Downregulation of miR-17-92 Cluster by PERK Fine-Tunes Unfolded Protein Response Mediated Apoptosis


 and
and 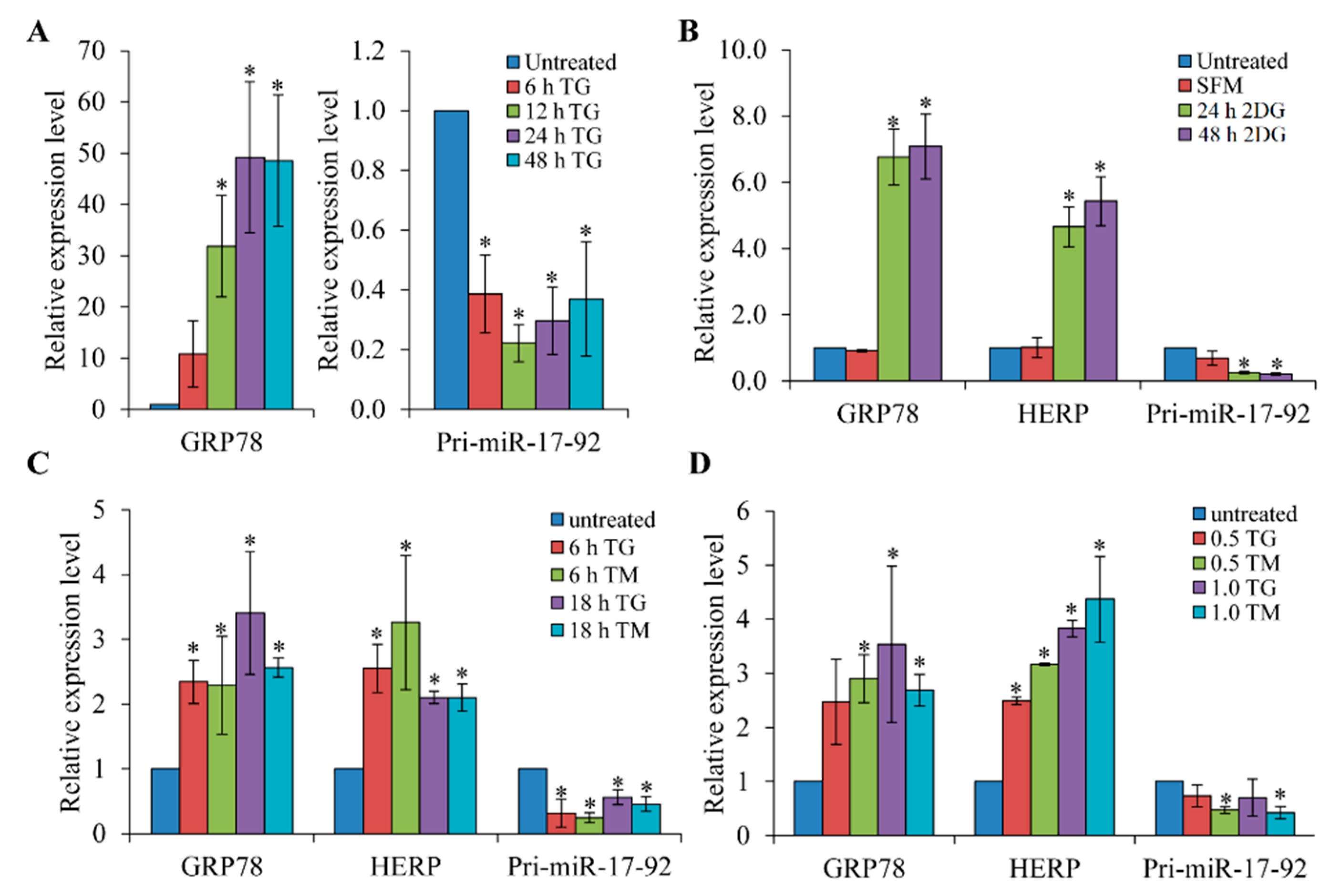{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Treatments
2.2. Plasmid Constructs
2.3. Generation of Stable Cell Lines
2.4. RNA Extraction, RT-PCR and Real Time RT-PCR
2.5. Measurement of miRNA Levels Using TaqMan qRT-PCR Assays
2.6. Luciferase Reporter Assays
2.7. Apoptotic Nuclei Measurements
2.8. Western Blotting
2.9. Statistical Analysis
3. Results
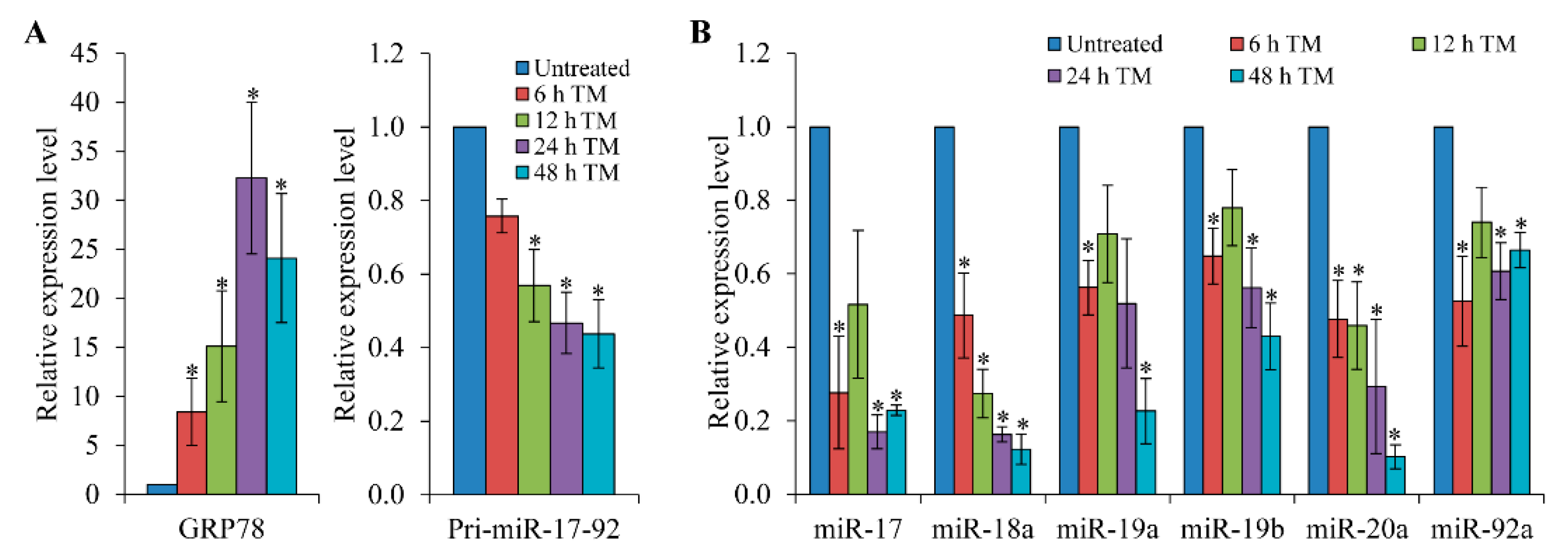
3.1. Expression of the pri-miR-17-92 is Down-Regulated upon Induction of ER Stress
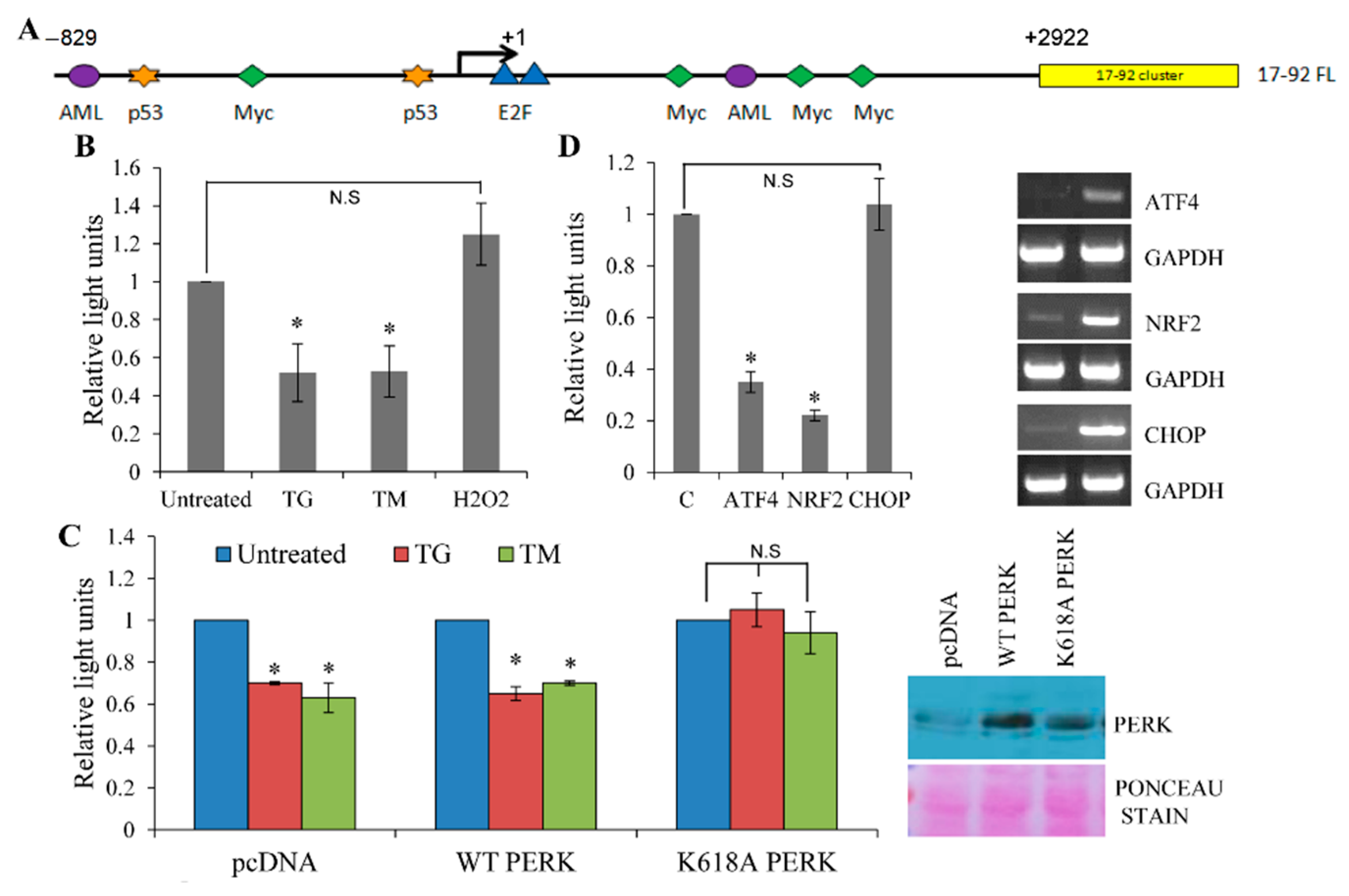
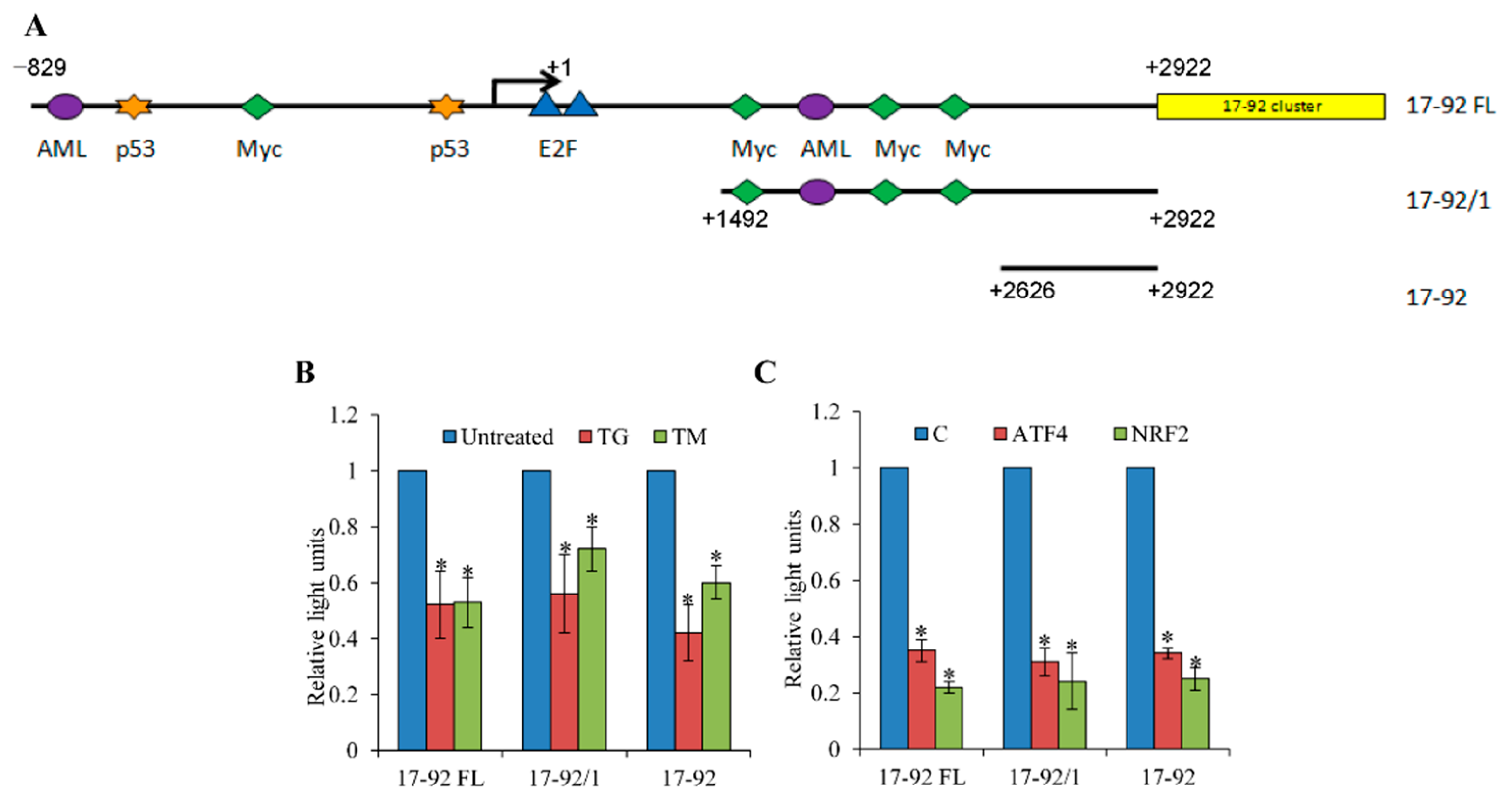
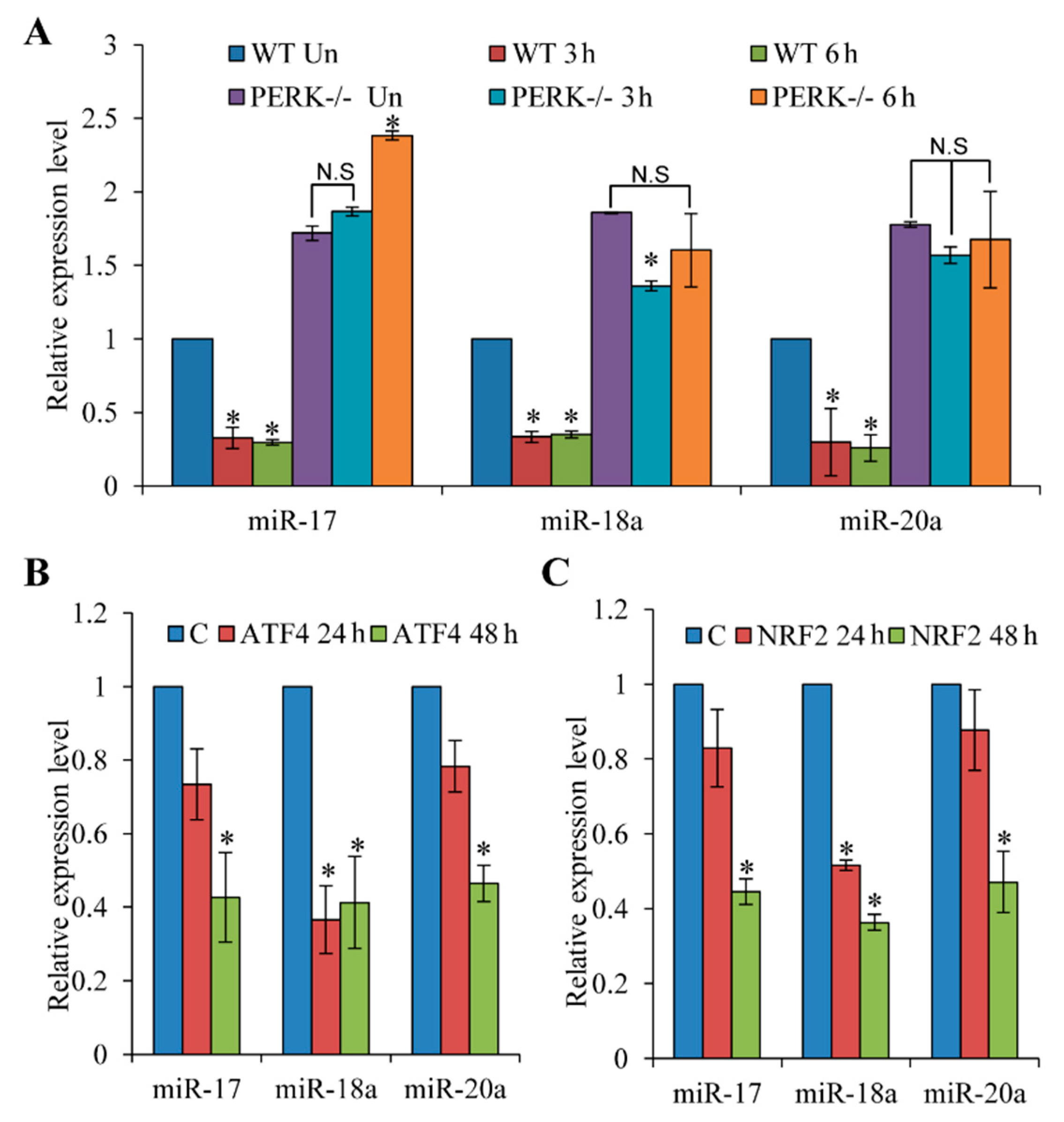
3.2. Repression of miR-17-92 Promoter during ER Stress is PERK-Dependent
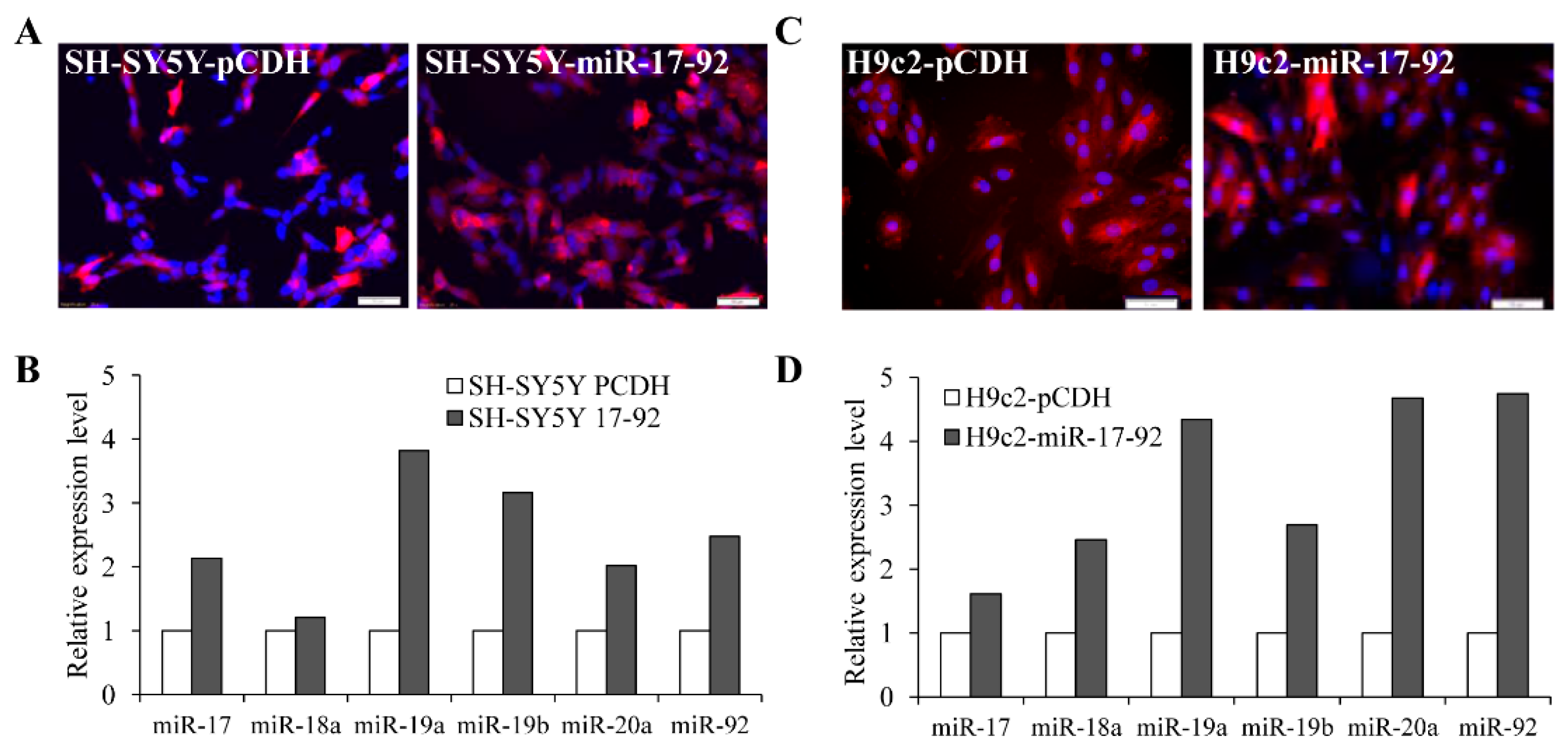
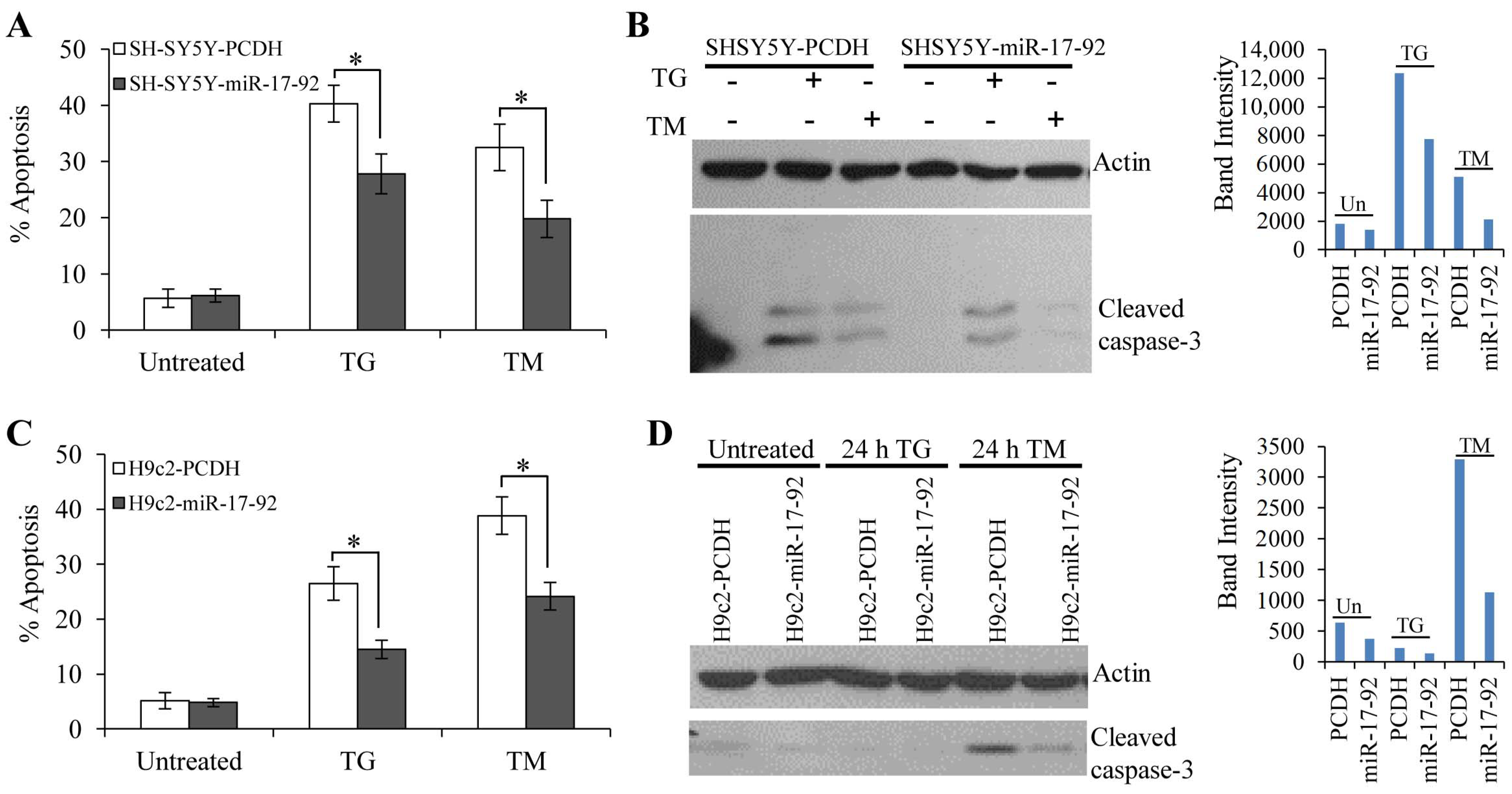
3.3. MiR-17-92 Cluster Protects Against UPR-Induced Cell Death
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Healy, S.J.; Gorman, A.M.; Mousavi-Shafaei, P.; Gupta, S.; Samali, A. Targeting the endoplasmic reticulum-stress response as an anticancer strategy. Eur. J. Pharmacol. 2009, 625, 234–246. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.M.; Barua, D.; Arabkari, V.; Islam, N.; Gupta, A.; Gupta, S. Hyperactivation of nuclear receptor coactivators induces PERK-dependent cell death. Oncotarget 2018, 9, 11707–11721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hetz, C.; Papa, F.R. The Unfolded Protein Response and Cell Fate Control. Mol. Cell 2018, 69, 169–181. [Google Scholar] [CrossRef] [Green Version]
- Harding, H.P.; Zhang, Y.; Bertolotti, A.; Zeng, H.; Ron, D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol. Cell 2000, 5, 897–904. [Google Scholar] [CrossRef]
- Kobayashi, M.; Yamamoto, M. Molecular mechanisms activating the Nrf2-Keap1 pathway of antioxidant gene regulation. Antioxid. Redox Signal. 2005, 7, 385–394. [Google Scholar] [CrossRef]
- Cullinan, S.B.; Zhang, D.; Hannink, M.; Arvisais, E.; Kaufman, R.J.; Diehl, J.A. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol. Cell Biol. 2003, 23, 7198–7209. [Google Scholar] [CrossRef] [Green Version]
- Hetz, C.; Martinon, F.; Rodriguez, D.; Glimcher, L.H. The unfolded protein response: Integrating stress signals through the stress sensor IRE1alpha. Physiol. Rev. 2011, 91, 1219–1243. [Google Scholar] [CrossRef]
- Barua, D.; Gupta, A.; Gupta, S. Targeting the IRE1-XBP1 axis to overcome endocrine resistance in breast cancer: Opportunities and challenges. Cancer Lett. 2020, 486, 29–37. [Google Scholar] [CrossRef]
- Ha, M.; Kim, V.N. Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–524. [Google Scholar] [CrossRef]
- Ameres, S.L.; Zamore, P.D. Diversifying microRNA sequence and function. Nat. Rev. Mol. Cell Biol. 2013, 14, 475–488. [Google Scholar] [CrossRef] [PubMed]
- Gregory, R.I.; Chendrimada, T.P.; Cooch, N.; Shiekhattar, R. Human RISC couples microRNA biogenesis and posttranscriptional gene silencing. Cell 2005, 123, 631–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valencia-Sanchez, M.A.; Liu, J.; Hannon, G.J.; Parker, R. Control of translation and mRNA degradation by miRNAs and siRNAs. Genes Dev. 2006, 20, 515–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwal, V.; Bell, G.W.; Nam, J.W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. Elife 2015, 4, e05005. [Google Scholar] [CrossRef] [PubMed]
- Read, D.E.; Gupta, A.; Cawley, K.; Gupta, S. Regulation of ER Stress Responses by microRNAs. In Endoplasmic Reticulum Stress in Health and Disease; Agostinis, P., Afshin, S., Eds.; Springer: Heidelberg, Germany, 2012; pp. 143–161. [Google Scholar] [CrossRef]
- Read, D.E.; Gupta, A.; Ladilov, Y.; Samali, A.; Gupta, S. miRNA signature of unfolded protein response in H9c2 rat cardiomyoblasts. Cell Biosci. 2014, 4, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartoszewska, S.; Kochan, K.; Madanecki, P.; Piotrowski, A.; Ochocka, R.; Collawn, J.F.; Bartoszewski, R. Regulation of the unfolded protein response by microRNAs. Cell Mol. Biol. Lett. 2013, 18, 555–578. [Google Scholar] [CrossRef] [Green Version]
- Cawley, K.; Logue, S.E.; Gorman, A.M.; Zeng, Q.; Patterson, J.; Gupta, S.; Samali, A. Disruption of microRNA biogenesis confers resistance to ER stress-induced cell death upstream of the mitochondrion. PLoS ONE 2013, 8, e73870. [Google Scholar] [CrossRef]
- Chitnis, N.; Pytel, D.; Diehl, J.A. UPR-inducible miRNAs contribute to stressful situations. Trends Biochem. Sci. 2013, 38, 447–452. [Google Scholar] [CrossRef] [Green Version]
- Mendell, J.T. miRiad roles for the miR-17-92 cluster in development and disease. Cell 2008, 133, 217–222. [Google Scholar] [CrossRef] [Green Version]
- Xiang, J.; Wu, J. Feud or Friend? The Role of the miR-17-92 Cluster in Tumorigenesis. Curr. Genom. 2010, 11, 129–135. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Huang, J.; Yang, N.; Greshock, J.; Megraw, M.S.; Giannakakis, A.; Liang, S.; Naylor, T.L.; Barchetti, A.; Ward, M.R.; et al. microRNAs exhibit high frequency genomic alterations in human cancer. Proc. Natl. Acad. Sci. USA 2006, 103, 9136–9141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hossain, M.M.; Sultana, A.; Barua, D.; Islam, M.N.; Gupta, A.; Gupta, S. Differential expression, function and prognostic value of miR-17-92 cluster in ER-positive and triple-negative breast cancer. Cancer. Treat. Res. Commun. 2020, 25, 100224. [Google Scholar] [CrossRef] [PubMed]
- Aguda, B.D.; Kim, Y.; Piper-Hunter, M.G.; Friedman, A.; Marsh, C.B. MicroRNA regulation of a cancer network: Consequences of the feedback loops involving miR-17-92, E2F, and Myc. Proc. Natl. Acad. Sci. USA 2008, 105, 19678–19683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, H.L.; Xue, G.; Mei, Q.; Wang, Y.Z.; Ding, F.X.; Liu, M.F.; Lu, M.H.; Tang, Y.; Yu, H.Y.; Sun, S.H. Repression of the miR-17-92 cluster by p53 has an important function in hypoxia-induced apoptosis. EMBO J. 2009, 28, 2719–2732. [Google Scholar] [CrossRef] [Green Version]
- Fontana, L.; Pelosi, E.; Greco, P.; Racanicchi, S.; Testa, U.; Liuzzi, F.; Croce, C.M.; Brunetti, E.; Grignani, F.; Peschle, C. MicroRNAs 17-5p-20a-106a control monocytopoiesis through AML1 targeting and M-CSF receptor upregulation. Nat. Cell Biol. 2007, 9, 775–787. [Google Scholar] [CrossRef]
- He, L.; Thomson, J.M.; Hemann, M.T.; Hernando-Monge, E.; Mu, D.; Goodson, S.; Powers, S.; Cordon-Cardo, C.; Lowe, S.W.; Hannon, G.J.; et al. A microRNA polycistron as a potential human oncogene. Nature 2005, 435, 828–833. [Google Scholar] [CrossRef]
- Ventura, A.; Young, A.G.; Winslow, M.M.; Lintault, L.; Meissner, A.; Erkeland, S.J.; Newman, J.; Bronson, R.T.; Crowley, D.; Stone, J.R.; et al. Targeted deletion reveals essential and overlapping functions of the miR-17 through 92 family of miRNA clusters. Cell 2008, 132, 875–886. [Google Scholar] [CrossRef] [Green Version]
- Fontana, L.; Fiori, M.E.; Albini, S.; Cifaldi, L.; Giovinazzi, S.; Forloni, M.; Boldrini, R.; Donfrancesco, A.; Federici, V.; Giacomini, P.; et al. Antagomir-17-5p abolishes the growth of therapy-resistant neuroblastoma through p21 and BIM. PLoS ONE 2008, 3, e2236. [Google Scholar] [CrossRef] [Green Version]
- Woods, K.; Thomson, J.M.; Hammond, S.M. Direct regulation of an oncogenic micro-RNA cluster by E2F transcription factors. J. Biol. Chem. 2007, 282, 2130–2134. [Google Scholar] [CrossRef] [Green Version]
- Puthalakath, H.; O’Reilly, L.A.; Gunn, P.; Lee, L.; Kelly, P.N.; Huntington, N.D.; Hughes, P.D.; Michalak, E.M.; McKimm-Breschkin, J.; Motoyama, N.; et al. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell 2007, 129, 1337–1349. [Google Scholar] [CrossRef] [Green Version]
- Xiao, C.; Srinivasan, L.; Calado, D.P.; Patterson, H.C.; Zhang, B.; Wang, J.; Henderson, J.M.; Kutok, J.L.; Rajewsky, K. Lymphoproliferative disease and autoimmunity in mice with increased miR-17-92 expression in lymphocytes. Nat. Immunol. 2008, 9, 405–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samali, A.; Fitzgerald, U.; Deegan, S.; Gupta, S. Methods for monitoring endoplasmic reticulum stress and the unfolded protein response. Int. J. Cell Biol. 2010, 2010, 830307. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Hossain, M.M.; Miller, N.; Kerin, M.; Callagy, G.; Gupta, S. NCOA3 coactivator is a transcriptional target of XBP1 and regulates PERK-eIF2alpha-ATF4 signalling in breast cancer. Oncogene 2016, 35, 5860–5871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, A.; Read, D.E.; Gupta, S. Assays for induction of the unfolded protein response and selective activation of the three major pathways. Methods Mol. Biol. 2015, 1292, 19–38. [Google Scholar] [CrossRef]
- Ogata, M.; Hino, S.; Saito, A.; Morikawa, K.; Kondo, S.; Kanemoto, S.; Murakami, T.; Taniguchi, M.; Tanii, I.; Yoshinaga, K.; et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol. Cell Biol. 2006, 26, 9220–9231. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, Y.; Larkin, D.; Lara-Lemus, R.; Ramos-Castaneda, J.; Liu, M.; Arvan, P. Endoplasmic reticulum (ER) chaperone regulation and survival of cells compensating for deficiency in the ER stress response kinase, PERK. J. Biol. Chem. 2008, 283, 17020–17029. [Google Scholar] [CrossRef] [Green Version]
- Buytaert, E.; Matroule, J.Y.; Durinck, S.; Close, P.; Kocanova, S.; Vandenheede, J.R.; de Witte, P.A.; Piette, J.; Agostinis, P. Molecular effectors and modulators of hypericin-mediated cell death in bladder cancer cells. Oncogene 2008, 27, 1916–1929. [Google Scholar] [CrossRef] [Green Version]
- Verfaillie, T.; Rubio, N.; Garg, A.D.; Bultynck, G.; Rizzuto, R.; Decuypere, J.P.; Piette, J.; Linehan, C.; Gupta, S.; Samali, A.; et al. PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ. 2012, 19, 1880–1891. [Google Scholar] [CrossRef] [Green Version]
- Martin, D.; Rojo, A.I.; Salinas, M.; Diaz, R.; Gallardo, G.; Alam, J.; De Galarreta, C.M.; Cuadrado, A. Regulation of heme oxygenase-1 expression through the phosphatidylinositol 3-kinase/Akt pathway and the Nrf2 transcription factor in response to the antioxidant phytochemical carnosol. J. Biol. Chem. 2004, 279, 8919–8929. [Google Scholar] [CrossRef] [Green Version]
- McMahon, M.; Samali, A.; Chevet, E. Regulation of the unfolded protein response by noncoding RNA. Am. J. Physiol. Cell Physiol. 2017, 313, C243–C254. [Google Scholar] [CrossRef] [Green Version]
- Upton, J.P.; Wang, L.; Han, D.; Wang, E.S.; Huskey, N.E.; Lim, L.; Truitt, M.; McManus, M.T.; Ruggero, D.; Goga, A.; et al. IRE1alpha cleaves select microRNAs during ER stress to derepress translation of proapoptotic Caspase-2. Science 2012, 338, 818–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, K.; Liu, H.; Song, Z.; Jiang, Y.; Kim, H.; Samavati, L.; Nguyen, H.M.; Yang, Z.Q. The UPR Transducer IRE1 Promotes Breast Cancer Malignancy by Degrading Tumor Suppressor microRNAs. iScience 2020, 23, 101503. [Google Scholar] [CrossRef]
- Sun, H.; Lin, D.C.; Guo, X.; Kharabi Masouleh, B.; Gery, S.; Cao, Q.; Alkan, S.; Ikezoe, T.; Akiba, C.; Paquette, R.; et al. Inhibition of IRE1alpha-driven pro-survival pathways is a promising therapeutic application in acute myeloid leukemia. Oncotarget 2016, 7, 18736–18749. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Hossain, M.M.; Read, D.E.; Hetz, C.; Samali, A.; Gupta, S. PERK regulated miR-424(322)-503 cluster fine-tunes activation of IRE1 and ATF6 during Unfolded Protein Response. Sci. Rep. 2015, 5, 18304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McQuiston, A.; Diehl, J.A. Recent insights into PERK-dependent signaling from the stressed endoplasmic reticulum. F1000Res 2017, 6, 1897. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Read, D.E.; Deepti, A.; Cawley, K.; Gupta, A.; Oommen, D.; Verfaillie, T.; Matus, S.; Smith, M.A.; Mott, J.L.; et al. Perk-dependent repression of miR-106b-25 cluster is required for ER stress-induced apoptosis. Cell Death Dis. 2012, 3, e333. [Google Scholar] [CrossRef]
- Watson, P.J.; Fairall, L.; Schwabe, J.W. Nuclear hormone receptor co-repressors: Structure and function. Mol. Cell Endocrinol. 2012, 348, 440–449. [Google Scholar] [CrossRef]
- Wortel, I.M.N.; van der Meer, L.T.; Kilberg, M.S.; van Leeuwen, F.N. Surviving Stress: Modulation of ATF4-Mediated Stress Responses in Normal and Malignant Cells. Trends Endocrinol. Metab. 2017, 28, 794–806. [Google Scholar] [CrossRef]
- Karpinski, B.A.; Morle, G.D.; Huggenvik, J.; Uhler, M.D.; Leiden, J.M. Molecular cloning of human CREB-2: An ATF/CREB transcription factor that can negatively regulate transcription from the cAMP response element. Proc. Natl. Acad. Sci. USA 1992, 89, 4820–4824. [Google Scholar]
- Liang, G.; Hai, T. Characterization of human activating transcription factor 4, a transcriptional activator that interacts with multiple domains of cAMP-responsive element-binding protein (CREB)-binding protein. J. Biol. Chem. 1997, 272, 24088–24095. [Google Scholar]
- Ferguson, J.; Smith, M.; Zudaire, I.; Wellbrock, C.; Arozarena, I. Glucose availability controls ATF4-mediated MITF suppression to drive melanoma cell growth. Oncotarget 2017, 8, 32946–32959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soda, T.; Frank, C.; Ishizuka, K.; Baccarella, A.; Park, Y.U.; Flood, Z.; Park, S.K.; Sawa, A.; Tsai, L.H. DISC1-ATF4 transcriptional repression complex: Dual regulation of the cAMP-PDE4 cascade by DISC1. Mol. Psychiatry 2013, 18, 898–908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kansanen, E.; Kuosmanen, S.M.; Leinonen, H.; Levonen, A.L. The Keap1-Nrf2 pathway: Mechanisms of activation and dysregulation in cancer. Redox Biol. 2013, 1, 45–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, P.; Rojo de la Vega, M.; Sammani, S.; Mascarenhas, J.B.; Kerins, M.; Dodson, M.; Sun, X.; Wang, T.; Ooi, A.; Garcia, J.G.N.; et al. RPA1 binding to NRF2 switches ARE-dependent transcriptional activation to ARE-NRE-dependent repression. Proc. Natl. Acad. Sci. USA 2018, 115, E10352–E10361. [Google Scholar] [CrossRef] [Green Version]
- Svoronos, A.A.; Engelman, D.M.; Slack, F.J. OncomiR or Tumor Suppressor? The Duplicity of MicroRNAs in Cancer. Cancer Res. 2016, 76, 3666–3670. [Google Scholar] [CrossRef] [Green Version]
- Mestdagh, P.; Bostrom, A.K.; Impens, F.; Fredlund, E.; Van Peer, G.; De Antonellis, P.; von Stedingk, K.; Ghesquiere, B.; Schulte, S.; Dews, M.; et al. The miR-17-92 microRNA cluster regulates multiple components of the TGF-beta pathway in neuroblastoma. Mol. Cell 2010, 40, 762–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Huang, Z.P.; Seok, H.Y.; Ding, J.; Kataoka, M.; Zhang, Z.; Hu, X.; Wang, G.; Lin, Z.; Wang, S.; et al. mir-17-92 cluster is required for and sufficient to induce cardiomyocyte proliferation in postnatal and adult hearts. Circ. Res. 2013, 112, 1557–1566. [Google Scholar] [CrossRef]
- Ottman, R.; Levy, J.; Grizzle, W.E.; Chakrabarti, R. The other face of miR-17-92a cluster, exhibiting tumor suppressor effects in prostate cancer. Oncotarget 2016, 7, 73739. [Google Scholar] [CrossRef] [Green Version]
- Ma, H.; Pan, J.S.; Jin, L.X.; Wu, J.; Ren, Y.D.; Chen, P.; Xiao, C.; Han, J. MicroRNA-17~92 inhibits colorectal cancer progression by targeting angiogenesis. Cancer Lett. 2016, 376, 293–302. [Google Scholar] [CrossRef]
- Pitale, P.M.; Gorbatyuk, O.; Gorbatyuk, M. Neurodegeneration: Keeping ATF4 on a Tight Leash. Front. Cell Neurosci. 2017, 11, 410. [Google Scholar] [CrossRef] [Green Version]
- Lange, P.S.; Chavez, J.C.; Pinto, J.T.; Coppola, G.; Sun, C.W.; Townes, T.M.; Geschwind, D.H.; Ratan, R.R. ATF4 is an oxidative stress-inducible, prodeath transcription factor in neurons in vitro and in vivo. J. Exp. Med. 2008, 205, 1227–1242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, J.L.; Flockhart, R.; Veal, G.J.; Lovat, P.E.; Redfern, C.P. Regulation of endoplasmic reticulum stress-induced cell death by ATF4 in neuroectodermal tumor cells. J. Biol. Chem. 2010, 285, 6091–6100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Bonay, M.; Deramaudt, T.B. Nrf2: New insight in cell apoptosis. Cell Death Dis. 2015, 6, e1897. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Read, D.E.; Gupta, A.; Cawley, K.; Fontana, L.; Agostinis, P.; Samali, A.; Gupta, S. Downregulation of miR-17-92 Cluster by PERK Fine-Tunes Unfolded Protein Response Mediated Apoptosis. Life 2021, 11, 30. https://doi.org/10.3390/life11010030
Read DE, Gupta A, Cawley K, Fontana L, Agostinis P, Samali A, Gupta S. Downregulation of miR-17-92 Cluster by PERK Fine-Tunes Unfolded Protein Response Mediated Apoptosis. Life. 2021; 11(1):30. https://doi.org/10.3390/life11010030
Chicago/Turabian StyleRead, Danielle E., Ananya Gupta, Karen Cawley, Laura Fontana, Patrizia Agostinis, Afshin Samali, and Sanjeev Gupta. 2021. "Downregulation of miR-17-92 Cluster by PERK Fine-Tunes Unfolded Protein Response Mediated Apoptosis" Life 11, no. 1: 30. https://doi.org/10.3390/life11010030