Pathogenic APC Variants in Latvian Familial Adenomatous Polyposis Patients

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Methods
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Jasperson, K.W.; Patel, S.G.; Ahnen, D.J. APC-Associated Polyposis Conditions; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- McKusick-Nathans Institute of Genetic Medicine. Online Mendelian Inheritance in Man, OMIM® n.d. Available online: https://omim.org/ (accessed on 16 June 2019).
- Papp, J.; Kovacs, M.E.; Matrai, Z.; Orosz, E.; Kásler, M.; Børresen-Dale, A.L.; Olah, E. Contribution of APC and MUTYH mutations to familial adenomatous polyposis susceptibility in Hungary. Fam. Cancer 2016, 15, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Bethesda (MD). Gene. Natl Libr Med (US), Natl Cent Biotechnol Information; 2004. Available online: https://www.ncbi.nlm.nih.gov/gene/ (accessed on 16 June 2019).
- Talseth-Palmer, B.A. The genetic basis of colonic adenomatous polyposis syndromes. Hered. Cancer Clin. Pract. 2017, 15, 5. [Google Scholar] [CrossRef] [PubMed]
- Borosenko, V.; Irmejs, A.; Melbărde-Gorkusa, I.; Gardovskis, A.; Pavărs, M.; Vanags, A.; Trofimovics, G.; Miklasevics, E.; Gardovskis, J. Initial results of colorectal polyposis research in Latvia. Anticancer Res. 2009, 29, 711–715. [Google Scholar] [PubMed]
- Fokkema, I.F.A.C.; Taschner, P.E.M.; Schaafsma, G.C.P.; Celli, J.; Laros, J.F.J.; den Dunnen, J.T. LOVD v.2.0: The next generation in gene variant databases. Hum. Mutat. 2011, 32, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Araujo, L.F.; Molfetta, G.A.; Vincenzi, O.C.; Huber, J.; Teixeira, L.A.; Ferraz, V.E.; Silva, W.A. Molecular basis of familial adenomatous polyposis in the southeast of Brazil: Identification of six novel mutations. Int. J. Biol. Mark. 2019, 34, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Leoz, M.L.; Carballal, S.; Moreira, L.; Ocaña, T.; Balaguer, F. The genetic basis of familial adenomatous polyposis and its implications for clinical practice and risk management. Appl. Clin. Genet. 2015, 8, 95–107. [Google Scholar] [CrossRef] [PubMed]
- UniProt Consortium. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar] [CrossRef] [PubMed]
- Heinen, C.D. Genotype to phenotype: Analyzing the effects of inherited mutations in colorectal cancer families. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2010, 693, 32–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyoshi, Y.; Ando, H.; Nagase, H.; Nishisho, I.; Horii, A.; Miki, Y.; Mori, T.; Utsunomiya, J.; Baba, S.; Petersen, G. Germ-line mutations of the APC gene in 53 familial adenomatous polyposis patients. Proc. Natl. Acad. Sci. USA 1992, 89, 4452–4456. [Google Scholar] [CrossRef] [PubMed]
- Sherry, S.T.; Ward, M.H.; Kholodov, M.; Baker, J.; Phan, L.; Smigielski, E.M.; Sirotkin, K. dbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef] [PubMed]
- Wildeman, M.; van Ophuizen, E.; den Dunnen, J.T.; Taschner, P.E.M. Improving sequence variant descriptions in mutation databases and literature using the Mutalyzer sequence variation nomenclature checker. Hum. Mutat. 2008, 29, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Unicancer Cancer Genetic Group. The UMD-APC Mutations Database n.d. Available online: http://www.umd.be/APC/ (accessed on 17 June 2019).
- Chung, D.C.; Mino, M.; Shannon, K.M. Case 34-2003. N. Engl. J. Med. 2003, 349, 1750–1760. [Google Scholar] [CrossRef] [PubMed]
- De Queiroz Rossanese, L.B.; De Lima Marson, F.A.; Ribeiro, J.D.; Coy, C.S.R.; Bertuzzo, C.S. APC germline mutations in families with familial adenomatous polyposis. Oncol. Rep. 2013, 30, 2081–2088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedl, W.; Aretz, S. Familial Adenomatous Polyposis: Experience from a Study of 1164 Unrelated German Polyposis Patients. Hered. Cancer Clin. Pract. 2005, 3, 95. [Google Scholar] [CrossRef] [PubMed]
- Plawski, A.; Slomski, R. APC gene mutations causing familial adenomatous polyposis in Polish patients. J. Appl. Genet. 2008, 49, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Torrezan, G.T.; Da Silva, F.C.C.; Santos, É.M.M.; Krepischi, A.C.V.; Achatz, M.I.W.; Junior, S.A.; Rossi, B.M.; Carraro, D.M. Mutational spectrum of the APC and MUTYH genes and genotype-phenotype correlations in Brazilian FAP, AFAP, and MAP patients. Orphanet J. Rare Dis. 2013, 8, 54. [Google Scholar] [CrossRef] [PubMed]
- Vandrovcová, J.; Štekrová, J.; Kebrdlová, V.; Kohoutová, M. Molecular analysis of the APC and MYH genes in Czech families affected by FAP or multiple adenomas: 13 novel mutations. Hum. Mutat. 2004, 23, 397. [Google Scholar] [CrossRef] [PubMed]



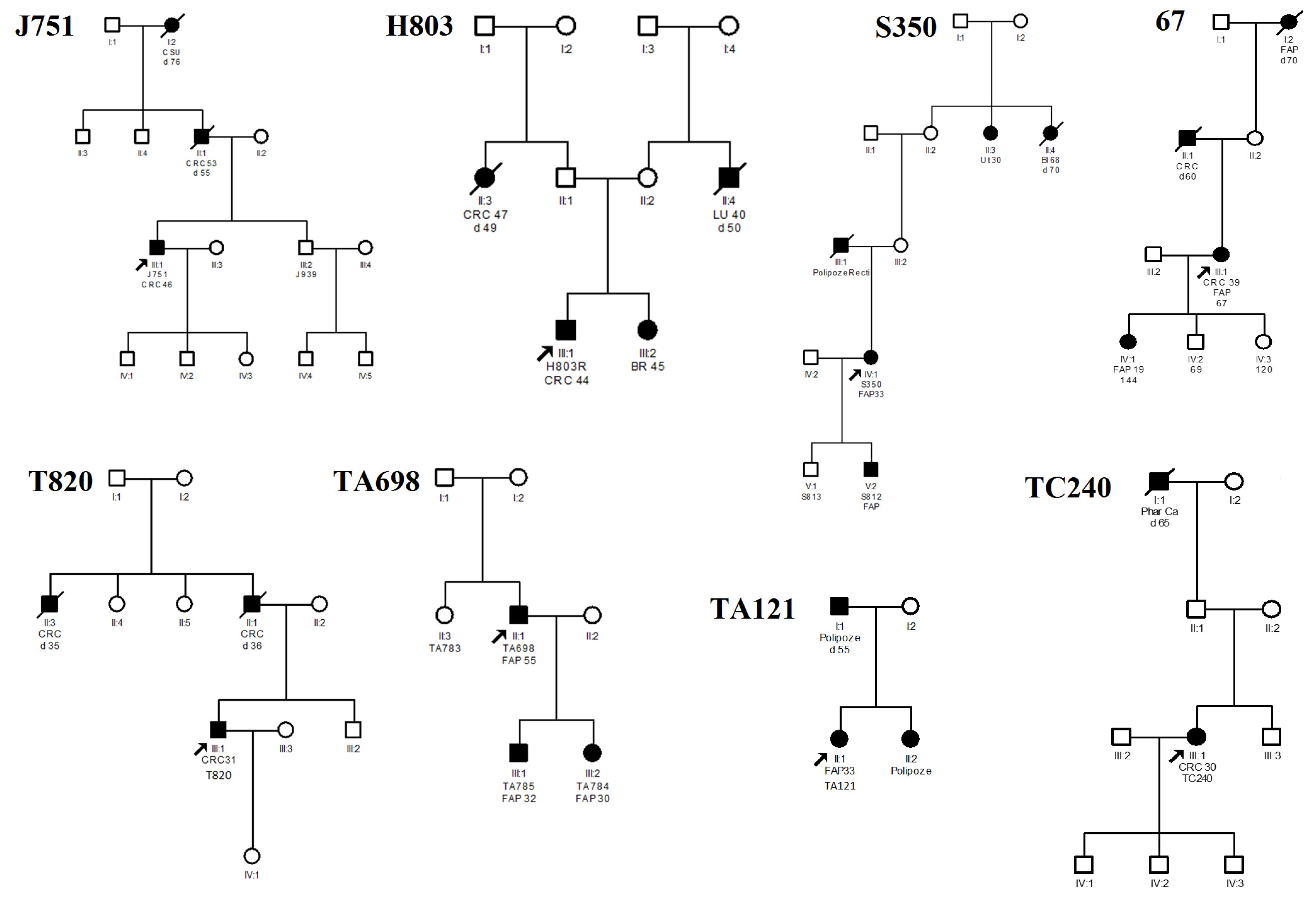{kind=link}
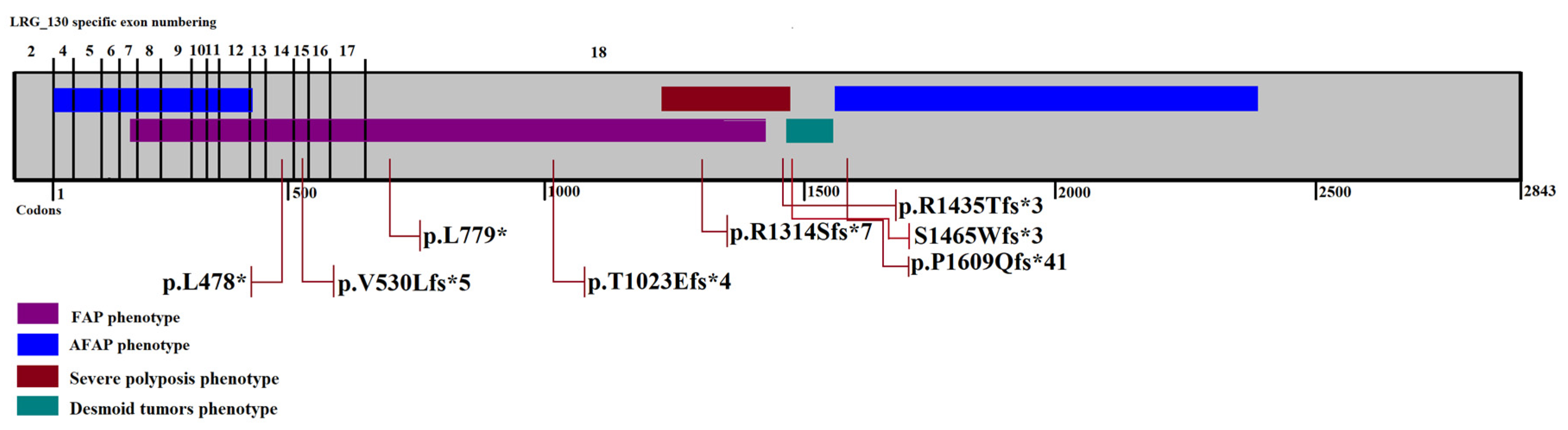{kind=link}
| Family (Proband) | Nucleotide Change 1 | AA Change | Type of Variant | Age of Diagnosis | Polyposis | CRC | Ref. |
|---|---|---|---|---|---|---|---|
| J751 | c.1433T>G | p.L478* | Nonsense | 46 | <100 | + | [14] |
| H803 | c.1586_1587insAT | p.V530Lfs*5 | Frameshift | 44 | Carpeted polyposis | + | novel |
| TA121 | c.2336delT | p.L779* | Frameshift | 33 | Carpeted polyposis | - | novel |
| S350 | c.3066_3067insGA | p.T1023Efs*4 | Frameshift | 33 | Carpeted polyposis | + | novel |
| 67 | c.3942delG | p.R1314Sfs*7 | Frameshift | 39 | Carpeted polyposis | + | [6] |
| T820 | c.4303_4304insC | p.R1435Tfs*3 | Frameshift | 31 | Carpeted polyposis | + | novel |
| TC240 | c.4393_4394delAG | S1465Wfs*3 | Frameshift | 30 | Diffuse polyposis | + | [13] |
| TA698 | c.4826delC | p.P1609Qfs*41 | Frameshift | 55 | <100 | - | [18] |
| Nucleotide Change 1 | Type of Variant | Criteria | Pathogenicity |
|---|---|---|---|
| c.1586_1587insAT | Frameshift | PVS1, PM1, PP1, PP3, PP4 | Pathogenic |
| c.2336delT | Frameshift | PVS1, PM1, PP1, PP3, PP4 | Pathogenic |
| c.3066_3067insGA | Frameshift | PVS1, PM1, PP1, PP3, PP4 | Pathogenic |
| c.4303_4304insC | Frameshift | PVS, PM1, PP1, PP3, PP4 | Pathogenic |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Daneberga, Z.; Berzina, D.; Borosenko, V.; Krumina, Z.; Kokaine-Sapovalova, L.; Gardovskis, A.; Berga-Svitina, E.; Gardovskis, J.; Miklasevics, E. Pathogenic APC Variants in Latvian Familial Adenomatous Polyposis Patients. Medicina 2019, 55, 612. https://doi.org/10.3390/medicina55100612
Daneberga Z, Berzina D, Borosenko V, Krumina Z, Kokaine-Sapovalova L, Gardovskis A, Berga-Svitina E, Gardovskis J, Miklasevics E. Pathogenic APC Variants in Latvian Familial Adenomatous Polyposis Patients. Medicina. 2019; 55(10):612. https://doi.org/10.3390/medicina55100612
Chicago/Turabian StyleDaneberga, Zanda, Dace Berzina, Viktors Borosenko, Zita Krumina, Linda Kokaine-Sapovalova, Andris Gardovskis, Egija Berga-Svitina, Janis Gardovskis, and Edvins Miklasevics. 2019. "Pathogenic APC Variants in Latvian Familial Adenomatous Polyposis Patients" Medicina 55, no. 10: 612. https://doi.org/10.3390/medicina55100612