino acids and phytohormones. J. App. Microbiol. 2006, 101, 1076–1086. [Google Scholar] [CrossRef] [PubMed]
- Aschenbrenner, I.A.; Cernava, T.; Berg, G.; Grube, M. Understanding microbial multi-species symbioses. Front. Microbiol. 2016, 7, 180. [Google Scholar] [CrossRef] [PubMed]
- Grube, M.; Berg, G. Microbial consortia of bacteria and fungi with focus on the lichen symbiosis. Fungal Biol. Rev. 2009, 23, 72–85. [Google Scholar] [CrossRef]
- Noël, A.; Garnier, A.; Clément, M.; Rouaud, I.; Sauvager, A.; Bousarghin, L.; Vásquez-Ocmín, P.; Maciuk, A.; Tomasi, S. Lichen-associated bacteria transform antibacterial usnic acid to products of lower antibiotic activity. Phytochemistry 2021, 181, 112535. [Google Scholar] [CrossRef] [PubMed]
- Francolini, I.; Norris, P.; Piozzi, A.; Donelli, G.; Stoodley, P. Usnic acid, A natural antimicrobial agent able to inhibit bacterial biofilm formation on polymer surfaces. Antimicrob. Agents Chemother. 2004, 48, 4360–4365. [Google Scholar] [CrossRef]
- Ingólfsdóttir, K. Usnic acid. Phytochemistry 2002, 61, 729–736. [Google Scholar] [CrossRef]
- Sierra, M.A.; Danko, D.C.; Sandoval, T.A.; Pishchany, G.; Moncada, B.; Kolter, R.; Mason, C.E.; Zambrano, M.M. The microbiomes of seven lichen genera reveal host specificity, a reduced core community and potential as source of antimicrobials. Front. Microbiol. 2020, 11, 398. [Google Scholar] [CrossRef]
- Cardinale, M.; Puglia, A.M.; Grube, M. Molecular analysis of lichen-associated bacterial communities, lichen-associated bacterial communities. FEMS Microbiol. Ecol. 2006, 57, 484–495. [Google Scholar] [CrossRef]
- Bjelland, T.; Grube, M.; Hoem, S.; Jørgensen, S.L.; Daae, F.L.; Thorseth, I.H.; Øvreås, L. Microbial metacommunities in the lichen–rock habitat. Environ. Microbiol. Rep. 2011, 3, 434–442. [Google Scholar] [CrossRef]
- Hodkinson, B.P.; Lutzoni, F. A microbiotic survey of lichen-associated bacteria reveals a new lineage from the Rhizobiales. Symbiosis 2009, 49, 163–180. [Google Scholar] [CrossRef]
- West, N.J.; Parrot, D.; Fayet, C.; Grube, M.; Tomasi, S.; Suzuki, M.T. Marine cyanolichens from different littoral zones are associated with distinct bacterial communities. PeerJ. 2018, 6, e5208. [Google Scholar] [CrossRef] [PubMed]
- Allen-Vercoe, E. Bringing the gut microbiota into focus through microbial culture, recent progress and future perspective. Curr. Opin. Microbiol. 2013, 16, 625–629. [Google Scholar] [CrossRef]
- Kaeberlein, T.; Lewis, K.; Epstein, S.S. Isolating ‘uncultivable’ microorganisms in pure culture in a simulated natural environment. Science 2002, 10, 1127–1129. [Google Scholar] [CrossRef] [PubMed]
- Epstein, S. The phenomenon of microbial uncultivability. Curr. Opin. Microbiol. 2013, 16, 636–642. [Google Scholar] [CrossRef]
- Chang, Y.; Hou, F.; Pan, Z.; Huang, Z.; Han, N.; Bin, L.; Deng, H.; Li, Z.; Ding, L.; Gao, H.; et al. Optimization of culturomics strategy in human fecal famples. Front. Microbiol. 2019, 10, 2891. [Google Scholar] [CrossRef]
- Biosca, E.G.; Flores, R.; Santander, R.D.; Díez-Gil, J.L.; Barreno, E. Innovative approaches using lichen enriched media to improve isolation and culturability of lichen associated bacteria. PLoS ONE 2016, 11, e0160328. [Google Scholar] [CrossRef]
- Ruibal, C.; Gueidan, C.; Selbmann, L.; Gorbushina, A.; Crous, P.; Groenewald, J.; Muggia, L.; Grube, M.; Isola, D.; Schoch, C.; et al. Phylogeny of rock-inhabiting fungi related to Dothideomycetes. Stud. Mycol. 2009, 64, 123–133. [Google Scholar] [CrossRef]
- Nienow, J.A.; Friedmann, E.I. Terrestrial lithophytic (rock) communities. In Antarctic Microbiol; Friedmann, E.I., Ed.; Wiley-Liss: New York, NY, USA, 1993; pp. 343–412. [Google Scholar]
- Maurya, A.P.; Rajkumari, J.; Pandey, P. Enrichment of antibiotic resistance genes (ARGs) in polyaromatic hydrocarbon-contaminated soils, a major challenge for environmental health. Environ. Sci. Pollut. Res. Int. 2021, 28, 12178–12189. [Google Scholar] [CrossRef]
- Vingataramin, L.; Frost, E.H. A single protocol for extraction of gDNA from bacteria and yeast. BioTech 2015, 58, 120–125. [Google Scholar] [CrossRef]
- Bates, D.; Mächler, M.; Bolker, B.; Walker, S. Fitting linear mixed-effects models Uusing lme4. J. Stat. Softw. 2015, 67, 1–48. [Google Scholar] [CrossRef]
- Lenth, R.V. Least-squares Means, the R package lsmeans. J. Stat. Softw. 2016, 69, 1–33. [Google Scholar] [CrossRef]
- Ginestet, C. Ggplot2, elegant graphics for data analysis. J. R. Stat. Soc. Ser. A-Stat. Soc. 2011, 174, 245. [Google Scholar] [CrossRef]
- Wickham, H. Reshaping data with the reshape package. J. Stat. Softw. 2007, 21, 1–20. [Google Scholar] [CrossRef]
- Ces Marées Noires qui ont Marqué la Bretagne. France 3 Bretagne. Available online: https://france3-regions.francetvinfo.fr/bretagne/ces-marees-noires-qui-ont-marque-bretagne-1214399.html (accessed on 6 January 2022).
- Grube, M.; Cernava, T.; Soh, J.; Fuchs, S.; Aschenbrenner, I.; Lassek, C.; Wegner, U.; Becher, D.; Riedel, K.; Sensen, C.W.; et al. Exploring functional contexts of symbiotic sustain within lichen-associated bacteria by comparative omics. ISME J. 2015, 9, 412–424. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-hit, a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef]
- Harding, T.; Jungblut, A.D.; Lovejoy, C.; Vincent, W.F. Microbes in high arctic snow and implications for the cold Bbiosphere. Appl. Environ. Microbiol. 2011, 77, 3234–3243. [Google Scholar] [CrossRef]
- Jeong, S.W.; Yang, J.E.; Choi, Y.J. Isolation and characterization of a yellow xanthophyll pigment-producing marine bacterium; Erythrobacter sp. SDW2 strain, in coastal seawater. Mar. Drugs 2022, 20, 73. [Google Scholar] [CrossRef]
- Hu, Y.; MacMillan, J.B. Erythrazoles A–B., Cytotoxic benzothiazoles from a marine-derived Erythrobacter sp. Org. Lett. 2011, 13, 6580–6583. [Google Scholar] [CrossRef]
- Parrot, D.; Antony-Babu, S.; Intertaglia, L.; Grube, M.; Tomasi, S.; Suzuki, M.T. Littoral lichens as a novel source of potentially bioactive Actinobacteria. Sci. Rep. 2015, 5, 15839. [Google Scholar] [CrossRef]
- Qu, J.-H.; Hui, M.; Qu, J.-Y.; Wang, F.-F.; Li, H.-F.; Hu, Y.-S.; Luo, Y.; Cai, J.P. Geodermatophilus taihuensis sp. nov., Isolated from the interfacial sediment of a eutrophic lake. Int. J. Syst. Evol. Microbiol. 2013, 63, 4108–4112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.-F.; Qu, J.-H.; Yang, J.-S.; Li, Z.-J.; Yuan, H.-L. Paracoccus chinensis sp. nov., Isolated from sediment of a reservoir. Int. J. Syst. Evol. Microbiol. 2009, 59, 2670–2674. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.W.; Lee, J.M.; Seo, J.P.; Schumann, P.; Kim, S.J.; Lee, S.D. Phycicola gilvus gen. nov., sp. nov., an actinobacterium isolated from living seaweed. Int. J. Syst. Evol. Microbiol. 2008, 58, 1318–1323. [Google Scholar] [CrossRef] [PubMed]
- Gundlapally, S.R.; Ara, S.; Sisinthy, S. Draft genome of Kocuria polaris CMS 76or(T) isolated from cyanobacterial mats; McMurdo Dry Valley; Antarctica, an insight into CspA family of proteins from Kocuria polaris CMS 76or(T). Arch. Microbiol. 2015, 197, 1019–1026. [Google Scholar] [CrossRef]
- Miyazawa, D.; Thanh, L.T.H.; Tani, A.; Shintani, M.; Loc, N.H.; Hatta, T.; Kimbara, K. Isolation and characterization of genes responsible for naphthalene degradation from thermophilic naphthalene degrader; Geobacillus sp. JF8. Microorganisms 2020, 8, 44. [Google Scholar] [CrossRef] [PubMed]
- Nazina, T.N.; Sokolova, D.S.; Grigoryan, A.A.; Shestakova, N.M.; Mikhailova, E.M.; Poltaraus, A.B.; Tourova, T.P.; Lysenko, A.M.; Osipov, G.A.; Belyaev, S.S. Geobacillus jurassicus sp. nov., a new thermophilic bacterium isolated from a high-temperature petroleum reservoir; and the validation of the Geobacillus species. Syst. Appl. Microbiol. 2005, 28, 43–53. [Google Scholar] [CrossRef]
- Pan, L.; Tang, X.; Li, C.; Yu, G.; Wang, Y. Biodegradation of sulfamethazine by an isolated thermophile–Geobacillus sp. S-07. World J. Microbiol. Biotechnol. 2017, 33, 85. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, X.; Liu, J.; Li, R.; Shen, B. Isolation of a thermophilic bacterium, Geobacillus sp. SH-1, capable of degrading aliphatic hydrocarbons and naphthalene simultaneously, and identification of its naphthalene degrading pathway. Bio. Technol. 2012, 124, 83–89. [Google Scholar] [CrossRef]
- Hong, S.H.; Ryu, H.; Kim, J.; Cho, K.-S. Rhizoremediation of diesel-contaminated soil using the plant growth-promoting rhizobacterium Gordonia sp. S2RP-17. Biodegradation 2011, 22, 593–601. [Google Scholar] [CrossRef]
- Wang, Y.; Zhan, W.; Ren, Q.; Cheng, S.; Wang, J.; Ma, X.; Zhang, C.; Wang, Y. Biodegradation of di-(2-ethylhexyl) phthalate by a newly isolated Gordonia sp. and its application in the remediation of contaminated soils. Sci. Total Environ. 2019, 689, 645–651. [Google Scholar] [CrossRef]
- Kalita, M.; Chutia, M.; Jha, D.K.; Subrahmanyam, G. Mechanistic understanding of Gordonia sp. in biodesulfurization of organosulfur compounds. Curr. Microbiol. 2022, 79, 82. [Google Scholar] [CrossRef] [PubMed]
- Zebrowska, J.; Witkowska, M.; Struck, A.; Laszuk, P.E.; Raczuk, E.; Ponikowska, M.; Skowron, P.M.; Zylicz-Stachula, A. Antimicrobial potential of the genera Geobacillus and Parageobacillus; as well as endolysins biosynthesized by their bacteriophages. Antibiotics 2022, 11, 242. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.-C.; Busse, H.-J.; Liu, H.-C.; Zhou, Y.-G.; Schinner, F.; Margesin, R. Sphingomonas glacialis sp. nov., A psychrophilic bacterium isolated from alpine glacier cryoconite. Int. J. Syst. Evol. Microbiol. 2011, 61, 587–591. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Göker, M. TYGS is an automated high-throughput platform for state-of-the-art genome-based taxonomy. Nat. Commun. 2019, 10, 2182. [Google Scholar] [CrossRef] [PubMed]
- Reddy, G.S.N.; Matsumoto, G.I.; Schumann, P.; Stackebrandt, E.; Shivaji, S. Psychrophilic pseudomonads from Antarctica, Pseudomonas antarctica sp. nov., Pseudomonas meridiana sp. nov. and Pseudomonas proteolytica sp. nov. Int. J. Syst. Evol. Microbiol. 2004, 54, 713–719. [Google Scholar] [CrossRef]
- Lee, J.; Cho, Y.-J.; Yang, J.Y.; Jung, Y.-J.; Hong, S.G.; Kim, O.-S. Complete genome sequence of Pseudomonas antarctica PAMC 27494; a bacteriocin-producing psychrophile isolated from Antarctica. J. Biotechnol. 2017, 259, 15–18. [Google Scholar] [CrossRef]
- Schlusselhuber, M.; Girard, L.; Cousin, F.J.; Lood, C.; De Mot, R.; Goux, D.; Desmasures, N. Pseudomonas crudilactis sp. nov., Isolated from raw milk in France. Antonie Van Leeuwenhoek 2021, 114, 719–730. [Google Scholar] [CrossRef]
- Jin, X.-F.; Kim, J.-K.; Liu, Q.-M.; Kang, M.-S.; He, D.; Jin, F.-X.; Kim, S.C.; Im, W.T. Sphingomonas ginsenosidivorax sp. nov., with the ability to transform ginsenosides. Antonie Van Leeuwenhoek 2013, 103, 1359–1367. [Google Scholar] [CrossRef]
- Kachur, K.; Suntres, Z.E. The antimicrobial properties of ginseng and ginseng extracts. Expert. Rev. Anti. Infect. Ther. 2016, 14, 81–94. [Google Scholar] [CrossRef]
- Feldgarden, M.; Brover, V.; Gonzalez-Escalona, N.; Frye, J.G.; Haendiges, J.; Haft, D.H.; Hoffmann, M.; Pettengill, J.B.; Prasad, A.B.; Tillman, G.E.; et al. AMRFinderPlus and the reference gene catalog facilitate examination of the genomic links among antimicrobial resistance; stress response; and virulence. Sci. Rep. 2021, 11, 12728. [Google Scholar] [CrossRef]
- Miral, A.; Jargeat, P.; Mambu, L.; Rouaud, I.; Tranchimand, S.; Tomasi, S. Microbial community associated with the crustose lichen Rhizocarpon geographicum L. (DC.) living on oceanic seashore, a large source of diversity revealed by using multiple isolation methods. Environ. Microbiol. Rep. 2022. [Google Scholar] [CrossRef] [PubMed]
- Pankratov, T.A. Acidobacteria in microbial communities of the bog and tundra lichens. Microbiology 2012, 81, 51–58. [Google Scholar] [CrossRef]
- Dance, A. The search for microbial dark matter. Nature 2020, 582, 301–303. [Google Scholar] [CrossRef] [PubMed]
- Benson, D.R.; Silvester, W.B. Biology of Frankia strains; actinomycete symbionts of actinorhizal plants. Microbiol. Rev. 1993, 57, 293–319. [Google Scholar] [CrossRef]
- Biswas, R.; Majhi, A.K.; Sarkar, A. The role of arsenate reducing bacteria for their prospective application in arsenic contaminated groundwater aquifer system. Biocatal. Agri. Biotechnol. 2019, 20, 101218. [Google Scholar] [CrossRef]
- Campanini, B.; Pieroni, M.; Raboni, S.; Bettati, S.; Benoni, R.; Pecchini, C.; Costantino, G.; Mozzarelli, A. Inhibitors of the sulfur assimilation pathway in bacterial pathogens as enhancers of antibiotic therapy. Curr. Med. Chem. 2015, 22, 187–213. [Google Scholar] [CrossRef] [PubMed]
- Nikaido, H. Multidrug resistance in bacteria. Annu. Rev. Biochem. 2009, 78, 119–146. [Google Scholar] [CrossRef]
- Blair, J.M.A.; Piddock, L.J.V. Structure; function and inhibition of RND efflux pumps in Gram-negative bacteria, an update. Curr. Opin. Microbiol. 2009, 12, 512–519. [Google Scholar] [CrossRef]
- Rodríguez-Herva, J.J.; García, V.; Hurtado, A.; Segura, A.; Ramos, J.L. The ttgGHI solvent efflux pump operon of Pseudomonas putida DOT-T1E is located on a large self-transmissible plasmid. Environ. Microbiol. 2007, 9, 1550–1561. [Google Scholar] [CrossRef]
- Espinosa-Urgel, M.; Salido, A.; Ramos, J.L. Genetic analysis of functions involved in adhesion of Pseudomonas putida to seeds. J. Bacteriol. 2000, 182, 2363–2369. [Google Scholar] [CrossRef]
- Matilla, M.A.; Espinosa-Urgel, M.; Rodríguez-Herva, J.J.; Ramos, J.L.; Ramos-González, M.I. Genomic analysis reveals the major driving forces of bacterial life in the rhizosphere. Genome Biol. 2007, 8, R179. [Google Scholar] [CrossRef] [Green Version]
- Barabote, R.D.; Johnson, O.L.; Zetina, E.; San Francisco, S.K.; Fralick, J.A.; San Francisco, M.J.D. Erwinia chrysanthemi tolC is involved in resistance to antimicrobial plant chemicals and is essential for phytopathogenesis. J. Bacteriol. 2003, 185, 5772–5778. [Google Scholar] [CrossRef] [PubMed]
- González-Pasayo, R.; Martínez-Romero, E. Multiresistance genes of Rhizobium etli CFN42. Mol. Plant. Microbe. Interact. 2000, 13, 572–577. [Google Scholar] [CrossRef] [PubMed]
- Boustie, J.; Grube, M. Lichens—A promising source of bioactive secondary metabolites. Plant Genet. Res. 2005, 3, 273–287. [Google Scholar] [CrossRef]
- Kokubun, T.; Shiu, W.K.P.; Gibbons, S. Inhibitory activities of lichen-derived compounds against methicillin- and multidrug-resistant Staphylococcus aureus. Planta Med. 2007, 73, 176–179. [Google Scholar] [CrossRef] [PubMed]
- Hearn, E.M.; Dennis, J.J.; Gray, M.R.; Foght, J.M. Identification and characterization of the emhABC efflux system for polycyclic aromatic hydrocarbons in Pseudomonas fluorescens cLP6a. J. Bacteriol. 2003, 185, 6233–6240. [Google Scholar] [CrossRef]
- Das, N.; Kotoky, R.; Maurya, A.P.; Bhuyan, B.; Pandey, P. Paradigm shift in antibiotic-resistome of petroleum hydrocarbon contaminated soil. Sci. Total Environ. 2021, 757, 143777. [Google Scholar] [CrossRef]
- Bugg, T.; Foght, J.M.; Pickard, M.A.; Gray, M.R. Uptake and active efflux of polycyclic aromatic hydrocarbons by Pseudomonas fluorescens LP6a. Appl. Environ. Microbiol. 2000, 66, 5387–5392. [Google Scholar] [CrossRef]
- Adebusuyi, A.A.; Foght, J.M. An alternative physiological role for the EmhABC efflux pump in Pseudomonas fluorescens cLP6a. BMC Microbiol. 2011, 11, 252. [Google Scholar] [CrossRef]
- Cunningham, C.J.; Kuyukina, M.S.; Ivshina, B.I.; Konev, I.A.; Peshkur, A.T.; Knapp, W.C. Potential risks of antibiotic resistant bacteria and genes in bioremediation of petroleum hydrocarbon contaminated soils. Environ. Sci. Process. Impacts 2020, 22, 1110–1124. [Google Scholar] [CrossRef]
- Hemala, L.; Zhang, D.; Margesin, R. Cold-active antibacterial and antifungal activities and antibiotic resistance of bacteria isolated from an alpine hydrocarbon-contaminated industrial site. Res. Microbiol. 2014, 165, 447–456. [Google Scholar] [CrossRef]
- Máthé, I.; Benedek, T.; Táncsics, A.; Palatinszky, M.; Lányi, S.; Márialigeti, K. Diversity; activity; antibiotic and heavy metal resistance of bacteria from petroleum hydrocarbon contaminated soils located in Harghita County (Romania). Int. Biodeterior. Biodegrad. 2012, 73, 41–49. [Google Scholar] [CrossRef]
- Mohammadi, M.; Bagheri, L.; Badreldin, A.; Fatehi, P.; Pakzad, L.; Suntres, Z.; van Wijnen, A.J. Biological effects of gyrophoric acid and other lichen derived metabolites; on cell proliferation; apoptosis and cell signaling pathways. Chem.-Biol. Interact. 2022, 351, 109768. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Yang, Y.; Ma, L.; Ju, F.; Guo, F.; Tiedje, J.M.; Zhang, T. Metagenomic and network analysis reveal wide distribution and co-occurrence of environmental antibiotic resistance genes. ISME J. 2015, 9, 2490–2502. [Google Scholar] [CrossRef]
- Jones, K.C.; de Voogt, P. Persistent organic pollutants (POPs), state of the science. Environ. Pollut. 1999, 100, 209–221. [Google Scholar] [CrossRef]
- Walters, W.; Hyde, E.R.; Berg-Lyons, D.; Ackermann, G.; Humphrey, G.; Parada, A.; Gilbert, J.A.; Jansson, J.K.; Caporaso, J.G.; Fuhrman, J.A.; et al. Improved Bacterial 16S rRNA Gene (V4 and V4-5) and Fungal Internal Transcribed Spacer Marker Gene Primers for Microbial Community Surveys. mSystems 2015, 1, e00009-15. [Google Scholar] [CrossRef] [PubMed]
- Aramaki, T.; Blanc-Mathieu, R.; Endo, H.; Ohkubo, K.; Kanehisa, M.; Goto, S.; Ogata, H. KofamKOALA: KEGG Ortholog assignment based on profile HMM and adaptive score threshold. Bioinformatics 2020, 36, 2251–2252. [Google Scholar] [CrossRef] [PubMed]
- Machado, D.; Andrejev, S.; Tramontano, M.; Patil, K.R. Fast automated reconstruction of genome-scale metabolic models for microbial species and communities. Nucleic Acids Res. 2018, 6, 7542–7553. [Google Scholar] [CrossRef]
- King, Z.A.; Dräger, A.; Miller, P.; Federowicz, S.; Lerman, J.A.; Ebrahim, A.; Palsoon, B.A.; Lewis, N.E. BiGG Models: A platform for integrating, standardizing and sharing genome-scale models. Nucleic Acids Res. 2015, 44, 515–522. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bacterial Species | Isolation Medium Code | Medium Description |
|---|---|---|
| Aquaspirillum arcticum | 15B | Lichen–algal-based medium |
| Geobacillus sp. | 16B | Lichen–algal-based minimal medium |
| Jeongeupia chitinilytica | 15B | Lichen–algal-based medium |
| Klenkia taihuensis | 14B | Macroalgae-based medium |
| Kocuria polaris | 14B | Macroalgae-based medium |
| Microterricola gilva | 16B | Lichen–algal-based minimal medium |
| Paracoccus chinensis | 19F, 20F | Lichen–algal antibiotic based |
| Pseudomonas gessardii | 20F | Lichen–algal antibiotic based |
| Pseudomonas helmanticensis | 16B, 20F | Lichen–algal-based medium and lichen–algal antibiotic based |
| Roseomonas mucosa | 15B | Lichen–algal-based medium |
| Sinomonas sp. | 15B | Lichen–algal-based medium |
| Skermanella sp. | 14B | Macroalgae-based medium |
| Tardiphaga sp. | 14B | Macroalgae-based medium |
| uncultured Erythrobacter sp. | 16B | Lichen–algal-based minimal medium |
| uncultured Gordonia sp. | 16B | Lichen–algal-based minimal medium |
| uncultured Nakamurella sp. | 8B, 16B | TSA and lichen–algal-based minimal medium |
| Zafaria cholistanensis | 14B | Macroalgae-based medium |
| Lichen Population | Locality | % of Tolerant Strains for MTBE | % of Tolerant Strains for PFOA |
|---|---|---|---|
| BAUL | Baulon | na | 0.76 |
| CARO | Carolles | 1.01 | 1.2 |
| CROZ | Crozon | 1.07 | 0.76 |
| ITXA | Itxassou | 0.25 | 0.25 |
| MONT | Plounéour-Ménez | na | na |
| TREG | Trégastel | 0.7 | 0.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miral, A.; Kautsky, A.; Alves-Carvalho, S.; Cottret, L.; Guillerm-Erckelboudt, A.-Y.; Buguet, M.; Rouaud, I.; Tranchimand, S.; Tomasi, S.; Bartoli, C. Rhizocarpon geographicum Lichen Discloses a Highly Diversified Microbiota Carrying Antibiotic Resistance and Persistent Organic Pollutant Tolerance. Microorganisms 2022, 10, 1859. https://doi.org/10.3390/microorganisms10091859
Miral A, Kautsky A, Alves-Carvalho S, Cottret L, Guillerm-Erckelboudt A-Y, Buguet M, Rouaud I, Tranchimand S, Tomasi S, Bartoli C. Rhizocarpon geographicum Lichen Discloses a Highly Diversified Microbiota Carrying Antibiotic Resistance and Persistent Organic Pollutant Tolerance. Microorganisms. 2022; 10(9):1859. https://doi.org/10.3390/microorganisms10091859
Chicago/Turabian StyleMiral, Alice, Adam Kautsky, Susete Alves-Carvalho, Ludovic Cottret, Anne-Yvonne Guillerm-Erckelboudt, Manon Buguet, Isabelle Rouaud, Sylvain Tranchimand, Sophie Tomasi, and Claudia Bartoli. 2022. "Rhizocarpon geographicum Lichen Discloses a Highly Diversified Microbiota Carrying Antibiotic Resistance and Persistent Organic Pollutant Tolerance" Microorganisms 10, no. 9: 1859. https://doi.org/10.3390/microorganisms10091859