
Salvia miltiorrhiza Protects Endothelial Dysfunction against Mitochondrial Oxidative Stress


1
Department of Chinese Medicine, Kaohsiung Chang Gung Memorial Hospital and Chang Gung University College of Medicine, Kaohsiung 83342, Taiwan
2
Graduate Institute of Clinical Medical Sciences, College of Medicine, Chang Gung University, Kaohsiung 33302, Taiwan
3
College of Medicine, Kaohsiung Medical University, Kaohsiung 80708, Taiwan
4
College of Nursing, Fooyin University, Kaohsiung 83102, Taiwan
*
Author to whom correspondence should be addressed.
Life 2021, 11(11), 1257; https://doi.org/10.3390/life11111257
Submission received: 19 October 2021
/
Revised: 5 November 2021
/
Accepted: 16 November 2021
/
Published: 18 November 2021
(This article belongs to the Special Issue Mitochondrial Mechanisms of Oxidative Stress and Endothelial Dysfunction)
Abstract
:Salvia miltiorrhiza (SM) is a common traditional Chinese medicine used in the treatment of cardiovascular and cerebrovascular diseases. Endothelial dysfunction plays an important role in the pathology of cardiovascular diseases. Endothelial dysfunction may induce inflammation and change vascular tone and permeability. The main pathological mechanism of endothelial dysfunction is the formation of reactive oxygen species (ROS). Mitochondria are the main source of energy and can also produce large amounts of ROS. Recent studies have shown that extracts of SM have antioxidative, anti-inflammatory, and antithrombus properties. In this review, we discuss the mechanism of oxidative stress in the mitochondria, endothelial dysfunction, and the role of SM in these oxidative events.
1. Introduction
1.1. Reactive Oxygen Species and Oxidative Stress
Free radicals may damage human health. There are many oxidative agents in our environment (air, water, tobacco, alcohol, heavy or transition metals, drugs, industrial solvents, radiation, etc.) and also produced inside the human body. Oxidative stress occurs due to oxygen-related metabolic reactions. It originates from the disequilibrium between reactive oxygen species (ROS) formation and enzymatic and nonenzymatic antioxidants in living organisms [1].
ROS can be produced in compartments, such as the cytoplasm, mitochondria, peroxisomes, and endoplasmic reticulum [2]. While cells suffer from oxidative stress, ROS are produced from the respiratory chain, leading to electron transfer. The superoxide radical (O2•−), which dismutates from hydrogen peroxide (H2O2) and molecular oxygen (O2), is a toxic compound following ROS stimulation [3,4]. Oxidative stress causes cell damage through three mechanisms: lipid peroxidation of membranes, oxidative modification of proteins, and DNA damage [5]. In the challenge of physical stresses like genotoxic stress and viral infection, ROS and reactive nitrogen species (RNS), as the intracellular signal transducers, may lead to autophagy, which is a catabolic process by damaging organelles and recycling cellular components [6].
Because of this, the results might cause aging and many human diseases, such as cancer, cardiovascular disease, metabolic disease, and infectious disease [7].
1.2. Endothelium
The endothelium exists in blood vessels, lymphatic vessels, and cornea. In this review article, we mainly discuss the endothelium in blood vessels. The arterial vessel is outlined by three distinct layers. The first layer is the tunica intima, a single layer of squamous endothelial cells, covering the internal surface of vessels; the second layer is the tunica media, which comprises vascular smooth muscle cells (VSMCs); and the final layer is the tunica adventitia, an elastic lamina with terminal nerve fibers and surrounding connective tissue. The endothelium [8] plays an extensive variety of essential roles in the control of vascular function, not only as a barrier between blood and tissues but also as an endocrine organ. The functions of the endothelium are as follows: (1) maintaining the balance between coagulation and fibrinolysis to provide the proper hemostatic balance [9]; (2) regulating coagulation [10,11]; (3) platelet adhesion and aggregation; (4) inflammation [12], (5) leukocyte activation, adhesion, and transmigration [13,14]; (6) regulating the regional blood flow to maintain the vascular tone and growth [15]; and (7) control of cell proliferation and angiogenesis [16].
In the normal vascular endothelium, high levels of nitric oxide (NO) and prostacyclin (PGI2) [17] and low levels of ROS, uric acid, endothelin-1 (ET-1), and angiotensin II (Ang II) contribute to endothelium-dependent vasodilatation [18]. The levels of inflammation-related factors, such as C-reactive protein (CRP), tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), soluble intercellular adhesion molecule (sICAM), soluble vascular cell adhesion molecule (sVCAM), and E-selectin, are low in physical conditions as well. When it comes to the anticoagulative status in the endothelium, levels of von Willebrand factor (vWF), plasminogen activator inhibitor-1 (PAI-1), and P-selectin are low [19,20].
1.3. Mitochondria
Mitochondria are important for energy metabolism [21]. The mitochondrial respiratory chain, which takes place in the inner mitochondrial membrane, is vital for energy metabolism. It is made of five compounds (complex I, II, III, IV, and V) [22], which catalyze the phosphorylation of ADP to ATP by electron transfer between them [23]. In 1956, Denham Harman first mentioned that free radicals, as by-products of the normal metabolism of mitochondria, attack cell constituents [24]. The following studies also suggest that intracellular ROS production is caused by the mitochondria. The production of mitochondrial superoxide radicals occurs primarily at complex I (NADH) and complex III (ubiquinone-cytochrome c reductase) [25]. In complex I, the electron transport chain of NADH and FADH2 transfers electrons to oxygen and hydrogen, and ROS or RNS production might occur during these electron transport steps. Under the general metabolic status, complex III is the main place of ROS production [26].
Mitochondria are also involved in several cellular processes, such as signaling through mitochondrial ROS (mtROS) [27], regulation of calcium storage [28], steroid generation [29], cellular differentiation, mitophagy, and apoptosis [30]. These functions maintain and control the cell cycle, cell growth, and cell death. The cytosolic Ca2+, which is released by the endoplasmic reticulum, is internalized by mitochondria via the uniporter and released by Na+/Ca2+ or H+/Ca2+ exchangers [31]. In both the intermembrane space and the matrix of mitochondria, while Ca2+ is taken up, it regulates the activity of transporters, enzymes, and proteins involved in organelle metabolism [32]. Steroidogenesis is a multistep process for biosynthesis of steroid hormone from cholesterol. Mitochondria-associated endoplasmic reticulum membranes (MAMs) play important roles in regulating steriodogenesis and maintain many cellular functions, including lipid metabolism, calcium signaling, and apoptosis [33]. During the cell cycle, mitochondrial membrane permeabilization (MMP) is mainly the decisive event in cell death through the release of catabolic hydrolases and enzymes [30].
1.4. Salvia miltiorrhiza
Salvia miltiorrhiza (SM) belongs to the Lamiaceae family and is a traditional Chinese medicine (TCM), also known as Danshen, in its Chinese name. SM is widely used for the treatment of circulatory diseases, including cardiovascular and cerebrovascular diseases. It has multiple biological functions, such as antioxidative stress, anti-inflammation, and antithrombosis. Based on previous studies, there are more than 49 diterpenoid quinones, and over 36 water-soluble phenolic acids and 23 essential oil components have been identified and isolated from SM [34].
The predominant bioactive compounds in SM contain two major groups of chemicals. One group includes lipophilic compounds (terpenoids), such as tanshinone I (Tan I), tanshinone IIA (Tan IIA), acetyltanshinone IIA, cryptotanshinone, isocryptotanshinone, dihydrotanshinone, 15,16-dihydrotanshinone I, and miltirone. These terpenoids exhibit a spectrum of potential biological activities, including antioxidant, antibacterial, anti-inflammatory, antiatherogenic, neuroprotective, antitumor, and antidiabetic effects. The other group includes hydrophilic phenolic acids, such as caffeic acid, danshensu, salvianolic acid A (SalA), salvianolic acid B (SalB), lithospermic acid, and lithospermic acid B. These polyphenol structures protect the cardiovascular system via ROS scavenge, leukocyte–endothelial adherence reduction, aortic smooth muscle cell inflammation, and metalloproteinase expression inhibition, as well as competitive binding of salvianolic acids to target proteins to interrupt protein–protein interactions [35]. Our previous population-based studies also revealed that SM is the most commonly used TCM for ischemic heart disease [36] and ischemic stroke [37] treatment. Wang et al. [38] reviewed 39 clinical trials using SM treatment for cardiovascular diseases and the conclusions supported that SM had beneficial therapeutic properties for cardioprotective effects through different cell signaling pathways. Several reviews have also provided the same conclusions not only in cardiovascular disease [39,40] but also in metabolic syndrome patients [41].
Mitochondrial oxidative stress may lead to endothelial dysfunction, which is known to be associated with cardiovascular disease and aging. We review current studies in this field to show the mechanisms of SM in protecting endothelial dysfunction against mitochondrial oxidative stress.
2. Monograph of Mitochondrial Oxidative Stress, Endothelial Dysfunction, and Salvia miltiorrhiza
2.1. Mitochondrial Oxidative Stress
Mitochondria are one of the main sources of oxidative stress, as they use oxygen for energy production. ROS and RNS, generated by tightly regulated enzymes, are involved in many processes of normal physiology, such as signaling transferring pathways, induction of mitogenic response, and defense against pathogens. Excessive stimulation of NAD(P)H and the electron transport chain leads to the overproduction of ROS, which results in oxidative stress (Figure 1). Once oxidative stress is induced, it causes irreversible injury to proteins, lipids, and mitochondrial nucleic acid components, leading to the release of cytochrome c in the cytosol, which in turn causes apoptosis. These have been considered to be the main causes of many diseases, including neurodegenerative diseases [42], malignant tumor, ischemic heart disease, and diabetes [7].
Mitochondrial dysfunction triggered by any pathological situation can lead to a significant increase in ROS levels. For instance, hypoxia is the deficiency of oxygen, which is the terminal acceptor of electrons from the electron transport chain. Moreover, xenobiotics or their metabolic by-products can also lead to mitochondrial dysfunction through many kinds of mechanisms [43,44].
The basis of the free radial theory for aging in mitochondria is related to oxidative stress, which leads to mitochondrial DNA mutations. The accumulation of mitochondrial DNA mutations causes oxidative phosphorylation malfunction, and imbalanced antioxidant enzymes leads to overproduction of ROS and the formation of a vicious cycle [31]. Oxidative stress also deranges the mitochondrial respiratory chain, affects Ca2+ homeostasis [32,45], and influences membrane permeability and mitochondrial defense systems. It not only occurs in aging and age-related disorders, but also in cancer [5]. The by-products of lipid peroxidation can induce carcinogenesis. Mutations in complex I subunit dehydrogenase subunit 6 (ND6) increase the metastatic potential by producing excessive ROS, whereas an ND5 mutation enhances tumorigenesis by oxidative stress and Akt (also known as protein kinase B) activation [46,47]. Recent studies have also shown that increased mitochondrial fission is a pro-tumorigenic phenotype [44].
Cytochrome c oxidase (CcO), the terminal oxidase of the mitochondrial electron transport chain, is a highly regulated enzyme that is involved in mitochondrial oxidative metabolism and ATP synthesis. CcO dysfunction shows a consistently positive association with increased mitochondrial ROS production and cellular toxicity [43].
Mitochondrial membrane permeabilization (MMP) is an important event in pathological cell death induced by ischemia/reperfusion, xenobiotic intoxication, neurodegenerative disease, and viral infection. Inhibition of MMP constitutes an important strategy for the pharmaceutical prevention of unwarranted cell death. In contrast, the induction of MMP in tumor cells constitutes the goal of anticancer chemotherapy [30].
In cancer cells, quality control and biogenesis are often upregulated in mitochondria. Several pathologies and adverse environmental conditions disrupt mitochondrial function in multiple ways, such as MtDNA mutations, deletions, or impaired DNA replication. A few cancers produce oncogenic metabolites via mutations in nuclear-encoded mitochondrial tricarboxylic acid (TCA) cycle enzymes; in contrast, there is negative selection for pathogenic mitochondrial genome mutations [48].
2.2. Endothelial Dysfunction
Alterations in endothelial cells and vasculature play an important role in the pathogenesis of human vascular diseases, such as coronary heart disease, peripheral vascular disease, stroke, venous thrombosis, diabetes, tumor growth, and metastasis [20]. Endothelial dysfunction includes impaired vasodilation with decreased vascular repair capacity, increased oxidative stress, uric acid formation, increased lipid peroxide radicals, high levels of nitrotyrosine, enhanced procoagulant phenotype, enhanced proinflammatory phenotype, and increased endothelial microparticles and circulating endothelial cells [20]. Elevation of circulating endothelial cells (CECs) and reduction of endothelial progenitor cells (EPCs) are potential diagnostic biomarkers for endothelial dysfunction and they have been described in different kinds of cardiovascular diseases. CECs are mature cells that are shed from blood vessels and are rare in normally present but increased in endothelial dysfunction. The circulating endothelial progenitor cells, which arise from the bone marrow, are non-differentiated and immature endothelial cells, and contribute to the repair and renewal of damaged endothelium [49]. One study concluded that the CEC level is a more sensitive marker for vascular damage compared to the EPC level, which might be increased secondarily as a repair mechanism due to more severe vascular damage [50].
In the pathogenesis of atherosclerosis, multiple stimuli, such as oxidized low-density lipoproteins (LDLs), high glucose level, uric acid, and homocysteine, damage the integrity of the vascular endothelium, cause vessel leakage, increase leukocyte adhesion to the clammy endothelium, induce VSCM contraction, lead to endothelial nitric oxide synthase (eNOS) uncoupling, and decrease NO production [51].
Several studies showed an impairment of endothelial function in both macro- and microvascular complications due to hyperglycemia in both animal models and human participants [52,53,54]. Hyperglycemia results in oxidative stress via an increase of the production of ROS and RNS, and affects vascular homeostasis by impairing vasorelaxation and increasing vasoconstriction, eventually inducing endothelial dysfunction. Advanced glycation end products (AGEs) also induce decreased NO synthesis, eNOS expression, and increased ET-1 expression, leading to endothelial dysfunction [55,56]. The oxidative stress in endothelial cells is shown in Figure 2.
Under the situation of endothelial dysfunction, inflammatory and procoagulant biomarkers, such as IL-6, TNF-α, PAI-1, D-dimer, and vWF, are increased in diabetic patients with micro- and macrovascular complications, including cardiovascular disease or nephropathy [54]. These proinflammatory cytokines are also important for angiogenesis and migration of endothelial cells [57]. It is interesting that TNF-α regulates both pro- and antiangiogenic properties and interacts with two distinct transmembrane receptors: tumor necrosis factor receptor 1 (TNFR1) and tumor necrosis factor receptor 2 (TNFR2). TNFR1 is related to differentiation, cell death, and apoptosis, while TNFR2 cell maintains cell survival and proliferation. A recent report showed that human EPCs are immunosuppressive and this effect was TNF-α/TNFR2 dependent [58,59].
Endothelial dysfunction of vessels was characterized by increased blood levels of vascular endothelial growth factor (VEGF), endothelin-1 (ET-1), P-selectin (PSel), homocysteine (Hcys), nitrites (NO2), and cyclic guanosine monophosphate (cGMP), as well as decreased PGI2 values. These indices of increasing VEGF and ET-1 and decreasing PGI2 were observed in most lung cancer cases. Disturbances of vascular endothelial function were associated with the patient’s age, disease duration, morphological form, and lung cancer stage [60]. Angiotensin II (Ang II), as a powerful vasoconstrictor for controlling blood pressure and vascular remodeling, has also been shown to have important inflammatory and oxidative actions in the endothelium [61,62]. Ang II promotes apoptosis in endothelial cells by generating ROS, increasing NADPH oxidase (Nox) activity in endothelial cells, and enhancing superoxide production via AT1R and AT2R [63,64].
Endothelial cells are coupled with malignant tumor cells in almost every stage of the metastatic steps, including infiltration of cancer cells into the nearby tissue; endothelial transmigration, also calls “intravasation”; survival in the blood stream; and extravasation followed by colonization of the target organ. Dysfunctional blood vessels within the tumor are heterogeneous and highly permeable, resulting from the activity of factors, such as hypoxia and chronic growth factor stimulation [65]. Cancer metastasis and secondary tumor initiation largely depend on circulating tumor cells and vascular endothelial cell interactions. Endothelial glycocalyx (GCX) dysfunction may play a significant role in this process [65,66]. One study showed that microvascular endothelial dysfunction, as defined by a reactive hyperemia peripheral arterial tonometry index ≤2.0, was associated with an increase of over two times the risk of solid tumor cancer. Besides its known ability to predict cardiovascular disease, microvascular endothelial dysfunction may be a useful index for solid tumor cancer prediction [67].
2.3. Salvia miltiorrhiza and Mitochondrial Oxidative Stress
Various oxidases, including xanthine oxidase, uncoupled NOS, cytochrome P450 enzymes, and mitochondrial and NADPH oxidases (Nox), produce superoxide, which is toxic via univalent reduction [68,69]. Superoxide dismutases (SODs) are the major antioxidant enzymes that degrade superoxide to the more stable ROS, H2O2, which is then converted to water and oxygen by either catalase or glutathione peroxidase (GPx) [70]. There are three isoforms of SOD in mammals: cytoplasmic Cu/ZnSOD (SOD1), mitochondrial MnSOD (SOD2), and extracellular Cu/ZnSOD (SOD3) [71].
SM may reduce ROS production by inhibiting oxidases, reducing the production of superoxide, inhibiting the oxidative modification of low-density lipoproteins (LDLs), and ameliorating mitochondrial oxidative stress. SM also increases the activities of catalase, manganese SOD, GPx, and eNOS [72]. In a previous study, SM hydrophilic extract could reverse the induction of vascular endothelial growth factor (VEGF) expression by high glucose levels through mitigation of mitochondrial oxidative stress [73].
The Kelch-like ECH-associated protein 1 (Keap1)-nuclear factor erythroid 2-related factor 2 (Nrf2) system is central for mammalian cyto-protection against electrophilic and oxidative stress [74]. In acetaminophen-induced hepatocyte injury, one study found that the component salvianolic acid C exhibits a protective effect by ameliorating inflammatory response, caspase-mediated anti-apoptotic effects, and mitochondrial oxidative stress through inhibition of the Keap1/Nrf2/heme oxygenase-1 (HO-1) signaling axis [75].
Malondialdehyde (MDA) is one of the final products of polyunsaturated fatty acid peroxidation in the cells. An increasing MDA level is recognized as a relevant biomarker of oxidative stress with free radical overproduction [76,77]. In neurological areas, glutamate excitotoxicity is related to several diseases, including cerebral ischemia and neurodegenerative diseases. One study [78] revealed that Tan IIA could suppress glutamate-induced oxidative stress by reducing ROS levels and MDA, and by increasing the activities of SOD and catalase. Tan IIA prevents glutamate-induced mitochondrial dysfunction by enhancing the mitochondrial membrane potential and ATP content, and by decreasing the mitochondrial protein carbonyl content. Furthermore, Tan IIA can suppress glutamate-induced apoptosis through regulation of apoptosis-related protein expression, including an elevation of B cell lymphoma 2 (Bcl-2) protein levels, reduction of Bcl-2-associated X (Bax) and cleaved caspase-3 levels, and suppression of Jun N-terminal kinase (JNK)1/2, and furthermore, p38 mitogen-activated protein kinase (MAPK) activation [79].
Tan IIA administration resulted in a significant decrease in the mitochondrial fusion proteins, mitofusin (Mfn) 1/2 and Optic atrophy 1 (Opa1), as well as an increase in the fission protein dynamin-related protein 1 (Drp1) in human osteosarcoma cells [80].
2.4. Salvia miltiorrhiza in Endothelium Dysfunction
Dihydrotanshinone, cryptotanshinone, Tan I, and Tan IIA, as new acetylcholinesterase (AChE) inhibitors, have the potential to penetrate the blood–brain barrier and may be used to treat Alzheimer’s disease [81].
Dihydrotanshinone exerts a vasorelaxant effect by inhibiting Ca2+ influx in VSMCs, and it is independent of pathways involving the endothelium, muscarinic receptors, beta-adrenoceptors, adenylyl cyclase, and guanylyl cyclase [82].
Cryptotanshinone (CTS) possesses anti-inflammatory properties by suppressing the TNF-α-induced increase in endothelial permeability, monocyte adhesion, intercellular cell adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule-1 (VCAM-1), and monocyte chemoattractant protein-1 (MCP-1), and restoring NO production [83]. CTS significantly inhibited sodium-nitroprusside (SNP)-induced cell toxicity and the generation of ROS and RNS, and improved the mitochondrial membrane potential (MMP) in neuro-2a (N2a) cells. CTS significantly inhibited SNP-induced peroxidation of lipids and proteins and the expression of glutamate-cysteine ligase catalytic subunit (Gclc) mRNA. CTS elevated Akt and cyclic AMP response element-binding protein and further blocked SNP-induced activation of nuclear factor-kappa B (NF-κB), extracellular signal-regulated kinase (ERK)1/2, and JNK/MAPK pathways. Additionally, the increase in the mitochondrial Bax/Bcl-2 ratio, activation of cytosolic procaspase-3, and release of cytochrome c from mitochondria to the cytosol were significantly reduced by CTS [84].
Tan IIA reduced intracellular oxidative stress and increased NO generation by restoring high glucose-induced eNOS uncoupling by targeting the NADPH oxidase, heat shock protein 90 (HSP90) [85], GTP cyclohydrolase-1 (GTPCH1) [86], dihydrofolate reductase (DHFR) [87], and Phosphoinositide 3-kinases (PI3K) pathways [88,89]. Endothelial injury and subsequent atherogenic events may be provoked by chronic oxidative stressors like H2O2 and methylglyoxal. Tan IIA has the potential to stabilize atherosclerotic plaques by inhibiting LDL oxidation, monocyte adhesion to the endothelium, smooth muscle cell migration and proliferation, macrophage cholesterol accumulation, proinflammatory cytokine expression, and platelet aggregation [90]. In vivo, the atherosclerotic change region decreases by 3.5 times after Tan IIA treatment. Intracellular chloride channel 1 (CLIC1) is involved in the oxidative stress and inflammatory process. Tan IIA reduced MDA production, increased SOD activity, decreased TNF-α and IL-6 levels, and suppressed the expression of CLIC1, ICAM-1, and VCAM-1 in atherosclerotic mice. In vitro, the antioxidative and anti-inflammatory effects of Tan IIA were dose dependent, further confirming this result. Moreover, CLIC1 depletion abolished the Tan IIA-mediated decrease in ROS and MDA production in human umbilical vein endothelial cells (HUVECs). Additionally, Tan IIA inhibited both CLIC1 membrane translocation and the chloride ion concentration [91].
The endothelial protective effects of Tan IIA derivatives enhanced efficacy against H2O2-induced injury via Nrf2 activation and excellent hydrophilic activity [92]. CD31, also known as platelet endothelial cell adhesion molecule (PECAM-1) [93], was suppressed in Tan IIA-treated xenografts, indicating antineovascularization. Tan IIA facilitates Bcl-2 translocation to the mitochondrial outer membrane, prevents mitochondrial permeability transition pore opening, decreases cytochrome c release, prevents caspase-3 activation, and restrains apoptosis [94].
Ursolic acid, an aqueous extract of SM, also reduced the expression of the NADPH oxidase subunit Nox4 and suppressed the production of ROS in human endothelial cells [95]. The vasodilatation mechanisms of SM aqueous extract and salvianolic acid B were produced by the inhibition of Ca2+ influx in VSMCs. The opening of K+ channels had a minor contribution to their effects, but endothelium-dependent mechanisms were not involved [96].
Salvianolic acid A (SalA) treatment inhibited the toll-like receptor 4 and nuclear factor kappa B pathway, and prompted a lowering of proinflammatory mediators including IL-1β, IL-6, TNF-α, ICAM-1, and VCAM-1. In addition, SalA treatment significantly decreased oxidative stress by increasing antioxidant enzyme activity, upregulating the nuclear factor erythroid 2-related factor 2/heme oxygenase-1 pathway, and downregulating the expression of p47phox and p22phox in vivo. p47phox, known as neutrophil cytosol factor 1, relates to activation of NADPH oxidase and is required for atherosclerosis lesion progression in ApoE−/− mice [97,98]. p22phox, also known as the human neutrophil cytochrome b light chain, is an essential component of the membrane-associated enzyme phagocyte NADPH-oxidase and exists in endothelial and vascular smooth muscle cells. Furthermore, SalA suppressed oxidized LDL-induced expression of lectin-like oxidized LDL receptor-1, the phosphorylation of nuclear factor kappa B (p65), ICAM-1, and VCAM-1, and inhibited NADPH oxidase subunit 4-mediated ROS generation in HUVECs [99]. SalA protects HUVECs against tert-butyl hydroperoxide-induced oxidative injury via a mitochondria-dependent pathway [100]. SalA inhibits endothelial dysfunction and vascular remodeling in spontaneously hypertensive rats. Therefore, Sal A could be a potential drug therapy to prevent further targeted organ damage induced by vascular remodeling [101].
Salvianolic acid B (SalB) prevents oxidative stress-induced endothelial dysfunction by downregulating Nox-4, eNOS, and nicotinamide adenine dinucleotide phosphate (NADPH)-oxidase expression. During apoptosis, mitochondria take over multiple apoptotic signals. The Bcl-2 family of proteins regulate the promotion or inhibition of apoptosis [102]. Bax and Bak, as proapoptotic members, result in the release of cytochrome c from mitochondria [103], whereas Bcl-2 and Bcl-extra large (Bcl-xL) are the common antiapoptotic proteins that promote cell survival [104]. SalB decreased the Bax/Bcl-xL ratio and caspase-3 activation after H2O2 induction. One study revealed that activation of the mTOR/p70S6K/4EBP1 pathway is required for both SalB-mediated angiogenic and protective effects against oxidative stress-induced cell injury in human bone marrow-derived endothelial progenitor cells (BM-EPCs). SalB suppress mitogen-activated protein kinase 3 and 6 (MKK3/6)-p38 MAPK-ATF2 and ERK1/2 signaling pathways, reduces intracellular ROS levels and apoptosis, and further protects BM-EPCs against oxidative stress-related cell injury [105]. Sal B decreased the Ang II-induced elevation of arterial systolic blood pressure in mice, increased the impaired endothelium-dependent relaxation, and attenuated the endothelium-dependent over-contractions in both the aorta and renal arteries of Ang II-infused mice. Furthermore, Sal B treatment stabilized the elevating AT1 receptors, NADPH oxidase subunits (Nox-2 and Nox-4), and nitrotyrosine in the arteries of Ang II-infused mice or in Ang II-treated HUVECs [106] (Table 1).
3. Conclusions
Through this review, we found that SM has antioxidative, anti-inflammatory, and antithrombotic effects. SM can decrease ROS formation in the mitochondria, preventing endothelial cell dysfunction. Endothelial dysfunction may be related to many cardiovascular and cerebrovascular diseases, such as stroke, acute myocardial infarction, peripheral vascular disease, and Alzheimer’s disease. Both lipophilic and hydrophilic components of SM can protect the endothelium against mitochondrial oxidative stress. More research is needed to discover the mechanism of SM in preventing oxidative stress in the endothelium.
Author Contributions
Conceptualization, Y.-C.H.; writing—original draft preparation, Y.-C.C.; revised the manuscript, I.-L.H., Y.-N.L., W.-L.H. and Y.-C.H. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Marrocco, I.; Altieri, F.; Peluso, I. Measurement and Clinical Significance of Biomarkers of Oxidative Stress in Humans. Oxidative Med. Cell. Longev. 2017, 2017, 1–32. [Google Scholar] [CrossRef]
- Forrester, S.J.; Kikuchi, D.S.; Hernandes, M.S.; Xu, Q.; Griendling, K.K. Reactive Oxygen Species in Metabolic and Inflammatory Signaling. Circ. Res. 2018, 122, 877–902. [Google Scholar] [CrossRef]
- Imlay, J.A. Cellular Defenses against Superoxide and Hydrogen Peroxide. Annu. Rev. Biochem. 2008, 77, 755–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiley, P.; Storz, G. Exploiting Thiol Modifications. PLoS Biol. 2004, 2, e400. [Google Scholar] [CrossRef] [PubMed]
- Kudryavtseva, A.V.; Krasnov, G.S.; Dmitriev, A.; Alekseev, B.Y.; Kardymon, O.; Sadritdinova, A.F.; Fedorova, M.S.; Pokrovsky, A.V.; Melnikova, N.V.; Kaprin, A.D.; et al. Mitochondrial dysfunction and oxidative stress in aging and cancer. Oncotarget 2016, 7, 44879–44905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filomeni, G.; De Zio, D.; Cecconi, F. Oxidative stress and autophagy: The clash between damage and metabolic needs. Cell Death Differ. 2015, 22, 377–388. [Google Scholar] [CrossRef] [Green Version]
- Bhat, A.H.; Dar, K.B.; Anees, S.; Zargar, M.A.; Masood, A.; Sofi, M.A.; Ganie, S.A. Oxidative stress, mitochondrial dysfunction and neurodegenerative diseases; a mechanistic insight. Biomed. Pharmacother. 2015, 74, 101–110. [Google Scholar] [CrossRef]
- Krüger-Genge, A.; Blocki, A.; Franke, R.-P.; Jung, F. Vascular Endothelial Cell Biology: An Update. Int. J. Mol. Sci. 2019, 20, 4411. [Google Scholar] [CrossRef] [Green Version]
- Michel, C.C.; Curry, F.E. Microvascular permeability. Physiol. Rev. 1999, 79, 703–761. [Google Scholar] [CrossRef]
- Stern, D.M.; Esposito, C.; Gerlach, H.; Gerlach, M.; Ryan, J.; Handley, D.; Nawroth, P. Endothelium and Regulation of Coagulation. Diabetes Care 1991, 14, 160–166. [Google Scholar] [CrossRef]
- Atherton, A.; Born, G.V. Quantitative investigations of the adhesiveness of circulating polymorphonuclear leucocytes to blood vessel walls. J. Physiol. 1972, 222, 447–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szmitko, P.E.; Wang, C.H.; Weisel, R.D.; de Almeida, J.R.; Anderson, T.J.; Verma, S. New markers of inflammation and endothelial cell activation: Part, I. Circulation 2003, 108, 1917–1923. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Shimizu, K.; Ogawa, F.; Yanaba, K.; Iwata, Y.; Muroi, E.; Takenaka, M.; Komura, K.; Hasegawa, M.; Fujimoto, M.; et al. Platelets Control Leukocyte Recruitment in a Murine Model of Cutaneous Arthus Reaction. Am. J. Pathol. 2010, 176, 259–269. [Google Scholar] [CrossRef] [Green Version]
- Smyth, S.S.; McEver, R.P.; Weyrich, A.S.; Morrell, C.N.; Hoffman, M.R.; Arepally, G.M.; French, P.A.; Dauerman, H.L.; Becker, R.L.; 2009 Platelet Colloquium Participants. Platelet functions beyond hemostasis. Journal of thrombosis and haemostasis. J. Thromb. Haemost. 2009, 7, 1759–1766. [Google Scholar] [CrossRef] [PubMed]
- Barton, M.; Baretella, O.; Meyer, M.R. Obesity and risk of vascular disease: Importance of endothelium-dependent vasoconstriction. Br. J. Pharmacol. 2012, 165, 591–602. [Google Scholar] [CrossRef] [PubMed]
- Michiels, C. Endothelial cell functions. J. Cell. Physiol. 2003, 196, 430–443. [Google Scholar] [CrossRef]
- Moncada, S.; Higgs, E.A.; Vane, J.R. Human arterial and venous tissues generate prostacyclin (prostaglandin x), a potent inhibitor of platelet aggregation. Lancet 1977, 1, 18–20. [Google Scholar] [CrossRef]
- Furchgott, R.F.; Zawadzki, J.V. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature 1980, 288, 373–376. [Google Scholar] [CrossRef]
- Quyyumi, A.A. Endothelial function in health and disease: New insights into the genesis of cardiovascular disease. Am. J. Med. 1998, 105, 32S–39S. [Google Scholar] [CrossRef]
- Rajendran, P.; Rengarajan, T.; Thangavel, J.; Nishigaki, Y.; Sakthisekaran, D.; Sethi, G.; Nishigaki, I. The vascular endothelium and human diseases. Int. J. Biol. Sci. 2013, 9, 1057–1069. [Google Scholar] [CrossRef] [Green Version]
- Ernster, L.; Schatz, G. Mitochondria: A historical review. J. Cell Biol. 1981, 91, 227s–255s. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-Q.; Yager, J.D.; Russo, J. Regulation of mitochondrial respiratory chain structure and function by estrogens/estrogen receptors and potential physiological/pathophysiological implications. Biochim. Biophys. Acta (BBA) Bioenerg. 2005, 1746, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Ghezzi, D.; Zeviani, M. Assembly factors of human mitochondrial respiratory chain complexes: Physiology and pathophysiology. Adv. Exp. Med. Biol. 2012, 748, 65–106. [Google Scholar] [PubMed]
- Harman, D. Aging: A Theory Based on Free Radical and Radiation Chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selivanov, V.A.; Votyakova, T.V.; Pivtoraiko, V.N.; Zeak, J.; Sukhomlin, T.; Trucco, M.; Roca, J.; Cascante, M. Reactive Oxygen Species Production by Forward and Reverse Electron Fluxes in the Mitochondrial Respiratory Chain. PLoS Comput. Biol. 2011, 7, e1001115. [Google Scholar] [CrossRef] [Green Version]
- Finkel, T.; Holbrook, N.J. Oxidants, oxidative stress and the biology of ageing. Nature 2000, 408, 239–247. [Google Scholar] [CrossRef]
- Li, X.; Fang, P.; Mai, J.; Choi, E.T.; Wang, H.; Yang, X.F. Targeting mitochondrial reactive oxygen species as novel therapy for inflammatory diseases and cancers. J. Hematol. Oncol. 2013, 6, 19. [Google Scholar] [CrossRef] [Green Version]
- Bravo-Sagua, R.; Parra, V.; López-Crisosto, C.; Díaz, P.; Quest, A.F.G.; Lavandero, S. Calcium Transport and Signaling in Mitochondria. Compr. Physiol. 2017, 7, 623–634. [Google Scholar]
- Miller, W.L.; Auchus, R.J. The Molecular Biology, Biochemistry, and Physiology of Human Steroidogenesis and Its Disorders. Endocr. Rev. 2011, 32, 81–151. [Google Scholar] [CrossRef] [Green Version]
- Kroemer, G.; Galluzzi, L.; Brenner, C. Mitochondrial Membrane Permeabilization in Cell Death. Physiol. Rev. 2007, 87, 99–163. [Google Scholar] [CrossRef]
- Wang, C.-H.; Wu, S.-B.; Wu, Y.-T.; Wei, Y.-H. Oxidative stress response elicited by mitochondrial dysfunction: Implication in the pathophysiology of aging. Exp. Biol. Med. 2013, 238, 450–460. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.; Pizzo, P.; Filadi, R. Calcium, mitochondria and cell metabolism: A functional triangle in bioenergetics. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 1068–1078. [Google Scholar] [CrossRef] [PubMed]
- Doghman-Bouguerra, M.; Lalli, E. The ER-mitochondria couple: In life and death from steroidogenesis to tumorigenesis. Mol. Cell. Endocrinol. 2017, 441, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Pang, H.; Wu, L.; Tang, Y.; Zhou, G.; Qu, C.; Duan, J.-A. Chemical Analysis of the Herbal Medicine Salviae miltiorrhizae Radix et Rhizoma (Danshen). Molecules 2016, 21, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, J.H.-C.; Hong, C.-Y. Salvianolic acids: Small compounds with multiple mechanisms for cardiovascular protection. J. Biomed. Sci. 2011, 18, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hung, Y.-C.; Tseng, Y.-J.; Hu, W.-L.; Chen, H.-J.; Li, T.-C.; Tsai, P.-Y.; Chen, H.-P.; Huang, M.-H.; Su, F.-Y. Demographic and Prescribing Patterns of Chinese Herbal Products for Individualized Therapy for Ischemic Heart Disease in Taiwan: Population-Based Study. PLoS ONE 2015, 10, e0137058. [Google Scholar]
- Hung, I.-L.; Hung, Y.-C.; Wang, L.-Y.; Hsu, S.-F.; Chen, H.-J.; Tseng, Y.-J.; Kuo, C.-E.; Hu, W.-L.; Li, T.-C. Chinese Herbal Products for Ischemic Stroke. Am. J. Chin. Med. 2015, 43, 1365–1379. [Google Scholar] [CrossRef]
- Wang, L.; Ma, R.; Liu, C.; Liu, H.; Zhu, R.; Guo, S.; Tang, M.; Li, Y.; Niu, J.; Fu, M.; et al. Salvia miltiorrhiza: A Potential Red Light to the Development of Cardiovascular Diseases. Curr. Pharm. Des. 2017, 23, 1077–1097. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.-M.; Xu, S.-W.; Liu, P. Salvia miltiorrhizaBurge (Danshen): A golden herbal medicine in cardiovascular therapeutics. Acta Pharmacol. Sin. 2018, 39, 802–824. [Google Scholar] [CrossRef] [Green Version]
- Ren, J.; Fu, L.; Nile, S.H.; Zhang, J.; Kai, G. Salvia miltiorrhiza in Treating Cardiovascular Diseases: A Review on Its Pharmacological and Clinical Applications. Front. Pharmacol. 2019, 10, 753. [Google Scholar] [CrossRef]
- Orgah, J.O.; He, S.; Wang, Y.; Jiang, M.; Wang, Y.; Orgah, E.A.; Duan, Y.; Zhao, B.; Zhang, B.; Han, J.; et al. Pharmacological potential of the combination of Salvia miltiorrhiza (Danshen) and Carthamus tinctorius (Honghua) for diabetes mellitus and its cardiovascular complications. Pharmacol. Res. 2020, 153, 104654. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, S.; Avadhani, N.G. Cytochrome c oxidase dysfunction in oxidative stress. Free Radic. Biol. Med. 2012, 53, 1252–1263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srinivasan, S.; Guha, M.; Kashina, A.; Avadhani, N.G. Mitochondrial dysfunction and mitochondrial dynamics-The cancer connection. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 602–614. [Google Scholar] [CrossRef] [PubMed]
- Buchholz, J.N.; Behringer, E.J.; Pottorf, W.J.; Pearce, W.J.; Vanterpool, C.K. Age-dependent changes in Ca2+ homeostasis in peripheral neurones: Implications for changes in function. Aging Cell 2007, 6, 285–296. [Google Scholar] [CrossRef] [Green Version]
- Sharma, L.K.; Fang, H.; Liu, J.; Vartak, R.; Deng, J.; Bai, Y. Mitochondrial respiratory complex I dysfunction promotes tumorigenesis through ROS alteration and AKT activation. Hum. Mol. Genet. 2011, 20, 4605–4616. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, K.; Takenaga, K.; Akimoto, M.; Koshikawa, N.; Yamaguchi, A.; Imanishi, H.; Nakada, K.; Honma, Y.; Hayashi, J.-I. ROS-Generating Mitochondrial DNA Mutations Can Regulate Tumor Cell Metastasis. Science 2008, 320, 661–664. [Google Scholar] [CrossRef] [Green Version]
- Zong, W.X.; Rabinowitz, J.D.; White, E. Mitochondria and Cancer. Mol. Cell 2016, 61, 667–676. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Malik, A.B.; Rehman, J. Endothelial progenitor cells and vascular repair. Curr. Opin. Hematol. 2014, 21, 224–228. [Google Scholar] [CrossRef] [Green Version]
- Farinacci, M.; Krahn, T.; Dinh, W.; Volk, H.D.; Düngen, H.D.; Wagner, J.; Konen MSc, T.; von Ahsen, O. Circulating endothelial cells as biomarker for cardiovascular diseases. Res. Pract. Thromb. Haemost. 2019, 3, 49–58. [Google Scholar] [CrossRef] [Green Version]
- Davignon, J.; Ganz, P. Role of Endothelial Dysfunction in Atherosclerosis. Circulation 2004, 109, III-27–III-32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grassi, D.; Desideri, G.; Necozione, S.; Ruggieri, F.; Blumberg, J.B.; Stornello, M.; Ferri, C. Protective Effects of Flavanol-Rich Dark Chocolate on Endothelial Function and Wave Reflection During Acute Hyperglycemia. Hypertension 2012, 60, 827–832. [Google Scholar] [CrossRef] [Green Version]
- Dhananjayan, R.; Koundinya, K.S.S.; Malati, T.; Kutala, V.K. Endothelial Dysfunction in Type 2 Diabetes Mellitus. Indian J. Clin. Biochem. 2016, 31, 372–379. [Google Scholar] [CrossRef] [Green Version]
- Domingueti, C.P.; Dusse, L.M.; Carvalho, M.; de Sousa, L.P.; Gomes, K.B.; Fernandes, A.P. Diabetes mellitus: The linkage between oxidative stress, inflammation, hypercoagulability and vascular complications. J. Diabetes Complicat. 2016, 30, 738–745. [Google Scholar] [CrossRef] [PubMed]
- Brownlee, M. The Pathobiology of Diabetic Complications: A Unifying Mechanism. Diabetes 2005, 54, 1615–1625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, R.; Kaur, M.; Singh, J. Endothelial dysfunction and platelet hyperactivity in type 2 diabetes mellitus: Molecular insights and therapeutic strategies. Cardiovasc. Diabetol. 2018, 17, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Prisco, A.R.; Hoffmann, B.R.; Kaczorowski, C.C.; McDermott-Roe, C.; Stodola, T.J.; Exner, E.C.; Greene, A.S. Tumor Necrosis Factor α Regulates Endothelial Progenitor Cell Migration via CADM1 and NF-kB. Stem Cells 2016, 34, 1922–1933. [Google Scholar] [CrossRef] [Green Version]
- Nouri Barkestani, M.; Shamdani, S.; Afshar Bakshloo, M.; Arouche, N.; Bambai, B.; Uzan, G.; Naserian, S. TNFα priming through its interaction with TNFR2 enhances endothelial progenitor cell immunosuppressive effect: New hope for their widespread clinical application. Cell Commun. Signal. 2021, 19, 1–16. [Google Scholar] [CrossRef]
- Naserian, S.; Abdelgawad, M.E.; Bakshloo, M.A.; Ha, G.; Arouche, N.; Cohen, J.L.; Salomon, B.; Uzan, G. The TNF/TNFR2 signaling pathway is a key regulatory factor in endothelial progenitor cell immunosuppressive effect. Cell Commun. Signal. 2020, 18, 1–14. [Google Scholar] [CrossRef]
- Dumanskiy, Y.V.; Stoliarova, O.Y.; Syniachenko, O.V.; Iegudina, E.D. Endothelial dysfunction of vessels at lung cancer. Exp. Oncol. 2015, 37, 277–2780. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Iwao, H. Molecular and cellular mechanisms of angiotensin II-mediated cardiovascular and renal diseases. Pharmacol. Rev. 2000, 52, 11–34. [Google Scholar] [PubMed]
- Dandona, P.; Dhindsa, S.; Ghanim, H.; Chaudhuri, A. Angiotensin II and inflammation: The effect of angiotensin-converting enzyme inhibition and angiotensin II receptor blockade. J. Hum. Hypertens. 2007, 21, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Dimmeler, S.; Rippmann, V.; Weiland, U.; Haendeler, J.; Zeiher, A.M. Angiotensin II Induces Apoptosis of Human Endothelial Cells. Circ. Res. 1997, 81, 970–976. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Barker, T.A.; Berk, B.C. Angiotensin II and the endothelium: Diverse signals and effects. Hypertension 2005, 45, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Blazejczyk, A.; Papiernik, D.; Porshneva, K.; Sadowska, J.; Wietrzyk, J. Endothelium and cancer metastasis: Perspectives for antimetastatic therapy. Pharmacol. Rep. 2015, 67, 711–718. [Google Scholar] [CrossRef] [PubMed]
- Mensah, S.A.; Nersesyan, A.A.; Harding, I.C.; Lee, C.I.; Tan, X.; Banerjee, S.; Niedre, M.; Torchilin, V.P.; Ebong, E.E. Flow-regulated endothelial glycocalyx determines metastatic cancer cell activity. FASEB J. 2020, 34, 6166–6184. [Google Scholar] [CrossRef] [Green Version]
- Toya, T.; Sara, J.D.; Corban, M.T.; Taher, R.; Godo, S.; Herrmann, J.; Lerman, L.O.; Lerman, A. Assessment of peripheral endothelial function predicts future risk of solid-tumor cancer. Eur. J. Prev. Cardiol. 2020, 27, 608–618. [Google Scholar] [CrossRef]
- Fridovich, I. Superoxide anion radical (O2-.), superoxide dismutases, and related matters. J. Biol. Chem. 1997, 272, 18515–18517. [Google Scholar] [CrossRef] [Green Version]
- Li, J.-M.; Shah, A.M. Differential NADPH- versus NADH-dependent superoxide production by phagocyte-type endothelial cell NADPH oxidase. Cardiovasc. Res. 2001, 52, 477–486. [Google Scholar] [CrossRef] [Green Version]
- Han, J.; Shuvaev, V.V.; Muzykantov, V.R. Catalase and superoxide dismutase conjugated with platelet-endothelial cell adhesion molecule antibody distinctly alleviate abnormal endothelial permeability caused by exogenous reactive oxygen species and vascular endothelial growth factor. J. Pharmacol. Exp. Ther. 2011, 338, 82–91. [Google Scholar] [CrossRef] [Green Version]
- Fukai, T.; Ushio-Fukai, M. Superoxide Dismutases: Role in Redox Signaling, Vascular Function, and Diseases. Antioxid. Redox Signal. 2011, 15, 1583–1606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.C.; Chang, Y.C.; Hu, W.L.; Hung, Y.C. Oxidative Stress and Salvia miltiorrhiza in Aging-Associated Cardiovascular Diseases. Oxidative Med. Cell. Longev. 2016, 2016, 4797102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, S.; Huo, D.; Wang, S.; Qian, Q. Inhibition of glucose-induced vascular endothelial growth factor expression by Salvia miltiorrhiza hydrophilic extract in human microvascular endothelial cells: Evidence for mitochondrial oxidative stress. J. Ethnopharmacol. 2011, 137, 985–991. [Google Scholar] [CrossRef] [PubMed]
- Horie, Y.; Suzuki, T.; Inoue, J.; Iso, T.; Wells, G.; Moore, T.W.; Mizushima, T.; Dinkova-Kostova, A.T.; Kasai, T.; Kamei, T.; et al. Molecular basis for the disruption of Keap1–Nrf2 interaction via Hinge & Latch mechanism. Commun. Biol. 2021, 4, 1–11. [Google Scholar]
- Wu, C.-T.; Deng, J.-S.; Huang, W.-C.; Shieh, P.-C.; Chung, M.-I.; Huang, G.-J. Salvianolic Acid C against Acetaminophen-Induced Acute Liver Injury by Attenuating Inflammation, Oxidative Stress, and Apoptosis through Inhibition of the Keap1/Nrf2/HO-1 Signaling. Oxidative Med. Cell. Longev. 2019, 2019, 1–13. [Google Scholar] [CrossRef]
- Del Rio, D.; Stewart, A.J.; Pellegrini, N. A review of recent studies on malondialdehyde as toxic molecule and biological marker of oxidative stress. Nutr. Metab. Cardiovasc. Dis. 2005, 15, 316–328. [Google Scholar] [CrossRef]
- Gaweł, S.; Wardas, M.; Niedworok, E.; Wardas, P. Malondialdehyde (MDA) as a lipid peroxidation marker. Wiad. Lek. 2004, 57, 453–455. [Google Scholar]
- Lingyun, X.; Han, W.; Wang, H.; Ding, F.; Xiao, L.; Shi, R.; Haifeng, L.; Huang, Z. Tanshinone IIA Inhibits Glutamate-Induced Oxidative Toxicity through Prevention of Mitochondrial Dysfunction and Suppression of MAPK Activation in SH-SY5Y Human Neuroblastoma Cells. Oxidative Med. Cell. Longev. 2017, 2017, 1–13. [Google Scholar]
- Wada, T.; Penninger, J.M. Mitogen-activated protein kinases in apoptosis regulation. Oncogene 2004, 23, 2838–2849. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.T.; Huang, C.C.; Huang, W.L.; Lin, T.K.; Liao, P.L.; Wang, P.W.; Liou, C.-W.; Chuang, J.-H. Tanshinone IIA induces intrinsic apoptosis in osteosarcoma cells both in vivo and in vitro associated with mitochondrial dysfunction. Sci. Rep. 2017, 7, 40382. [Google Scholar] [CrossRef] [Green Version]
- Ren, Y.; Houghton, P.J.; Hider, R.C.; Howes, M.-J.R. Novel Diterpenoid Acetylcholinesterase Inhibitors fromSalvia miltiorhiza. Planta Medica 2004, 70, 201–204. [Google Scholar]
- Lam, F.F.; Yeung, J.H.; Chan, K.M.; Or, P.M. Dihydrotanshinone, a lipophilic component of Salvia miltiorrhiza (danshen), relaxes rat coronary artery by inhibition of calcium channels. J. Ethnopharmacol. 2008, 119, 318–321. [Google Scholar] [CrossRef]
- Ahmad, Z.; Ng, C.T.; Fong, L.Y.; Bakar, N.A.; Hussain, N.H.; Ang, K.P.; Ee, G.C.L.; Hakim, M.N. Cryptotanshinone inhibits TNF-α-induced early atherogenic events in vitro. J. Physiol. Sci. 2016, 66, 213–220. [Google Scholar] [CrossRef]
- Mahesh, R.; Jung, H.W.; Kim, G.W.; Kim, Y.S.; Park, Y.K. Cryptotanshinone from Salviae miltiorrhizae radix inhibits sodium-nitroprusside-induced apoptosis in neuro-2a cells. Phytother. Res. 2012, 26, 1211–1219. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, J.K.A.; Ackerman, A.W.; Gross, E.R.; Stepp, D.W.; Shi, Y.; Fontana, J.T.; Baker, J.E.; Sessa, W.C. Heat Shock Protein 90 Mediates the Balance of Nitric Oxide and Superoxide Anion from Endothelial Nitric-oxide Synthase. J. Biol. Chem. 2001, 276, 17621–17624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Baumgardt, S.L.; Fang, J.; Shi, Y.; Qiao, S.; Bosnjak, Z.J.; Vásquez-Vivar, J.; Xia, Z.; Warltier, D.C.; Kersten, J.R.; et al. Transgenic overexpression of GTP cyclohydrolase 1 in cardiomyocytes ameliorates post-infarction cardiac remodeling. Sci. Rep. 2017, 7, 3093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chalupsky, K.; Cai, H. Endothelial dihydrofolate reductase: Critical for nitric oxide bioavailability and role in angiotensin II uncoupling of endothelial nitric oxide synthase. Proc. Natl. Acad. Sci. USA 2005, 102, 9056–9061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Z.-W.; Xie, X.-L.; Zhou, S.-F.; Li, C.G. Mechanism of reversal of high glucose-induced endothelial nitric oxide synthase uncoupling by tanshinone IIA in human endothelial cell line EA.hy926. Eur. J. Pharmacol. 2012, 697, 97–105. [Google Scholar] [CrossRef]
- Morello, F.; Perino, A.; Hirsch, E. Phosphoinositide 3-kinase signalling in the vascular system. Cardiovasc. Res. 2008, 82, 261–271. [Google Scholar] [CrossRef] [Green Version]
- Gao, S.; Liu, Z.; Li, H.; Little, P.J.; Liu, P.; Xu, S. Cardiovascular actions and therapeutic potential of tanshinone IIA. Atherosclerosis 2012, 220, 3–10. [Google Scholar] [CrossRef]
- Zhu, J.; Xu, Y.; Ren, G.; Hu, X.; Wang, C.; Yang, Z.; Li, Z.; Mao, W.; Lu, D. Tanshinone IIA Sodium sulfonate regulates antioxidant system, inflammation, and endothelial dysfunction in atherosclerosis by downregulation of CLIC1. Eur. J. Pharmacol. 2017, 815, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Bi, X.; Zhang, K.; He, L.; Gao, B.; Gu, Q.; Li, X.; Chen, J.; Wang, J. Synthesis and biological evaluation of tanshinone IIA derivatives as novel endothelial protective agents. Future Med. Chem. 2017, 9, 1073–1085. [Google Scholar] [CrossRef]
- Caligiuri, G. Mechanotransduction, immunoregulation, and metabolic functions of CD31 in cardiovascular pathophysiology. Cardiovasc. Res. 2019, 115, 1425–1434. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; He, H.; Qiao, Y.; Huang, J.; Wu, Z.; Xu, P.; Yin, D.; He, M. Tanshinone IIA Pretreatment Protects H9c2 Cells against Anoxia/Reoxygenation Injury: Involvement of the Translocation of Bcl-2 to Mitochondria Mediated by 14-3-3η. Oxidative Med. Cell. Longev. 2018, 2018, 3583921. [Google Scholar] [CrossRef] [Green Version]
- Steinkamp-Fenske, K.; Bollinger, L.; Völler, N.; Xu, H.; Yao, Y.; Bauer, R.; Förstermann, U.; Li, H. Ursolic acid from the Chinese herb Danshen (Salvia miltiorrhiza L.) upregulates eNOS and downregulates Nox4 expression in human endothelial cells. Atherosclerosis 2007, 195, e104–e111. [Google Scholar] [CrossRef]
- Lam, F.F.; Yeung, J.H.; Kwan, Y.W.; Chan, K.M.; Or, P.M. Salvianolic acid B, an aqueous component of danshen (Salvia miltiorrhiza), relaxes rat coronary artery by inhibition of calcium channels. Eur. J. Pharmacol. 2006, 553, 240–245. [Google Scholar] [CrossRef]
- El-Benna, J.; Dang, P.M.-C.; Gougerot-Pocidalo, M.-A.; Marie, J.-C. Fran p47phox, the phagocyte NADPH oxidase/NOX2 organizer: Structure, phosphorylation and implication in diseases. Exp. Mol. Med. 2009, 41, 217–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ago, T.; Nunoi, H.; Ito, T.; Sumimoto, H. Mechanism for phosphorylation-induced activation of the phagocyte NADPH oxidase protein p47(phox). Triple replacement of serines 303, 304, and 328 with aspartates disrupts the SH3 domain-mediated intramolecular interaction in p47(phox), thereby activating the oxidase. J. Biol. Chem. 1999, 274, 33644–33653. [Google Scholar] [PubMed] [Green Version]
- Song, Q.; Zhang, Y.; Han, X.; Zhang, Y.; Zhang, X.; Gao, Y.; Zhang, J.; Chu, L.; Zhao, S. Potential mechanisms underlying the protective effects of salvianic acid A against atherosclerosis in vivo and vitro. Biomed. Pharmacother. 2019, 109, 945–956. [Google Scholar] [CrossRef]
- Jia, D.; Li, T.; Chen, X.; Ding, X.; Chai, Y.; Chen, A.F.; Zhu, Z.; Zhang, C. Salvianic acid A sodium protects HUVEC cells against tert-butyl hydroperoxide induced oxidative injury via mitochondria-dependent pathway. Chem.-Biol. Interact. 2018, 279, 234–242. [Google Scholar] [CrossRef]
- Teng, F.; Yin, Y.; Cui, Y.; Deng, Y.; Li, D.; Cho, K.; Zhang, G.; Lu, A.; Wu, W.; Yang, M.; et al. Salvianolic acid A inhibits endothelial dysfunction and vascular remodeling in spontaneously hypertensive rats. Life Sci. 2016, 144, 86–93. [Google Scholar] [CrossRef]
- Adams, J.M.; Cory, S. The Bcl-2 Protein Family: Arbiters of Cell Survival. Science 1998, 281, 1322–1326. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.C.; Zong, W.-X.; Cheng, E.H.-Y.; Lindsten, T.; Panoutsakopoulou, V.; Ross, A.J.; Roth, K.A.; MacGregor, G.R.; Thompson, C.B.; Korsmeyer, S.J. Proapoptotic BAX and BAK: A Requisite Gateway to Mitochondrial Dysfunction and Death. Science 2001, 292, 727–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Opferman, J.T.; Kothari, A. Anti-apoptotic BCL-2 family members in development. Cell Death Differ. 2018, 25, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Jacobi, A.; Vater, C.; Zou, X.; Stiehler, M. Salvianolic acid B protects human endothelial progenitor cells against oxidative stress-mediated dysfunction by modulating Akt/mTOR/4EBP1, p38 MAPK/ATF2, and ERK1/2 signaling pathways. Biochem. Pharmacol. 2014, 90, 34–49. [Google Scholar] [CrossRef]
- Ling, W.C.; Liu, J.; Lau, C.W.; Murugan, D.D.; Mustafa, M.R.; Huang, Y. Treatment with salvianolic acid B restores endothelial function in angiotensin II-induced hypertensive mice. Biochem. Pharmacol. 2017, 136, 76–85. [Google Scholar] [CrossRef]
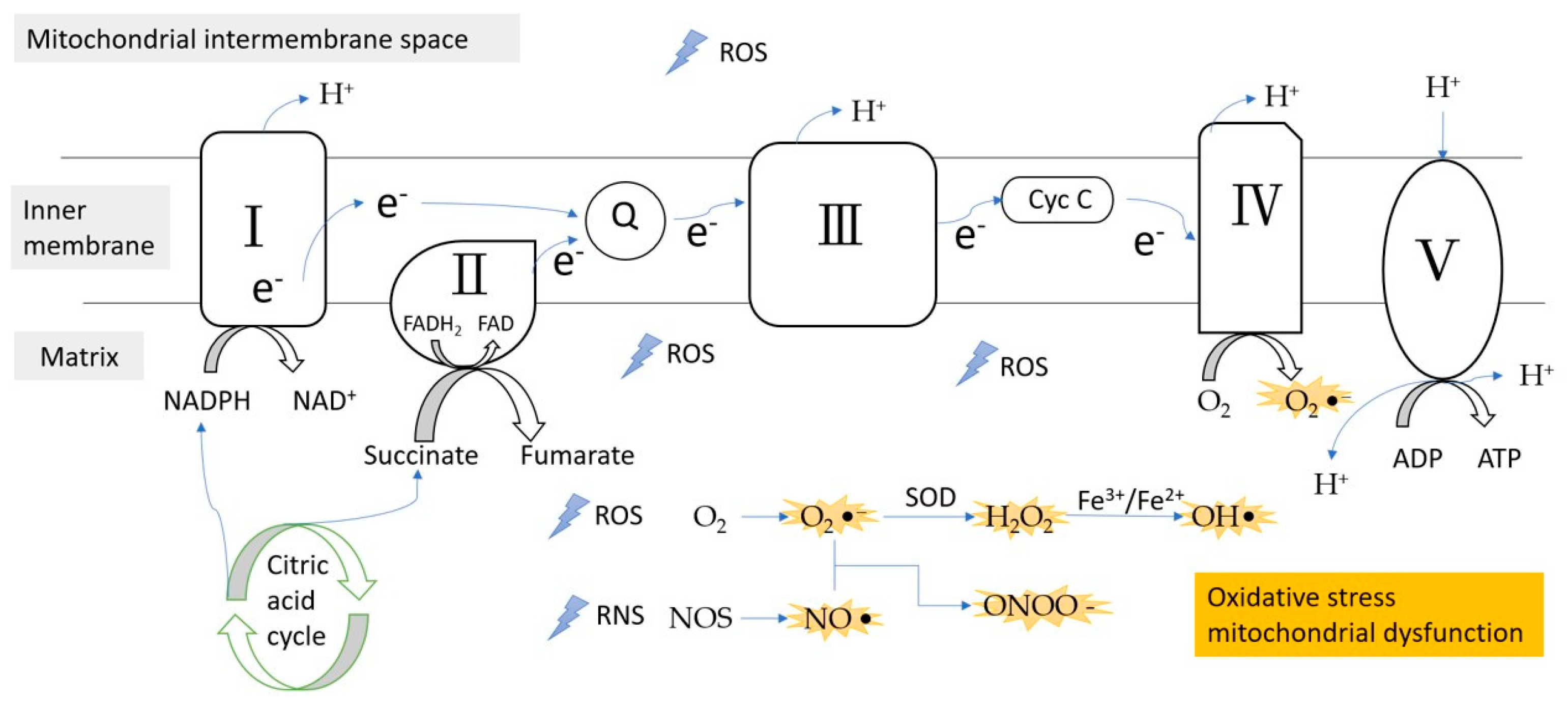
Figure 1.
ROS/RNS formation during the electron transfer chain in the inner membrane of mitochondria. Complex III is the main site of ROS production. ROS, reactive oxygen species; RNS, reactive nitrogen species; e−, electron; Q, coenzyme Q; Cyc C, cytochrome c; SOD, superoxidase dismutase.
Figure 1.
ROS/RNS formation during the electron transfer chain in the inner membrane of mitochondria. Complex III is the main site of ROS production. ROS, reactive oxygen species; RNS, reactive nitrogen species; e−, electron; Q, coenzyme Q; Cyc C, cytochrome c; SOD, superoxidase dismutase.

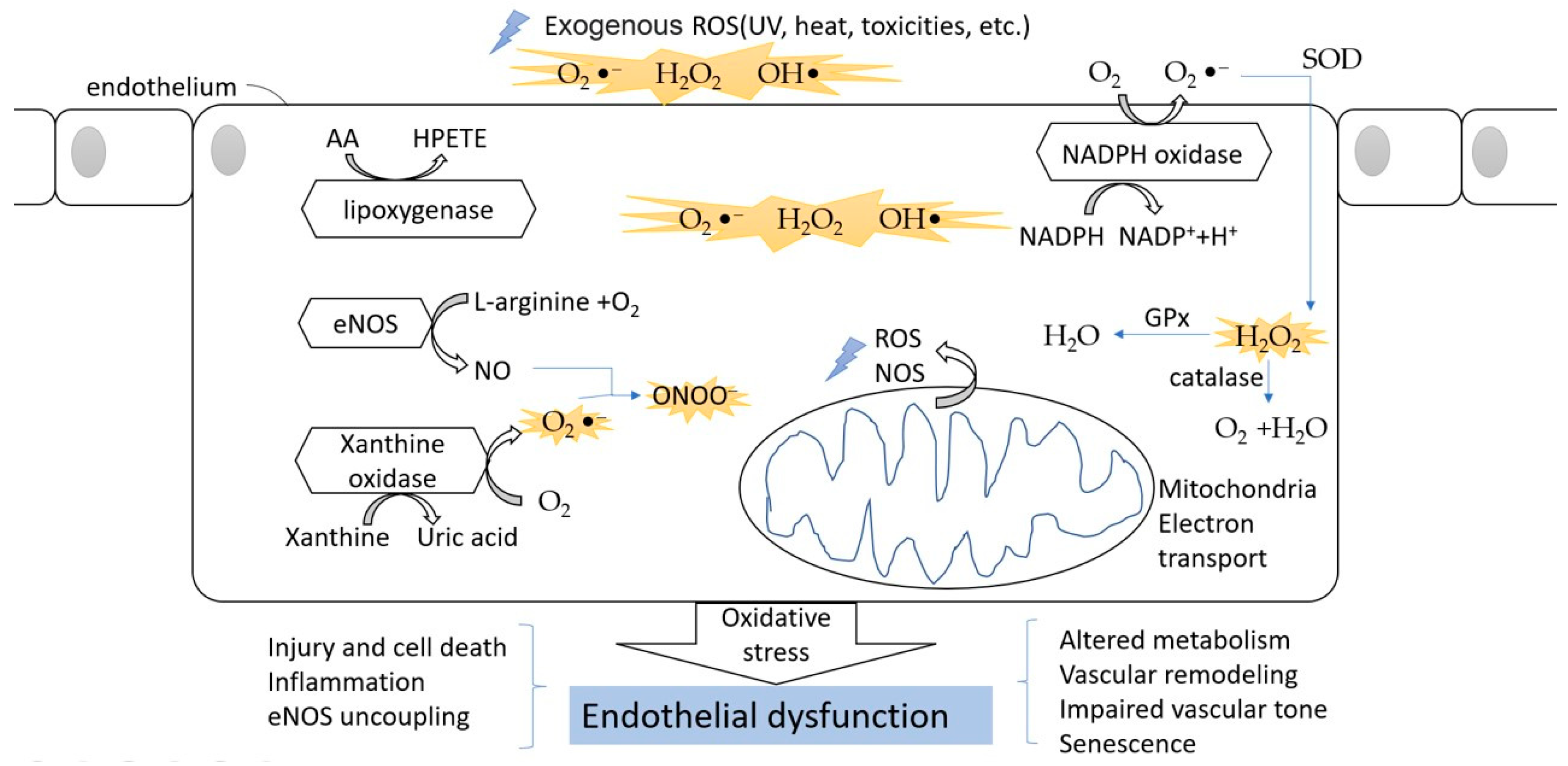
Figure 2.
The oxidative stress in endothelial cells. The ROS can divide between an exogenous source and internal source. In the endothelium, during the processes of NO formation, uric acid generation, AA metabolism, and NADPH oxidation, ROS are released into the cytoplasm. The oxidative stress finally leads to endothelial dysfunction and affects normal physical function.
Figure 2.
The oxidative stress in endothelial cells. The ROS can divide between an exogenous source and internal source. In the endothelium, during the processes of NO formation, uric acid generation, AA metabolism, and NADPH oxidation, ROS are released into the cytoplasm. The oxidative stress finally leads to endothelial dysfunction and affects normal physical function.


{kind=link}
{kind=link}
Table 1.
The antioxidative mechanisms of Salvia miltiorrhiza.
| Bioactive Compounds | Preventing Mechanism | Reference |
|---|---|---|
| SM | ↓ ROS, O2•− ⊝ oxidases, oxLDL, and ameliorating mitochondrial oxidative stress. ↑ catalase, SOD, GPx, and coupled eNOS | [72] |
| SM hydrophilic extract | ⊝ VEGF expression, Ca2+ influx in VSMC ⊕ vasorelaxant ameliorating mitochondrial oxidative stress | [73] |
| Salvianolic acid A | ↑ SOD, Nrf2/HO-1 pathway ↓ IL-1β, IL-6, TNF-α, ICAM-1, VCAM-1 ⊝ TLR4/NF-κB pathway, oxLDL, p47phox and p22phox, LOX-1, NF-κB p65 phosphorylation, Nox4 | [99] |
| Salvianolic acid B | ↓ Nox4, eNOS ⊝ Ca2+ influx in VSMC, Bax/Bcl-xL ratio, caspase-3, MKK3/6-p38 MAPK-ATF2 and ERK1/2 signaling pathways ⊕ vasorelaxant, mTOR/p70S6K/4EBP1 pathways | [96,105] |
| Salvianolic acid C | ↓ Inflammation, oxidative Stress, and apoptosis ⊝ Keap1/Nrf2/HO-1 signaling axis | [75] |
| Ursolic acid | ↓ Nox4, ROS | [95] |
| Tanshinone I | ⊝ AChE | [81] |
| Tanshinone IIA | ↑ Bcl-2, mitochondrial membrane potential and ATP, SOD, catalase, NO, ratio of BH4 to BH2, Drp1 ↓ ROS, Bax, caspase-3, eNOS uncoupling, atherosclerotic lesion, MDA, Mfn1/2 and Opa1, cytochrome c release ⊕ HSP90, GTPCH1, DHFR, Nrf2, 14-3-3η ⊝ JNK, p38 MAPK, AChE, O2•−, Nox4, PI3K, LDL oxidation, monocyte adhesion, SMC migration and proliferation, macrophage cholesterol accumulation, proinflammatory cytokine expression, platelet aggregation, CLIC1, ICAM-1, VCAM-1, CD31, mitochondrial permeability transition pore opening | [78,79,80,81,88,91,92,94] |
| Dihydrotanshinone | ⊝ Ca2+ influx in VSMC, AChE | [81,82] |
| Cryptotanshinone | ⊝ AChE, TNF-α ↓ endothelial permeability, monocyte adhesion, ICAM-1, VCAM-1 and MCP-1 ⊕ NO | [81,83] |
↑: increase; ↓: decrease; ⊝: inhibit; ⊕: promote SM, Salvia miltiorrhiza; ROS, reactive oxygen species; oxLDL, oxidized low-density lipoprotein; SOD, superoxide dismutase; GPx, glutathione peroxidase; VEGF, vascular endothelial growth factor; AChE, Acetylcholinesterase; BH4, tetrahydrobiopterin; BH2, 7,8-dihydrobiopterin; HSP90, heat-shock protein of 90 kDa; GTPCH1, GTP cyclohydrolase-1; DHFR, dihydrofolate reductase; VSMC, vascular smooth muscle cells; IL-1β, interleukin-1β; IL-6, interleukin-6; TNF-α, tumor necrosis factor-α; ICAM-1, intercellular cell adhesion molecule-1; VCAM-1, vascular cell adhesion molecule-1; TLR4/NF-κB, toll-like receptor 4/nuclear factor kappa B; LOX-1, Lectin-like oxidized low-density lipoprotein receptor-1; NO, nitric oxide; O2•−, superoxide; SMC, smooth muscle cell; MDA, malondialdehyde; CLIC1, Intracellular chloride channel 1; Nrf2, nuclear fac-tor (erythroid-derived 2)-like-2 factor; Drp1, dynamin-related protein 1; Opa1, Optic atrophy 1; Mfn1/2, Mitofusin 1/2.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Cheng, Y.-C.; Hung, I.-L.; Liao, Y.-N.; Hu, W.-L.; Hung, Y.-C. Salvia miltiorrhiza Protects Endothelial Dysfunction against Mitochondrial Oxidative Stress. Life 2021, 11, 1257. https://doi.org/10.3390/life11111257
AMA Style
Cheng Y-C, Hung I-L, Liao Y-N, Hu W-L, Hung Y-C. Salvia miltiorrhiza Protects Endothelial Dysfunction against Mitochondrial Oxidative Stress. Life. 2021; 11(11):1257. https://doi.org/10.3390/life11111257
Chicago/Turabian StyleCheng, Yu-Chen, I-Ling Hung, Yen-Nung Liao, Wen-Long Hu, and Yu-Chiang Hung. 2021. "Salvia miltiorrhiza Protects Endothelial Dysfunction against Mitochondrial Oxidative Stress" Life 11, no. 11: 1257. https://doi.org/10.3390/life11111257
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.