Structural, Mechanical, Anisotropic, and Thermal Properties of AlAs in oC12 and hP6 Phases under Pressure

Key Laboratory of Ministry of Education for Wide Band-Gap Semiconductor Materials and Devices, School of Microelectronics, Xidian University, Xi’an 710071, China
*
Author to whom correspondence should be addressed.
Materials 2018, 11(5), 740; https://doi.org/10.3390/ma11050740
Submission received: 30 March 2018
/
Revised: 26 April 2018
/
Accepted: 3 May 2018
/
Published: 7 May 2018
Abstract
:The structural, mechanical, anisotropic, and thermal properties of oC12-AlAs and hP6-AlAs under pressure have been investigated by employing first-principles calculations based on density functional theory. The elastic constants, bulk modulus, shear modulus, Young’s modulus, B/G ratio, and Poisson’s ratio for oC12-AlAs and hP6-AlAs have been systematically investigated. The results show that oC12-AlAs and hP6-AlAs are mechanically stable within the considered pressure. Through the study of lattice constants (a, b, and c) with pressure, we find that the incompressibility of oC12-AlAs and hP6-AlAs is the largest along the c-axis. At 0 GPa, the bulk modulus B of oC12-AlAs, hP6-AlAs, and diamond-AlAs are 76 GPa, 75 GPa, and 74 Gpa, respectively, indicating that oC12-AlAs and hP6-AlAs have a better capability of resistance to volume than diamond-AlAs. The pressure of transition from brittleness to ductility for oC12-AlAs and hP6-AlAs are 1.21 GPa and 2.11 GPa, respectively. The anisotropy of Young’s modulus shows that oC12-AlAs and hP6-AlAs have greater isotropy than diamond-AlAs. To obtain the thermodynamic properties of oC12-AlAs and hP6-AlAs, the sound velocities, Debye temperature, and minimum thermal conductivity at considered pressure were investigated systematically. At ambient pressure, oC12-AlAs (463 K) and hP6-AlAs (471 K) have a higher Debye temperature than diamond-AlAs (433 K). At T = 300 K, hP6-AlAs (0.822 W/cm·K−1) has the best thermal conductivity of the three phases, and oC12-AlAs (0.809 W/cm·K−1) is much close to diamond-AlAs (0.813 W/cm·K−1).
1. Introduction
Group III–V compound semiconductor materials are the “core” of solid-state light sources and power electronic devices because of their large band gap, high breakdown field, high thermal conductivity, high saturated electron drift velocity, strong radiation resistance, and superior performance [1,2,3,4,5,6,7]. They have broad application prospects in semiconductor lighting, new generation mobile communications, energy Internet, high-speed rail transportation, new energy vehicles, consumer electronics, and other fields, and it is hoped that these materials will break through the bottleneck of traditional semiconductor technology [8,9,10,11,12]. Among these compound semiconductors, GaN, AlN, AlP, and AlAs have been of considerable interest, because understanding their structural and electronic properties is crucial to semiconductor technological applications. First-principles calculations based on density functional theory (DFT) represent one of the most accurate microscopic theories in materials science. Advances in the accuracy and efficiency of first-principles electronic structure calculations play an increasingly important role in the prediction of material structures and properties.
In ref. [13], Mujica et al. provided a detailed review of the current known structures, high-pressure behavior, and theoretical work on the group III–V compound semiconductor materials. Under normal conditions, AlN and GaN crystallize in the wurtzite structure, and they may also form a zinc blende structure when the epitaxial growth technique is used [14]. The first-principles calculations confirm that the zinc blende structure is metastable, although it can lie close in enthalpy (<50 meV) to that of the stable wurtzite structure [15]. For orthorhombic GaN (Pnma-GaN), it was evaluated that GaN will have a direct band gap of 1.85 eV, and that Pnma-GaN is mechanically and dynamically stable at ambient pressure [16]. Using the CALYPSO (Crystal structure AnaLYsis by Particle Swarm Optimization) code, Liu et al. [17] investigated four novel AlN phases (Pmn21-AlN, Pbam-AlN, Pbca-AlN, and Cmcm-AlN), and proved that these four novel AlN phases are more favorable in thermodynamics than the rock-salt structure at ambient pressure, and can be transformed to the rock-salt structure under certain pressures. Using first-principles calculations, the four predicted novel AlN phases, which are wide direct band-gap semiconductors with band gaps of 5.95 (Pmn21-AlN), 5.99 (Pbam-AlN), 5.88 (Pbca-AlN), and 5.59 eV (Cmcm-AlN), were calculated in detail [18]. The phase stability, mechanical, and optoelectronic properties of bct-AlN (at ambient pressure) and h-AlN (at higher pressure) were investigated by Yang et al. [19]. Their investigation proved that bct-AlN is mechanically and dynamically stable at ambient pressure, that the h-AlN phase can be stabilized by increasing pressure, and that it is mechanically and dynamically stable at 10 GPa. For the AlP semiconductors with four novel AlN phases (Pmn21-AlP, Pbam-AlP, Pbca-AlP, and bct-AlP), the electronic properties are calculated by a hybrid functional [20]. All four of these novel AlN phases behave in a ductile manner, and the band gaps are 3.22 eV, 3.27 eV, 3.47 eV, and 3.04 eV for Pmn21-AlP, Pbam-AlP, Pbca-AlP, and bct-AlP, respectively. In 1983, Froyen and Cohen [21] investigated the static and structural properties of AlAs. In 1994, Greene et al. [22] studied the crystal structure of AlAs in a diamond anvil cell using energy-dispersive X-ray diffraction to 46 GPa; this study was the first experimental observation of AlAs transforming from the zinc blende structure to the NiAs structure, and the equilibrium transformation pressure was reported to be 7 ± 5 GPa. Liu et al. [23] proved that AlAs can transition from the zinc blende structure to the NiAs structure at 6.1 GPa, which is in agreement with Greene’s work. Mujica et al. [24] studied the phase stability of AlAs, including zinc blende, wurtzite, NaCl, CsCl, β-tin, NiAs, and sc16 structures, and proved that sc16-AlAs is not thermodynamically stable at any pressure, whereas CsCl-AlAs is thermodynamically stable at very high pressures. An ab initio total energy investigation of the high-pressure phase diagrams (including Cmcm and cinnabar) of AlAs was conducted by Mujica et al. [25] to prove that the Cmcm structure is stable within a certain pressure range, and that the cinnabar structures are not thermodynamically stable at any pressure. Srivastava et al. [26] studied the stability of the AlAs using the local density approximation and the generalized gradient approximation potential. Their study revealed that under the application of pressure, the zinc-blende structure first transforms to the wurtzite structure at 3.88 GPa.
Utilizing first-principles calculations, the theoretical and experimental research studies of AlAs in diamond, zinc-blende, NiAs, rock-salt, wurtzite, NaC1, CsCl, β-tin, NiAs, wurtzite, and sc16 structures have been performed [21,22,23,24,25,26]. Recently, Liu et al. [27] investigated the phase transformation and properties of three metastable phases of AlAs (hP6-, oC12-, and cI24-AlAs). The detailed physical properties of oC12-AlAs and hP6-AlAs with the change in pressure have not yet been determined. Therefore, this work presents the structural, mechanical, anisotropic, and thermal properties of oC12-AlAs and hP6-AlAs under pressure.
2. Materials and Methods
First-principles calculations, which were applied to the theoretical investigations on AlAs in oC12 and hP6 phases, were performed using density functional theory (DFT) [28,29] based on the Cambridge Series Total Energy Package (CASTEP) code [30]. All of the calculations were performed with the generalized gradient approximation (GGA) in the form of the Perdew–Burke–Ernzerhof (PBE) functional [31,32,33] for the exchange correlation potential. The Al-3s23p and As-4s24p3 were regarded as the valence electron structures. To ensure the precision at 1 meV, the plane-wave pseudopotential method was employed; the energy cut-off Ecut was 550 eV, and the k-point sampling of the Brillouin zones constructed using the Monkhorst–Pack scheme were 11 × 11 × 4, 6 × 10 × 4 grids for AlAs in hP6 and oC12 phases in a conventional cell. The Broyden–Fletcher–Goldfarb–Shanno (BFGS) [34] minimization was applied to the geometry optimization, and the thresholds of the converged structures were as follows: the total energy tolerance was 5 × 10−6 eV/atom; the maximum force on the atom was 0.01 eV/Å; the maximum ionic displacement was less than 5 × 10−4 Å; and the maximum stress was less than 0.02 GPa. The ultrasoft quasipotential method was used to describe the presence of tightly bound core electrons. In addition, the Voigt–Reuss–Hill approximation was employed to estimate the bulk modulus, shear modulus, and Young’s modulus. To obtain the elastic constants under various pressures, we consider the strains to be non-volume-conserving, because this method is consistent with our calculated elastic constants using the stress–strain coefficients.
3. Results and Discussion
3.1. Structural Properties
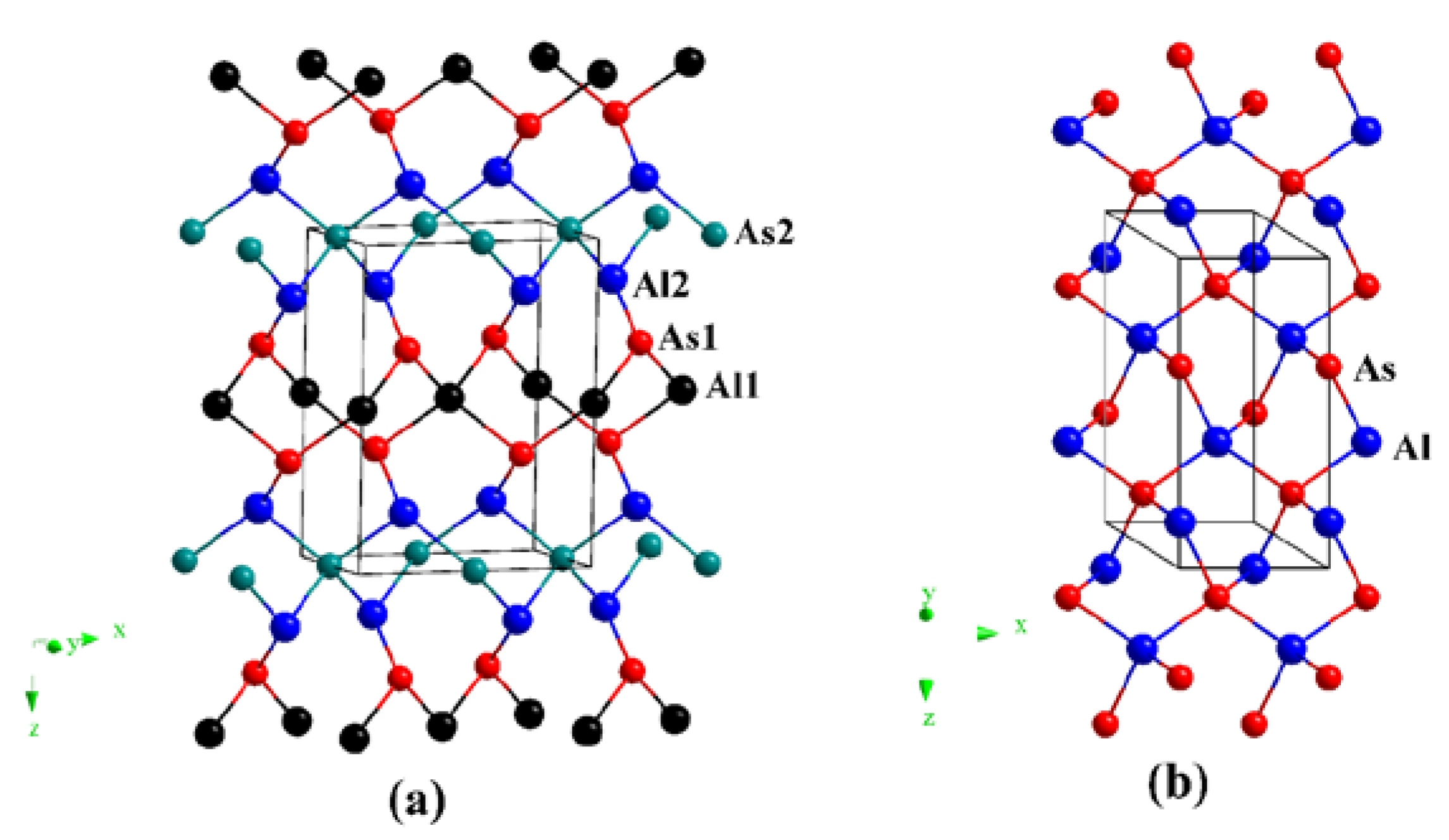
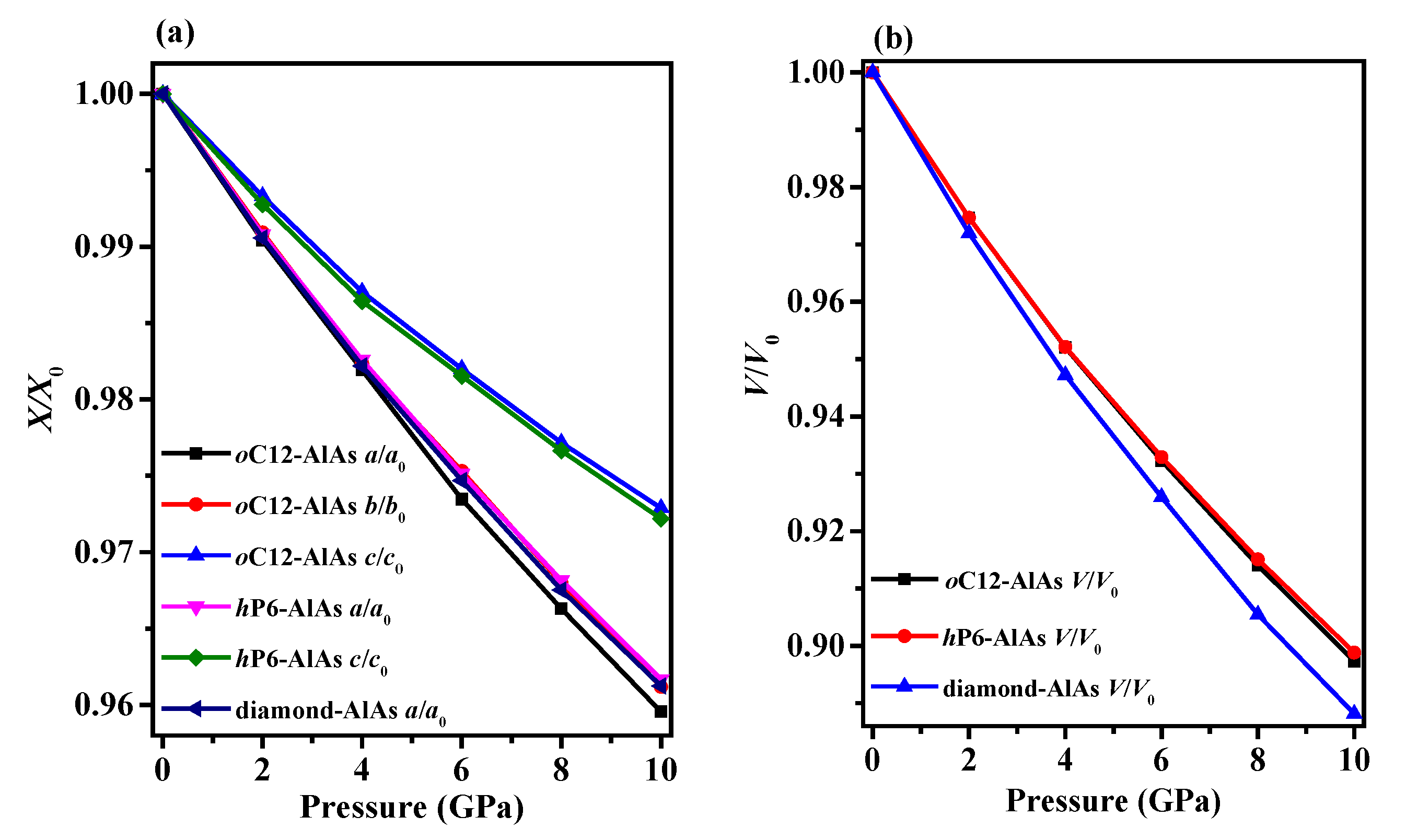
The crystal structures of oC12-AlAs and hP6-AlAs are shown in Figure 1. They were obtained with the lowest-energy structure at the same stoichiometry. The oC12-AlAs is a C-centered orthorhombic crystal system (space group C222) with 12 atoms per unit cell, including four Al atoms and four As atoms. At zero pressure, within the structure of oC12, four inequivalent atoms represented as Al1, Al2, As1, and As2 occupy the crystallographic 2d, 4k, 4k, and 2b sites in the conventional cell, respectively, which are 2d sites (0.00000, 0.00000, 0.50000), 4k sites (0.25000, 0.25000, 0.16735), 4k sites (0.25000, 0.25000, 0.66468) and 2b sites (0.50000, 0.00000, 0.00000). The hP6-AlAs is a primitive centered hexagonal structure (space group P6422) with six atoms per unit cell, including three Al atoms and three As atoms. At zero pressure, regarding the atomic positions of oC12-AlAs, Al atoms occupy the crystallographic 3c sites (0.50000, 0.00000, 1.00000), and As atoms occupy the crystallographic 3d sites (0.50000, 0.00000, 0.50000). All of the atoms in oC12-AlAs and hP6-AlAs combine to form Al–As bonds, indicating that no Al–Al (As–As) bonds were presented in the oC12 and hP6 structures. The equilibrium crystal lattice parameters of oC12-AlAs and hP6-AlAs at zero pressure are listed in Table 1; in addition, the optimized lattice parameters and experimental values for diamond-AlAs at zero pressure are also listed in Table 1. In this work, the lattice parameter of diamond-AlAs is 5.675 Å, which is consistent with the experimental value 5.661 Å [35], indicating that our results are valid and realistic. For oC12-AlAs, the lattice constants are a = 6.972 Å, b = 3.968 Å, and c = 9.108 Å; for hP6-AlAs, the lattice constants are a = b = 4.019 Å and c = 8.990 Å. For oC12-AlAs and hP6-AlAs, the lattice parameters a, b, and c in this work are also clearly consistent with those in the previous report [27], providing further evidence of the accuracy of our work. To compare the incompressibility of oC12-AlAs, hP6-AlAs, and diamond-AlAs under pressure, the lattice constants X/X0 compression and primitive cell volume V/V0 as functions of pressure are shown in Figure 2. As shown in Figure 2, both the lattice constants’ X/X0 compression and the primitive cell volume V/V0 have negative slopes, illustrating that as the pressure increases, decreases occur in the lattice constants and the primitive cell volume. In Figure 2a, for oC12-AlAs, the incompressibility along the b-axis is less than that along the c-axis, but it is larger than that along the a-axis. For hP6-AlAs, the incompressibility along the c-axis is larger than that along the a-axis (b-axis). The lattice constants’ ratios X/X0 clearly indicate the elastic anisotropy of both oC12-AlAs and hP6-AlAs. Along the a-axis, the incompressibility of diamond-AlAs is slightly less than that of hP6-AlAs, but it is larger than that of oC12-AlAs. Along the b-axis, the incompressibility of hP6-AlAs and oC12-AlAs are almost equal and both are larger than that of diamond-AlAs. Along the c-axis, the incompressibility of hP6-AlAs is less than that of oC12-AlAs, but it is larger than that of diamond-AlAs. From Figure 2b, we can see that the volume incompressibility is similar to that of a/a0, b/b0, and c/c0. The volume compressibility of oC12-AlAs is slightly larger than that of hP6-AlAs, but less than that of diamond-AlAs.
3.2. Mechanical Properties
The elastic constants of oC12-AlAs, hP6-AlAs, and diamond-AlAs at different pressures are listed in Table 2, which are used to analyze the mechanical stability. At zero pressure, the elastic constants of oC12-AlAs and hP6-AlAs in this work are in excellent agreement with the results of previous research [27], and the elastic constants of diamond-AlAs are also clearly consistent with the available experimental data [36], proving that our work is accurate and trustworthy. The orthorhombic phase has nine independent elastic constants (C11, C12, C13, C22, C23, C33, C44, C55, and C66). The mechanical stability criteria of the orthorhombic structure are shown below [37]:
The hexagonal structure has five independent elastic constants (C11, C12, C13, C33, and C44,). The mechanical stability criteria of the hexagonal phase are shown below [38]:
According to the abovementioned criteria, all of the independent elastic constants of oC12-AlAs and hP6-AlAs at different pressures are positive and satisfy the mechanical stability criteria, indicating that oC12-AlAs and hP6-AlAs are mechanically stable under the considered pressure. The elastic constants C11, C12, C13, C22, C23, and C33 increase with different rates under increasing pressure, whereas there is no apparent regular pattern in the changes of C44, C55, and C66. The elastic constants C11, C22, and C33 denote the resistance to linear compression along the a, b, and c axes, respectively. For example, oC12-AlAs has a larger C22 than C11, but it is smaller than C33, which manifested that its b-axis is less compressible than its a-axis, but more compressible than its c-axis. At ambient pressure, both oC12-AlAs and hP6-AlAs have larger C11, C22, and C33 than C11 of diamond-AlAs, which manifested that oC12-AlAs and hP6-AlAs have a stronger resistance to linear compression than diamond-AlAs. These results are consistent with the conclusions in the preceding part of this paper. In addition, all of the above three elastic constants (C11, C22, and C33) increase under increasing pressure, indicating that the greater the pressure, the more favorable the mechanical properties of oC12-AlAs and hP6-AlAs along the a, b, and c axes will be.
The elastic modulus, including bulk modulus B, shear modulus G, Young’s modulus E, and Poisson’s ratio v of oC12-AlAs and hP6-AlAs are listed in Table 2. The larger the values of B and G, the better the capability of resistance to volume and shape change. The Young’s modulus E and Poisson’s ratio v are obtained by the following expressions [39,40,41]: E = 9BG/(3B + G) and v = (3B − 2G)/[2(3B + G)]. The results of oC12-AlAs, hP6-AlAs, and diamond-AlAs at zero pressure are consistent with the values in references [27,36], which show that the results are reliable. The Young’s modulus can be applied to describe the corresponding tensile strain. The higher the value of E, the stiffer the materials, and the Poisson’s ratio can indicate the stability of a crystal against shear deformation. A larger Poisson ratio means better plasticity. As listed in Table 2, the values of bulk modulus B and Poisson’s ratio v for oC12-AlAs and hP6-AlAs increased at different rates under increasing pressure. At zero pressure, the bulk modulus B of oC12-AlAs, hP6-AlAs, and diamond-AlAs is 76 GPa, 75 GPa, and 74 GPa, respectively, which indicated that oC12-AlAs’s capability of resistance to volume change is the best, and diamond-AlAs’s is the weakest. Also, the shear modulus G of oC12-AlAs, hP6-AlAs, and diamond-AlAs (at zero pressure) is 44 GPa, 46 GPa, and 44 GPa, respectively, which shows that the capability of resistance to shape change of oC12-AlAs and diamond-AlAs are almost equal, and both are worse than that of hP6-AlAs. The oC12-AlAs and hP6-AlAs have close numerical values, both in bulk modulus and shear modulus, leading to oC12-AlAs and hP6-AlAs having similar values for both Young’s modulus and Poisson’s ratio. The maximum values of shear modulus G and Young’s modulus E (48 GPa and 124 GPa, respectively) for oC12-AlAs are at 6 GPa; the maximum values of shear modulus G and Young’s modulus E (51 GPa and 131 GPa, respectively) for oC12-AlAs are also at 6 GPa. All of the values of bulk modulus B and G of hP6-AlAs at high pressures (≥6 GPa) are larger than those of oC12-AlAs, proving that hP6-AlAs have a larger capability of resistance to volume and shape change than oC12-AlAs.
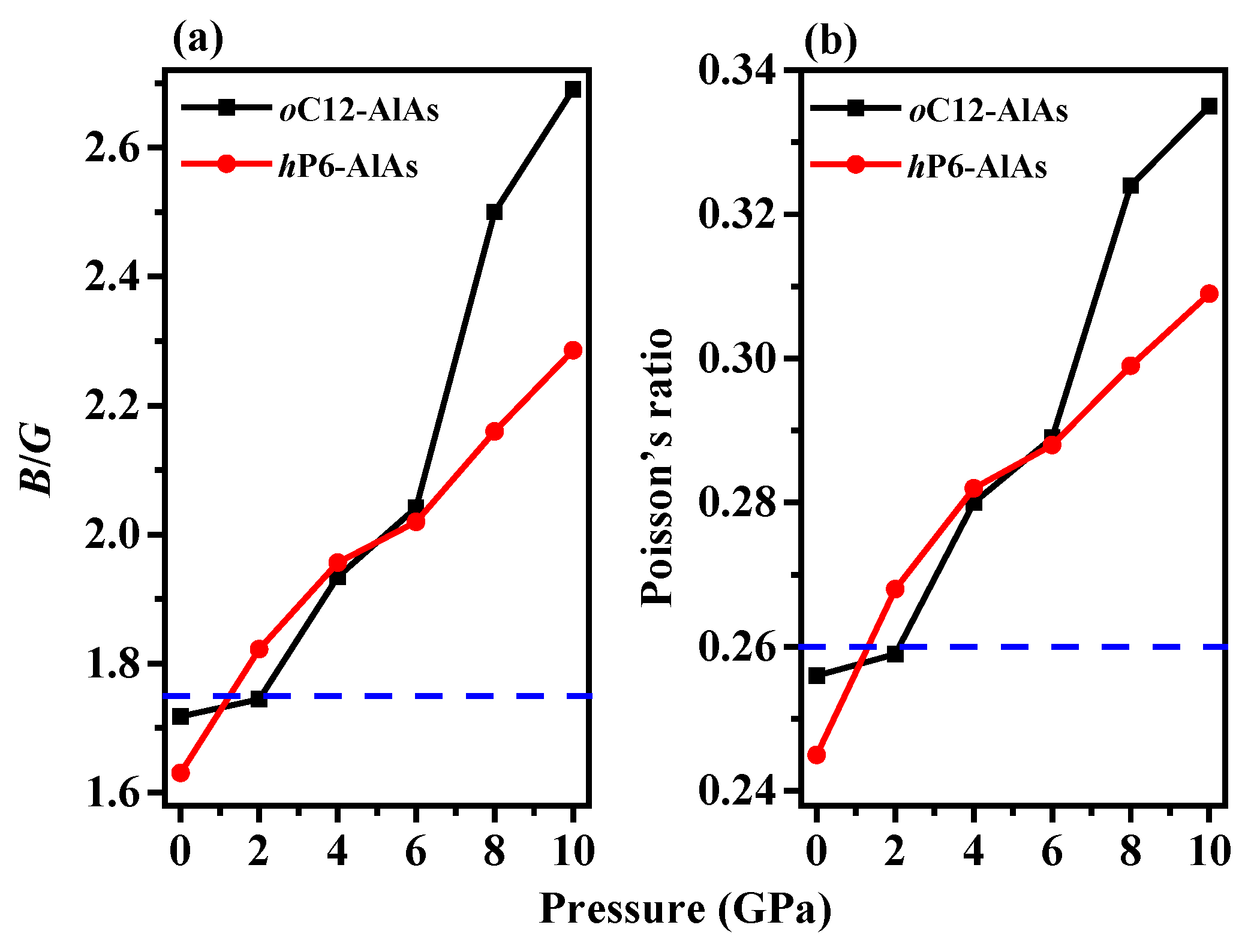
As proposed by Pugh, the brittle and ductile behavior of materials can be predicted by the ratio of bulk to shear modulus (B/G) [42]. The modulus ratio of B/G and value of v as functions of pressure are shown in Figure 3. The modulus ratio of B/G is an indication of the extent of the plastic range for a pure metal, with a high value of B/G (B/G > 1.75) being associated with malleability and a low value (B/G < 1.75) being associated with brittleness. In addition, the modulus ratio B/G is related to Poisson’s ratio v [42]: B/G = 2(1 + v)/[3(1 − 2v)] or v = (3B/G − 2)/(6B/G + 2). Therefore, Poisson’s ratio can also be used to quantify the malleability. A solid with a larger value of ν (v > 0.26) is ductile, whereas a solid with a lower value of ν is brittle. As shown in Figure 3, the modulus ratio B/G and Poisson’s ratio increase with pressure for both of the phases, indicating that the greater the pressure, the more favorable the ductile properties of oC12-AlAs and hP6-AlAs will be. The pressure of transition from brittleness to ductility are 1.21 GPa and 2.11 GPa for oC12-AlAs and hP6-AlAs, respectively.
3.3. Anisotropic Properties
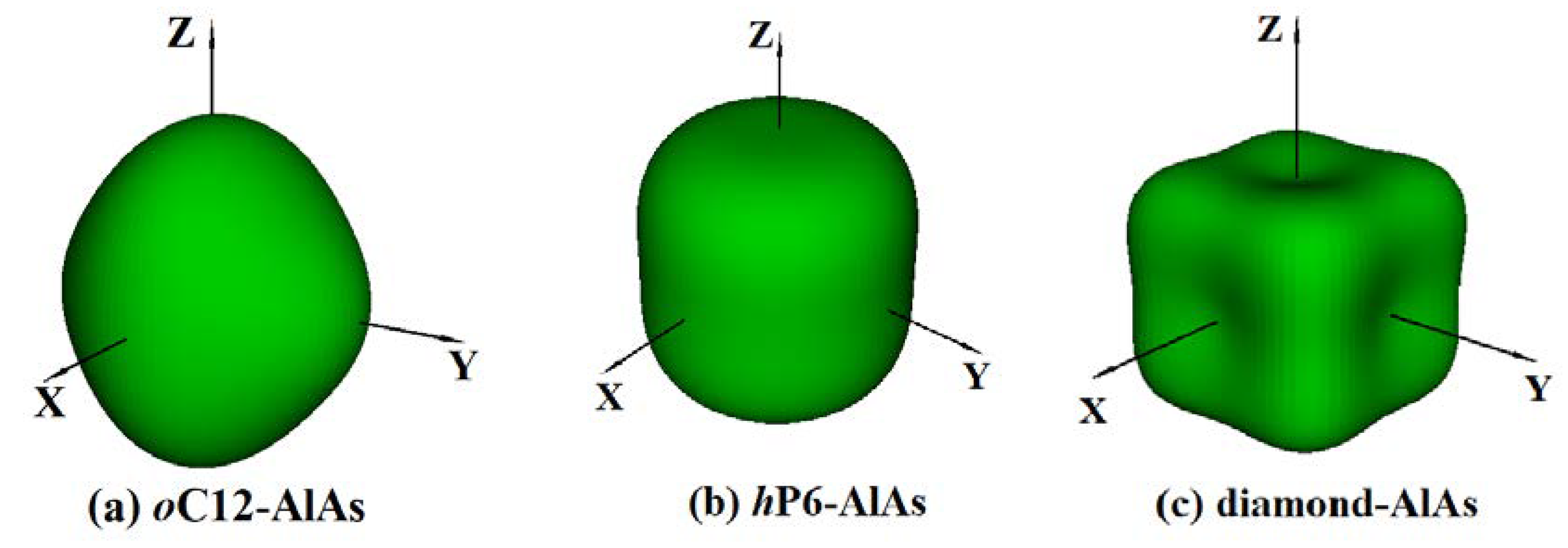
It is well-known that the anisotropy of elasticity is an important implication in engineering science and crystal physics. The three-dimensional (3D) surface construction is a valid method to describe the elastic anisotropy of a solid perfectly. For isotropic systems, the three-dimensional direction dependence will exhibit spherical symmetry, i.e., the physical, chemical, and other aspects of the nature of the materials will not change for different directions; the higher the spherical deviation, the higher the anisotropy content [43]. The 3D surface constructions of Young’s modulus E for oC12-AlAs, hP6-AlAs, and diamond-AlAs at zero pressure are shown in Figure 4 using ELAM codes (Elastic Anisotropy Measures) [44]. Obviously, the 3D surface constructions of the directional dependences of reciprocals of the Young’s modulus for the two novel materials are different because of their different crystal structures; these differences indicated that the Young’s modulus for these three phases show some mechanical anisotropy. Regarding the orthorhombic structure oC12 phase (Figure 4a), the 3D surface constructions of Young’s modulus along the x, y, and z-axis deviate from the spherical shape largely, i.e., the oC12-AlAs has high anisotropy in Young’s modulus. The 3D surface constructions of the Young’s modulus for the hexagonal structure hP6 phase (Figure 4b) has a smaller amount of deviation than that of the oC12 phase from the sphere, indicating that the Young’s modulus for the hP6 phase shows a larger isotropy than that of the oC12-AlAs. The maximum value (minimum value) of oC12-AlAs, hP6-AlAs, and diamond-AlAs is 121 GPa (100 GPa), 124 GPa (104 GPa), and 132 GPa (83 GPa), respectively. The Emax/Emin (oC12-AlAs) = 1.21, Emax/Emin (hP6-AlAs) = 1.19, and Emax/Emin (diamond-AlAs) = 1.59 show that diamond-AlAs has the greatest anisotropy, and hP6-AlAs has the least anisotropy of the three structures.
Following the procedure of Brugger [45], the single-crystal elastic constants can be applied to calculate the phase velocities of pure transverse and longitudinal modes; these phase velocities can indicate the elastic anisotropy in these crystals [46]. The sound velocities in the directions of oC12-AlAs, hP6-AlAs, and diamond-AlAs at different pressures are listed in Table 3. For orthorhombic symmetry, the sound velocities in the directions are obtained by the following expression [47]:
For hexagonal symmetry, the sound velocities in the directions are obtained by the following expression [47]:
For cubic symmetry, the sound velocities in the directions are obtained by the following expression [47]:
where ρ is the density of AlAs; vl is the longitudinal sound velocity; and vt1 and vt2 are the first transverse mode and the second transverse mode, respectively. As indicated by the above Equation (9), for orthorhombic symmetry, C11, C22, and C33 determine the longitudinal sound velocities along the [100], [010], and [001] directions, respectively, and C44, C55, and C66 correspond to the transverse sound velocities. For hexagonal symmetry, C33 determines the longitudinal sound velocity along the [001] direction, and C11 and C44 correspond to the transverse sound velocities (Equation (10)). It is obvious that the longitudinal sound velocities for oC12-AlAs along different directions increase with increasing pressure, and that the first and second transverse modes first increase and reach the maximum value at 6 GPa before decreasing. For hP6-AlAs, the longitudinal sound velocities changes in the [100] direction with no trend, whereas in the [001] direction, it first increases and reaches the maximum value at 8 GPa before decreasing. The changes of the first and second transverse sound velocities of hP6-AlAs also exhibit this initial increase and subsequent decrease regularity, but they reached their maximum values at different pressures. At 0 GPa, for oC12-AlAs (hP6-AlAs), the lowest sound velocity is 3151 m/s (3245 m/s), the highest sound velocity is 5999 m/s (5890 m/s), and the maximum (minimum) value of the sound velocity in the diamond phase is 6397 m/s (3305 m/s). In addition, at 10 GPa, the lowest sound velocity is 2711 m/s (3269 m/s), and the highest sound velocity is 6590 m/s (6225 m/s). The change trend is related to the corresponding elastic constants and indicates the elastic anisotropy of AlAs in different structures or under different pressures.
3.4. Thermal Properties
The sound velocity and Debye temperature (ΘD) are two fundamental parameters for evaluating the chemical bonding characteristics and thermal properties of materials in materials science. The Debye temperature is obtained by the following expressions [48,49]:
where h is Planck’s constant, kB is Boltzmann’s constant, NA is Avogadro’s number, n is the number of atoms in the molecule, M is the molecular weight, and ρ is the density. vm is the average sound velocity, which can be obtained by the following expression [48]:
where vl and vt are the longitudinal and transverse sound velocities, respectively, which can be obtained from Navier’s equation [50]:
The density, sound velocity, and Debye temperature for oC12-AlAs and hP6-AlAs are listed in Table 4. The Debye temperature for diamond-AlAs in this work is in excellent agreement with the results of previous research. Usually for materials, the higher the Debye temperature, the greater the hardness. With the increase in pressure, the Debye temperature for oC12-AlAs and hP6-AlAs increases first and then decreases, with the maximum (484 K and 498 K, respectively) at 6 GPa. That is, at 6 GPa, the hardness becomes best, and the bonds become strongest. At ambient pressure, the Debye temperatures are 463 K, 471 K, and 433 K for oC12-AlAs, hP6-AlAs, and diamond-AlAs respectively, indicating that oC12-AlAs and hP6-AlAs have higher Debye temperature than diamond-AlAs. The longitudinal and transverse sound velocities of oC12-AlAs are similar to those of hP6-AlAs, because oC12-AlAs and hP6-AlAs have the similar elastic moduli. The density and vl both increase with increasing pressure. With the increase in the pressure, the transverse sound velocities and average sound velocity show a non-monotonic increase or decrease. The average sound velocities of hP6-AlAs at different pressure are relatively large, exceeding 3600 m/s, which is slightly greater than those of oC12-AlAs (3400 m/s). In addition, at ambient pressure, the sound velocities are 3672 m/s, 3746 m/s, and 3562 m/s for oC12-AlAs, hP6-AlAs, and diamond-AlAs, respectively.
The thermal conductivity is the physical property of a material that conducts heat. Thermal conductivity can be used to determine the maximum power of a semiconductor device to operate and determine the efficiency of a semiconductor for thermoelectric energy conversion. The minimum thermal conductivity κmin can be calculated by Cahill’s model theory, which is expressed as follows [52]:
where N is the number of atoms in a conventional volume, and vl and vt are the longitudinal and transverse sound velocities, respectively. To obtain the minimum thermal conductivity with the temperature changes, Cahill et al. found that κmin as a function of temperature can be expressed by the following expression [51]:
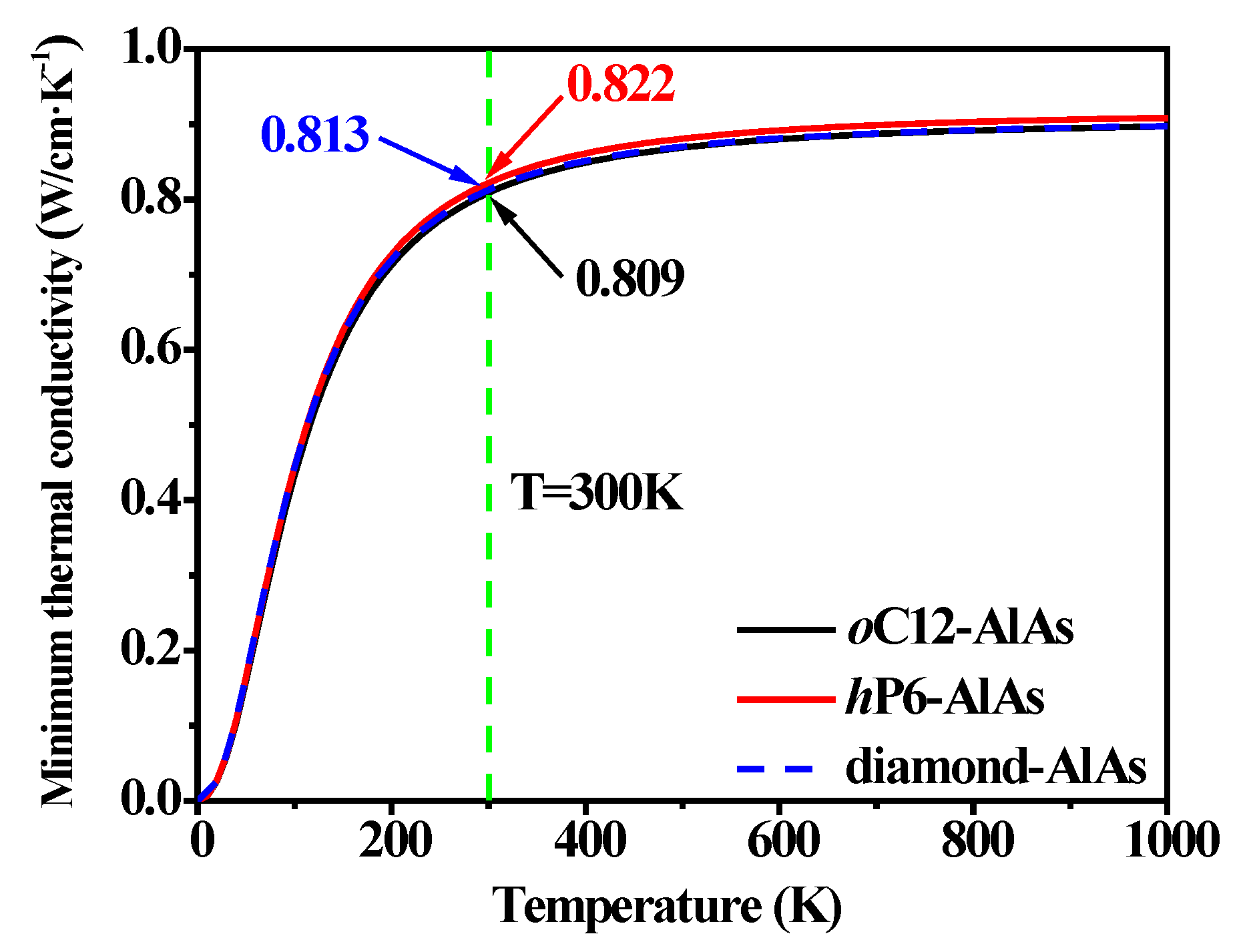
where vi represents the three acoustic modes (two transverse and one longitudinal). Θi is the cutoff frequency for each polarization expressed in K, Θi = vi[h/(2πkB)] (6π2n)1/3, and n is the number density of atoms. The temperature dependence of the minimum thermal conductivity for oC12-AlAs, hP6-AlAs, and diamond-AlAs at ambient pressure are shown in Figure 5. The thermal conductivities of oC12-AlAs, hP6-AlAs, and diamond-AlAs increase with the increase in temperature, and finally reach the corresponding stable value after 500 K. In the whole temperature range, the values of their minimum thermal conductivity κmin are similar to each other. Among two AlAs isomers and diamond-AlAs, hP6-AlAs possesses the highest value, which is slightly higher than that of oC12-AlAs and diamond-AlAs, and the data curves of oC12-AlAs and diamond-AlAs almost coincide. At T = 300 K, the minimum thermal conductivity κmin of oC12-AlAs, hP6-AlAs, and diamond-AlAs are 0.809 W/cm·K−1, 0.822 W/cm·K−1, and 0.813 W/cm·K−1, respectively. All of these investigation results illustrate that hP6-AlAs has the best thermal conductivity of the three structures, and oC12-AlAs is much close to diamond-AlAs.
4. Conclusions
In summary, the structural, mechanical, anisotropic, and thermal properties of oC12-AlAs, hP6-AlAs, and diamond-AlAs have been systematically investigated by employing first-principles calculations. The lattice constants ratios a/a0, b/b0, c/c0, and V/V0 show that the incompressibility of oC12-AlAs, and hP6-AlAs is largest along the c-axis, and the incompressibility of oC12-AlAs is slightly less than that of hP6-AlAs, but larger than diamond-AlAs. Also, the C11, C22, and C33 increase under increasing pressure, indicating that the greater the pressure, the more favorable the mechanical properties of oC12-AlAs and hP6-AlAs along the a, b, and c axes. At ambient pressure, the calculated bulk modulus and shear modulus have indicated that oC12-AlAs and hP6-AlAs have a better capability of resistance to volume and shape change than diamond-AlAs. According to the values of B/G and v, it is found that the pressures of transition from the brittle to ductile for oC12-AlAs, and hP6-AlAs are 1.21 GPa and 2.11 GPa, respectively. Detailed analyses of the anisotropy factors (Young’s modulus, Poisson’s ratio, and sound velocities, etc.) demonstrate that AlAs in diamond phase has the greatest anisotropy of the three phases. Furthermore, the Debye temperature and the minimum thermal conductivity at considered pressure were also investigated in this paper. At ambient pressure, the Debye temperature of oC12-AlAs, hP6-AlAs, and diamond-AlAs are 463 K, 471 K, and 433 K respectively. At T = 300 K, the minimum thermal conductivity κmin of oC12-AlAs, hP6-AlAs, and diamond-AlAs are 0.809 W/cm·K−1, 0.822 W/cm·K−1, and 0.813 W/cm·K−1 respectively, indicating that AlAs in oC12 and hP6 phases have favorable thermal conductivity.
Author Contributions
Q.F. designed the project; W.Z., C.C., Q.F., and Y.S. performed the calculations; W.Z., C.C., Y.S., Q.F. and Y.Y. analyzed the results, W.Z., C.C. and Y.S. wrote the manuscript.
Funding
This research was funded by [National Natural Science Foundation of China] grant number [61474089].
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 61474089).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Schroten, E.; Goossens, A.; Schoonman, J. Photo and electroreflectance of cubic boron phosphide. J. Appl. Phys. 1998, 83, 1660–1663. [Google Scholar] [CrossRef]
- Durandurdu, M. Pressure-induced phase transition of zinc-blende AlN: An ab initio molecular dynamics study. J. Phys. Chem. Solids 2008, 69, 2894–2897. [Google Scholar] [CrossRef]
- Gomez, H.; Taylor, T.R.; Neumark, D.M. Anion photoelectron spectroscopy of aluminum phosphide clusters. J. Phys. Chem. A 2001, 105, 6886–6893. [Google Scholar] [CrossRef]
- Bowen, P.; Highfield, J.G.; Mocellin, A.; Ring, T.A. Degradation of aluminum nitride powder in an aqueous environmet. J. Am. Ceram. Soc. 1990, 73, 724–728. [Google Scholar] [CrossRef]
- Takahashi, N.; Matsumoto, Y.; Nakamura, T. Investigations of structure and morphology of the AlN nano-pillar crystal films prepared by halide chemical vapor deposition under atmospheric pressure. J. Phys. Chem. Solids 2006, 67, 665–668. [Google Scholar] [CrossRef]
- Golikova, O.A. Boron and Boron-based semiconductors. Phys. Status Solidi A 1979, 51, 31–40. [Google Scholar] [CrossRef]
- Fan, Q.Y.; Chai, C.C.; Wei, Q.; Yang, Y.T. Thermodynamic, elastic, elastic anisotropy and minimum thermal conductivity of β-GaN under high temperature. Chin. J. Phys. 2017, 55, 400–411. [Google Scholar] [CrossRef]
- Adachi, S. GaAs, AlAs, and AlxGa1−x As: Material parameters for use in research and device applications. J. Appl. Phys. 1985, 58, R1–R29. [Google Scholar] [CrossRef]
- Guo, L. Structural, energetic, and electronic properties of hydrogenated aluminum arsenide clusters. J. Nanopart. Res. 2011, 13, 2029–2039. [Google Scholar] [CrossRef]
- Shen, L.Y.; Xu, X.S.; Lu, W.; Shi, B. Aluminum nitride shaping by non-aqueous gelcasting of low-viscosity and high solid-loading slurry. Ceram. Int. 2016, 42, 5569–5574. [Google Scholar] [CrossRef]
- Chen, B.C.; Ho, C.Y.; Wen, M.Y.; Chen, C.S.; Ma, C.; Tsai, Y.H. Ultrashort-laser-pulse machining characteristics of aluminum nitride and aluminum oxide. Ceram. Int. 2015, 41, S191–S196. [Google Scholar] [CrossRef]
- McNeil, L.E.; Grimsditch, M.; French, R.H. Vibrational spectroscopy of aluminum nitride. J. Am. Ceram. Soc. 1993, 76, 1132–1136. [Google Scholar] [CrossRef]
- Mujica, A.; Rubio, A.; Munoz, A.; Needs, R.J. High-pressure phases of group-IV, III-V, and II-VI compounds. Rev. Mod. Phys. 2003, 75, 863. [Google Scholar] [CrossRef]
- Lei, T.; Fanciulli, M.; Molnar, R.J.; Moustakas, T.D.; Graham, R.J.; Scanlon, J. Epitaxial growth of zinc blende and wurtzitic gallium nitride thin films on (001) silicon. Appl. Phys. Lett. 1991, 59, 944–946. [Google Scholar] [CrossRef]
- Serrano, J.; Rubio, A.; Hernández, E.; Muñoz, A.; Mujica, A. Theoretical study of the relative stability of structural phases in group-III nitrides at high pressures. Phys. Rev. B 2000, 62, 16612. [Google Scholar] [CrossRef]
- Fan, Q.Y.; Chai, C.C.; Wei, Q.; Yang, J.H.; Zhou, P.K.; Zhang, D.Y.; Yang, Y.T. A New Phase of GaN. J. Chem. 2016, 2016, 8612892. [Google Scholar] [CrossRef]
- Liu, C.; Hu, M.; Luo, K.; Cui, L.; Yu, D.L.; Zhao, Z.S.; He, J.L. Novel high-pressure phases of AlN: A first-principles study. Comput. Mater. Sci. 2016, 117, 496–501. [Google Scholar] [CrossRef]
- Yang, R.K.; Zhu, C.S.; Wei, Q.; Du, Z.A. First-principles study of the properties of four predicted novel phases of AlN. J. Phys. Chem. Solids 2017, 104, 68–78. [Google Scholar] [CrossRef]
- Yang, R.K.; Zhu, C.S.; Wei, Q.; Du, Z. Phase stability, mechanical and optoelectronic properties of two novel phases of AlN. Mod. Phys. Lett. B 2017, 31, 1750201. [Google Scholar] [CrossRef]
- Yang, R.K.; Zhu, C.S.; Wei, Q.; Zhang, D.Y. First-principles study on phases of AlP. Solid State Commun. 2017, 267, 23–28. [Google Scholar] [CrossRef]
- Froyen, S.; Cohen, M.L. Structural properties of III-V zinc-blende semiconductors under pressure. Phys. Rev. B 1983, 28, 3258. [Google Scholar] [CrossRef]
- Greene, R.G.; Luo, H.; Li, T.; Ruoff, A.L. Phase transformation of AlAs to NiAs structure at high pressure. Phys. Rev. Lett. 1994, 72, 2045. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.C.; Lu, Z.W.; Klein, B.M. Pressure-induced phase transformations in AlAs: Comparison between ab initio theory and experiment. Phys. Rev. B 1995, 51, 5678. [Google Scholar] [CrossRef]
- Mujica, A.; Needs, R.J.; Munoz, A. First-principles pseudopotential study of the phase stability of the III-V semiconductors GaAs and AlAs. Phys. Rev. B 1995, 2, 8881. [Google Scholar] [CrossRef]
- Mujica, A.; Rodríguez-Hernández, P.; Radescu, S.; Needs, R.J.; Munoz, A. AlX (X = As, P, Sb) compounds under pressure. Phys. Status Solidi B 1999, 211, 39–43. [Google Scholar] [CrossRef]
- Srivastava, A.; Tyagi, N. High pressure behavior of AlAs nanocrystals: The first-principle study. High Press. Res. 2012, 32, 43–47. [Google Scholar] [CrossRef]
- Liu, C.; Ma, M.D.; Yuan, X.H.; Sun, H.; Ying, P.; Xu, B.; Zhao, Z.S.; He, J.L. Metastable phases, phase transformation and properties of AlAs based on first-principle study. Comput. Mater. Sci. 2017, 128, 337–342. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. 1964, 136, B864. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133. [Google Scholar] [CrossRef]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.I.; Refson, K.; Payne, M.C. First principles methods using CASTEP. Z. Krist.-Cryst. Mater. 2005, 220, 567–570. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed]
- Krukau, A.V.; Vydrov, O.A.; Izmaylov, A.F.; Scuseria, G.E. Influence of the exchange screening parameter on the performance of screened hybrid functionals. J. Chem. Phys. 2006, 125, 224106. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.Y.; Chai, C.C.; Wei, Q.; Yang, Y.T. Two novel silicon phases with direct band gaps. Phys. Chem. Chem. Phys. 2016, 18, 12905–12913. [Google Scholar] [CrossRef] [PubMed]
- Pfrommer, B.G.; Côté, M.; Louie, S.G.; Cohen, M.L. Relaxation of crystals with the quasi-Newton method. J. Comput. Chem. 1997, 131, 233–240. [Google Scholar] [CrossRef]
- Wasilewski, Z.R.; Dion, M.M.; Lockwood, D.J.; Poole, P.; Streater, R.W.; SpringThorpe, A.J. Determination of AlxGa(1-x) As composition: The MBE perspective. J. Cryst. Growth 1997, 175, 238–243. [Google Scholar] [CrossRef]
- Gehrsitz, S.; Sigg, H.; Herres, N.; Bachem, K.; Köhler, K.; Reinhart, F.K. Compositional dependence of the elastic constants and the lattice parameter of AlxGa1−xAs. Phys. Rev. B 1999, 60, 11601. [Google Scholar] [CrossRef]
- Nye, J.F. Physical Properties of Crystals; Oxford University Press: Oxford, UK, 1985. [Google Scholar]
- Wu, Z.J.; Zhao, E.J.; Xiang, H.P.; Hao, X.F.; Liu, X.J.; Meng, J. Crystal structures and elastic properties of superhard IrN2 and IrN3 from first principles. Phys. Rev. B 2007, 76, 054115. [Google Scholar] [CrossRef]
- Hill, R. The elastic behaviour of a crystalline aggregate. Proc. Phys. Soc. A 1952, 65, 349. [Google Scholar] [CrossRef]
- Fan, Q.Y.; Chai, C.C.; Wei, Q.; Yang, Y.T.; Yang, Q.; Chen, P.Y.; Xing, M.J.; Zhang, J.Q.; Yao, R. Prediction of novel phase of silicon and Si-Ge alloys. J. Solid State Chem. 2016, 233, 471–483. [Google Scholar] [CrossRef]
- Fan, Q.Y.; Chai, C.C.; Wei, Q.; Zhou, P.K.; Zhang, J.Q.; Yang, Y.T. Si96: A new silicon allotrope with interesting physical properties. Materials 2016, 9, 284. [Google Scholar] [CrossRef] [PubMed]
- Pugh, S.F. XCII. Relations between the elastic moduli and the plastic properties of polycrystalline pure metals. Philos. Mag. 1954, 45, 823–843. [Google Scholar] [CrossRef]
- Hu, W.C.; Liu, Y.; Li, D.J.; Zeng, X.Q.; Xu, C.S. First-principles study of structural and electronic properties of C14-type Laves phase Al2Zr and Al2Hf. Comput. Mater. Sci. 2014, 83, 27–34. [Google Scholar] [CrossRef]
- Marmier, A.; Lethbridge, Z.A.; Walton, R.I.; Smith, C.W.; Parker, S.C.; Evans, K.E. ElAM: A computer program for the analysis and representation of anisotropic elastic properties. Comput. Phys. Commun. 2010, 181, 2102–2115. [Google Scholar] [CrossRef] [Green Version]
- Brugger, K. Determination of third-order elastic coefficients in crystals. J. Appl. Phys. 1965, 36, 768–773. [Google Scholar] [CrossRef]
- Sun, L.; Gao, Y.M.; Xiao, B.; Li, Y.F.; Wang, G.L. Anisotropic elastic and thermal properties of titanium borides by first-principles calculations. J. Alloys Compd. 2013, 579, 457–467. [Google Scholar] [CrossRef]
- Duan, Y.H.; Sun, Y.; Peng, M.J.; Zhou, S.G. Anisotropic elastic properties of the Ca-Pb compounds. J. Alloys Compd. 2014, 595, 14–21. [Google Scholar] [CrossRef]
- Anderson, O.L. A simplified method for calculating the Debye temperature from elastic constants. J. Phys. Chem. Solids 1963, 24, 909–917. [Google Scholar] [CrossRef]
- Fan, Q.Y.; Wei, Q.; Yan, H.Y.; Zhang, M.G.; Zhang, Z.X.; Zhang, J.Q.; Zhang, D.Y. Elastic and electronic properties of Pbca-BN: First-principles calculations. Comput. Mater. Sci. 2014, 85, 80–87. [Google Scholar] [CrossRef]
- Fan, Q.Y.; Wei, Q.; Chai, C.C.; Zhang, M.G.; Yan, H.Y.; Zhang, Z.X.; Zhang, J.Q.; Zhang, D.Y. Elastic and electronic properties of Imm2- and I4-m2-BCN. Comput. Mater. Sci. 2015, 97, 6–13. [Google Scholar] [CrossRef]
- Grimmeiss, H.G.; Monema, B. Temperature dependence of the refractive index of AlAs and AlP. Phys. Status Solidi A 1971, 5, 109–114. [Google Scholar] [CrossRef]
- Sichel, E.K.; Pankove, J.I. Thermal conductivity of GaN, 25–360 K. J. Phys. Chem. Solids 1977, 38, 330. [Google Scholar] [CrossRef]
Figure 1.
The crystal structures of AlAs in the oC12 phase (a) and hP6 phase (b).

Figure 2.
The lattice constants X/X0 compression (a) and primitive cell volume V/V0 (b) as functions of pressure for oC12-AlAs, hP6-AlAs, and diamond-AlAs.
Figure 2.
The lattice constants X/X0 compression (a) and primitive cell volume V/V0 (b) as functions of pressure for oC12-AlAs, hP6-AlAs, and diamond-AlAs.

Figure 3.
The ratio of bulk to shear modulus (B/G) (a) and Poisson’s ratio (b) of oC12-AlAs, hP6-AlAs, and diamond-AlAs as a function of pressure.
Figure 3.
The ratio of bulk to shear modulus (B/G) (a) and Poisson’s ratio (b) of oC12-AlAs, hP6-AlAs, and diamond-AlAs as a function of pressure.

Figure 4.
The three-dimensional (3D) surface constructions of Young’s modulus E for oC12-AlAs, hP6-AlAs, and diamond-AlAs at ambient pressure.
Figure 4.
The three-dimensional (3D) surface constructions of Young’s modulus E for oC12-AlAs, hP6-AlAs, and diamond-AlAs at ambient pressure.

Figure 5.
Temperature dependence of the minimum thermal conductivity for oC12-AlAs, hP6-AlAs, and diamond-AlAs at ambient pressure.
Figure 5.
Temperature dependence of the minimum thermal conductivity for oC12-AlAs, hP6-AlAs, and diamond-AlAs at ambient pressure.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
The lattice parameters a, b, c (in Å) of AlAs in the oC12, hP6, and diamond phases. PBE: Perdew–Burke–Ernzerhof.
Table 1.
The lattice parameters a, b, c (in Å) of AlAs in the oC12, hP6, and diamond phases. PBE: Perdew–Burke–Ernzerhof.
| Materials | PBE | Exp. | ||
|---|---|---|---|---|
| a | b | c | a | |
| diamond-AlAs | 5.675 | 5.661 1 | ||
| oC12-AlAs | 6.972 | 3.968 | 9.108 | |
| 6.975 2 | 3.977 2 | 9.094 2 | ||
| hP6-AlAs | 4.019 | 8.990 | ||
| 4.026 2 | 8.973 2 | |||
Table 2.
The elastic constants (in GPa) and the elastic modulus (in GPa) of oC12-AlAs, hP6-AlAs, and diamond-AlAs under pressure.
Table 2.
The elastic constants (in GPa) and the elastic modulus (in GPa) of oC12-AlAs, hP6-AlAs, and diamond-AlAs under pressure.
| Materials | P | C11 | C12 | C13 | C22 | C23 | C33 | C44 | C55 | C66 | B | G | E | v |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| oC12-AlAs | 0 | 120 | 41 | 50 | 130 | 45 | 145 | 40 | 52 | 42 | 76 | 44 | 111 | 0.257 |
| 0 1 | 127 | 39 | 45 | 121 | 52 | 153 | 47 | 38 | 43 | 74 | 43 | 108 | 0.257 | |
| 2 | 133 | 48 | 59 | 138 | 51 | 155 | 47 | 58 | 44 | 82 | 47 | 118 | 0.259 | |
| 4 | 137 | 56 | 65 | 145 | 59 | 163 | 44 | 55 | 41 | 89 | 46 | 118 | 0.280 | |
| 6 | 146 | 63 | 74 | 154 | 67 | 177 | 47 | 59 | 45 | 98 | 48 | 124 | 0.289 | |
| 8 | 156 | 68 | 82 | 161 | 73 | 186 | 33 | 41 | 45 | 105 | 42 | 111 | 0.324 | |
| 10 | 162 | 77 | 91 | 169 | 80 | 195 | 41 | 33 | 44 | 113 | 42 | 112 | 0.335 | |
| hP6-AlAs | 0 | 127 | 42 | 49 | 140 | 53 | 43 | 75 | 46 | 115 | 0.245 | |||
| 0 1 | 126 | 38 | 51 | 147 | 44 | 75 | 44 | 110 | 0.256 | |||||
| 2 | 133 | 50 | 56 | 147 | 50 | 42 | 82 | 45 | 114 | 0.268 | ||||
| 4 | 140 | 57 | 64 | 158 | 52 | 41 | 90 | 46 | 118 | 0.282 | ||||
| 6 | 165 | 62 | 75 | 171 | 53 | 51 | 103 | 51 | 131 | 0.288 | ||||
| 8 | 165 | 67 | 82 | 182 | 53 | 49 | 108 | 50 | 130 | 0.299 | ||||
| 10 | 171 | 75 | 86 | 174 | 53 | 48 | 112 | 49 | 128 | 0.309 | ||||
| diamond-AlAs | 0 | 116 | 53 | 55 | 74 | 44 | 110 | 0.252 | ||||||
| 0 2 | 120 | 57 | 57 | 78 | 45 |
Table 3.
The sound velocities along different directions of oC12-AlAs, hP6-AlAs, and diamond-AlAs under pressure.
Table 3.
The sound velocities along different directions of oC12-AlAs, hP6-AlAs, and diamond-AlAs under pressure.
| Materials | Directions | Pressure | ||||||
|---|---|---|---|---|---|---|---|---|
| 0 | 2 | 4 | 6 | 8 | 10 | |||
| oC12-AlAs | [100] | [100]vl | 5464 | 5675 | 5691 | 5813 | 5948 | 6007 |
| [010]vt1 | 3232 | 3264 | 3113 | 3227 | 3194 | 3130 | ||
| [001]vt2 | 3597 | 3747 | 3606 | 3696 | 3049 | 2711 | ||
| [010] | [010]vl | 5687 | 5780 | 5855 | 5971 | 6042 | 6135 | |
| [100]vt1 | 3232 | 3264 | 3113 | 3227 | 3194 | 3130 | ||
| [001]vt2 | 3154 | 3373 | 3225 | 3298 | 2736 | 3022 | ||
| [001] | [001]vl | 6006 | 6126 | 6208 | 6401 | 6494 | 6590 | |
| [100]vt1 | 3597 | 3747 | 3606 | 3696 | 3049 | 2711 | ||
| [010]vt2 | 3154 | 3373 | 3225 | 3298 | 2736 | 3022 | ||
| hP6-AlAs | [100] | [100]vl | 3243 | 3166 | 3129 | 3450 | 3333 | 3269 |
| [010]vt1 | 5607 | 5667 | 5747 | 6176 | 6116 | 6171 | ||
| [001]vt2 | 3622 | 3475 | 3502 | 3500 | 3466 | 3436 | ||
| [001] | [001]vl | 5887 | 5958 | 6105 | 6287 | 6424 | 6225 | |
| [100]vt1 | 3622 | 3475 | 3502 | 3500 | 3466 | 3436 | ||
| [010]vt2 | 3622 | 3475 | 3502 | 3500 | 3466 | 3436 | ||
| diamond-AlAs | [100] | [100]vl | 5676 | |||||
| [010]vt1 | 3909 | |||||||
| [001]vt2 | 3909 | |||||||
| [110] | [110]vl | 6225 | ||||||
| [10]vt1 | 4183 | |||||||
| [001]vt2 | 3837 | |||||||
| [111] | [111]vl | 6397 | ||||||
| [11]vt1 | 3305 | |||||||
| [11]vt2 | 3305 | |||||||
Table 4.
The density (ρ in g/cm3), sound velocity (vl, vt, vm, in m/s), and Debye temperature (ΘD in K) for oC12-AlAs, hP6-AlAs, and diamond-AlAs under pressure.
Table 4.
The density (ρ in g/cm3), sound velocity (vl, vt, vm, in m/s), and Debye temperature (ΘD in K) for oC12-AlAs, hP6-AlAs, and diamond-AlAs under pressure.
| Materials | Pressure | ρ (g/cm3) | vl | vt | vm | ΘD |
|---|---|---|---|---|---|---|
| oC12-AlAs | 0 | 4.02 | 5781 | 3305 | 3672 | 463 |
| 2 | 4.13 | 5916 | 3372 | 3748 | 476 | |
| 4 | 4.23 | 5960 | 3297 | 3673 | 473 | |
| 6 | 4.32 | 6123 | 3333 | 3718 | 484 | |
| 8 | 4.41 | 6044 | 3087 | 3458 | 457 | |
| 10 | 4.49 | 6135 | 3058 | 3432 | 458 | |
| hP6-AlAs | 0 | 4.04 | 5812 | 3376 | 3746 | 471 |
| 2 | 4.14 | 5856 | 3297 | 3668 | 468 | |
| 4 | 4.24 | 5975 | 3294 | 3671 | 474 | |
| 6 | 4.33 | 6287 | 3433 | 3829 | 498 | |
| 8 | 4.41 | 6293 | 3367 | 3761 | 494 | |
| 10 | 4.49 | 6284 | 3303 | 3694 | 490 | |
| diamond-AlAs | 0 | 3.60 | 6068 | 3495 | 3880 | 433 |
| 0 1 | 3.73 | 450 |
1 Ref [51].
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Zhang, W.; Chai, C.; Song, Y.; Fan, Q.; Yang, Y. Structural, Mechanical, Anisotropic, and Thermal Properties of AlAs in oC12 and hP6 Phases under Pressure. Materials 2018, 11, 740. https://doi.org/10.3390/ma11050740
AMA Style
Zhang W, Chai C, Song Y, Fan Q, Yang Y. Structural, Mechanical, Anisotropic, and Thermal Properties of AlAs in oC12 and hP6 Phases under Pressure. Materials. 2018; 11(5):740. https://doi.org/10.3390/ma11050740
Chicago/Turabian StyleZhang, Wei, Changchun Chai, Yanxing Song, Qingyang Fan, and Yintang Yang. 2018. "Structural, Mechanical, Anisotropic, and Thermal Properties of AlAs in oC12 and hP6 Phases under Pressure" Materials 11, no. 5: 740. https://doi.org/10.3390/ma11050740
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.