Altered Brain Metabolome Is Associated with Memory Impairment in the rTg4510 Mouse Model of Tauopathy

 , , ,
, , , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Behavioral Tests
2.3. Preparation of Brain Extracts
2.4. Metabolomics Study
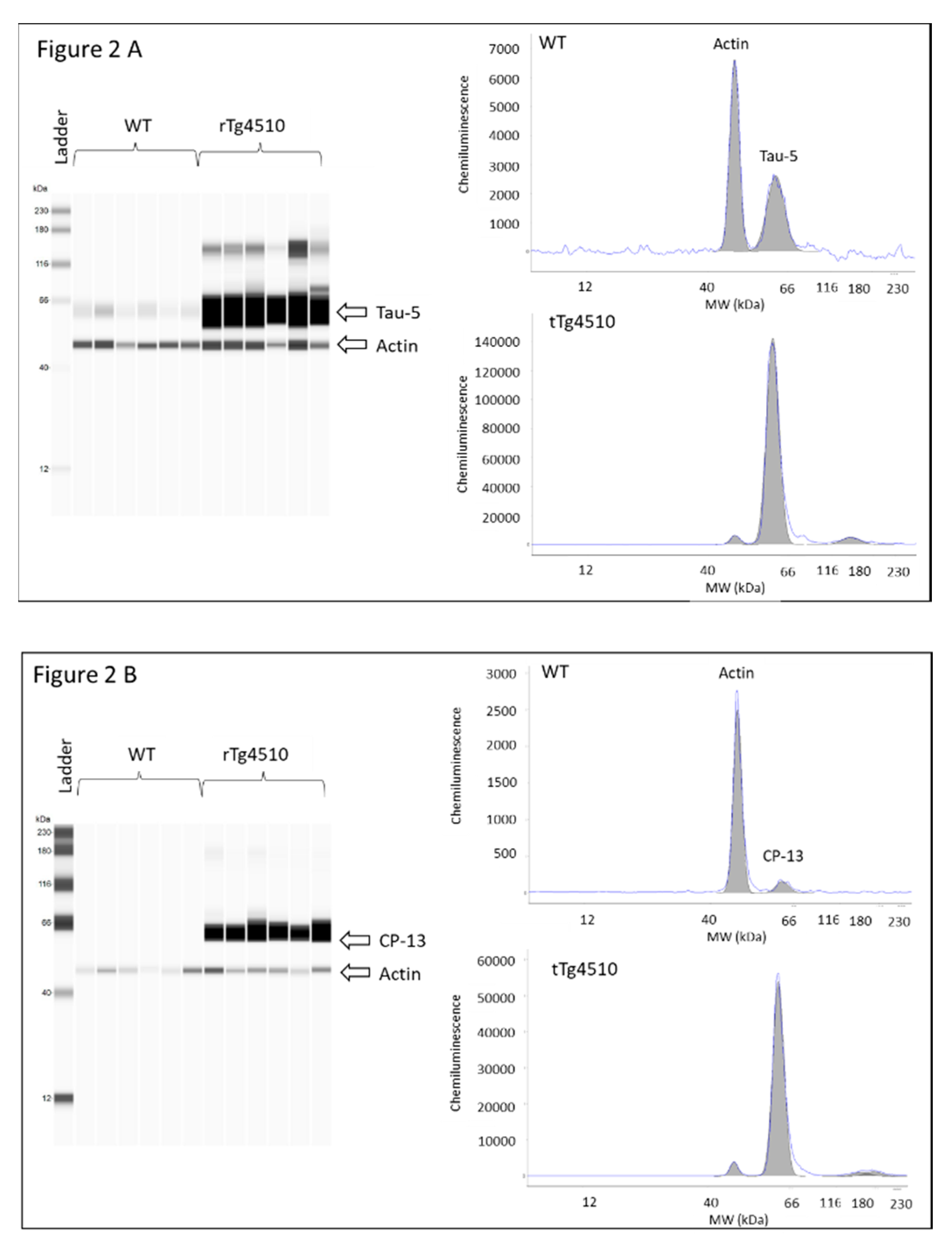
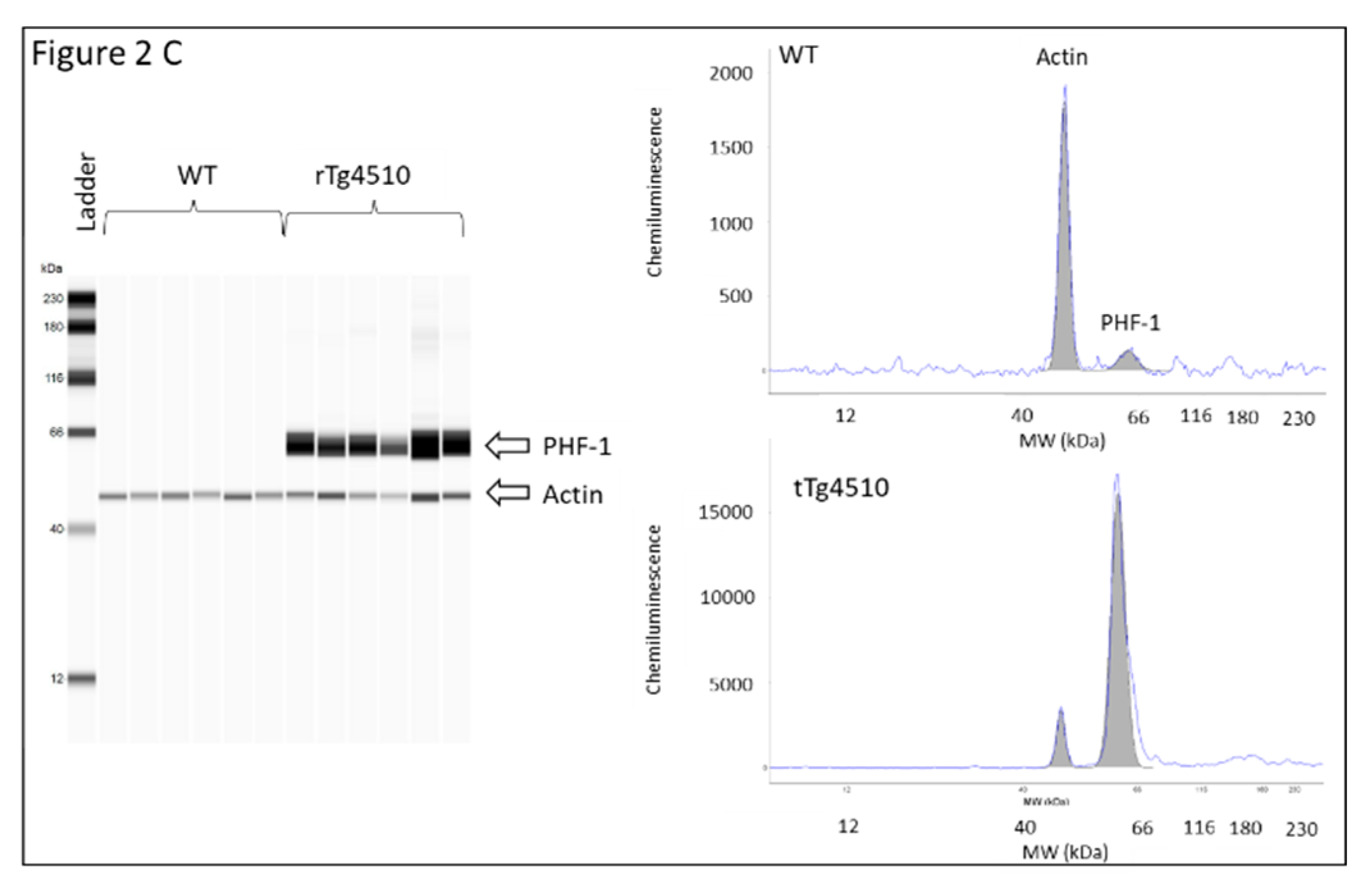
2.5. Determination of Protein Expression
2.6. Statistical Analysis
3. Results
3.1. Memory Impairment in the Transgenic Mice Model rTg4510 of Tauopathy
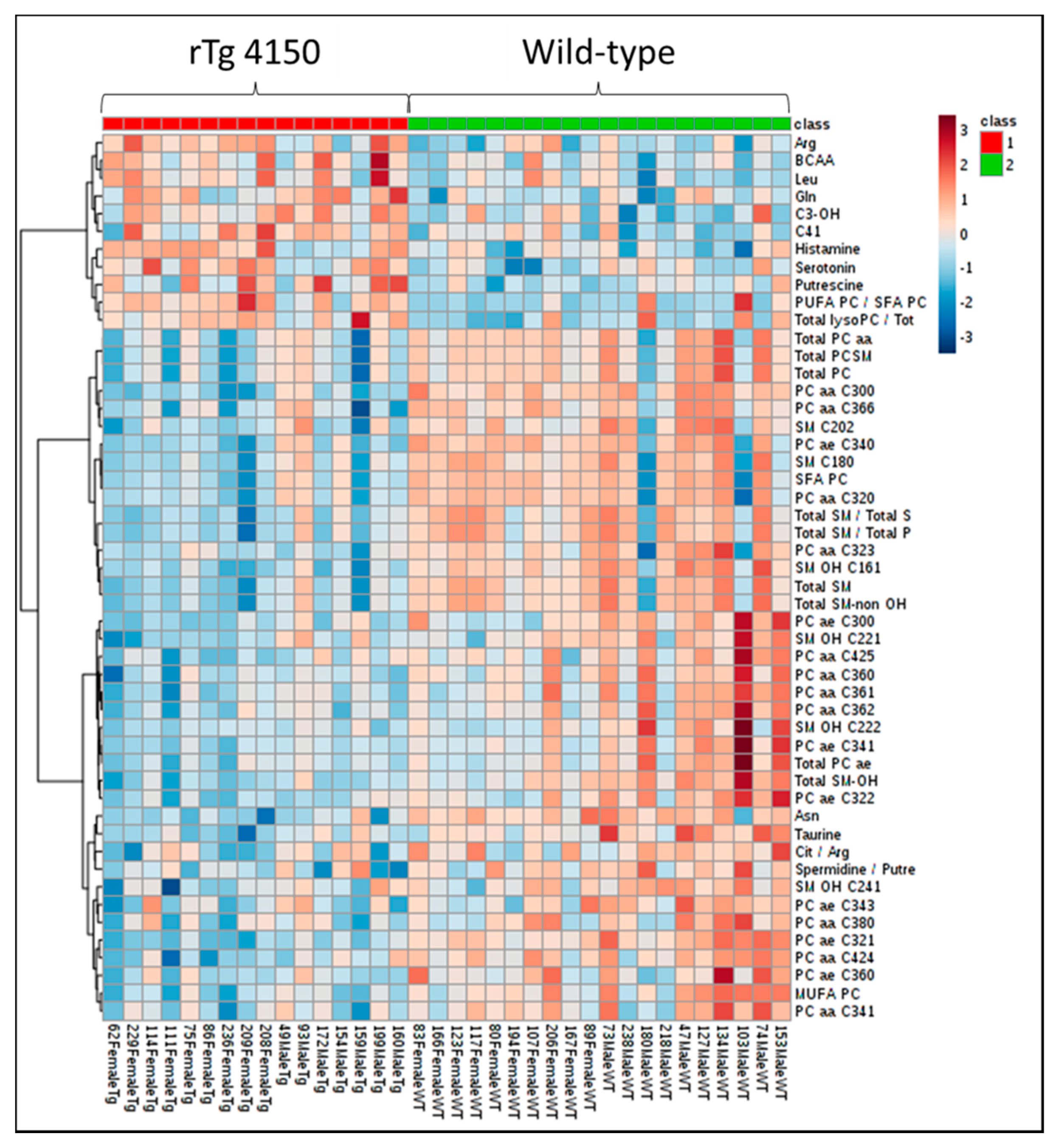
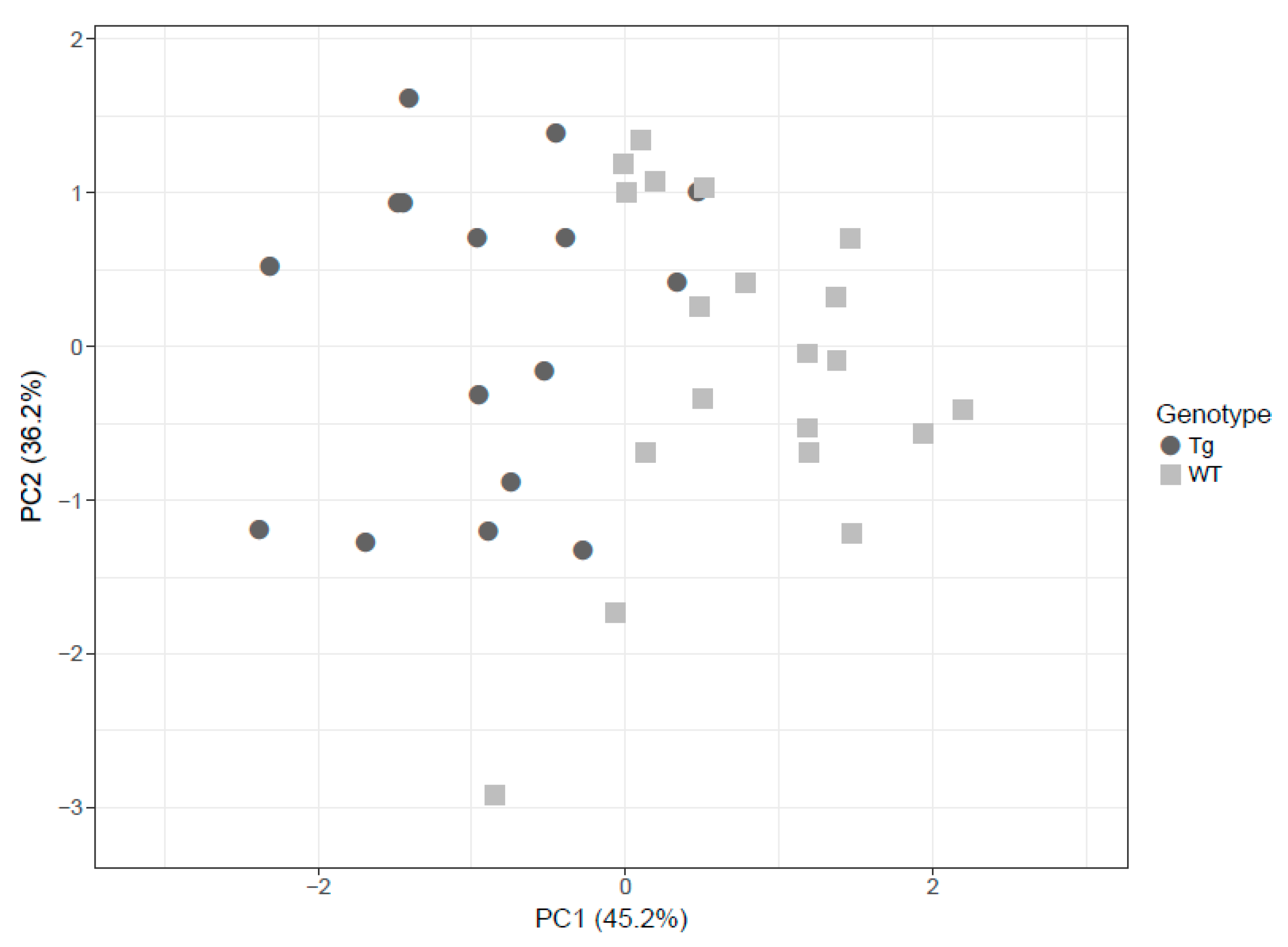
3.2. Altered Brain Metabolome Profile in rTg4510 Mice
3.3. Brain Metabolic Profile and Memory Impairment Associations
3.4. Brain Glutamine, Serotonin and SM C18:0 Are Predictors of Memory Impairment in rTg4510 Mice
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Aβ | amyloid beta |
| AD | Alzheimer’s disease |
| APP | amyloid precursor protein |
| CSF | cerebrospinal fluid |
| FTDP | frontotemporal dementia |
| LC | liquid chromatography |
| Lyso-PC | lysophosphatidylcholines |
| MAPT | mutant microtubule associated protein tau |
| MRM | multiple reaction monitoring |
| MWM | Morris Water Maze |
| NFT | neurofibrillary tangles |
| PCA | principal component analysis |
| PCR | protein chain reaction |
| PCs | phosphatidylcholines |
| PS1 | presenilin 1 |
| p-Tau | phosphorylated Tau |
| SPHs | sphingolipids |
| SM | sphingomyelin |
| WT | wild type |
References
- Prince, M.; Ali, G.C.; Guerchet, M.; Prina, A.M.; Albanese, E.; Wu, Y.T. Recent global trends in the prevalence and incidence of dementia, and survival with dementia. Alzheimers Res. Ther. 2016, 8, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blennow, K.; de Leon, M.J.; Zetterberg, H. Alzheimer’s disease. Lancet 2006, 368, 387–403. [Google Scholar] [CrossRef]
- Selkoe, D.J. Cell biology of protein misfolding: The examples of Alzheimer’s and Parkinson’s diseases. Nat. Cell Biol. 2004, 6, 1054–1061. [Google Scholar] [CrossRef] [PubMed]
- Skovronsky, D.M.; Lee, V.M.; Trojanowski, J.Q. Neurodegenerative diseases: New concepts of pathogenesis and their therapeutic implications. Annu. Rev. Pathol. 2006, 1, 151–170. [Google Scholar] [CrossRef]
- Gonzalez-Dominguez, R.; Garcia-Barrera, T.; Vitorica, J.; Gomez-Ariza, J.L. Application of metabolomics based on direct mass spectrometry analysis for the elucidation of altered metabolic pathways in serum from the APP/PS1 transgenic model of Alzheimer’s disease. J. Pharm. Biomed. Anal. 2015, 107, 378–385. [Google Scholar] [CrossRef]
- Graham, S.F.; Chevallier, O.P.; Roberts, D.; Holscher, C.; Elliott, C.T.; Green, B.D. Investigation of the human brain metabolome to identify potential markers for early diagnosis and therapeutic targets of Alzheimer’s disease. Anal. Chem. 2013, 85, 1803–1811. [Google Scholar] [CrossRef]
- Hu, Z.P.; Browne, E.R.; Liu, T.; Angel, T.E.; Ho, P.C.; Chan, E.C. Metabonomic profiling of TASTPM transgenic Alzheimer’s disease mouse model. J. Proteome Res. 2012, 11, 5903–5913. [Google Scholar] [CrossRef]
- Hughes, D.I.; Polgar, E.; Shehab, S.A.; Todd, A.J. Peripheral axotomy induces depletion of the vesicular glutamate transporter VGLUT1 in central terminals of myelinated afferent fibres in the rat spinal cord. Brain Res. 2004, 1017, 69–76. [Google Scholar] [CrossRef]
- Pan, X.; Nasaruddin, M.B.; Elliott, C.T.; McGuinness, B.; Passmore, A.P.; Kehoe, P.G.; Holscher, C.; McClean, P.L.; Graham, S.F.; Green, B.D. Alzheimer’s disease-like pathology has transient effects on the brain and blood metabolome. Neurobiol. Aging 2016, 38, 151–163. [Google Scholar] [CrossRef] [Green Version]
- Salek, R.M.; Xia, J.; Innes, A.; Sweatman, B.C.; Adalbert, R.; Randle, S.; McGowan, E.; Emson, P.C.; Griffin, J.L. A metabolomic study of the CRND8 transgenic mouse model of Alzheimer’s disease. Neurochem. Int. 2010, 56, 937–947. [Google Scholar] [CrossRef]
- Trushina, E.; Nemutlu, E.; Zhang, S.; Christensen, T.; Camp, J.; Mesa, J.; Siddiqui, A.; Tamura, Y.; Sesaki, H.; Wengenack, T.M.; et al. Defects in mitochondrial dynamics and metabolomic signatures of evolving energetic stress in mouse models of familial Alzheimer’s disease. PLoS ONE 2012, 7, e32737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramsden, M.; Kotilinek, L.; Forster, C.; Paulson, J.; McGowan, E.; SantaCruz, K.; Guimaraes, A.; Yue, M.; Lewis, J.; Carlson, G.; et al. Age-dependent neurofibrillary tangle formation, neuron loss, and memory impairment in a mouse model of human tauopathy (P301L). J. Neurosci. 2005, 25, 10637–10647. [Google Scholar] [CrossRef] [PubMed]
- Santacruz, K.; Lewis, J.; Spires, T.; Paulson, J.; Kotilinek, L.; Ingelsson, M.; Guimaraes, A.; DeTure, M.; Ramsden, M.; McGowan, E.; et al. Tau suppression in a neurodegenerative mouse model improves memory function. Science 2005, 309, 476–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spires, T.L.; Orne, J.D.; SantaCruz, K.; Pitstick, R.; Carlson, G.A.; Ashe, K.H.; Hyman, B.T. Region-specific dissociation of neuronal loss and neurofibrillary pathology in a mouse model of tauopathy. Am. J. Pathol. 2006, 168, 1598–1607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beckonert, O.; Keun, H.C.; Ebbels, T.M.; Bundy, J.; Holmes, E.; Lindon, J.C.; Nicholson, J.K. Metabolic profiling, metabolomic and metabonomic procedures for NMR spectroscopy of urine, plasma, serum and tissue extracts. Nat. Protoc. 2007, 2, 2692–2703. [Google Scholar] [CrossRef]
- Fiehn, O. Metabolomics--the link between genotypes and phenotypes. Plant Mol. Biol. 2002, 48, 155–171. [Google Scholar] [CrossRef]
- Jiang, Y.; Zhu, Z.; Shi, J.; An, Y.; Zhang, K.; Wang, Y.; Li, S.; Jin, L.; Ye, W.; Cui, M.; et al. Metabolomics in the Development and Progression of Dementia: A Systematic Review. Front. Neurosci. 2019, 13, 343. [Google Scholar] [CrossRef] [Green Version]
- Jimenez-Jimenez, F.J.; Molina, J.A.; Gomez, P.; Vargas, C.; de Bustos, F.; Benito-Leon, J.; Tallon-Barranco, A.; Orti-Pareja, M.; Gasalla, T.; Arenas, J. Neurotransmitter amino acids in cerebrospinal fluid of patients with Alzheimer’s disease. J. Neural Transm. 1998, 105, 269–277. [Google Scholar] [CrossRef]
- Nkuipou-Kenfack, E.; Duranton, F.; Gayrard, N.; Argiles, A.; Lundin, U.; Weinberger, K.M.; Dakna, M.; Delles, C.; Mullen, W.; Husi, H.; et al. Assessment of metabolomic and proteomic biomarkers in detection and prognosis of progression of renal function in chronic kidney disease. PLoS ONE 2014, 9, e96955. [Google Scholar] [CrossRef] [Green Version]
- Wang-Sattler, R.; Yu, Z.; Herder, C.; Messias, A.C.; Floegel, A.; He, Y.; Heim, K.; Campillos, M.; Holzapfel, C.; Thorand, B.; et al. Novel biomarkers for pre-diabetes identified by metabolomics. Mol. Syst. Biol. 2012, 8, 615. [Google Scholar] [CrossRef]
- Metsalu, T.; Vilo, J. ClustVis: A web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res. 2015, 43, W566–W570. [Google Scholar] [CrossRef]
- Barrier, L.; Fauconneau, B.; Noel, A.; Ingrand, S. Ceramide and Related-Sphingolipid Levels Are Not Altered in Disease-Associated Brain Regions of APP and APP/PS1 Mouse Models of Alzheimer’s Disease: Relationship with the Lack of Neurodegeneration? Int. J. Alzheimers Dis. 2010, 2011, 920958. [Google Scholar] [PubMed] [Green Version]
- Sontag, J.M.; Nunbhakdi-Craig, V.; Montgomery, L.; Arning, E.; Bottiglieri, T.; Sontag, E. Folate deficiency induces in vitro and mouse brain region-specific downregulation of leucine carboxyl methyltransferase-1 and protein phosphatase 2A B(alpha) subunit expression that correlate with enhanced tau phosphorylation. J. Neurosci. 2008, 28, 11477–11487. [Google Scholar] [CrossRef] [PubMed]
- Berger, Z.; Roder, H.; Hanna, A.; Carlson, A.; Rangachari, V.; Yue, M.; Wszolek, Z.; Ashe, K.; Knight, J.; Dickson, D.; et al. Accumulation of pathological tau species and memory loss in a conditional model of tauopathy. J. Neurosci. 2007, 27, 3650–3662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janus, C. Conditionally inducible tau mice--Designing a better mouse model of neurodegenerative diseases. Genes Brain Behav. 2008, 7 (Suppl. 1), 12–27. [Google Scholar] [CrossRef] [PubMed]
- Spires, T.L.; Meyer-Luehmann, M.; Stern, E.A.; McLean, P.J.; Skoch, J.; Nguyen, P.T.; Bacskai, B.J.; Hyman, B.T. Dendritic spine abnormalities in amyloid precursor protein transgenic mice demonstrated by gene transfer and intravital multiphoton microscopy. J. Neurosci. 2005, 25, 7278–7287. [Google Scholar] [CrossRef]
- Yue, M.; Hanna, A.; Wilson, J.; Roder, H.; Janus, C. Sex difference in pathology and memory decline in rTg4510 mouse model of tauopathy. Neurobiol. Aging 2011, 32, 590–603. [Google Scholar] [CrossRef]
- Wasek, B.; Arning, E.; Bottiglieri, T. The use of microwave irradiation for quantitative analysis of neurotransmitters in the mouse brain. J. Neurosci. Methods 2018, 307, 188–193. [Google Scholar] [CrossRef]
- Kosicek, M.; Zetterberg, H.; Andreasen, N.; Peter-Katalinic, J.; Hecimovic, S. Elevated cerebrospinal fluid sphingomyelin levels in prodromal Alzheimer’s disease. Neurosci. Lett. 2012, 516, 302–305. [Google Scholar] [CrossRef]
- Han, X.; Rozen, S.; Boyle, S.H.; Hellegers, C.; Cheng, H.; Burke, J.R.; Welsh-Bohmer, K.A.; Doraiswamy, P.M.; Kaddurah-Daouk, R. Metabolomics in early Alzheimer’s disease: Identification of altered plasma sphingolipidome using shotgun lipidomics. PLoS ONE 2011, 6, e21643. [Google Scholar] [CrossRef]
- Li, N.J.; Liu, W.T.; Li, W.; Li, S.Q.; Chen, X.H.; Bi, K.S.; He, P. Plasma metabolic profiling of Alzheimer’s disease by liquid chromatography/mass spectrometry. Clin. Biochem. 2010, 43, 992–997. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Xie, S.; Yang, J.; Yang, Q.; Xu, G. Structural identification of human blood phospholipids using liquid chromatogrpahy/quadrupole-linear ion trap mass spectrometry. Anal. Chim. Acta 2004, 525, 10. [Google Scholar] [CrossRef]
- Chan, R.B.; Oliveira, T.G.; Cortes, E.P.; Honig, L.S.; Duff, K.E.; Small, S.A.; Wenk, M.R.; Shui, G.; Di Paolo, G. Comparative lipidomic analysis of mouse and human brain with Alzheimer disease. J. Biol. Chem. 2012, 287, 2678–2688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cutler, R.G.; Kelly, J.; Storie, K.; Pedersen, W.A.; Tammara, A.; Hatanpaa, K.; Troncoso, J.C.; Mattson, M.P. Involvement of oxidative stress-induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2004, 101, 2070–2075. [Google Scholar] [CrossRef] [Green Version]
- Grimm, M.O.; Grimm, H.S.; Patzold, A.J.; Zinser, E.G.; Halonen, R.; Duering, M.; Tschape, J.A.; De Strooper, B.; Muller, U.; Shen, J.; et al. Regulation of cholesterol and sphingomyelin metabolism by amyloid-beta and presenilin. Nat. Cell Biol. 2005, 7, 1118–1123. [Google Scholar] [CrossRef]
- Dekker, A.D.; Coppus, A.M.; Vermeiren, Y.; Aerts, T.; van Duijn, C.M.; Kremer, B.P.; Naude, P.J.; Van Dam, D.; De Deyn, P.P. Serum MHPG strongly predicts conversion to Alzheimer’s disease in behaviorally characterized subjects with Down syndrome. J. Alzheimers Dis. 2015, 43, 871–891. [Google Scholar] [CrossRef] [Green Version]
- Meneses, A.; Liy-Salmeron, G. Serotonin and emotion, learning and memory. Rev. Neurosci. 2012, 23, 543–553. [Google Scholar] [CrossRef]
- Gonzalez-Dominguez, R.; Garcia-Barrera, T.; Gomez-Ariza, J.L. Using direct infusion mass spectrometry for serum metabolomics in Alzheimer’s disease. Anal. Bioanal. Chem. 2014, 406, 7137–7148. [Google Scholar] [CrossRef]
- Storga, D.; Vrecko, K.; Birkmayer, J.G.; Reibnegger, G. Monoaminergic neurotransmitters, their precursors and metabolites in brains of Alzheimer patients. Neurosci. Lett. 1996, 203, 29–32. [Google Scholar] [CrossRef]
- Danbolt, N.C. Glutamate uptake. Prog. Neurobiol. 2001, 65, 1–105. [Google Scholar] [CrossRef]
- Kulijewicz-Nawrot, M.; Sykova, E.; Chvatal, A.; Verkhratsky, A.; Rodriguez, J.J. Astrocytes and glutamate homoeostasis in Alzheimer’s disease: A decrease in glutamine synthetase, but not in glutamate transporter-1, in the prefrontal cortex. ASN Neuro 2013, 5, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Olabarria, M.; Noristani, H.N.; Verkhratsky, A.; Rodriguez, J.J. Age-dependent decrease in glutamine synthetase expression in the hippocampal astroglia of the triple transgenic Alzheimer’s disease mouse model: Mechanism for deficient glutamatergic transmission? Mol. Neurodegener. 2011, 6, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, S.R. Changes in the cellular distribution of glutamine synthetase in Alzheimer’s disease. J. Neurosci. Res. 2001, 66, 972–980. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Jiménez, F.J.; Molina, J.A.; Vargas, C.; Gómez, P.; Navarro, J.A.; Benito-León, J.; Ortí-Pareja, M.; Gasalla, T.; Cisneros, E.; Arenas, J. Neurotransmitter Amino Acids in Cerebrospinal Fluid of Patients with Parkinson’s Disease. J. Neurol Sci. 1996, 141, 39–44. [Google Scholar] [CrossRef]
- Pomara, N.; Singh, R.; Deptula, D.; Chou, J.C.; Schwartz, M.B.; LeWitt, P.A. Glutamate and other CSF amino acids in Alzheimer’s disease. Am. J. Psychiatry 1992, 149, 251–254. [Google Scholar] [PubMed]
- Martinez, M.; Frank, A.; Diez-Tejedor, E.; Hernanz, A. Amino acid concentrations in cerebrospinal fluid and serum in Alzheimer’s disease and vascular dementia. J. Neural Transm. Park. Dis. Dement. Sect. 1993, 6, 1–9. [Google Scholar] [CrossRef]
- Tohgi, H.; Abe, T.; Takahashi, S.; Kimura, M. A selective reduction of excitatory amino acids in cerebrospinal fluid of patients with Alzheimer type dementia compared with vascular dementia of the Binswanger type. Neurosci. Lett. 1992, 141, 5–8. [Google Scholar] [CrossRef]
- Degrell, I.; Hellsing, K.; Nagy, E.; Niklasson, F. Amino acid concentrations in cerebrospinal fluid in presenile and senile dementia of Alzheimer type and multi-infarct dementia. Arch. Gerontol. Geriatr. 1989, 9, 123–135. [Google Scholar] [CrossRef]
- Manyevitch, R.; Protas, M.; Scarpiello, S.; Deliso, M.; Bass, B.; Nanajian, A.; Chang, M.; Thompson, S.M.; Khoury, N.; Gonnella, R.; et al. Evaluation of Metabolic and Synaptic Dysfunction Hypotheses of Alzheimer’s Disease (AD): A Meta-Analysis of CSF Markers. Curr. Alzheimer Res. 2018, 15, 164–181. [Google Scholar] [CrossRef]
- Smith, C.C.; Bowen, D.M.; Francis, P.T.; Snowden, J.S.; Neary, D. Putative amino acid transmitters in lumbar cerebrospinal fluid of patients with histologically verified Alzheimer’s dementia. J. Neurol. Neurosurg. Psychiatry 1985, 48, 469–471. [Google Scholar] [CrossRef]
- Andersen, J.V.; Christensen, S.K.; Aldana, B.I.; Nissen, J.D.; Tanila, H.; Waagepetersen, H.S. Alterations in Cerebral Cortical Glucose and Glutamine Metabolism Precedes Amyloid Plaques in the APPswe/PSEN1dE9 Mouse Model of Alzheimer’s Disease. Neurochem. Res. 2017, 42, 1589–1598. [Google Scholar] [CrossRef] [PubMed]
- Madeira, C.; Vargas-Lopes, C.; Brandao, C.O.; Reis, T.; Laks, J.; Panizzutti, R.; Ferreira, S.T. Elevated Glutamate and Glutamine Levels in the Cerebrospinal Fluid of Patients With Probable Alzheimer’s Disease and Depression. Front. Psychiatry 2018, 9, 561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Balayssac, S.; Déjean, S.; Lalande, V.; Gilard, M.; Malet-Martino, M. A toolbox to explore NMR metabolomic data sets using the R environment. Chemom. Intell. Lab. Syst. 2013, 126, 9. [Google Scholar] [CrossRef]
- Dedeoglu, A.; Choi, J.K.; Cormier, K.; Kowall, N.W.; Jenkins, B.G. Magnetic resonance spectroscopic analysis of Alzheimer’s disease mouse brain that express mutant human APP shows altered neurochemical profile. Brain Res. 2004, 1012, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Kanawati, B.; Liu, L.; Witting, M.; Li, M.; Huang, J.; Schmitt-Kopplin, P.; Cai, Z. Ultrahigh resolution mass spectrometry-based metabolic characterization reveals cerebellum as a disturbed region in two animal models. Talanta 2014, 118, 45–53. [Google Scholar] [CrossRef]
- Wang, H.; Lian, K.; Han, B.; Wang, Y.; Kuo, S.H.; Geng, Y.; Qiang, J.; Sun, M.; Wang, M. Age-related alterations in the metabolic profile in the hippocampus of the senescence-accelerated mouse prone 8: A spontaneous Alzheimer’s disease mouse model. J. Alzheimers Dis. 2014, 39, 841–848. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Dominguez, R.; Garcia-Barrera, T.; Vitorica, J.; Gomez-Ariza, J.L. Region-sp ecific metabolic alterations in the brain of the APP/PS1 transgenic mice of Alzheimer’s disease. Biochim. Biophys. Acta 2014, 1842, 2395–2402. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.; Liu, H.; Kanawati, B.; Liu, L.; Dong, J.; Li, M.; Huang, J.; Schmitt-Kopplin, P.; Cai, Z. Hippocampal metabolomics using ultrahigh-resolution mass spectrometry reveals neuroinflammation from Alzheimer’s disease in CRND8 mice. Anal. Bioanal. Chem. 2013, 405, 5105–5117. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolite | WT | rTg4510 | t.stat | p-Value | FDR |
|---|---|---|---|---|---|
| Glycerophospholipids | |||||
| PC aa C30:0 | 3.71 ± 0.54 | 2.47 ± 0.45 | −7.5081 | <0.0001 | 0.0000012 |
| PC aa C32:0 | 101.9 ± 22.4 | 75.5 ± 15.0 | −3.2926 | 0.0023 | 0.0144960 |
| PC ae C32:1 | 1.66 ± 0.28 | 1.11 ± 0.147 | −8.0568 | <0.0001 | 0.0000005 |
| PC aa C42:4 | 0.146 ± 0.02 | 0.113 ± 0.018 | −5.0941 | <0.0001 | 0.0002996 |
| PC aa C32:3 | 0.228 ± 0.050 | 0.178 ± 0.022 | −3.3024 | 0.0023 | 0.0144960 |
| PC aa C34:1 | 146.9 ± 9.2 | 136.9 ± 7.1 | −3.577 | 0.0011 | 0.0082230 |
| PC aa C36:6 | 0.295 ± 0.037 | 0.239 ± 0.045 | −4.0477 | 0.0003 | 0.0032613 |
| PC ae C32:2 | 0.183 ± 0.037 | 0.143 ± 0.015 | −4.4828 | 0.0001 | 0.0014143 |
| PC aa C36:0 | 1.66 ± 0.94 | 0.815 ± 0.266 | −4.3839 | 0.0001 | 0.0017034 |
| PC ae C30:0 | 0.144 ± 0.024 | 0.118 ± 0.011 | −4.2498 | 0.0002 | 0.0021383 |
| PC ae C34:0 | 1.23 ± 0.35 | 0.687 ± 0.244 | −4.9941 | <0.0001 | 0.0003672 |
| PC ae C34:3 | 0.154 ± 0.025 | 0.13 ± 0.026 | −2.9554 | 0.0056 | 0.0264610 |
| PC ae C36:0 | 0.358 ± 0.075 | 0.296 ± 0.036 | −3.1638 | 0.0033 | 0.0171870 |
| PC aa C36:1 | 62.9 ± 10.3 | 51.1 ± 5.4 | −4.2241 | 0.0002 | 0.0021763 |
| PC ae C34:1 | 9.87 ± 2.93 | 7.33 ± 0.62 | −4.0578 | 0.0003 | 0.0032613 |
| PC aa C42:5 | 0.180 ± 0.039 | 0.141 ± 0.020 | −3.8824 | 0.0005 | 0.0044484 |
| PC aa C36:2 | 41.7 ± 7.9 | 34.1 ± 3.8 | −3.8131 | 0.0006 | 0.0050945 |
| PC aa C38:0 | 0.340 ± 0.044 | 0.301 ± 0.036 | −2.9493 | 0.0057 | 0.0264610 |
| PC ae C30:1 | 0.047 ± 0.012 | 0.038 ± 0.008 | −2.9132 | 0.0063 | 0.0284540 |
| PC ae C36:1 | 3.54 ± 1.56 | 2.52 ± 0.46 | −2.8434 | 0.0075 | 0.0333190 |
| PC aa C28:1 | 0.093 ± 0.026 | 0.0763 ± 0.007 | −2.7483 | 0.0095 | 0.0399800 |
| Amino acids | |||||
| Arg | 11.1 ± 3.1 | 15.8 ± 4.3 | 3.8875 | 0.0004 | 0.0044484 |
| Asn | 24.7 ± 2.8 | 19.4 ± 2.5 | −5.9583 | 0.00001 | 0.0000563 |
| Gln | 1739 ± 295 | 2162 ± 435 | 3.379 | 0.0018 | 0.0123810 |
| Tau | 579 ± 29 | 541 ± 22 | −4.3707 | 0.0001 | 0.0017034 |
| Leu | 14.3 ± 4.0 | 20.0 ± 7.2 | 3.2244 | 0.0028 | 0.0153300 |
| Pro | 23.4 ± 3.8 | 27.4 ± 4.8 | 2.7764 | 0.0089 | 0.0379620 |
| Trp | 5.56 ± 1.21 | 6.90 ± 1.77 | 2.6795 | 0.0113 | 0.0457270 |
| Val | 26.3 ± 4.4 | 31.4 ± 6.8 | 2.7782 | 0.0088 | 0.0379620 |
| Hist | 2.18 ± 0.57 | 3.0 ± 0.78 | 3.4083 | 0.0017 | 0.0118830 |
| Biogenic Amines | |||||
| Putrescine | 1.26 ± 0.20 | 1.65 ± 0.51 | 3.0893 | 0.0040 | 0.0191700 |
| Serotonin | 1.33 ± 0.26 | 1.66 ± 0.32 | 3.3716 | 0.0019 | 0.0123810 |
| Sphingolipids | |||||
| SM OH C16:1 | 0.220 ± 0.038 | 0.154 ± 0.024 | −6.3622 | <0.0001 | 0.0000225 |
| SM OH C22:1 | 0.090 ± 0.038 | 0.059 ± 0.020 | −3.5462 | 0.0012 | 0.0086668 |
| SM OH C22:2 | 0.122 ± 0.091 | 0.066 ± 0.012 | −3.1426 | 0.0035 | 0.0174490 |
| SM OH C24:1 | 0.134 ± 0.020 | 0.112 ± 0.021 | −3.1212 | 0.0037 | 0.0180030 |
| SM C26:1 | 0.05 ± 0.03 | 0.032 ± 0.015 | −2.7332 | 0.0099 | 0.0407570 |
| SM C18:0 | 30.4 ± 8.2 | 20.8 ± 5.2 | −3.6524 | 0.0009 | 0.0074096 |
| SM C20:2 | 0.123 ± 0.029 | 0.091 ± 0.024 | −3.603 | 0.0010 | 0.0079330 |
| Acylcarnitines | |||||
| C3-OH | 0.181 ± 0.02 | 0.2 ± 0.016 | 3.2767 | 0.0024 | 0.0147300 |
| C4:1 | 0.016 ± 0.002 | 0.019 ± 0.003 | 3.1925 | 0.0030 | 0.0162960 |
| Metabolite | lysoPC a C16:0 | lysoPC a C17:0 | PC aa C30:0 | PC aa C32:0 | PC aa C32:2 | PC aa C32:3 | PC aa C34:1 | PC aa C36:6 | PC aa C42:4 | PC ae C32:1 | PC ae C34:0 | PC ae C36:0 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| % Path in target | ρ | −0.423 | −0.422 | 0.477 | 0.451 | 0.348 | 0.358 | 0.346 | 0.358 | 0.447 | 0.449 | 0.499 | 0.468 |
| p-value | 0.010 | 0.010 | 0.003 | 0.006 | 0.038 | 0.032 | 0.039 | 0.032 | 0.006 | 0.006 | 0.002 | 0.004 | |
| Amino acids | Ala | Asn | Gln | Met | Orn | ||||||||
| % Path in target | ρ | 0.404 | 0.387 | −0.443 | 0.35 | −0.354 | |||||||
| p-value | 0.014 | 0.020 | 0.007 | 0.037 | 0.034 | ||||||||
| Biogenic amines | Ac-Orn | Putrescine | Serotonin | ||||||||||
| % Path in target | ρ | −0.338 | −0.49 | −0.54 | |||||||||
| p-value | 0.044 | 0.002 | 0.001 | ||||||||||
| Sphyngolipids | SM (OH) C16:1 | SM C18:0 | |||||||||||
| % Path in target | ρ | 0.415 | 0.44 | ||||||||||
| p-value | 0.012 | 0.007 | |||||||||||
| Acylcarnitines | C10:1 | C10:2 | C16:2 | C5-OH (C3-DC-M) | C5:1-DC | ||||||||
| % Path in target | ρ | −0.376 | 0.348 | −0.378 | −0.363 | 0.336 | |||||||
| p-value | 0.024 | 0.037 | 0.023 | 0.030 | 0.045 | ||||||||
| Model | Metabolite | β | p-Value | R2 |
|---|---|---|---|---|
| Gln | −0.403 | 0.004 | 0.521 | |
| Serotonin | −0.325 | 0.021 | ||
| SM C18:0 | 0.380 | 0.007 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tondo, M.; Wasek, B.; Escola-Gil, J.C.; de Gonzalo-Calvo, D.; Harmon, C.; Arning, E.; Bottiglieri, T. Altered Brain Metabolome Is Associated with Memory Impairment in the rTg4510 Mouse Model of Tauopathy. Metabolites 2020, 10, 69. https://doi.org/10.3390/metabo10020069
Tondo M, Wasek B, Escola-Gil JC, de Gonzalo-Calvo D, Harmon C, Arning E, Bottiglieri T. Altered Brain Metabolome Is Associated with Memory Impairment in the rTg4510 Mouse Model of Tauopathy. Metabolites. 2020; 10(2):69. https://doi.org/10.3390/metabo10020069
Chicago/Turabian StyleTondo, Mireia, Brandi Wasek, Joan Carles Escola-Gil, David de Gonzalo-Calvo, Clinton Harmon, Erland Arning, and Teodoro Bottiglieri. 2020. "Altered Brain Metabolome Is Associated with Memory Impairment in the rTg4510 Mouse Model of Tauopathy" Metabolites 10, no. 2: 69. https://doi.org/10.3390/metabo10020069