
Targeting Both Autophagy and Immunotherapy in Breast Cancer Treatment
 , ,
, ,
Abstract
:1. Introduction
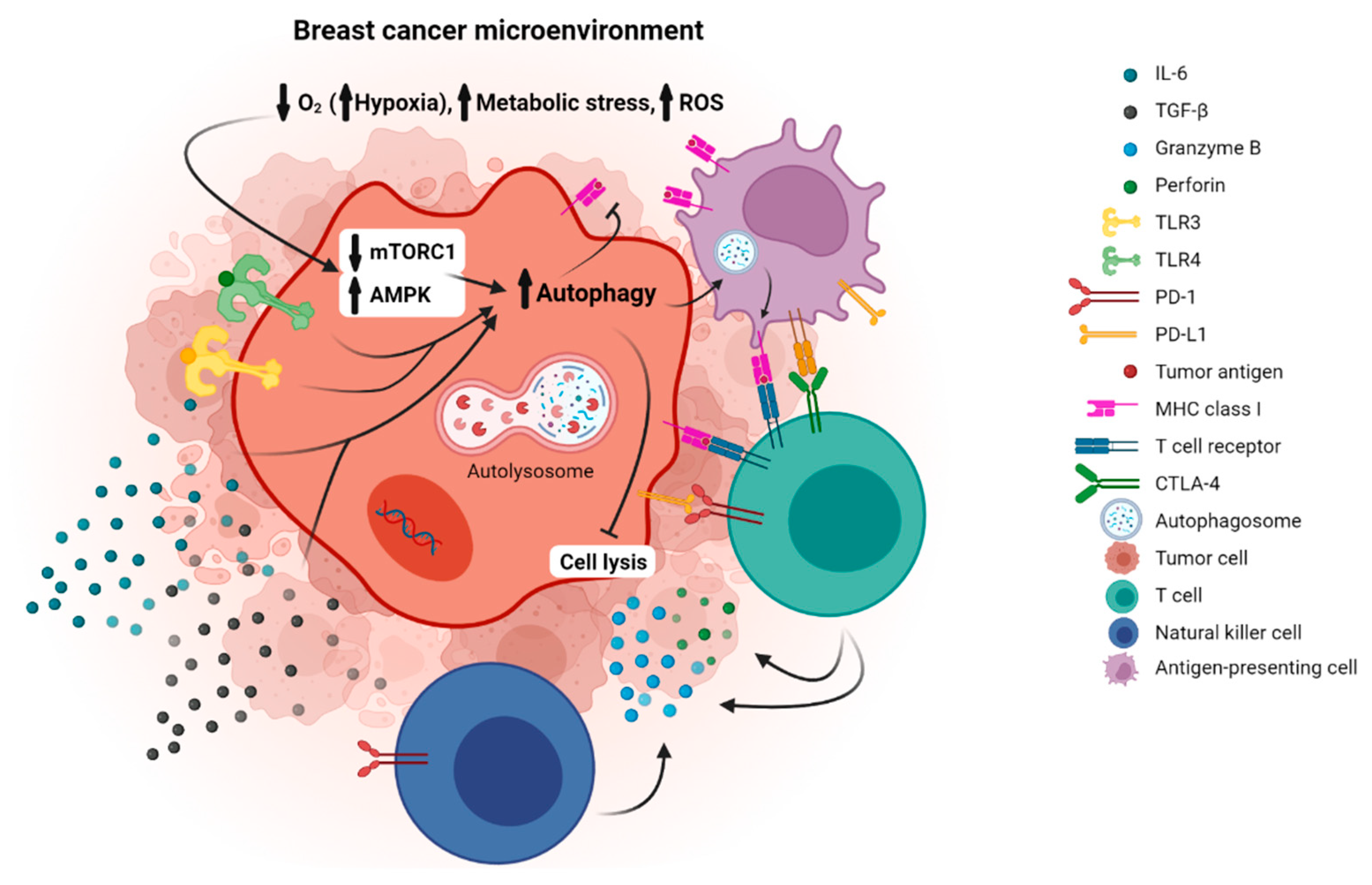
2. Autophagy and Breast Cancer
3. Immune Checkpoint Molecules and Breast Cancer
4. Immune Checkpoint Molecules and Autophagy in Breast Cancer
4.1. PD1/PD-L1 and Autophagy in Breast Cancer
4.2. CTLA-4 and Autophagy in Breast Cancer
5. Other Pathways of Immunoregulation and Autophagy in Breast Cancer
5.1. Tumor Promotion

{kind=link}
| ClinicalTrials.gov ID | Intervention | Study Phase | Location | Status | Start Date | Completion Date | Participants | Condition | Details | Primary Outcome | Results |
|---|---|---|---|---|---|---|---|---|---|---|---|
| NCT00765765 | HCQ + Ixabepilone | Phase 1–2 Non-randomized Open label | USA | terminated | February 2009 | December 2011 | 6 | Metastatic Breast Cancer | Dose escalation from 200 mg po qd to 200 mg po bid | Assess the antitumor activity, measured by tumor response rate, in patients who receive this regimen as a third-line treatment | The study was closed early due to slow accrual. Insufficient data were collected to analyze this outcome measure |
| NCT01292408 | HCQ | Phase 2 Open Label | Nether-lands | Unknown | January 2011 | - | 20 | Breast Cancer | Included patients with core-biopsy proven invasive adenocarcinoma of the breast | Detect differences in endogenous hypoxia markers (CA9, PAI-1, VEGF) and autophagy (LC3b) before and after treatment with HCQ. | - |
| NCT01446016 | CQ + Taxan/ Taxotere/ Abraxane/ Ixabepilone | Phase 2 Non-randomized Open label | USA | Completed | Sept 2011 | March 2019 | 47 | Advanced or Metastatic Breast Cancer | CQ in combination with Taxane or Taxane-like chemo agents in the treatment of patients with advanced or metastatic breast cancer who have failed anthracycline chemo base therapy | To determine the anti-tumor activity of the combination of CQ + Taxane or Taxane-like chemo agents (Paclitaxel, Docetaxel, Abraxane, Ixabepilone) measured by overall response rate | The overall response rate was 45.16%, the combination was well tolerated with only 13.15% of patients experiencing Grade ≥ 3 adverse events. |
| NCT02333890 | CQ vs. Placebo (prior to surgery) | Phase 2 Randomized double-blind placebo-controlled | Canada | Completed | July 2015 | March 2018 | 60 | Invasive Breast Cancer | Included patients with newly diagnosed histologically confirmed primary invasive breast cancer whowere not undergoing any treatment while awaiting surgery in the next 2–6 weeks | Effect of a brief course of CQ on tumour proliferation and apoptosis based on Ki67 and TUNEL assays | No significant differences between the CQ or placebo arms in Ki67 index pre- and post-drug treatment. Adverse effects were minimal and all classified as grade 1. The effects were significant enough to cause nearly 15% of patients to discontinue therapy |
| NCT01023477 | CQ | Phase 1–2 Non-randomized Open label | USA | Completed | December 2009 | October 2016 | 12 | Ductal Carcinoma In Situ (DCIS) | CQ standard dose (500 mg/week) or CQ low dose (250 mg/week) for 1 month prior to surgical removal of the tumor. | Tumor response evaluated by RECIST criteria as measured by breast MRI | Measurable reduction in proliferation of DCIS lesions and enhancement of immune cell migration into the duct |
| NCT02414776 | HCQ + hormonal therapy | Phase 1–2 Non-randomized Open label | USA | Terminated | July 2014 | November 2015 | 3 | ER+ Metastatic Breast Cancer | - | Number of Participants with Adverse Events as a Measure of the safety profile of orally administered HCQ with hormonal therapy | - |
| NCT04523857 | Abemaciclib + HCQ vs. Abemaciclib | Phase 2 Randomized Open label | USA | Not yet recruiting | April 2021 | - | 66 | Invasive Breast Cancer | Rate of protocol defined “severe toxicity” during cycle 1 (4 weeks) of combination HCQ 600 mg BID and Abema (at 100 mg and 150 mg BID) in a safety cohort of 6 patients at each dose of Abema | Incidence of treatment-emergent adverse events, Frequency of “clearance” of bone marrow DTCs by arm after 6 cycles of study treatment. | - |
| NCT03032406 | HCQ + Everolimus vs. Everolimus vs. HCQ | Phase 2 Pilot Randomized | USA | Recruiting | January 2017 | - | 60 | Breast Cancer Stage IIB | Histologically-confirmed, primary, invasive breast cancer diagnosed within 5 years of study entry. | Number of Adverse Events | - |
| NCT04316169 | Abemaciclib + HCQ | Phase 1 Non-randomized Open Label | USA | Not yet recruiting | July 2021 | - | 44 | HR+/Her 2- Advanced Breast Cancer | Arm A: Abemaciclib + HCQ 200 mg b.i.d. Arm B. Abemaciclib + HCQ 400 mg b.i.d. Arm C: Abemaciclib + HCQ 600 mg b.i.d. Arm D: Abemaciclib + HCQ + endocrine therapy | To determine safety and tolerability of HCQ + abemaciclib and HCQ + abemaciclib + endocrine therapy in HR+/Her2- Advanced Breast Cancer | - |
| NCT03774472 | HCQ + Palbociclib + Letrozole | Phase 1–2 Open label | USA | recruiting | August 2018 | - | 54 | Advanced, metastatic (stage IV) Breast Cancer (phase I) Early stage (stage I-III) Breast Cancer (phase II) | This phase I/II trial studies the side effects and best dose of HCQ when given together with palbociclib and letrozole before surgery in treating participants with estrogen receptor positive, HER2 negative breast cancer. | Incidence of adverse events, Change in breast tumor proliferation index (Ki67), Change in autophagy, Change in senescence, Change in cell cycle control, Change in proportion of patients achieving tumoral complete cell cycle arrest | - |
| Studies | Anti-Tumor Medication | Autophagy Inhibitor | Tumor Cells | Comparison | Results |
|---|---|---|---|---|---|
| Lefort, 2014 [83] | Cyclophosphamide + Adriamycin (AC) | Chloroquine (CQ) | MDA-MB-231 human breast cancer cells | AC vs. AC + CQ | The combined group experienced an additive tumor growth inhibition of 41% compared to AC treatment alone and a reduction in lung metastases |
| Liang, 2016 [84] | Carboplatin | Chloroquine (CQ) | SUM159 cells breast cancer cells | Carb vs. Carb + CQ | Carb + CQ reduced tumor growth, decreased mitochondrial metabolic activity, decreased cell viability, and increased levels of LC3b-II and p62 demonstrating that CQ can successfully inhibit autophagy induced by carboplatin |
| Shoemaker, 1978 [85] | 5-Fluorouracil (5-FU) | Chloroquine (CQ) | C3HBA breast cancer cells | 5-FU + CQ vs. control, 5-FU vs. 5-FU + CQ | The 5-FU + CQ group had significantly reduced tumor size compared to control group and 5-FU group |
| Shoemaker, 1979 [86] | 6-Propylthiouracil (PTU) + 5-Fluorouracil (5-FU) | Chloroquine (CQ) | C3HBA breast cancer cells | 5-FU + PTU + CQ vs. control group | Significant reduction in tumor growth compared to control group |
| Loehberg, 2012 [29] | Everolimus | Chloroquine (CQ) | MCF7 breast cancer cells | Everolimus + CQ vs. control group | The combined treatment group showed significant weight (4.1-fold) and size (4.6-fold) reduction compared to control |
| Seront, 2013 [87] | Rapamycin | Chloroquine (CQ) | MDA-MB-231 and MCF-7 breast cancer cells | Rapamycin vs. Rapamycin + CQ vs. CQ | When combined with CQ, rapamycin did not further alter tumor progression in either model cancer cell type, suggesting that potential rapamycin-induced autophagy was not playing a critical role in these tumors. Tumor growth reduction was observed only in mice with large, hypoxic mammary tumors |
| Dragowska, 2013 [88] | Gefitinib | Hydroxychloroquine (HCQ) | JIMT-1 breast cancer cells | Gefitinib vs. HCQ vs. Gefitinib + HCQ | Notably, when gefitinib was used in combination with HCQ there was a significant 58% reduction in tumor volume compared to vehicle-treated controls |
| Cufi, 2013 [89] | Trastuzumab | Chloroquine (CQ) | JIMT-1 breast cancer cells | Trastuzumab vs. CQ vs. Trastuzumab + CQ | The tumor size in the combination group was drastically reduced in a synergistic manner compared to control and monotherapy groups |
| Ratikan, 2013 [90] | Radiotherapy | Chloroquine (CQ) | MCaK breast cancer cells | Radiotherapy + CQ | Chloroquine blocked radiation-induced autophagy and drove MCaK cells into a more rapid apoptotic and more immunogenic form of cell death |
| Thomas, 2012 [91] | Nelfinavir + Celecoxib | Chloroquine (CQ) | MDA-MB-468 and MCF-7 breast cancer cells | Nelfinavir + Celecoxib + CQ | Synergistic enhancement of tumor cell killing by ERSA compounds, particularly in triple-negative breast cancer (TNBC) cells. |
| ClinicalTrials.gov ID | Intervention | Study Phase | Condition | Sample Size | Completion Year | OS (Median) | PFS (Median) | ORR |
|---|---|---|---|---|---|---|---|---|
| KEYNOTE-012 (NCT01848834) | Pembrolizumab | Phase Ib | Metastatic PD-L1 + TNBC | 32 | 2016 | 11.2 mo | 1.9 mo | 16% |
| KEYNOTE-086 (NCT02447003) | Pembrolizumab | Phase II | Advanced PD-L1 + TNBC | 170 | 2019 | 9 mo | 2 mo | 5% |
| KEYNOTE-028 (NCT02054806) | Pembrolizumab | Phase Ib | Metastatic PD L1 + BC | 25 | 2021 | 8.6 mo | - | 12% |
| KEYNOTE-150 (NCT02513472) | Pembrolizumab + Eribulin mesylate | Phase Ib/II | Metastatic TNBC with or without previous chemotherapy | 167 | 2019 | 16.1 mo | 4.1 mo | 23% |
| TOPACIO (NCT02657889) | Niraparib + Pembrolizumab | Phase II | TNBC | 55 | 2018 | - | - | 18.2% |
| PANACEA/ KEYNOTE-014 (NCT02129556) | Pembrolizumab | Phase II | HER2+ BC which has progressed on trastuzumab | 52 | 2017 | Estimated 65% at 12 mo in PD-L1+ | 12% at 12 mo in PD-L1 + p | 15% (PD-L1 + pop) |
| KEYNOTE-119 (NCT02555657) | Pembrolizumab vs. Chemotherapy (capecitabine, eribulin, gemcitabine, vinorelbine) | Phase III | Metastatic TNBC | 622 | 2019 | 9.9 vs. 10.8 mo | 2.1 vs. 3.4 mo | 18% vs. 9% |
| KEYNOTE-355 NCT02819518 | Pembrolizumab + Chemotherapy vs. Placebo + Chemotherapy | Phase III | TNBC | 882 | 2021 | - | 7.5 vs. 5.6 mo | - |
| NCT01375842 | Atezolizumab | Phase I | Advanced TNBC | 116 | 2018 | 17.6 mo | 1.4 mo | 24% in 1st-line treatment, 6% > 1 previous treatments |
| NCT01633970 | Atezolizumab + Nab-Paclitaxel | Phase Ib | TNBC (stage IV or locally recurrent) | 33 | 2020 | 14.7 mo | 5.5 mo | 39% |
| KATE2 (NCT02924883) | Trastuzumab emtansine + Atezolizumab vs. Trastuzumab emtansine + Placebo | Phase II | HER2+ Locally Advanced/Metastatic BC with Prior Trastuzumab and Taxane Based Therapy | 202 | 2017 | - | 8.2 vs. 6.8 mo | 46% vs. 44% |
| IMpassion130 (NCT02425891) | Atezolizumab + Nab-paclitaxel vs. Placebo + Nab-paclitaxel | Phase III | Metastatic TNBC | 910 | 2019 | 21 mo vs. 18.7 mo | 7.2 mo vs 5.5 mo | 56% vs. 46% |
| IMpassion131 (NCT03125902) | Atezolizumab + Paclitaxel vs. Placebo + Paclitaxel | Phase III | TNBC (advanced or metastatic) | 651 | 2019 | 18.1 vs. 22.8 mo | 5.7 vs. 5.6 mo | 49% vs. 41% |
| JAVELIN (NCT01772004) | Avelumab | Phase I | Metastatic Breast Cancer (MBC) | 168 | 2019 | 8.4 mo | 1.4 mo | 3% |
| TONIC (NCT02499367) | Nivolumab + Radiation therapy/Doxorubicin/Cyclophosphamide/Cisplatin | Phase II | TNBC | 66 | Active, not recruiting | - | - | 20% |
5.2. Tumor Suppression
5.3. “Triple Negative” Breast Cancer (TNBC)
6. Autophagy, Immunotherapy, and Other Types of Treatment in Breast Cancer
6.1. Hormonal Therapy
6.2. Chemotherapy
6.3. Radiation Therapy
7. Conclusions

Author Contributions
Funding
Conflicts of Interest
References
- DeSantis, C.E.; Ma, J.; Goding Sauer, A.; Newman, L.A.; Jemal, A. Breast cancer statistics, 2017, racial disparity in mortality by state. CA Cancer J. Clin. 2017, 67, 439–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McPherson, K.; Steel, C.M.; Dixon, J.M. ABC of breast diseases. Breast cancer-epidemiology, risk factors, and genetics. BMJ 2000, 321, 624–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darby, S.; McGale, P.; Correa, C.; Taylor, C.; Arriagada, R.; Clarke, M.; Cutter, D.; Davies, C.; Ewertz, M.; Godwin, J.; et al. Effect of radiotherapy after breast-conserving surgery on 10-year recurrence and 15-year breast cancer death: Meta-analysis of individual patient data for 10,801 women in 17 randomised trials. Lancet 2011, 378, 1707–1716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef]
- Planes-Laine, G.; Rochigneux, P.; Bertucci, F.; Chrétien, A.S.; Viens, P.; Sabatier, R.; Gonçalves, A. PD-1/PD-L1 Targeting in Breast Cancer: The First Clinical Evidences Are Emerging. A Literature Review. Cancers 2019, 11, 1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, S.; Schmid, P.; Rugo, H.S.; Winer, E.P.; Loirat, D.; Awada, A.; Cescon, D.W.; Iwata, H.; Campone, M.; Nanda, R.; et al. Pembrolizumab monotherapy for previously treated metastatic triple-negative breast cancer: Cohort A of the phase II KEYNOTE-086 study. Ann. Oncol. 2019, 30, 397–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanmeerbeek, I.; Sprooten, J.; De Ruysscher, D.; Tejpar, S.; Vandenberghe, P.; Fucikova, J.; Spisek, R.; Zitvogel, L.; Kroemer, G.; Galluzzi, L.; et al. Trial watch: Chemotherapy-induced immunogenic cell death in immuno-oncology. Oncoimmunology 2020, 9, 1703449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deter, R.L.; Baudhuin, P.; De Duve, C. Participation of lysosomes in cellular autophagy induced in rat liver by glucagon. J. Cell Biol. 1967, 35, C11–C16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Duve, C.; Pressman, B.C.; Gianetto, R.; Wattiaux, R.; Appelmans, F. Tissue fractionation studies. 6. Intracellular distribution patterns of enzymes in rat-liver tissue. Biochem. J. 1955, 60, 604–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romero, M.A.; Bayraktar Ekmekcigil, O.; Bagca, B.G.; Avci, C.B.; Sabitaliyevich, U.Y.; Zhenisovna, T.G.; Aras, A.; Farooqi, A.A. Role of Autophagy in Breast Cancer Development and Progression: Opposite Sides of the Same Coin. Adv. Exp. Med. Biol. 2019, 1152, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Marinković, M.; Šprung, M.; Buljubašić, M.; Novak, I. Autophagy Modulation in Cancer: Current Knowledge on Action and Therapy. Oxid. Med. Cell. Longev. 2018, 2018, 8023821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lisiak, N.; Toton, E.; Rybczynska, M. Autophagy as a Potential Therapeutic Target in Breast Cancer Treatment. Curr. Cancer Drug Targets 2018, 18, 629–639. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rybstein, M.D.; Bravo-San Pedro, J.M.; Kroemer, G.; Galluzzi, L. The autophagic network and cancer. Nat. Cell Biol. 2018, 20, 243–251. [Google Scholar] [CrossRef]
- Chen, P.; Cescon, M.; Bonaldo, P. Autophagy-mediated regulation of macrophages and its applications for cancer. Autophagy 2014, 10, 192–200. [Google Scholar] [CrossRef]
- Qu, X.; Yu, J.; Bhagat, G.; Furuya, N.; Hibshoosh, H.; Troxel, A.; Rosen, J.; Eskelinen, E.L.; Mizushima, N.; Ohsumi, Y.; et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Investig. 2003, 112, 1809–1820. [Google Scholar] [CrossRef] [Green Version]
- Yun, C.W.; Lee, S.H. The Roles of Autophagy in Cancer. Int. J. Mol. Sci. 2018, 19, 3466. [Google Scholar] [CrossRef] [Green Version]
- Anding, A.L.; Baehrecke, E.H. Cleaning House: Selective Autophagy of Organelles. Dev. Cell 2017, 41, 10–22. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; He, S.; Ma, B. Autophagy and autophagy-related proteins in cancer. Mol. Cancer 2020, 19, 12. [Google Scholar] [CrossRef]
- Mathew, R.; Karantza-Wadsworth, V.; White, E. Role of autophagy in cancer. Nat. Rev. Cancer 2007, 7, 961–967. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Davis, T.; Loos, B.; Sishi, B.; Huisamen, B.; Strijdom, H.; Engelbrecht, A.M. Autophagy is essential for the maintenance of amino acids and ATP levels during acute amino acid starvation in MDAMB231 cells. Cell Biochem. Funct. 2018, 36, 65–79. [Google Scholar] [CrossRef] [PubMed]
- Ahn, C.H.; Jeong, E.G.; Lee, J.W.; Kim, M.S.; Kim, S.H.; Kim, S.S.; Yoo, N.J.; Lee, S.H. Expression of beclin-1, an autophagy-related protein, in gastric and colorectal cancers. Apmis 2007, 115, 1344–1349. [Google Scholar] [CrossRef] [PubMed]
- Cocco, S.; Leone, A.; Piezzo, M.; Caputo, R.; Di Lauro, V.; Di Rella, F.; Fusco, G.; Capozzi, M.; Gioia, G.D.; Budillon, A.; et al. Targeting Autophagy in Breast Cancer. Int. J. Mol. Sci. 2020, 21, 7836. [Google Scholar] [CrossRef] [PubMed]
- Duan, L.; Motchoulski, N.; Danzer, B.; Davidovich, I.; Shariat-Madar, Z.; Levenson, V.V. Prolylcarboxypeptidase regulates proliferation, autophagy, and resistance to 4-hydroxytamoxifen-induced cytotoxicity in estrogen receptor-positive breast cancer cells. J. Biol. Chem. 2011, 286, 2864–2876. [Google Scholar] [CrossRef] [Green Version]
- Cook, K.L.; Wärri, A.; Soto-Pantoja, D.R.; Clarke, P.A.; Cruz, M.I.; Zwart, A.; Clarke, R. Hydroxychloroquine inhibits autophagy to potentiate antiestrogen responsiveness in ER+ breast cancer. Clin. Cancer Res. 2014, 20, 3222–3232. [Google Scholar] [CrossRef] [Green Version]
- Amaral, C.; Augusto, T.V.; Tavares-da-Silva, E.; Roleira, F.M.F.; Correia-da-Silva, G.; Teixeira, N. Hormone-dependent breast cancer: Targeting autophagy and PI3K overcomes Exemestane-acquired resistance. J. Steroid Biochem. Mol. Biol. 2018, 183, 51–61. [Google Scholar] [CrossRef]
- Chen, G.; Ding, X.F.; Bouamar, H.; Pressley, K.; Sun, L.Z. Everolimus induces G1 cell cycle arrest through autophagy-mediated protein degradation of cyclin D1 in breast cancer cells. Am. J. Physiol. Cell Physiol. 2019, 317, C244–C252. [Google Scholar] [CrossRef]
- Loehberg, C.R.; Strissel, P.L.; Dittrich, R.; Strick, R.; Dittmer, J.; Dittmer, A.; Fabry, B.; Kalender, W.A.; Koch, T.; Wachter, D.L.; et al. Akt and p53 are potential mediators of reduced mammary tumor growth by cloroquine and the mTOR inhibitor RAD001. Biochem. Pharmacol. 2012, 83, 480–488. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, J.; Qiu, Y.; Jin, M.; Chen, X.; Fan, G.; Wang, R.; Kong, D. ZSTK474, a specific class I phosphatidylinositol 3-kinase inhibitor, induces G1 arrest and autophagy in human breast cancer MCF-7 cells. Oncotarget 2016, 7, 19897–19909. [Google Scholar] [CrossRef] [Green Version]
- Vijayaraghavan, S.; Karakas, C.; Doostan, I.; Chen, X.; Bui, T.; Yi, M.; Raghavendra, A.S.; Zhao, Y.; Bashour, S.I.; Ibrahim, N.K.; et al. CDK4/6 and autophagy inhibitors synergistically induce senescence in Rb positive cytoplasmic cyclin E negative cancers. Nat. Commun. 2017, 8, 15916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chittaranjan, S.; Bortnik, S.; Dragowska, W.H.; Xu, J.; Abeysundara, N.; Leung, A.; Go, N.E.; DeVorkin, L.; Weppler, S.A.; Gelmon, K.; et al. Autophagy inhibition augments the anticancer effects of epirubicin treatment in anthracycline-sensitive and -resistant triple-negative breast cancer. Clin. Cancer Res. 2014, 20, 3159–3173. [Google Scholar] [CrossRef] [Green Version]
- Arnaout, A.; Robertson, S.J.; Pond, G.R.; Lee, H.; Jeong, A.; Ianni, L.; Kroeger, L.; Hilton, J.; Coupland, S.; Gottlieb, C.; et al. A randomized, double-blind, window of opportunity trial evaluating the effects of chloroquine in breast cancer patients. Breast Cancer Res. Treat. 2019, 178, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, A.H. Introduction to checkpoint inhibitors and cancer immunotherapy. Immunol. Rev. 2017, 276, 5–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chikuma, S.; Kanamori, M.; Mise-Omata, S.; Yoshimura, A. Suppressors of cytokine signaling: Potential immune checkpoint molecules for cancer immunotherapy. Cancer Sci. 2017, 108, 574–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, J.C.; Zhang, X.; Fedorov, A.A.; Nathenson, S.G.; Almo, S.C. Structural basis for co-stimulation by the human CTLA-4/B7-2 complex. Nature 2001, 410, 604–608. [Google Scholar] [CrossRef] [PubMed]
- Karandikar, N.J.; Vanderlugt, C.L.; Walunas, T.L.; Miller, S.D.; Bluestone, J.A. CTLA-4: A negative regulator of autoimmune disease. J. Exp. Med. 1996, 184, 783–788. [Google Scholar] [CrossRef] [PubMed]
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992, 11, 3887–3895. [Google Scholar] [CrossRef] [PubMed]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef] [Green Version]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef] [Green Version]
- Wolchok, J.D.; Neyns, B.; Linette, G.; Negrier, S.; Lutzky, J.; Thomas, L.; Waterfield, W.; Schadendorf, D.; Smylie, M.; Guthrie, T., Jr.; et al. Ipilimumab monotherapy in patients with pretreated advanced melanoma: A randomised, double-blind, multicentre, phase 2, dose-ranging study. Lancet Oncol. 2010, 11, 155–164. [Google Scholar] [CrossRef]
- Robert, C.; Thomas, L.; Bondarenko, I.; O’Day, S.; Weber, J.; Garbe, C.; Lebbe, C.; Baurain, J.F.; Testori, A.; Grob, J.J.; et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N. Engl. J. Med. 2011, 364, 2517–2526. [Google Scholar] [CrossRef] [Green Version]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Agata, Y.; Kawasaki, A.; Nishimura, H.; Ishida, Y.; Tsubata, T.; Yagita, H.; Honjo, T. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int. Immunol. 1996, 8, 765–772. [Google Scholar] [CrossRef] [Green Version]
- Dong, H.; Zhu, G.; Tamada, K.; Chen, L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat. Med. 1999, 5, 1365–1369. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Strome, S.E.; Salomao, D.R.; Tamura, H.; Hirano, F.; Flies, D.B.; Roche, P.C.; Lu, J.; Zhu, G.; Tamada, K.; et al. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat. Med. 2002, 8, 793–800. [Google Scholar] [CrossRef]
- Carter, L.; Fouser, L.A.; Jussif, J.; Fitz, L.; Deng, B.; Wood, C.R.; Collins, M.; Honjo, T.; Freeman, G.J.; Carreno, B.M. PD-1:PD-L inhibitory pathway affects both CD4+ and CD8+ T cells and is overcome by IL-2. Eur. J. Immunol. 2002, 32, 634–643. [Google Scholar] [CrossRef]
- Iwai, Y.; Ishida, M.; Tanaka, Y.; Okazaki, T.; Honjo, T.; Minato, N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc. Natl. Acad. Sci. USA 2002, 99, 12293–12297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwai, Y.; Hamanishi, J.; Chamoto, K.; Honjo, T. Cancer immunotherapies targeting the PD-1 signaling pathway. J. Biomed. Sci. 2017, 24, 26. [Google Scholar] [CrossRef]
- Gatti-Mays, M.E.; Balko, J.M.; Gameiro, S.R.; Bear, H.D.; Prabhakaran, S.; Fukui, J.; Disis, M.L.; Nanda, R.; Gulley, J.L.; Kalinsky, K.; et al. If we build it they will come: Targeting the immune response to breast cancer. NPJ Breast Cancer 2019, 5, 37. [Google Scholar] [CrossRef] [Green Version]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Diéras, V.; Hegg, R.; Im, S.A.; Shaw Wright, G.; et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef] [PubMed]
- Adams, S.; Gatti-Mays, M.E.; Kalinsky, K.; Korde, L.A.; Sharon, E.; Amiri-Kordestani, L.; Bear, H.; McArthur, H.L.; Frank, E.; Perlmutter, J.; et al. Current Landscape of Immunotherapy in Breast Cancer: A Review. JAMA Oncol. 2019, 5, 1205–1214. [Google Scholar] [CrossRef] [PubMed]
- Budczies, J.; Bockmayr, M.; Denkert, C.; Klauschen, F.; Lennerz, J.K.; Györffy, B.; Dietel, M.; Loibl, S.; Weichert, W.; Stenzinger, A. Classical pathology and mutational load of breast cancer-integration of two worlds. J. Pathol. Clin. Res. 2015, 1, 225–238. [Google Scholar] [CrossRef] [Green Version]
- Luen, S.; Virassamy, B.; Savas, P.; Salgado, R.; Loi, S. The genomic landscape of breast cancer and its interaction with host immunity. Breast 2016, 29, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Kassardjian, A.; Shintaku, P.I.; Moatamed, N.A. Expression of immune checkpoint regulators, cytotoxic T lymphocyte antigen 4 (CTLA-4) and programmed death-ligand 1 (PD-L1), in female breast carcinomas. PLoS ONE 2018, 13, e0195958. [Google Scholar] [CrossRef] [Green Version]
- De Souza, A.S.C.; Gonçalves, L.B.; Lepique, A.P.; de Araujo-Souza, P.S. The Role of Autophagy in Tumor Immunology-Complex Mechanisms That May Be Explored Therapeutically. Front. Oncol. 2020, 10, 603661. [Google Scholar] [CrossRef]
- White, E. The role for autophagy in cancer. J. Clin. Investig. 2015, 125, 42–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rebecca, V.W.; Amaravadi, R.K. Emerging strategies to effectively target autophagy in cancer. Oncogene 2016, 35, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Mowers, E.E.; Sharifi, M.N.; Macleod, K.F. Autophagy in cancer metastasis. Oncogene 2017, 36, 1619–1630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenific, C.M.; Thorburn, A.; Debnath, J. Autophagy and metastasis: Another double-edged sword. Curr. Opin. Cell Biol. 2010, 22, 241–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.; Lee, J.Y.; Wei, H.; Tanabe, O.; Engel, J.D.; Morrison, S.J.; Guan, J.L. FIP200 is required for the cell-autonomous maintenance of fetal hematopoietic stem cells. Blood 2010, 116, 4806–4814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Rucker, E.B., 3rd; Zhou, B.P. Autophagy regulation in the development and treatment of breast cancer. Acta Biochim. Biophys. Sin. 2016, 48, 60–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathew, R.; Karp, C.M.; Beaudoin, B.; Vuong, N.; Chen, G.; Chen, H.Y.; Bray, K.; Reddy, A.; Bhanot, G.; Gelinas, C.; et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell 2009, 137, 1062–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Zhang, Y.; Chen, Y.; Qian, J.; Zhang, X.; Yu, K. A Novel mTORC1/2 Inhibitor (MTI-31) Inhibits Tumor Growth, Epithelial-Mesenchymal Transition, Metastases, and Improves Antitumor Immunity in Preclinical Models of Lung Cancer. Clin. Cancer Res. 2019, 25, 3630–3642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lastwika, K.J.; Wilson, W., 3rd; Li, Q.K.; Norris, J.; Xu, H.; Ghazarian, S.R.; Kitagawa, H.; Kawabata, S.; Taube, J.M.; Yao, S.; et al. Control of PD-L1 Expression by Oncogenic Activation of the AKT-mTOR Pathway in Non-Small Cell Lung Cancer. Cancer Res 2016, 76, 227–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, C.A.; Gupta, H.B.; Sareddy, G.; Pandeswara, S.; Lao, S.; Yuan, B.; Drerup, J.M.; Padron, A.; Conejo-Garcia, J.; Murthy, K.; et al. Tumor-Intrinsic PD-L1 Signals Regulate Cell Growth, Pathogenesis, and Autophagy in Ovarian Cancer and Melanoma. Cancer Res. 2016, 76, 6964–6974. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Wu, W.K.K.; Gao, J.; Li, Z.; Dong, B.; Lin, X.; Li, Y.; Li, Y.; Gong, J.; Qi, C.; et al. Autophagy inhibition enhances PD-L1 expression in gastric cancer. J. Exp. Clin. Cancer Res. 2019, 38, 140. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Kim, F.J.; Maher, C.M. Sigma1 Pharmacology in the Context of Cancer. Handb. Exp. Pharmacol. 2017, 244, 237–308. [Google Scholar] [CrossRef]
- Thomas, J.D.; Longen, C.G.; Oyer, H.M.; Chen, N.; Maher, C.M.; Salvino, J.M.; Kania, B.; Anderson, K.N.; Ostrander, W.F.; Knudsen, K.E.; et al. Sigma1 Targeting to Suppress Aberrant Androgen Receptor Signaling in Prostate Cancer. Cancer Res. 2017, 77, 2439–2452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrock, J.M.; Spino, C.M.; Longen, C.G.; Stabler, S.M.; Marino, J.C.; Pasternak, G.W.; Kim, F.J. Sequential cytoprotective responses to Sigma1 ligand-induced endoplasmic reticulum stress. Mol. Pharmacol. 2013, 84, 751–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maher, C.M.; Thomas, J.D.; Haas, D.A.; Longen, C.G.; Oyer, H.M.; Tong, J.Y.; Kim, F.J. Small-Molecule Sigma1 Modulator Induces Autophagic Degradation of PD-L1. Mol. Cancer Res. 2018, 16, 243–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, H.; Valk, E.; Leung, R.; Rudd, C.E. CTLA-4 activation of phosphatidylinositol 3-kinase (PI 3-K) and protein kinase B (PKB/AKT) sustains T-cell anergy without cell death. PLoS ONE 2008, 3, e3842. [Google Scholar] [CrossRef] [Green Version]
- Alissafi, T.; Banos, A.; Boon, L.; Sparwasser, T.; Ghigo, A.; Wing, K.; Vassilopoulos, D.; Boumpas, D.; Chavakis, T.; Cadwell, K.; et al. Tregs restrain dendritic cell autophagy to ameliorate autoimmunity. J. Clin. Investig. 2017, 127, 2789–2804. [Google Scholar] [CrossRef] [Green Version]
- Shukla, S.A.; Bachireddy, P.; Schilling, B.; Galonska, C.; Zhan, Q.; Bango, C.; Langer, R.; Lee, P.C.; Gusenleitner, D.; Keskin, D.B.; et al. Cancer-Germline Antigen Expression Discriminates Clinical Outcome to CTLA-4 Blockade. Cell 2018, 173, 624–633.e8. [Google Scholar] [CrossRef] [Green Version]
- Chu, E.C.; Tarnawski, A.S. PTEN regulatory functions in tumor suppression and cell biology. Med. Sci. Monit. 2004, 10, Ra235–Ra241. [Google Scholar]
- Peng, W.; Chen, J.Q.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M.T.; Xu, C.; McKenzie, J.A.; Zhang, C.; Liang, X.; et al. Loss of PTEN Promotes Resistance to T Cell-Mediated Immunotherapy. Cancer Discov. 2016, 6, 202–216. [Google Scholar] [CrossRef] [Green Version]
- Cuervo, A.M.; Dice, J.F. A receptor for the selective uptake and degradation of proteins by lysosomes. Science 1996, 273, 501–503. [Google Scholar] [CrossRef]
- Wang, R.; Liu, Y.; Liu, L.; Chen, M.; Wang, X.; Yang, J.; Gong, Y.; Ding, B.S.; Wei, Y.; Wei, X. Tumor cells induce LAMP2a expression in tumor-associated macrophage for cancer progression. EBioMedicine 2019, 40, 118–134. [Google Scholar] [CrossRef] [Green Version]
- Akalay, I.; Janji, B.; Hasmim, M.; Noman, M.Z.; Thiery, J.P.; Mami-Chouaib, F.; Chouaib, S. EMT impairs breast carcinoma cell susceptibility to CTL-mediated lysis through autophagy induction. Autophagy 2013, 9, 1104–1106. [Google Scholar] [CrossRef] [PubMed]
- Baginska, J.; Viry, E.; Berchem, G.; Poli, A.; Noman, M.Z.; van Moer, K.; Medves, S.; Zimmer, J.; Oudin, A.; Niclou, S.P.; et al. Granzyme B degradation by autophagy decreases tumor cell susceptibility to natural killer-mediated lysis under hypoxia. Proc. Natl. Acad. Sci. USA 2013, 110, 17450–17455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefort, S.; Joffre, C.; Kieffer, Y.; Givel, A.M.; Bourachot, B.; Zago, G.; Bieche, I.; Dubois, T.; Meseure, D.; Vincent-Salomon, A.; et al. Inhibition of autophagy as a new means of improving chemotherapy efficiency in high-LC3B triple-negative breast cancers. Autophagy 2014, 10, 2122–2142. [Google Scholar] [CrossRef] [PubMed]
- Liang, D.H.; Choi, D.S.; Ensor, J.E.; Kaipparettu, B.A.; Bass, B.L.; Chang, J.C. The autophagy inhibitor chloroquine targets cancer stem cells in triple negative breast cancer by inducing mitochondrial damage and impairing DNA break repair. Cancer Lett. 2016, 376, 249–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shoemaker, J.P. Fifty-five percent complete remission of mammary carcinoma in mice with 5-fluorouracil and chloroquine. Cancer Res. 1978, 38, 2700–2702. [Google Scholar]
- Shoemaker, J.P.; Dagher, R.K. Remissions of mammary adenocarcinoma in hypothyroid mice given 5-fluorouracil and chloroquine phosphate. J. Natl. Cancer Inst. 1979, 62, 1575–1578. [Google Scholar]
- Seront, E.; Boidot, R.; Bouzin, C.; Karroum, O.; Jordan, B.F.; Gallez, B.; Machiels, J.P.; Feron, O. Tumour hypoxia determines the potential of combining mTOR and autophagy inhibitors to treat mammary tumours. Br. J. Cancer 2013, 109, 2597–2606. [Google Scholar] [CrossRef] [Green Version]
- Dragowska, W.H.; Weppler, S.A.; Wang, J.C.; Wong, L.Y.; Kapanen, A.I.; Rawji, J.S.; Warburton, C.; Qadir, M.A.; Donohue, E.; Roberge, M.; et al. Induction of autophagy is an early response to gefitinib and a potential therapeutic target in breast cancer. PLoS ONE 2013, 8, e76503. [Google Scholar] [CrossRef] [Green Version]
- Cufí, S.; Vazquez-Martin, A.; Oliveras-Ferraros, C.; Corominas-Faja, B.; Cuyàs, E.; López-Bonet, E.; Martin-Castillo, B.; Joven, J.; Menendez, J.A. The anti-malarial chloroquine overcomes primary resistance and restores sensitivity to trastuzumab in HER2-positive breast cancer. Sci. Rep. 2013, 3, 2469. [Google Scholar] [CrossRef] [Green Version]
- Ratikan, J.A.; Sayre, J.W.; Schaue, D. Chloroquine engages the immune system to eradicate irradiated breast tumors in mice. Int. J. Radiat. Oncol. Biol. Phys. 2013, 87, 761–768. [Google Scholar] [CrossRef]
- Thomas, S.; Sharma, N.; Golden, E.B.; Cho, H.; Agarwal, P.; Gaffney, K.J.; Petasis, N.A.; Chen, T.C.; Hofman, F.M.; Louie, S.G.; et al. Preferential killing of triple-negative breast cancer cells in vitro and in vivo when pharmacological aggravators of endoplasmic reticulum stress are combined with autophagy inhibitors. Cancer Lett. 2012, 325, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr. Opin. Cell Biol. 2010, 22, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Wong, P.M.; Puente, C.; Ganley, I.G.; Jiang, X. The ULK1 complex: Sensing nutrient signals for autophagy activation. Autophagy 2013, 9, 124–137. [Google Scholar] [CrossRef]
- Chollat-Namy, M.; Ben Safta-Saadoun, T.; Haferssas, D.; Meurice, G.; Chouaib, S.; Thiery, J. The pharmalogical reactivation of p53 function improves breast tumor cell lysis by granzyme B and NK cells through induction of autophagy. Cell Death Dis. 2019, 10, 695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ladoire, S.; Enot, D.; Senovilla, L.; Ghiringhelli, F.; Poirier-Colame, V.; Chaba, K.; Semeraro, M.; Chaix, M.; Penault-Llorca, F.; Arnould, L.; et al. The presence of LC3B puncta and HMGB1 expression in malignant cells correlate with the immune infiltrate in breast cancer. Autophagy 2016, 12, 864–875. [Google Scholar] [CrossRef] [Green Version]
- Maycotte, P.; Gearheart, C.M.; Barnard, R.; Aryal, S.; Mulcahy Levy, J.M.; Fosmire, S.P.; Hansen, R.J.; Morgan, M.J.; Porter, C.C.; Gustafson, D.L.; et al. STAT3-mediated autophagy dependence identifies subtypes of breast cancer where autophagy inhibition can be efficacious. Cancer Res. 2014, 74, 2579–2590. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Tanikawa, T.; Kryczek, I.; Xia, H.; Li, G.; Wu, K.; Wei, S.; Zhao, L.; Vatan, L.; Wen, B.; et al. Aerobic Glycolysis Controls Myeloid-Derived Suppressor Cells and Tumor Immunity via a Specific CEBPB Isoform in Triple-Negative Breast Cancer. Cell Metab. 2018, 28, 87–103.e6. [Google Scholar] [CrossRef] [Green Version]
- Curry, A.; Khatri, I.; Kos, O.; Zhu, F.; Gorczynski, R. Importance of CD200 expression by tumor or host cells to regulation of immunotherapy in a mouse breast cancer model. PLoS ONE 2017, 12, e0171586. [Google Scholar] [CrossRef] [Green Version]
- Shi, M.; Yao, Y.; Han, F.; Li, Y.; Li, Y. MAP1S controls breast cancer cell TLR5 signaling pathway and promotes TLR5 signaling-based tumor suppression. PLoS ONE 2014, 9, e86839. [Google Scholar] [CrossRef] [Green Version]
- Xie, R.; Nguyen, S.; McKeehan, K.; Wang, F.; McKeehan, W.L.; Liu, L. Microtubule-associated protein 1S (MAP1S) bridges autophagic components with microtubules and mitochondria to affect autophagosomal biogenesis and degradation. J. Biol. Chem. 2011, 286, 10367–10377. [Google Scholar] [CrossRef] [Green Version]
- Altinoz, M.A.; Ozpinar, A.; Alturfan, E.E.; Elmaci, I. Vinorelbine’s anti-tumor actions may depend on the mitotic apoptosis, autophagy and inflammation: Hypotheses with implications for chemo-immunotherapy of advanced cancers and pediatric gliomas. J. Chemother. 2018, 30, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Aqbi, H.F.; Tyutyunyk-Massey, L.; Keim, R.C.; Butler, S.E.; Thekkudan, T.; Joshi, S.; Smith, T.M.; Bandyopadhyay, D.; Idowu, M.O.; Bear, H.D.; et al. Autophagy-deficient breast cancer shows early tumor recurrence and escape from dormancy. Oncotarget 2018, 9, 22113–22122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garg, A.D.; Dudek, A.M.; Ferreira, G.B.; Verfaillie, T.; Vandenabeele, P.; Krysko, D.V.; Mathieu, C.; Agostinis, P. ROS-induced autophagy in cancer cells assists in evasion from determinants of immunogenic cell death. Autophagy 2013, 9, 1292–1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baghdadi, M.; Yoneda, A.; Yamashina, T.; Nagao, H.; Komohara, Y.; Nagai, S.; Akiba, H.; Foretz, M.; Yoshiyama, H.; Kinoshita, I.; et al. TIM-4 glycoprotein-mediated degradation of dying tumor cells by autophagy leads to reduced antigen presentation and increased immune tolerance. Immunity 2013, 39, 1070–1081. [Google Scholar] [CrossRef] [Green Version]
- Zamame Ramirez, J.A.; Romagnoli, G.G.; Falasco, B.F.; Gorgulho, C.M.; Sanzochi Fogolin, C.; Dos Santos, D.C.; Junior, J.P.A.; Lotze, M.T.; Ureshino, R.P.; Kaneno, R. Blocking drug-induced autophagy with chloroquine in HCT-116 colon cancer cells enhances DC maturation and T cell responses induced by tumor cell lysate. Int. Immunopharmacol. 2020, 84, 106495. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giannopoulos, S.; Bozkus, C.C.; Zografos, E.; Athanasiou, A.; Bongiovanni, A.M.; Doulaveris, G.; Bakoyiannis, C.N.; Theodoropoulos, G.E.; Zografos, G.C.; Witkin, S.S.; et al. Targeting Both Autophagy and Immunotherapy in Breast Cancer Treatment. Metabolites 2022, 12, 966. https://doi.org/10.3390/metabo12100966
Giannopoulos S, Bozkus CC, Zografos E, Athanasiou A, Bongiovanni AM, Doulaveris G, Bakoyiannis CN, Theodoropoulos GE, Zografos GC, Witkin SS, et al. Targeting Both Autophagy and Immunotherapy in Breast Cancer Treatment. Metabolites. 2022; 12(10):966. https://doi.org/10.3390/metabo12100966
Chicago/Turabian StyleGiannopoulos, Spyridon, Cansu Cimen Bozkus, Eleni Zografos, Aikaterini Athanasiou, Ann Marie Bongiovanni, Georgios Doulaveris, Chris N. Bakoyiannis, Georgios E. Theodoropoulos, Georgios C. Zografos, Steven S. Witkin, and et al. 2022. "Targeting Both Autophagy and Immunotherapy in Breast Cancer Treatment" Metabolites 12, no. 10: 966. https://doi.org/10.3390/metabo12100966