
Functional Annotations of Paralogs: A Blessing and a Curse


Abstract
:1. Introduction
2. Protein Function and Evolution
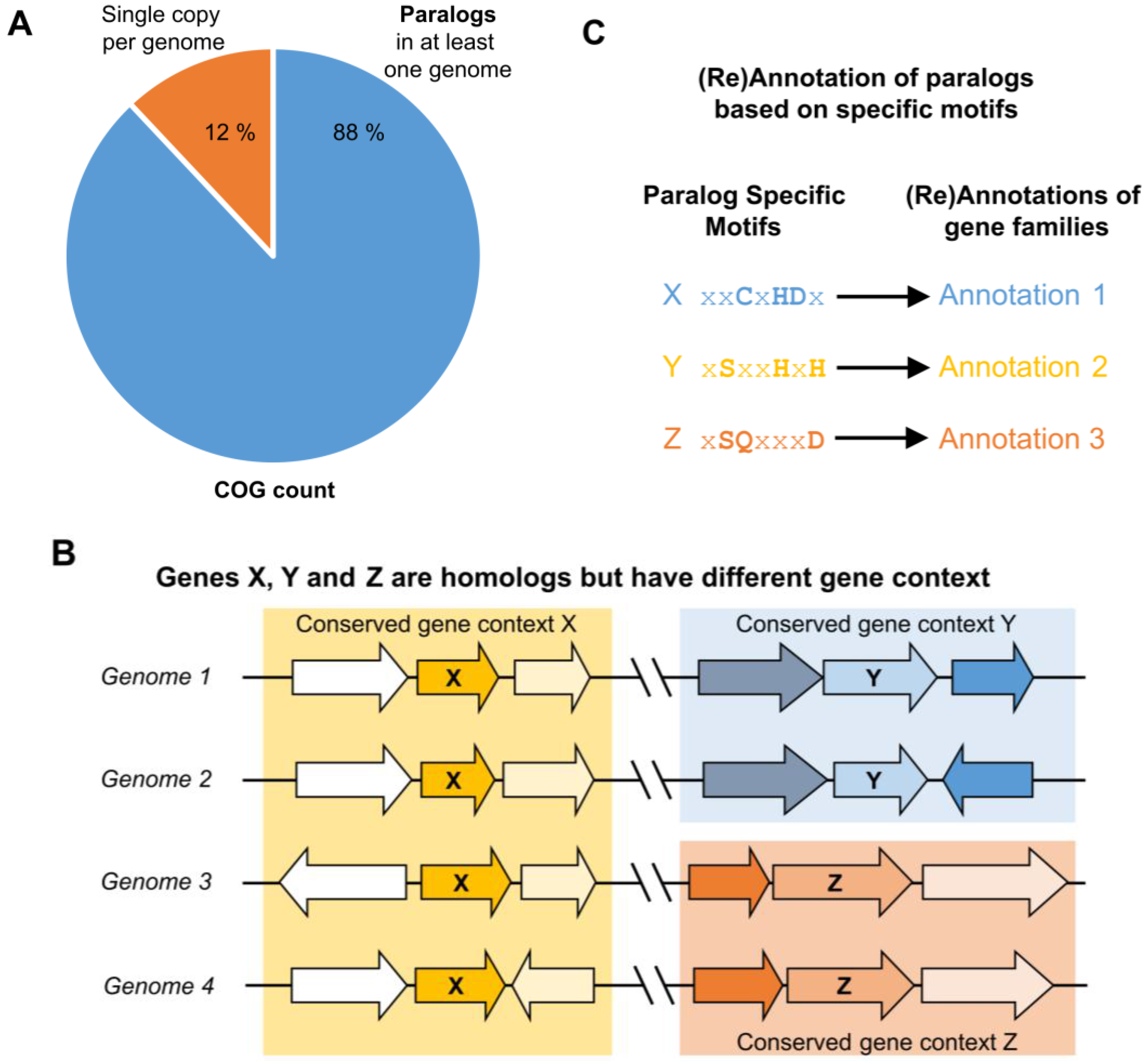
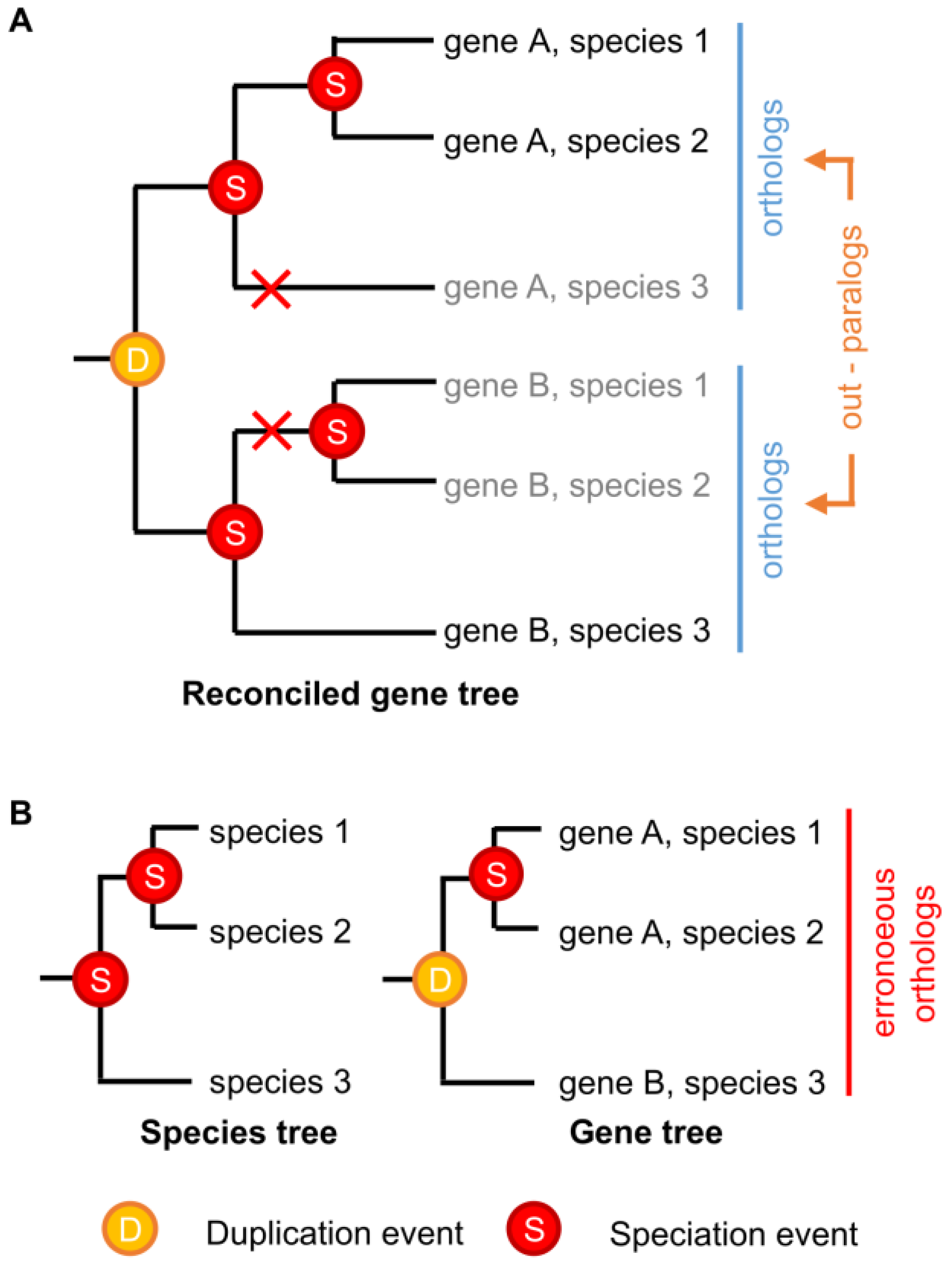
3. Identifying Orthologs and Paralogs in Practice
4. Lessons from Comparative Genomics
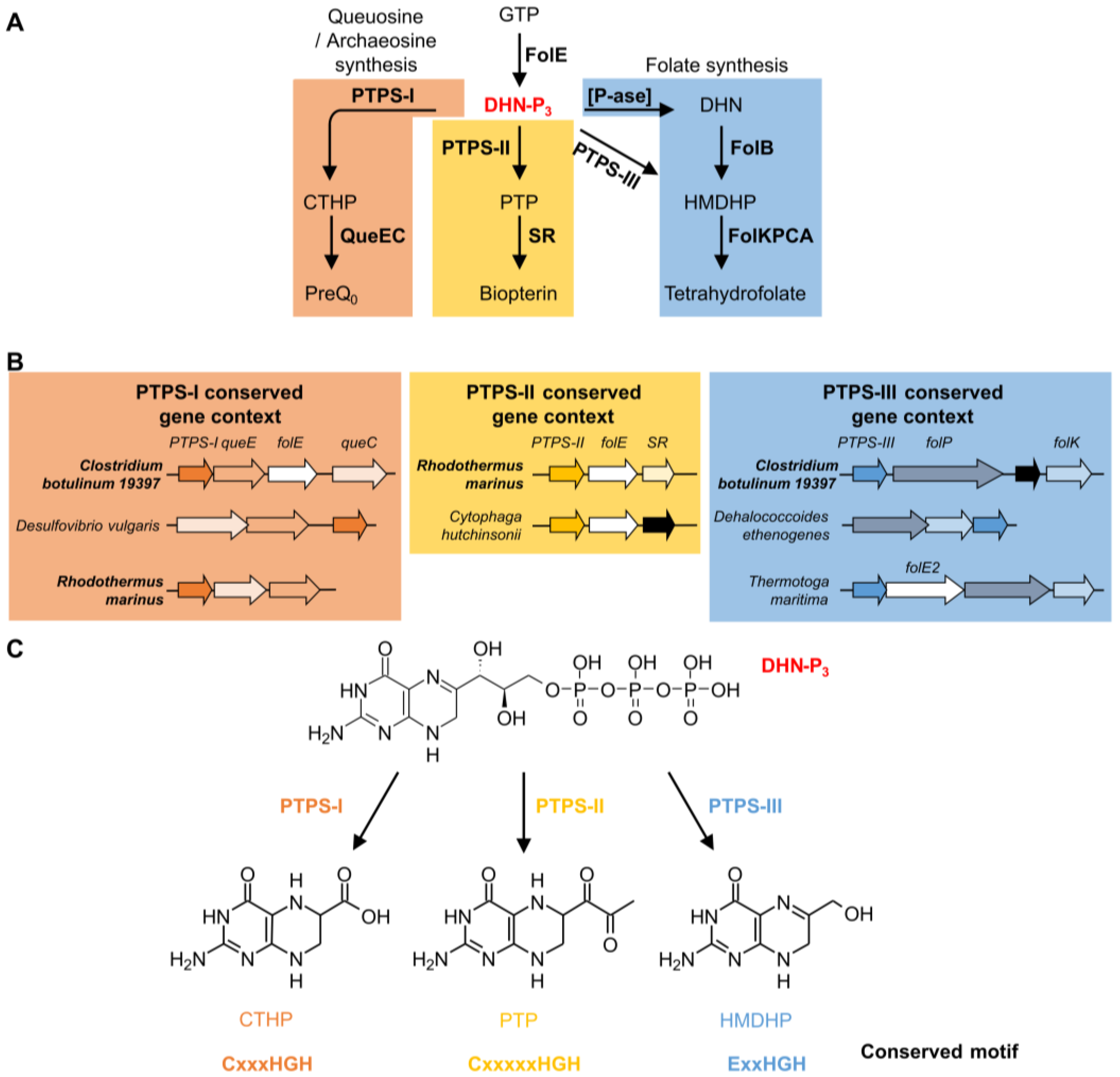
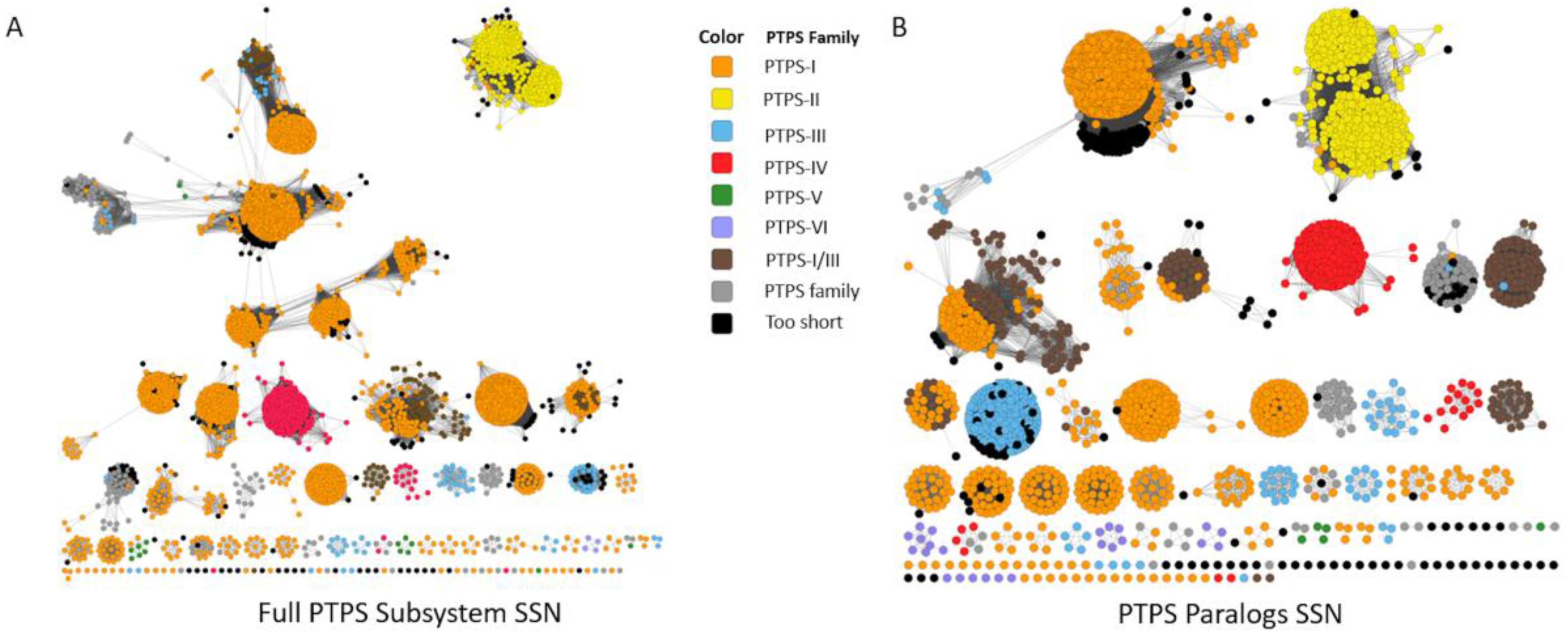
5. The COG0720 Case Study
6. Current Methods for Paralog Annotation in Genome Annotation Pipelines
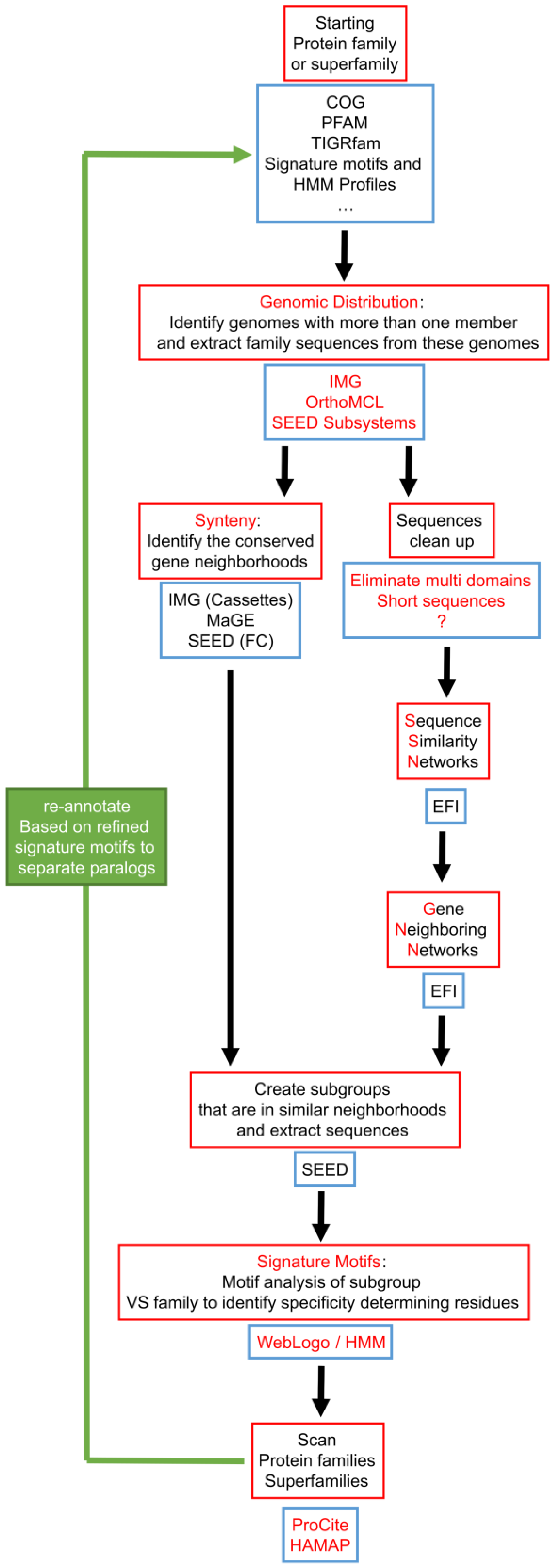
7. Integration of Tools for Paralog Separation in a Workflow
8. Materials and Methods
8.1. Bioinformatic Analyses
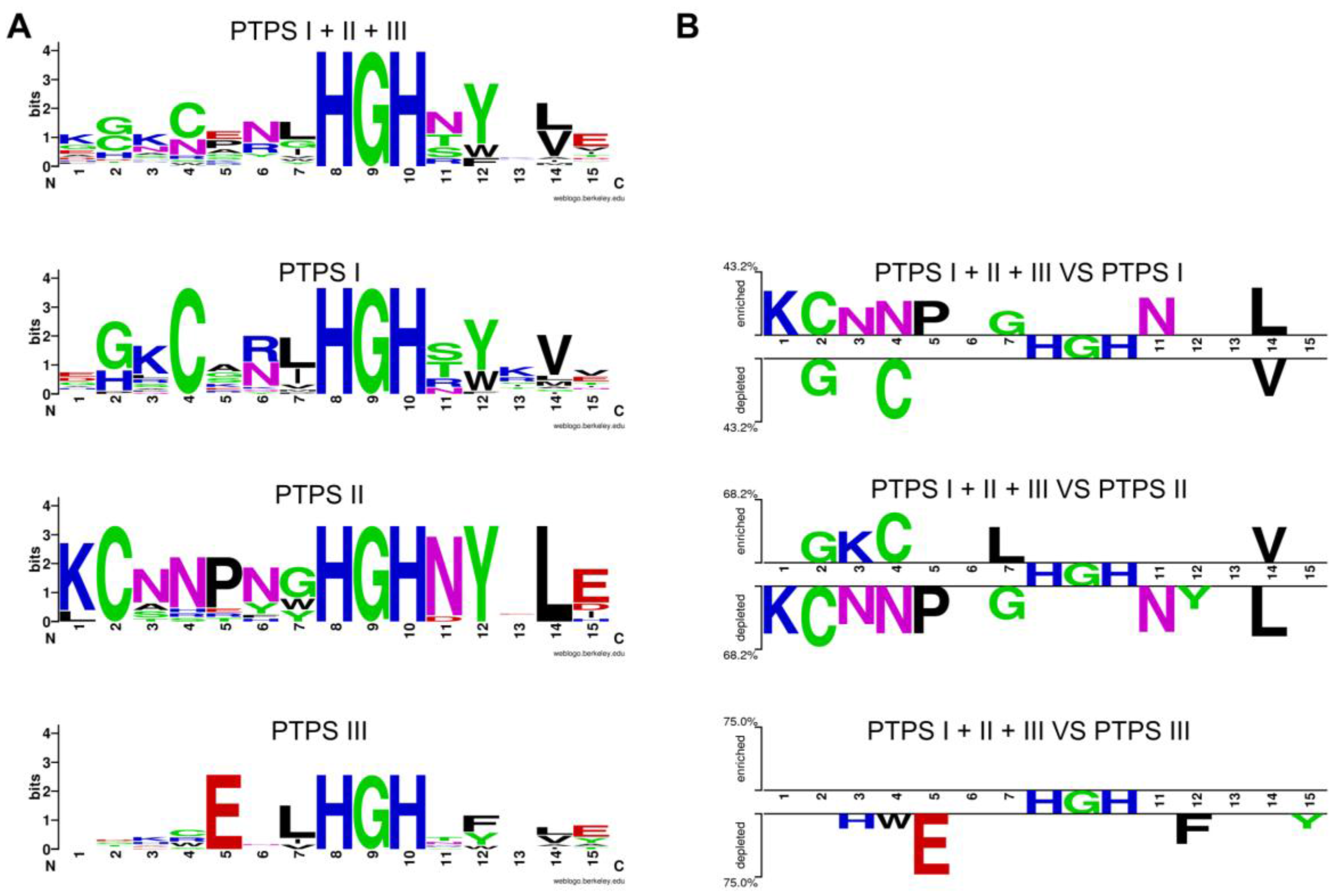
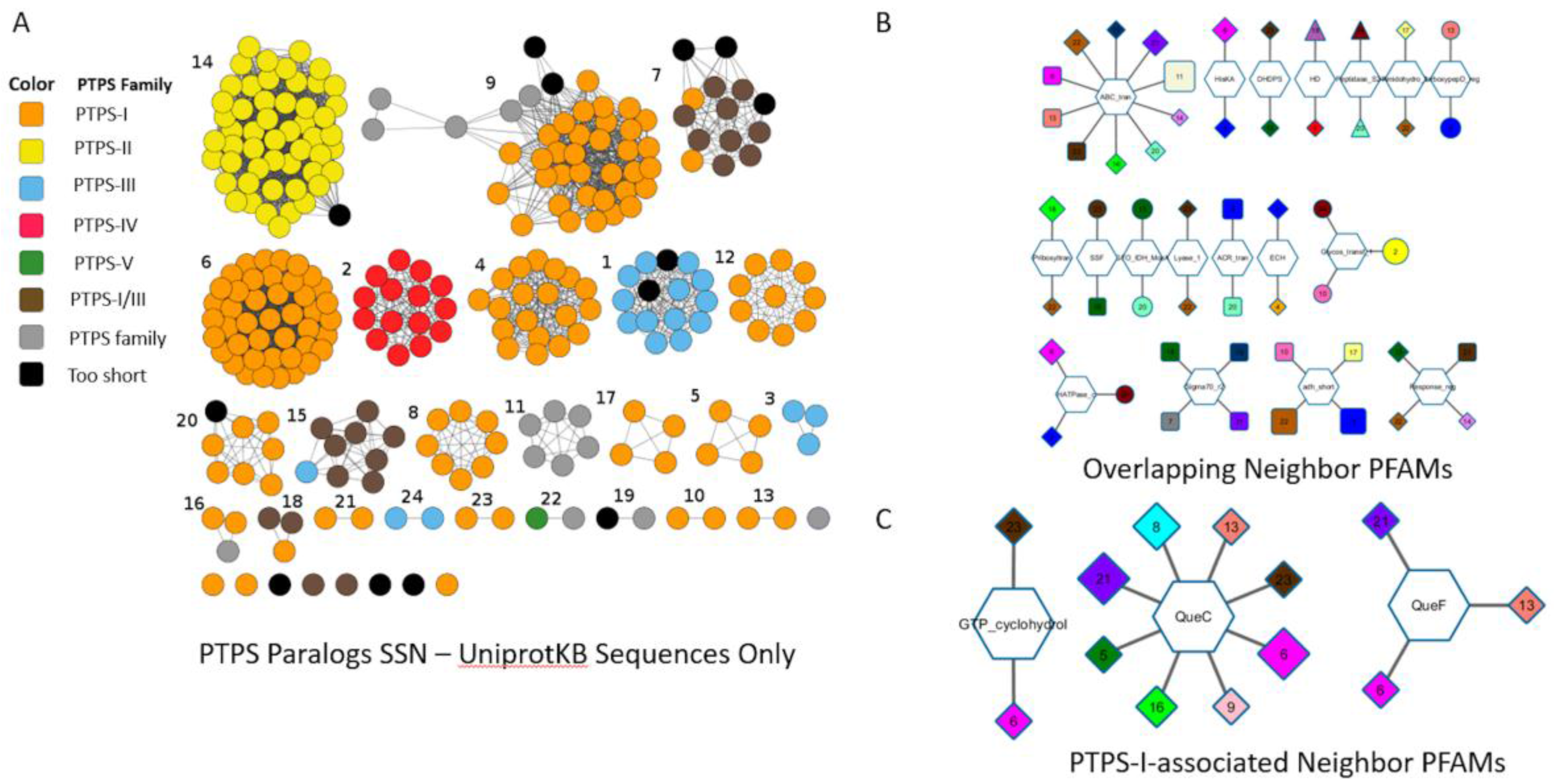
8.2. Sequence Similarity Network (SSN) and Neighborhood
9. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix

References
- Reddy, T.B.K.; Thomas, A.D.; Stamatis, D.; Bertsch, J.; Isbandi, M.; Jansson, J.; Mallajosyula, J.; Pagani, I.; Lobos, E.A.; Kyrpides, N.C. The Genomes OnLine Database (GOLD) v.5: A metadata management system based on a four level (meta)genome project classification. Nucleic Acids Res. 2015, 43, D1099–D1106. [Google Scholar] [CrossRef] [PubMed]
- Lasken, R.S.; McLean, J.S. Recent advances in genomic DNA sequencing of microbial species from single cells. Nat. Rev. Genet. 2014, 15, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Shendure, J.; Ji, H. Next-generation DNA sequencing. Nat. Biotechnol. 2008, 26, 1135–1145. [Google Scholar] [CrossRef] [PubMed]
- Mellis, I.A.; Raj, A. Half dozen of one, six billion of the other: What can small- and large-scale molecular systems biology learn from one another? Genome Res. 2015, 25, 1466–1472. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.A. On the interpretation of χ2 from contingency tables, and the calculation of P. J. R. Stat. Soc. 1922, 85, 87. [Google Scholar] [CrossRef]
- Larntz, K. Small-sample comparisons of exact levels for chi-squared goodness-of-fit statistics. J. Am. Stat. Assoc. 1978, 73, 253–263. [Google Scholar] [CrossRef]
- Klimke, W.; O’Donovan, C.; White, O.; Brister, J.R.; Clark, K.; Fedorov, B.; Mizrachi, I.; Pruitt, K.D.; Tatusova, T. Solving the Problem: Genome Annotation Standards before the Data Deluge. Stand. Genom. Sci. 2011, 5, 168–193. [Google Scholar] [CrossRef] [PubMed]
- Brent, R. Genomic biology. Cell 2000, 100, 169–183. [Google Scholar] [CrossRef]
- Davidson, D.; Baldock, R. Bioinformatics beyond sequence: Mapping gene function in the embryo. Nat. Rev. Genet. 2001, 2, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Murali, T.M. Computationally Driven Experimental Biology. Computer 2012, 45, 22–23. [Google Scholar]
- Tritt, A.; Eisen, J.A.; Facciotti, M.T.; Darling, A.E. An integrated pipeline for de novo assembly of microbial genomes. PLoS ONE 2012, 7, e42304. [Google Scholar] [CrossRef] [PubMed]
- Dunitz, M.I.; Lang, J.M.; Jospin, G.; Darling, A.E.; Eisen, J.A.; Coil, D.A. Swabs to genomes: A comprehensive workflow. PeerJ 2015, 3, e960. [Google Scholar] [CrossRef] [PubMed]
- Overbeek, R.; Olson, R.; Pusch, G.D.; Olsen, G.J.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Parrello, B.; Shukla, M.; et al. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res. 2014, 42, D206–D214. [Google Scholar] [CrossRef] [PubMed]
- Crappé, J.; Ndah, E.; Koch, A.; Steyaert, S.; Gawron, D.; de Keulenaer, S.; de Meester, E.; de Meyer, T.; van Criekinge, W.; van Damme, P.; Menschaert, G. Proteoformer: Deep proteome coverage through ribosome profiling and MS integration. Nucleic Acids Res. 2015, 43, e29. [Google Scholar] [CrossRef] [PubMed]
- Siezen, R.J.; van Hijum, S.A.F.T. Genome (re-)annotation and open-source annotation pipelines. Microb. Biotechnol. 2010, 3, 362–369. [Google Scholar] [CrossRef] [PubMed]
- Overmars, L.; Siezen, R.J.; Francke, C. A Novel Quality Measure and Correction Procedure for the Annotation of Microbial Translation Initiation Sites. PLoS ONE 2015, 10, e0133691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, I.-M.A.; Markowitz, V.M.; Chu, K.; Anderson, I.; Mavromatis, K.; Kyrpides, N.C.; Ivanova, N.N. Improving microbial genome annotations in an integrated database context. PLoS ONE 2013, 8, e54859. [Google Scholar] [CrossRef] [PubMed]
- Bastian, F.B.; Chibucos, M.C.; Gaudet, P.; Giglio, M.; Holliday, G.L.; Huang, H.; Lewis, S.E.; Niknejad, A.; Orchard, S.; Poux, S.; et al. The Confidence Information Ontology: A step towards a standard for asserting confidence in annotations. Database (Oxford) 2015, 2015, bav043. [Google Scholar] [CrossRef] [PubMed]
- Óhéigeartaigh, S.S.; Armisén, D.; Byrne, K.P.; Wolfe, K.H. SearchDOGS bacteria, software that provides automated identification of potentially missed genes in annotated bacterial genomes. J. Bacteriol. 2014, 196, 2030–2042. [Google Scholar] [CrossRef] [PubMed]
- Bork, P.; Bairoch, A. Go hunting in sequence databases but watch out for the traps. Trends Genet. 1996, 12, 425–427. [Google Scholar] [CrossRef]
- Schnoes, A.M.; Brown, S.D.; Dodevski, I.; Babbitt, P.C. Annotation error in public databases: Misannotation of molecular function in enzyme superfamilies. PLoS Comput. Biol. 2009, 5, e1000605. [Google Scholar] [CrossRef] [PubMed]
- Anton, B.P.; Kasif, S.; Roberts, R.J.; Steffen, M. Objective: Biochemical function. Front. Genet. 2014, 5, 210. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Ye, Y.; Ng, M.K.; Ho, S.-S.; Shi, R. Collective prediction of protein functions from protein-protein interaction networks. BMC Bioinform. 2014, 15 (Suppl. 2), S9. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, F.; Oesterhelt, D. A manual curation strategy to improve genome annotation: Application to a set of haloarchael genomes. Life (Basel, Switzerland) 2015, 5, 1427–1444. [Google Scholar] [CrossRef] [PubMed]
- Poux, S.; Magrane, M.; Arighi, C.N.; Bridge, A.; O’Donovan, C.; Laiho, K. UniProt Consortium Expert curation in UniProtKB: A case study on dealing with conflicting and erroneous data. Database (Oxford) 2014, 2014, bau016. [Google Scholar] [CrossRef] [PubMed]
- Brenner, S.E. Errors in genome annotation. Trends Genet. 1999, 15, 132–133. [Google Scholar] [CrossRef]
- Bell, M.J.; Collison, M.; Lord, P. Can inferred provenance and its visualisation be used to detect erroneous annotation? A case study using UniProtKB. PLoS ONE 2013, 8, e75541. [Google Scholar] [CrossRef] [PubMed]
- Poptsova, M.S.; Gogarten, J.P. Using comparative genome analysis to identify problems in annotated microbial genomes. Microbiology 2010, 156, 1909–1917. [Google Scholar] [CrossRef] [PubMed]
- Radivojac, P.; Clark, W.T.; Oron, T.R.; Schnoes, A.M.; Wittkop, T.; Sokolov, A.; Graim, K.; Funk, C.; Verspoor, K.; Ben-Hur, A.; et al. A large-scale evaluation of computational protein function prediction. Nat. Methods 2013, 10, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Gillis, J.; Pavlidis, P. Characterizing the state of the art in the computational assignment of gene function: Lessons from the first critical assessment of functional annotation (CAFA). BMC Bioinform. 2013, 14 (Suppl. 3), S15. [Google Scholar] [CrossRef]
- Kahanda, I.; Funk, C.S.; Ullah, F.; Verspoor, K.M.; Ben-Hur, A. A close look at protein function prediction evaluation protocols. Gigascience 2015, 4, 41. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Galperin, M.Y. Sequence—Evolution—Function; Springer US: Boston, MA, USA, 2003. [Google Scholar]
- Lee, D.; Redfern, O.; Orengo, C. Predicting protein function from sequence and structure. Nat. Rev. Mol. Cell Biol. 2007, 8, 995–1005. [Google Scholar] [CrossRef] [PubMed]
- Percudani, R.; Carnevali, D.; Puggioni, V. Ureidoglycolate hydrolase, amidohydrolase, lyase: How errors in biological databases are incorporated in scientific papers and vice versa. Database (Oxford) 2013, 2013, bat071. [Google Scholar] [CrossRef] [PubMed]
- Mao, F.; Su, Z.; Olman, V.; Dam, P.; Liu, Z.; Xu, Y. Mapping of orthologous genes in the context of biological pathways: An application of integer programming. Proc. Natl. Acad. Sci. USA 2006, 103, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Bork, P.; Koonin, E.V. Predicting functions from protein sequences—Where are the bottlenecks? Nat. Genet. 1998, 18, 313–318. [Google Scholar] [CrossRef] [PubMed]
- Green, M.L.; Karp, P.D. Genome annotation errors in pathway databases due to semantic ambiguity in partial EC numbers. Nucleic Acids Res. 2005, 33, 4035–4039. [Google Scholar] [CrossRef] [PubMed]
- Devos, D.; Valencia, A. Intrinsic errors in genome annotation. Trends Genet. 2001, 17, 429–431. [Google Scholar] [CrossRef]
- Promponas, V.J.; Iliopoulos, I.; Ouzounis, C.A. Annotation inconsistencies beyond sequence similarity-based function prediction—Phylogeny and genome structure. Stand. Genom. Sci. 2015, 10, 108. [Google Scholar] [CrossRef] [PubMed]
- Dornfeld, C.; Weisberg, A.J.; K C, R.; Dudareva, N.; Jelesko, J.G.; Maeda, H.A. Phylobiochemical characterization of class-Ib aspartate/prephenate aminotransferases reveals evolution of the plant arogenate phenylalanine pathway. Plant Cell 2014, 26, 3101–3114. [Google Scholar] [CrossRef] [PubMed]
- Verdel-Aranda, K.; López-Cortina, S.T.; Hodgson, D.A.; Barona-Gómez, F. Molecular annotation of ketol-acid reductoisomerases from Streptomyces reveals a novel amino acid biosynthesis interlock mediated by enzyme promiscuity. Microb. Biotechnol. 2015, 8, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.D.; Babbitt, P.C. New insights about enzyme evolution from large scale studies of sequence and structure relationships. J. Biol. Chem. 2014, 289, 30221–30228. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Orengo, C.A. Protein function annotation using protein domain family resources. Methods 2016, 93, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Barona-Gómez, F. Re-annotation of the sequence > annotation: Opportunities for the functional microbiologist. Microb. Biotechnol. 2015, 8, 2–4. [Google Scholar] [CrossRef] [PubMed]
- Van Lanen, S.G.; Reader, J.S.; Swairjo, M.A.; de Crécy-Lagard, V.; Lee, B.; Iwata-Reuyl, D. From cyclohydrolase to oxidoreductase: Discovery of nitrile reductase activity in a common fold. Proc. Natl. Acad. Sci. USA 2005, 102, 4264–4269. [Google Scholar] [CrossRef] [PubMed]
- Phillips, G.; Swairjo, M.A.; Gaston, K.W.; Bailly, M.; Limbach, P.A.; Iwata-Reuyl, D.; de Crécy-Lagard, V. Diversity of archaeosine synthesis in crenarchaeota. ACS Chem. Biol. 2012, 7, 300–305. [Google Scholar] [CrossRef] [PubMed]
- Pribat, A.; Blaby, I.K.; Lara-Núñez, A.; Gregory, J.F.; de Crécy-Lagard, V.; Hanson, A.D. FolX and FolM are essential for tetrahydromonapterin synthesis in Escherichia coli and Pseudomonas aeruginosa. J. Bacteriol. 2010, 192, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Gerdes, S.; El Yacoubi, B.; Bailly, M.; Blaby, I.K.; Blaby-Haas, C.E.; Jeanguenin, L.; Lara-Núñez, A.; Pribat, A.; Waller, J.C.; Wilke, A.; et al. Synergistic use of plant-prokaryote comparative genomics for functional annotations. BMC Genom. 2011, 12 (Suppl. 1), S2. [Google Scholar] [CrossRef] [PubMed]
- Bailly, M.; de Crécy-Lagard, V. Predicting the pathway involved in post-translational modification of elongation factor P in a subset of bacterial species. Biol. Direct 2010, 5, 3. [Google Scholar] [CrossRef] [PubMed]
- Waller, J.C.; Alvarez, S.; Naponelli, V.; Lara-Nuñez, A.; Blaby, I.K.; da Silva, V.; Ziemak, M.J.; Vickers, T.J.; Beverley, S.M.; Edison, A.S.; et al. A role for tetrahydrofolates in the metabolism of iron-sulfur clusters in all domains of life. Proc. Natl. Acad. Sci. USA 2010, 107, 10412–10417. [Google Scholar] [CrossRef] [PubMed]
- De Crécy-Lagard, V.; Forouhar, F.; Brochier-Armanet, C.; Tong, L.; Hunt, J.F. Comparative genomic analysis of the DUF71/COG2102 family predicts roles in diphthamide biosynthesis and B12 salvage. Biol. Direct 2012, 7, 32. [Google Scholar] [CrossRef] [PubMed]
- Adams, N.E.; Thiaville, J.J.; Proestos, J.; Juárez-Vázquez, A.L.; McCoy, A.J.; Barona-Gómez, F.; Iwata-Reuyl, D.; de Crécy-Lagard, V.; Maurelli, A.T. Promiscuous and adaptable enzymes fill “holes” in the tetrahydrofolate pathway in Chlamydia species. mBio 2014, 5, e01378–e013714. [Google Scholar] [CrossRef] [PubMed]
- De Crécy-Lagard, V.; El Yacoubi, B.; de la Garza, R.D.; Noiriel, A.; Hanson, A.D. Comparative genomics of bacterial and plant folate synthesis and salvage: Predictions and validations. BMC Genom. 2007, 8, 245. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, K.; Blaby, I.K.; Thiaville, P.C.; Majumder, M.; Grosjean, H.; Yuan, Y.A.; Gupta, R.; de Crécy-Lagard, V. The archaeal COG1901/DUF358 SPOUT-methyltransferase members, together with pseudouridine synthase Pus10, catalyze the formation of 1-methylpseudouridine at position 54 of tRNA. RNA 2012, 18, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Phillips, G.; Grochowski, L.L.; Bonnett, S.; Xu, H.; Bailly, M.; Blaby-Haas, C.; El Yacoubi, B.; Iwata-Reuyl, D.; White, R.H.; de Crécy-Lagard, V. Functional promiscuity of the COG0720 family. ACS Chem. Biol. 2012, 7, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Haas, C.E.; Rodionov, D.A.; Kropat, J.; Malasarn, D.; Merchant, S.S.; de Crécy-Lagard, V. A subset of the diverse COG0523 family of putative metal chaperones is linked to zinc homeostasis in all kingdoms of life. BMC Genom. 2009, 10, 470. [Google Scholar] [CrossRef] [PubMed]
- Pribat, A.; Blaby, I.K.; Lara-Núñez, A.; Jeanguenin, L.; Fouquet, R.; Frelin, O.; Gregory, J.F.; Philmus, B.; Begley, T.P.; de Crécy-Lagard, V.; et al. A 5-formyltetrahydrofolate cycloligase paralog from all domains of life: Comparative genomic and experimental evidence for a cryptic role in thiamin metabolism. Funct. Integr. Genom. 2011, 11, 467–478. [Google Scholar] [CrossRef] [PubMed]
- Galperin, M.Y.; Makarova, K.S.; Wolf, Y.I.; Koonin, E.V. Expanded microbial genome coverage and improved protein family annotation in the COG database. Nucleic Acids Res. 2015, 43, D261–D269. [Google Scholar] [CrossRef] [PubMed]
- Lan, N.; Montelione, G.T.; Gerstein, M. Ontologies for proteomics: Towards a systematic definition of structure and function that scales to the genome level. Curr. Opin. Chem. Biol. 2003, 7, 44–54. [Google Scholar] [CrossRef]
- Lan, N.; Jansen, R.; Gerstein, M. Toward a systematic definition of protein function that scales to the genome level: Defining function in terms of interactions. IEEE Proc. 2002, 90, 1848–1858. [Google Scholar]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef] [PubMed]
- Reference Genome Group of the Gene Ontology Consortium. The Gene Ontology’s Reference Genome Project: A unified framework for functional annotation across species. PLoS Comput. Biol. 2009, 5, e1000431. [Google Scholar]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Reed, J.L.; Famili, I.; Thiele, I.; Palsson, B.O. Towards multidimensional genome annotation. Nat. Rev. Genet. 2006, 7, 130–141. [Google Scholar] [CrossRef] [PubMed]
- Yandell, M.; Ence, D. A beginner’s guide to eukaryotic genome annotation. Nat. Rev. Genet. 2012, 13, 329–342. [Google Scholar] [CrossRef] [PubMed]
- Richardson, E.J.; Watson, M. The automatic annotation of bacterial genomes. Brief. Bioinform. 2013, 14, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Jensen, L.J.; Ussery, D.W.; Brunak, S. Functionality of system components: Conservation of protein function in protein feature space. Genome Res. 2003, 13, 2444–2449. [Google Scholar] [CrossRef] [PubMed]
- Pereira, C.; Denise, A.; Lespinet, O. A meta-approach for improving the prediction and the functional annotation of ortholog groups. BMC Genom. 2014, 15 (Suppl. 6), S16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, D.P.; Krishnamurthy, N.; Sjölander, K. Automated protein subfamily identification and classification. PLoS Comput. Biol. 2007, 3, e160. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, B.E.; Jordan, M.I.; Srouji, J.R.; Brenner, S.E. Genome-scale phylogenetic function annotation of large and diverse protein families. Genome Res. 2011, 21, 1969–1980. [Google Scholar] [CrossRef] [PubMed]
- Fitch, W.M. Distinguishing homologous from analogous proteins. Syst. Biol. 1970, 19, 99–113. [Google Scholar] [CrossRef]
- Altenhoff, A.M.; Studer, R.A.; Robinson-Rechavi, M.; Dessimoz, C. Resolving the ortholog conjecture: Orthologs tend to be weakly, but significantly, more similar in function than paralogs. PLoS Comput. Biol. 2012, 8, e1002514. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Mackey, A.J.; Stoeckert, C.J.; Roos, D.S. OrthoMCL-DB: Querying a comprehensive multi-species collection of ortholog groups. Nucleic Acids Res. 2006, 34, D363–D368. [Google Scholar] [CrossRef] [PubMed]
- Altenhoff, A.M.; Škunca, N.; Glover, N.; Train, C.-M.; Sueki, A.; Piližota, I.; Gori, K.; Tomiczek, B.; Müller, S.; Redestig, H.; et al. The OMA orthology database in 2015: Function predictions, better plant support, synteny view and other improvements. Nucleic Acids Res. 2015, 43, D240–D249. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Cepas, J.; Szklarczyk, D.; Forslund, K.; Cook, H.; Heller, D.; Walter, M.C.; Rattei, T.; Mende, D.R.; Sunagawa, S.; Kuhn, M.; et al. eggNOG 4.5: A hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 2016, 44, D286–D293. [Google Scholar] [CrossRef] [PubMed]
- Gerlt, J.A.; Babbitt, P.C. Can sequence determine function? Genome Biol. 2000, 1, S0005. [Google Scholar] [CrossRef] [PubMed]
- Jensen, R.A. Orthologs and paralogs—We need to get it right. Genome Biol. 2001, 2, S1002. [Google Scholar] [CrossRef]
- Studer, R.A.; Robinson-Rechavi, M. How confident can we be that orthologs are similar, but paralogs differ? Trends Genet. 2009, 25, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Nehrt, N.L.; Clark, W.T.; Radivojac, P.; Hahn, M.W. Testing the ortholog conjecture with comparative functional genomic data from mammals. PLoS Comput. Biol. 2011, 7, e1002073. [Google Scholar] [CrossRef] [PubMed]
- Gharib, W.H.; Robinson-Rechavi, M. When orthologs diverge between human and mouse. Brief. Bioinform. 2011, 12, 436–441. [Google Scholar] [CrossRef] [PubMed]
- Gabaldón, T.; Koonin, E.V. Functional and evolutionary implications of gene orthology. Nat. Rev. Genet. 2013, 14, 360–366. [Google Scholar] [CrossRef] [PubMed]
- Gout, J.-F.; Lynch, M. Maintenance and loss of duplicated genes by dosage subfunctionalization. Mol. Biol. Evol. 2015, 32, 2141–2148. [Google Scholar] [CrossRef] [PubMed]
- Papp, B.; Pál, C.; Hurst, L.D. Dosage sensitivity and the evolution of gene families in yeast. Nature 2003, 424, 194–197. [Google Scholar] [CrossRef] [PubMed]
- Gout, J.-F.; Kahn, D.; Duret, L.; Paramecium Post-Genomics Consortium. The relationship among gene expression, the evolution of gene dosage, and the rate of protein evolution. PLoS Genet. 2010, 6, e1000944. [Google Scholar] [CrossRef]
- Qian, W.; Liao, B.-Y.; Chang, A.Y.-F.; Zhang, J. Maintenance of duplicate genes and their functional redundancy by reduced expression. Trends Genet. 2010, 26, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.T.Y.; Pang, Y.L.J.; Deng, W.; Babu, I.R.; Dyavaiah, M.; Begley, T.J.; Dedon, P.C. Reprogramming of tRNA modifications controls the oxidative stress response by codon-biased translation of proteins. Nat. Commun. 2012, 3, 937. [Google Scholar] [CrossRef] [PubMed]
- Fillinger, S.; Boschi-Muller, S.; Azza, S.; Dervyn, E.; Branlant, G.; Aymerich, S. Two glyceraldehyde-3-phosphate dehydrogenases with opposite physiological roles in a nonphotosynthetic bacterium. J. Biol. Chem. 2000, 275, 14031–14037. [Google Scholar] [CrossRef] [PubMed]
- Rusin, L.Y.; Lyubetskaya, E.V.; Gorbunov, K.Y.; Lyubetsky, V.A. Reconciliation of gene and species trees. BioMed Res. Int. 2014, 2014, 642089. [Google Scholar] [CrossRef] [PubMed]
- Szöllősi, G.J.; Tannier, E.; Daubin, V.; Boussau, B. The inference of gene trees with species trees. Syst. Biol. 2015, 64, e42–e62. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.-C.; Rasmussen, M.D.; Bansal, M.S.; Kellis, M. Most parsimonious reconciliation in the presence of gene duplication, loss, and deep coalescence using labeled coalescent trees. Genome Res. 2014, 24, 475–486. [Google Scholar] [CrossRef] [PubMed]
- Doyon, J.-P.; Ranwez, V.; Daubin, V.; Berry, V. Models, algorithms and programs for phylogeny reconciliation. Brief. Bioinform. 2011, 12, 392–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arvestad, L.; Berglund, A.-C.; Lagergren, J.; Sennblad, B. Bayesian gene/species tree reconciliation and orthology analysis using MCMC. Bioinformatics 2003, 19 (Suppl. 1), i7–i15. [Google Scholar] [CrossRef] [PubMed]
- Vernot, B.; Stolzer, M.; Goldman, A.; Durand, D. Reconciliation with non-binary species trees. J. Comput. Biol. 2008, 15, 981–1006. [Google Scholar] [CrossRef] [PubMed]
- Kolaczkowski, B.; Thornton, J.W. Long-branch attraction bias and inconsistency in Bayesian phylogenetics. PLoS ONE 2009, 4, e7891. [Google Scholar] [CrossRef] [PubMed]
- Kolaczkowski, B.; Thornton, J.W. Performance of maximum parsimony and likelihood phylogenetics when evolution is heterogeneous. Nature 2004, 431, 980–984. [Google Scholar] [CrossRef] [PubMed]
- Hahn, M.W. Bias in phylogenetic tree reconciliation methods: Implications for vertebrate genome evolution. Genome Biol. 2007, 8, R141. [Google Scholar] [CrossRef] [PubMed]
- Jeffroy, O.; Brinkmann, H.; Delsuc, F.; Philippe, H. Phylogenomics: The beginning of incongruence? Trends Genet. 2006, 22, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, B.E.; Jordan, M.I.; Muratore, K.E.; Brenner, S.E. Protein molecular function prediction by Bayesian phylogenomics. PLoS Comput. Biol. 2005, 1, e45. [Google Scholar] [CrossRef] [PubMed]
- Sahraeian, S.M.; Luo, K.R.; Brenner, S.E. SIFTER search: A web server for accurate phylogeny-based protein function prediction. Nucleic Acids Res. 2015, 43, W141–W147. [Google Scholar] [CrossRef] [PubMed]
- Giribet, G. Efficient tree searches with available algorithms. Evol. Bioinform. Online 2007, 3, 341–356. [Google Scholar] [PubMed]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Howe, K.; Bateman, A.; Durbin, R. QuickTree: Building huge Neighbour-Joining trees of protein sequences. Bioinformatics 2002, 18, 1546–1547. [Google Scholar] [CrossRef] [PubMed]
- Hillis, D.M. Approaches for assessing phylogenetic accuracy. Syst. Biol. 1995, 44, 3–16. [Google Scholar] [CrossRef]
- Cotton, J.A. Analytical methods for detecting paralogy in molecular datasets. Methods Enzymol. 2005, 395, 700–724. [Google Scholar] [PubMed]
- Lechner, M.; Hernandez-Rosales, M.; Doerr, D.; Wieseke, N.; Thévenin, A.; Stoye, J.; Hartmann, R.K.; Prohaska, S.J.; Stadler, P.F. Orthology detection combining clustering and synteny for very large datasets. PLoS ONE 2014, 9, e105015. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, D.M.; Wolf, Y.I.; Mushegian, A.R.; Koonin, E.V. Computational methods for Gene Orthology inference. Brief. Bioinform. 2011, 12, 379–391. [Google Scholar] [CrossRef] [PubMed]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A.; Koonin, E.V. The COG database: A tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef] [PubMed]
- Kuzniar, A.; van Ham, R.C.H.J.; Pongor, S.; Leunissen, J.A.M. The quest for orthologs: Finding the corresponding gene across genomes. Trends Genet. 2008, 24, 539–551. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.N.K.; Liu, L.; Pearl, D.; Edwards, S.V. Tangled trees: The challenge of inferring species trees from coalescent and noncoalescent genes. Methods Mol. Biol. 2012, 856, 3–28. [Google Scholar] [PubMed]
- Gerlt, J.A.; Bouvier, J.T.; Davidson, D.B.; Imker, H.J.; Sadkhin, B.; Slater, D.R.; Whalen, K.L. Enzyme Function Initiative-Enzyme Similarity Tool (EFI-EST): A web tool for generating protein sequence similarity networks. Biochim. Biophys. Acta 2015, 1854, 1019–1037. [Google Scholar] [CrossRef] [PubMed]
- Cantarel, B.L.; Morrison, H.G.; Pearson, W. Exploring the relationship between sequence similarity and accurate phylogenetic trees. Mol. Biol. Evol. 2006, 23, 2090–2100. [Google Scholar] [CrossRef] [PubMed]
- Kelly, S.; Maini, P.K. DendroBLAST: Approximate phylogenetic trees in the absence of multiple sequence alignments. PLoS ONE 2013, 8, e58537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trachana, K.; Forslund, K.; Larsson, T.; Powell, S.; Doerks, T.; von Mering, C.; Bork, P. A phylogeny-based benchmarking test for orthology inference reveals the limitations of function-based validation. PLoS ONE 2014, 9, e111122. [Google Scholar] [CrossRef] [PubMed]
- Swofford, D.L.; Waddell, P.J.; Huelsenbeck, J.P.; Foster, P.G.; Lewis, P.O.; Rogers, J.S. Bias in phylogenetic estimation and its relevance to the choice between parsimony and likelihood methods. Syst. Biol. 2001, 50, 525–539. [Google Scholar] [CrossRef] [PubMed]
- Tatusov, R.L.; Koonin, E.V.; Lipman, D.J. A genomic perspective on protein families. Science 1997, 278, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Overbeek, R.; Larsen, N.; Pusch, G.D.; D’Souza, M.; Selkov, E.; Kyrpides, N.; Fonstein, M.; Maltsev, N.; Selkov, E. WIT: Integrated system for high-throughput genome sequence analysis and metabolic reconstruction. Nucleic Acids Res. 2000, 28, 123–125. [Google Scholar] [CrossRef] [PubMed]
- Overbeek, R.; Fonstein, M.; D’Souza, M.; Pusch, G.D.; Maltsev, N. Use of contiguity on the chromosome to predict functional coupling. In Silico Biol. 1999, 1, 93–108. [Google Scholar] [PubMed]
- Overbeek, R.; Begley, T.; Butler, R.M.; Choudhuri, J.V.; Chuang, H.-Y.; Cohoon, M.; de Crécy-Lagard, V.; Diaz, N.; Disz, T.; Edwards, R.; et al. The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res. 2005, 33, 5691–5702. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Osterman, A.; Overbeek, R.; Godzik, A. Automatic detection of subsystem/pathway variants in genome analysis. Bioinformatics 2005, 21 (Suppl. 1), i478–i486. [Google Scholar] [CrossRef] [PubMed]
- Liberal, R.; Pinney, J.W. Simple topological properties predict functional misannotations in a metabolic network. Bioinformatics 2013, 29, i154–i161. [Google Scholar] [CrossRef] [PubMed]
- Osterman, A.; Overbeek, R. Missing genes in metabolic pathways: A comparative genomics approach. Curr. Opin. Chem. Biol. 2003, 7, 238–251. [Google Scholar] [CrossRef]
- Earnshaw, W.C. Deducing protein function by forensic integrative cell biology. PLoS Biol. 2013, 11, e1001742. [Google Scholar] [CrossRef] [PubMed]
- Hanson, A.D.; Pribat, A.; Waller, J.C.; de Crécy-Lagard, V. “Unknown” proteins and “orphan” enzymes: The missing half of the engineering parts list—And how to find it. Biochem. J. 2010, 425, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, M.; Thompson, M.; Fierro, J.; Bowers, P. Computational method to assign microbial genes to pathways. J. Cell. Biochem. Suppl. 2001, 84 (Suppl. 37), 106–109. [Google Scholar] [CrossRef] [PubMed]
- Dandekar, T.; Snel, B.; Huynen, M.; Bork, P. Conservation of gene order: A fingerprint of proteins that physically interact. Trends Biochem. Sci. 1998, 23, 324–328. [Google Scholar] [CrossRef]
- Yanai, I.; Mellor, J.C.; de Lisi, C. Identifying functional links between genes using conserved chromosomal proximity. Trends Genet. 2002, 18, 176–179. [Google Scholar] [CrossRef]
- Price, M.N.; Huang, K.H.; Arkin, A.P.; Alm, E.J. Operon formation is driven by co-regulation and not by horizontal gene transfer. Genome Res. 2005, 15, 809–819. [Google Scholar] [CrossRef] [PubMed]
- Ream, D.C.; Bankapur, A.R.; Friedberg, I. An event-driven approach for studying gene block evolution in bacteria. Bioinformatics 2015, 31, 2075–2083. [Google Scholar] [CrossRef] [PubMed]
- Junier, I.; Rivoire, O. Conserved units of co-expression in bacterial genomes: An evolutionary insight into transcriptional regulation. PLoS ONE 2016, 11, e0155740. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, J.G.; Roth, J.R. Selfish operons: Horizontal transfer may drive the evolution of gene clusters. Genetics 1996, 143, 1843–18460. [Google Scholar] [PubMed]
- Henry, C.S.; Lerma-Ortiz, C.; Gerdes, S.Y.; Mullen, J.D.; Colasanti, R.; Zhukov, A.; Frelin, O.; Thiaville, J.J.; Zallot, R.; Niehaus, T.D.; et al. Systematic identification and analysis of frequent gene fusion events in metabolic pathways. BMC Genom. 2016, 17, 473. [Google Scholar] [CrossRef] [PubMed]
- Green, M.L.; Karp, P.D. Using genome-context data to identify specific types of functional associations in pathway/genome databases. Bioinformatics 2007, 23, i205–i211. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Hagelsieb, G. The power of operon rearrangements for predicting functional associations. Comput. Struct. Biotechnol. J. 2015, 13, 402–406. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef] [PubMed]
- Overbeek, R.; Fonstein, M.; D’Souza, M.; Pusch, G.D.; Maltsev, N. The use of gene clusters to infer functional coupling. Proc. Natl. Acad. Sci. USA 1999, 96, 2896–2901. [Google Scholar] [CrossRef] [PubMed]
- Dehal, P.S.; Joachimiak, M.P.; Price, M.N.; Bates, J.T.; Baumohl, J.K.; Chivian, D.; Friedland, G.D.; Huang, K.H.; Keller, K.; Novichkov, P.S.; et al. MicrobesOnline: An integrated portal for comparative and functional genomics. Nucleic Acids Res. 2010, 38, D396–D400. [Google Scholar] [CrossRef] [PubMed]
- Oberto, J. SyntTax: A web server linking synteny to prokaryotic taxonomy. BMC Bioinform. 2013, 14, 4. [Google Scholar] [CrossRef] [PubMed]
- Vallenet, D.; Labarre, L.; Rouy, Z.; Barbe, V.; Bocs, S.; Cruveiller, S.; Lajus, A.; Pascal, G.; Scarpelli, C.; Médigue, C. MaGe: A microbial genome annotation system supported by synteny results. Nucleic Acids Res. 2006, 34, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Goyer, A.; Hasnain, G.; Frelin, O.; Ralat, M.A.; Gregory, J.F.; Hanson, A.D. A cross-kingdom Nudix enzyme that pre-empts damage in thiamin metabolism. Biochem. J. 2013, 454, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Klaus, S.M.J.; Wegkamp, A.; Sybesma, W.; Hugenholtz, J.; Gregory, J.F.; Hanson, A.D. A nudix enzyme removes pyrophosphate from dihydroneopterin triphosphate in the folate synthesis pathway of bacteria and plants. J. Biol. Chem. 2005, 280, 5274–5280. [Google Scholar] [CrossRef] [PubMed]
- McLennan, A.G. The Nudix hydrolase superfamily. Cell. Mol. Life Sci. 2006, 63, 123–143. [Google Scholar] [CrossRef] [PubMed]
- Gunawardana, D.; Likic, V.A.; Gayler, K.R. A comprehensive bioinformatics analysis of the Nudix superfamily in Arabidopsis thaliana. Comp. Funct. Genom. 2009, 2009, 820381. [Google Scholar] [CrossRef] [PubMed]
- Piovesan, D.; Giollo, M.; Leonardi, E.; Ferrari, C.; Tosatto, S.C.E. INGA: Protein function prediction combining interaction networks, domain assignments and sequence similarity. Nucleic Acids Res. 2015, 43, W134–W140. [Google Scholar] [CrossRef] [PubMed]
- Bastard, K.; Smith, A.A.T.; Vergne-Vaxelaire, C.; Perret, A.; Zaparucha, A.; de Melo-Minardi, R.; Mariage, A.; Boutard, M.; Debard, A.; Lechaplais, C.; et al. Revealing the hidden functional diversity of an enzyme family. Nat. Chem. Biol. 2014, 10, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Pandya, C.; Liu, C.; Al-Obaidi, N.F.; Wang, M.; Zheng, L.; Toews Keating, S.; Aono, M.; Love, J.D.; Evans, B.; et al. Panoramic view of a superfamily of phosphatases through substrate profiling. Proc. Natl. Acad. Sci. USA 2015, 112, E1974–E1983. [Google Scholar] [CrossRef] [PubMed]
- Akiva, E.; Brown, S.; Almonacid, D.E.; Barber, A.E.; Custer, A.F.; Hicks, M.A.; Huang, C.C.; Lauck, F.; Mashiyama, S.T.; Meng, E.C.; et al. The Structure-Function Linkage Database. Nucleic Acids Res. 2014, 42, D521–D530. [Google Scholar] [CrossRef] [PubMed]
- Furnham, N.; Sillitoe, I.; Holliday, G.L.; Cuff, A.L.; Rahman, S.A.; Laskowski, R.A.; Orengo, C.A.; Thornton, J.M. FunTree: A resource for exploring the functional evolution of structurally defined enzyme superfamilies. Nucleic Acids Res. 2012, 40, D776–D782. [Google Scholar] [CrossRef] [PubMed]
- Furnham, N.; Dawson, N.L.; Rahman, S.A.; Thornton, J.M.; Orengo, C.A. Large-scale analysis exploring evolution of catalytic machineries and mechanisms in enzyme superfamilies. J. Mol. Biol. 2016, 428, 253–267. [Google Scholar] [CrossRef] [PubMed]
- Thöny, B.; Auerbach, G.; Blau, N. Tetrahydrobiopterin biosynthesis, regeneration and functions. Biochem. J. 2000, 347, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Inoue, Y.; Kawasaki, Y.; Harada, T.; Hatakeyama, K.; Kagamiyama, H. Purification and cDNA cloning of rat 6-pyruvoyl-tetrahydropterin synthase. J. Biol. Chem. 1991, 266, 20791–20796. [Google Scholar] [PubMed]
- Kong, J.S.; Kang, J.-Y.; Kim, H.L.; Kwon, O.-S.; Lee, K.H.; Park, Y.S. 6-Pyruvoyltetrahydropterin synthase orthologs of either a single or dual domain structure are responsible for tetrahydrobiopterin synthesis in bacteria. FEBS Lett. 2006, 580, 4900–4904. [Google Scholar] [CrossRef] [PubMed]
- Dittrich, S.; Mitchell, S.L.; Blagborough, A.M.; Wang, Q.; Wang, P.; Sims, P.F.G.; Hyde, J.E. An atypical orthologue of 6-pyruvoyltetrahydropterin synthase can provide the missing link in the folate biosynthesis pathway of malaria parasites. Mol. Microbiol. 2008, 67, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Pribat, A.; Jeanguenin, L.; Lara-Núñez, A.; Ziemak, M.J.; Hyde, J.E.; de Crécy-Lagard, V.; Hanson, A.D. 6-pyruvoyltetrahydropterin synthase paralogs replace the folate synthesis enzyme dihydroneopterin aldolase in diverse bacteria. J. Bacteriol. 2009, 191, 4158–4165. [Google Scholar] [CrossRef] [PubMed]
- McCarty, R.M.; Somogyi, A.; Bandarian, V. Escherichia coli QueD is a 6-carboxy-5,6,7,8-tetrahydropterin synthase. Biochemistry 2009, 48, 2301–2303. [Google Scholar] [CrossRef] [PubMed]
- Reader, J.S.; Metzgar, D.; Schimmel, P.; de Crécy-Lagard, V. Identification of four genes necessary for biosynthesis of the modified nucleoside queuosine. J. Biol. Chem. 2004, 279, 6280–6285. [Google Scholar] [CrossRef]
- Markowitz, V.M.; Chen, I.-M.A.; Palaniappan, K.; Chu, K.; Szeto, E.; Pillay, M.; Ratner, A.; Huang, J.; Woyke, T.; Huntemann, M.; et al. IMG 4 version of the integrated microbial genomes comparative analysis system. Nucleic Acids Res. 2014, 42, D560–D567. [Google Scholar] [CrossRef]
- Markowitz, V.M.; Chen, I.-M.A.; Chu, K.; Pati, A.; Ivanova, N.N.; Kyrpides, N.C. Ten years of maintaining and expanding a microbial genome and metagenome analysis system. Trends Microbiol. 2015, 23, 730–741. [Google Scholar] [CrossRef] [PubMed]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid annotations using subsystems technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef] [PubMed]
- Brettin, T.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Olsen, G.J.; Olson, R.; Overbeek, R.; Parrello, B.; Pusch, G.D.; et al. RASTtk: A modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci. Rep. 2015, 5, 8365. [Google Scholar] [CrossRef] [PubMed]
- Tatusova, T.; DiCuccio, M.; Badretdin, A.; Chetvernin, V.; Ciufo, S.; Li, W. Prokaryotic genome annotation pipeline. In The NCBI Handbook [Internet]; National Center for Biotechnology Information (US): Bethesda, MD, USA, 2013. [Google Scholar]
- Kersey, P.J.; Allen, J.E.; Armean, I.; Boddu, S.; Bolt, B.J.; Carvalho-Silva, D.; Christensen, M.; Davis, P.; Falin, L.J.; Grabmueller, C.; et al. Ensembl Genomes 2016: More genomes, more complexity. Nucleic Acids Res. 2016, 44, D574–D580. [Google Scholar] [CrossRef] [PubMed]
- Angiuoli, S.V.; Gussman, A.; Klimke, W.; Cochrane, G.; Field, D.; Garrity, G.; Kodira, C.D.; Kyrpides, N.; Madupu, R.; Markowitz, V.; et al. Toward an online repository of Standard Operating Procedures (SOPs) for (meta)genomic annotation. OMICS 2008, 12, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Huntemann, M.; Ivanova, N.N.; Mavromatis, K.; Tripp, H.J.; Paez-Espino, D.; Palaniappan, K.; Szeto, E.; Pillay, M.; Chen, I.-M.A.; Pati, A.; et al. The standard operating procedure of the DOE-JGI Microbial Genome Annotation Pipeline (MGAP v.4). Stand. Genom. Sci. 2015, 10, 86. [Google Scholar] [CrossRef] [PubMed]
- Mavromatis, K.; Ivanova, N.N.; Chen, I.-M.A.; Szeto, E.; Markowitz, V.M.; Kyrpides, N.C. The DOE-JGI Standard Operating Procedure for the Annotations of Microbial Genomes. Stand. Genom. Sci. 2009, 1, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Markowitz, V.M.; Chen, I.-M.A.; Palaniappan, K.; Chu, K.; Szeto, E.; Grechkin, Y.; Ratner, A.; Anderson, I.; Lykidis, A.; Mavromatis, K.; et al. The integrated microbial genomes system: An expanding comparative analysis resource. Nucleic Acids Res. 2010, 38, D382–D390. [Google Scholar] [CrossRef] [PubMed]
- Meyer, F.; Overbeek, R.; Rodriguez, A. FIGfams: Yet another set of protein families. Nucleic Acids Res. 2009, 37, 6643–6654. [Google Scholar] [CrossRef] [PubMed]
- Pedruzzi, I.; Rivoire, C.; Auchincloss, A.H.; Coudert, E.; Keller, G.; de Castro, E.; Baratin, D.; Cuche, B.A.; Bougueleret, L.; Poux, S.; et al. HAMAP in 2015: Updates to the protein family classification and annotation system. Nucleic Acids Res. 2015, 43, D1064–D1070. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, A.; Chang, H.-Y.; Daugherty, L.; Fraser, M.; Hunter, S.; Lopez, R.; McAnulla, C.; McMenamin, C.; Nuka, G.; Pesseat, S.; et al. The InterPro protein families database: The classification resource after 15 years. Nucleic Acids Res. 2015, 43, D213–D221. [Google Scholar] [CrossRef] [PubMed]
- Vilella, A.J.; Severin, J.; Ureta-Vidal, A.; Heng, L.; Durbin, R.; Birney, E. EnsemblCompara GeneTrees: Complete, duplication-aware phylogenetic trees in vertebrates. Genome Res. 2009, 19, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Crooks, G.E.; Hon, G.; Chandonia, J.-M.; Brenner, S.E. WebLogo: A sequence logo generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef] [PubMed]
- Vacic, V.; Iakoucheva, L.M.; Radivojac, P. Two Sample Logo: A graphical representation of the differences between two sets of sequence alignments. Bioinformatics 2006, 22, 1536–1537. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.D.; Gerlt, J.A.; Seffernick, J.L.; Babbitt, P.C. A gold standard set of mechanistically diverse enzyme superfamilies. Genome Biol. 2006, 7, R8. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Markowitz, V.M.; Chen, I.M.A.; Palaniappan, K.; Chu, K.; Szeto, E.; Grechkin, Y.; Ratner, A.; Jacob, B.; Huang, J.; Williams, P.; et al. IMG: The integrated microbial genomes database and comparative analysis system. Nucleic Acids Res. 2012, 40, D115–D122. [Google Scholar] [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- Wattam, A.R.; Abraham, D.; Dalay, O.; Disz, T.L.; Driscoll, T.; Gabbard, J.L.; Gillespie, J.J.; Gough, R.; Hix, D.; Kenyon, R.; et al. PATRIC, the bacterial bioinformatics database and analysis resource. Nucleic Acids Res. 2014, 42, D581–D591. [Google Scholar] [CrossRef] [PubMed]
- Larsson, A. AliView: A fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 2014, 30, 3276–3278. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Sakai, A.; Zhang, X.; Vetting, M.W.; Kumar, R.; Hillerich, B.; San Francisco, B.; Solbiati, J.; Steves, A.; Brown, S.; et al. Prediction and characterization of enzymatic activities guided by sequence similarity and genome neighborhood networks. eLife 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| COG 1 | Paralog Subfamilies | Ref. | ||
|---|---|---|---|---|
| 0780 and 0302 | QueF (Queuosine synthesis) | FolE (Tetrahydrofolate synthesis) | QueF-Like (Aracheosine synthesis) | [45,46] |
| 1539 | FolX (Tetrahydromonapterin synthesis) | FolB (Tetrahydrofolate synthesis) | [47] | |
| 1028 and 0262 | FolM (Tetrahydromonapterin synthesis) | FadG (fatty acid synthesis) | FolA (Tetrahydrofolate synthesis) | [47] |
| 0009 | YciO (Unknown function) | YrdC/TsaC (t6A synthesis) | [48] | |
| 1509 | YjeK (Protein modification) | LamB (Lysine degradation) | [49] | |
| 2269 and 1190 | YjeA (Protein modification) | LysRS (Protein synthesis) | [49] | |
| 0354 and 0404 | YgfZ (Iron-sulfur cluster repair) | GcvT (One carbon metabolism) | [50] | |
| 2102 | Dph6 (Diphtamide synthesis) | DUF71-B12 group (function unknown, B12 salvage) | [51] | |
| 5424 | PqqC (PQQ synthesis) | CT610 (Para-aminobenzoate synthesis) | [52] | |
| 1478 | CofE (F420 synthesis) | CT611 (Tetrahydrofolate synthesis) | [53] | |
| 1901 | TrmY Archaeal m1Psi54 methylase | Bacterial unknown methylase | [54] | |
| 0720 | PTPS-I (QueD, Queuosine synthesis) | PTPS-II (Biopterin synthesis) | PTPS-III (Folate synthesis) | [55] |
| 0523 | 15 subfamilies identified | [56] | ||
| 0212 | 5-formyltetrahydrofolate cycloligase (5-FCL) | Thiamin metabolism | [57] | |
| Domain | Genome Count with COG0720 | COG0720 Gene Count | Genomes with COG0720 Paralogs | Total Genomes |
|---|---|---|---|---|
| Archaea | 561 | 698 | 134 | 771 |
| Bacteria | 636 | 821 | 164 | 1056 |
| Eukaryota | 60 | 65 | 5 | 220 |
| Annotation | Database Identifier | Annotation | Database Identifier | Annotation |
|---|---|---|---|---|
| NCBI annotation | WP_011011332.1 | 6-carboxy-5,6,7,8-tetrahydropterin synthase (NCBI Reference Sequence) | WP_011012422.1 | 6-pyruvoyltetrahydropterin synthase (NCBI Reference Sequence) |
| Ensemble bacteria | AAL80343 | putative 6-pyruvoyl tetrahydrobiopterin synthase (PF0219) | AAL81402 | hypothetical protein (PF1278) |
| PATRIC (uses the RAST annotation pipeline) | fig186497.12.peg.227 | Queuosine biosynthesis QueD, PTPS-I | fig186497.12.peg.1340 | Folate biosynthesis protein PTPS-III, catalyzes a reaction that bypasses dihydroneopterin aldolase (FolB) |
| putative 6-pyruvoyl tetrahydrobiopterin synthase | hypothetical protein | |||
| MaGe | PF0219 | putative 6-pyruvoyl tetrahydrobiopterin synthase automatic/finished | PF1278 | hypothetical protein automatic/finished |
| NCBI RefSeq Annotation: putative 6-pyruvoyl tetrahydrobiopterin synthase | NCBI RefSeq Annotation: hypothetical protein | |||
| TrEMBL annotation: Putative 6-pyruvoyl tetrahydrobiopterin synthase | TrEMBL annotation: Dihydroneopterin monophosphate aldolase | |||
| IMG | 638172701 2 | preQ(0) biosynthesis protein QueD | 638173858 2 | hypothetical protein |
| 2625830234 3 | 6-pyruvoyltetrahydropterin/6-carboxytetrahydropterin synthase | 2625831353 3 | 6-pyruvoyltetrahydropterin/6-carboxytetrahydropterin synthase |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zallot, R.; Harrison, K.J.; Kolaczkowski, B.; De Crécy-Lagard, V. Functional Annotations of Paralogs: A Blessing and a Curse. Life 2016, 6, 39. https://doi.org/10.3390/life6030039
Zallot R, Harrison KJ, Kolaczkowski B, De Crécy-Lagard V. Functional Annotations of Paralogs: A Blessing and a Curse. Life. 2016; 6(3):39. https://doi.org/10.3390/life6030039
Chicago/Turabian StyleZallot, Rémi, Katherine J. Harrison, Bryan Kolaczkowski, and Valérie De Crécy-Lagard. 2016. "Functional Annotations of Paralogs: A Blessing and a Curse" Life 6, no. 3: 39. https://doi.org/10.3390/life6030039