The Patterns and Puzzles of Genetic Diversity of Endangered Freshwater Mussel Unio crassus Philipsson, 1788 Populations from Vistula and Neman Drainages (Eastern Central Europe)


 , ,
, ,
Abstract
:1. Introduction
- (i)
- test the correlation between the genetic differentiation and geographic isolation of U. crassus populations using a hierarchical approach,
- (ii)
- examine the distribution of genetic variation among populations of U. crassus at multiple spatial scales (within rivers, among rivers within drainages, and across drainages),
- (iii)
- test the role of current vs historical gene flow into the distribution of U. crassus genetic diversity on the basis of populations representing currently isolated Neman and Vistula drainages (Central Europe) with the reported ancient connection,
- (iv)
- asses the phylogenetic affinities among U. crassus populations.
2. Materials and Methods
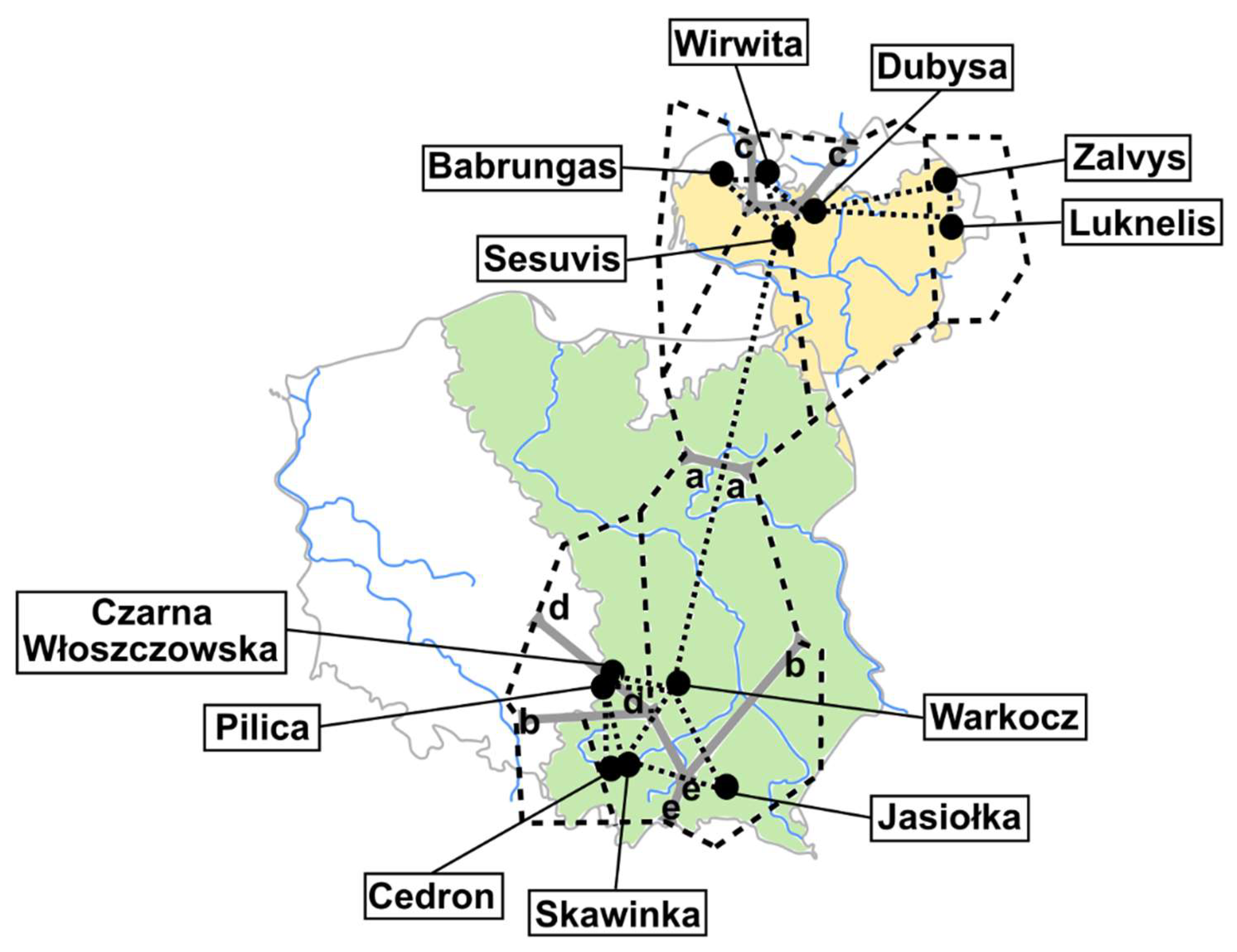
2.1. Sample Collection and Identification
2.2. Extraction, PCR Amplification and Sequencing
2.3. DNA Sequence Analysis
3. Results
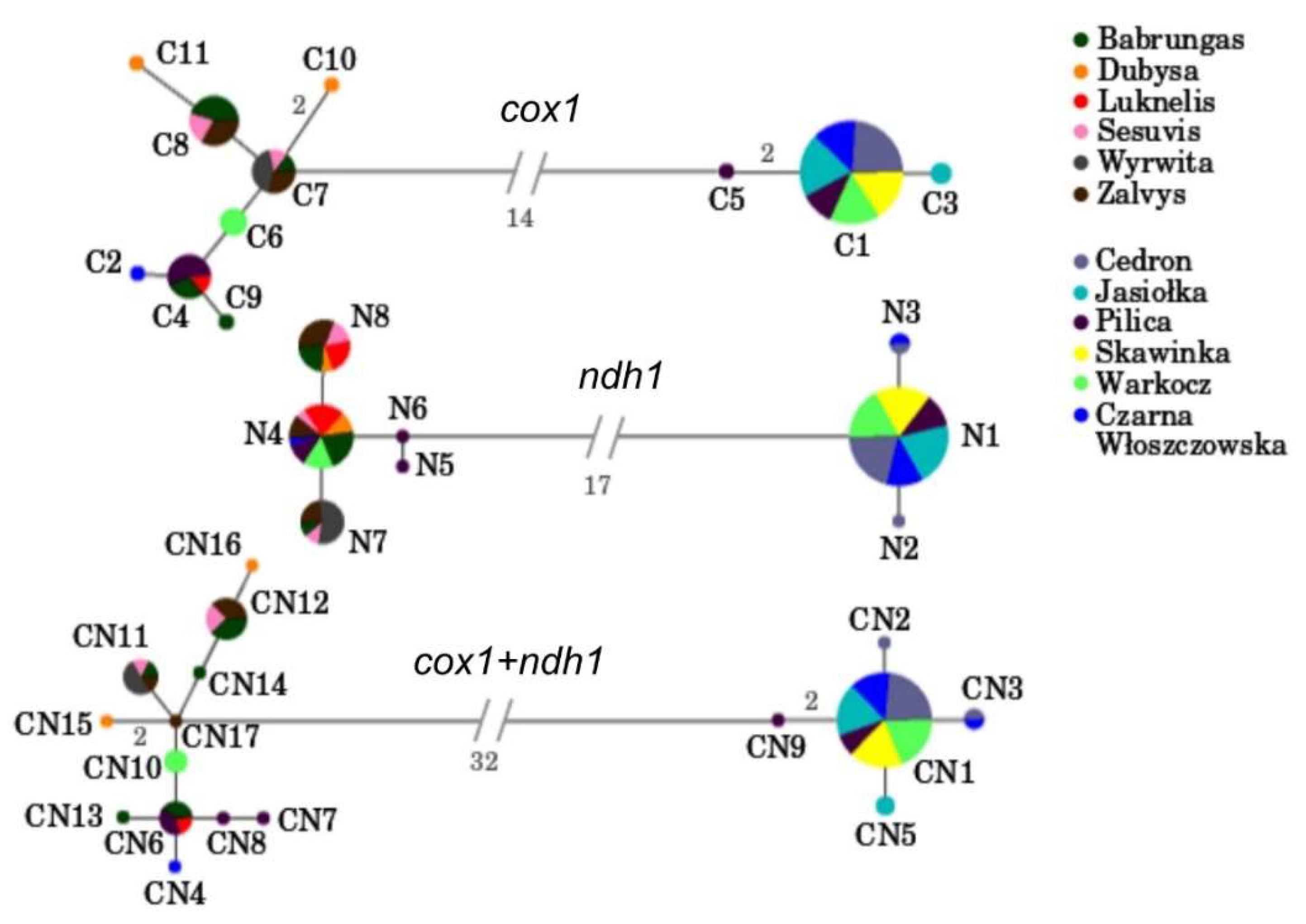
3.1. Mitochondrial Gene Regions
3.2. Within Population Variability
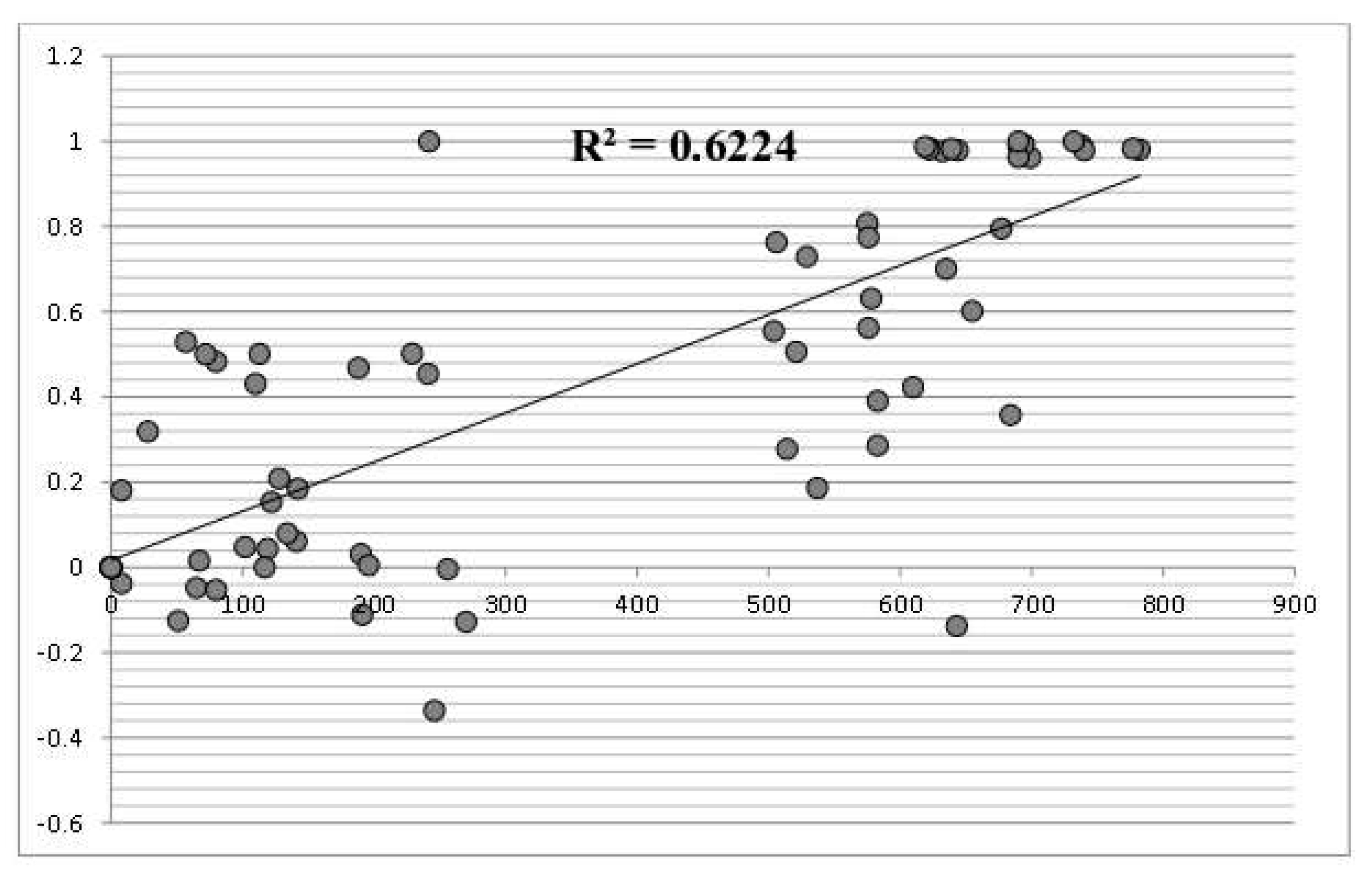
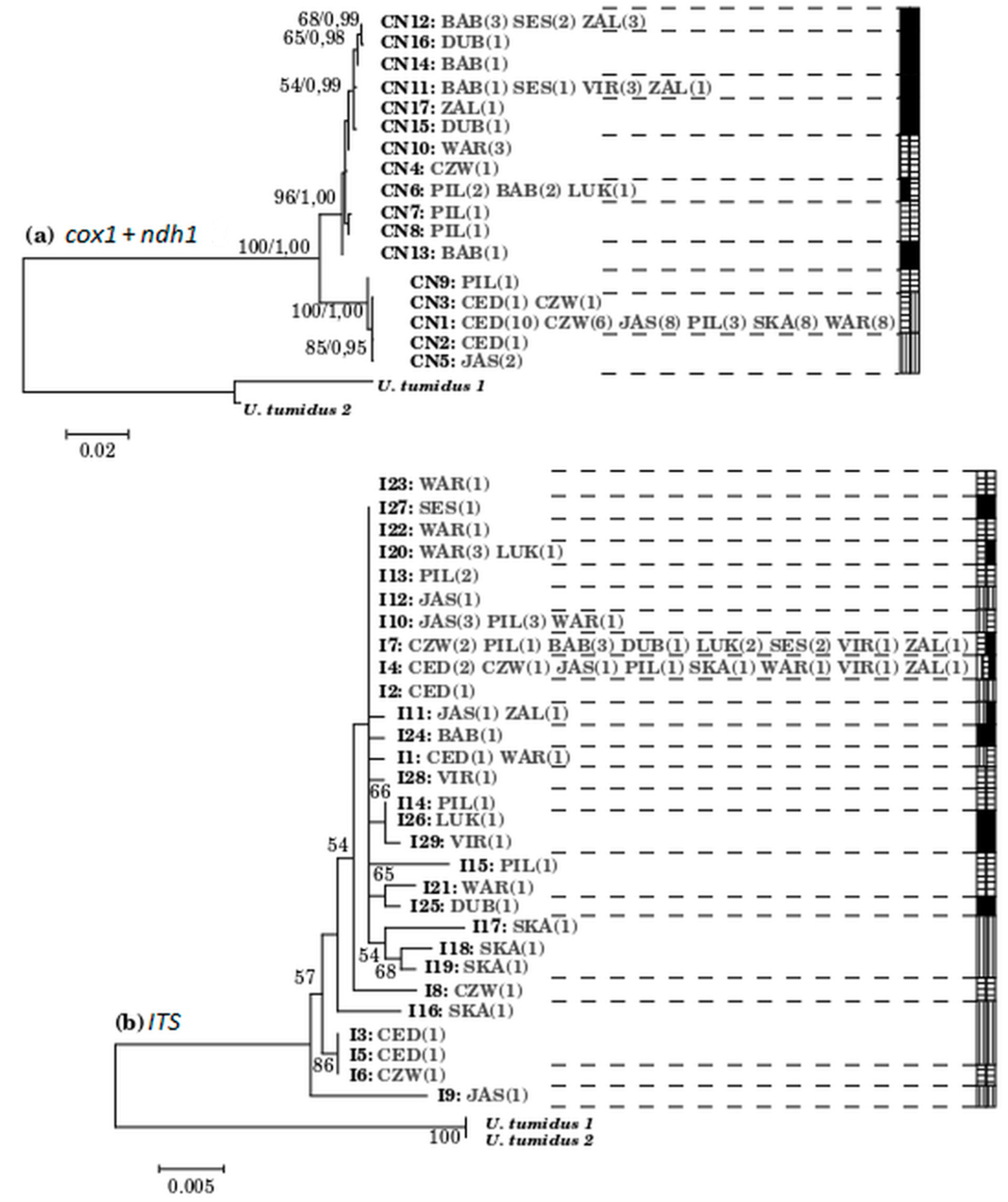
3.3. Among Population Structure
3.4. The Entire ITS Region of the Nuclear Ribosomal DNA
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Graf, D.L.; Cummings, K.S. Actual and Alleged Freshwater Mussels (Mollusca: Bivalvia: Unionoida) from Madagascar and the Mascarenes, with Description of a New Genus, Germainaia. Proc. Acad. Nat. Sci. Philadelphia 2009, 158, 221–238. [Google Scholar] [CrossRef]
- IUCN 2020. The IUCN Red List of Threatened Species. Version 2020-2. Available online: https://www.iucnredlist.org (accessed on 21 July 2020).
- Machordom, A.; Araujo, R.; Erpenbeck, D.; Ramos, M.-Á. Phylogeography and conservation genetics of endangered European Margaritiferidae (Bivalvia: Unionoidea). Biol. J. Linn. Soc. 2003, 78, 235–252. [Google Scholar] [CrossRef] [Green Version]
- Lydeard, C.; Cowie, R.H.; Ponder, W.F.; Bogan, A.E.; Bouchet, P.; Clark, S.A.; Cummings, K.S.; Frest, T.J.; Gargominy, O.; Herbert, D.G.; et al. The Global Decline of Nonmarine Mollusks. Bioscience 2004, 54, 321. [Google Scholar] [CrossRef]
- Geist, J.; Kuehn, R. Genetic diversity and differentiation of central European freshwater pearl mussel (Margaritifera margaritifera L.) populations: Implications for conservation and management. Mol. Ecol. 2005, 14, 425–439. [Google Scholar] [CrossRef] [PubMed]
- Geist, J.; Kuehn, R. Host-parasite interactions in oligotrophic stream ecosystems: The roles of life-history strategy and ecological niche. Mol. Ecol. 2008, 17, 997–1008. [Google Scholar] [CrossRef] [PubMed]
- Douda, K.; Horký, P.; Bílý, M. Host limitation of the thick-shelled river mussel: Identifying the threats to declining affiliate species. Anim. Conserv. 2012, 15, 536–544. [Google Scholar] [CrossRef]
- Taeubert, J.E.; Martinez, A.M.P.; Gum, B.; Geist, J. The relationship between endangered thick-shelled river mussel (Unio crassus) and its host fishes. Biol. Conserv. 2012, 155, 94–103. [Google Scholar] [CrossRef]
- Ćmiel, A.M.; Zając, K.; Lipińska, A.M.; Zając, T. Glochidial infestation of fish by the endangered thick-shelled river mussel Unio crassus. Aquat. Conserv. Mar. Freshw. Ecosyst. 2018, 28, 535–544. [Google Scholar] [CrossRef]
- Killeen, I.J.; Seddon, M.B.; Holmes, A.M. Molluscan Conservation: A Strategy for the 21st Century. J. Conchol. 1998, Spec. Publ, 320. [Google Scholar]
- Araujo, R.; Gómez, I.; Machordom, A. The identity and biology of Unio mancus Lamarck, 1819 (= U. elongatulus) (Bivalvia: Unionidae) in the Iberian Peninsula. J. Molluscan Stud. 2005, 71, 25–31. [Google Scholar] [CrossRef] [Green Version]
- Kallersjo, M.; von Proschwitz, T.; Lundberg, S.; Eldenas, P.; Erseus, C. Evaluation of ITS rDNA as a complement to mitochondrial gene sequences for phylogenetic studies in freshwater mussels: An example using Unionidae from north-western Europe. Zool. Scr. 2005, 34, 415–424. [Google Scholar] [CrossRef]
- Zając, K.; Zając, T.A.; Adamski, P.; Bielański, W.; Ćmiel, A.M.; Lipińska, A.M. Dispersal and mortality of translocated thick-shelled river mussel Unio crassus Philipsson, 1788 adults revealed by radio tracking. Aquat. Conserv. Mar. Freshw. Ecosyst. 2019, 29, 331–340. [Google Scholar] [CrossRef]
- Zając, K.; Zając, T.A. Seasonal patterns in the developmental rate of glochidia in the endangered thick-shelled river mussel, Unio crassus Philipsson, 1788. Hydrobiologia 2020. [Google Scholar] [CrossRef]
- Araujo, R.; Toledo, C.; Machordom, A. Redescription of Unio gibbus, A West Palaearctic Freshwater Mussel with Hookless Glochidia. Malacologia 2009, 51, 131–141. [Google Scholar] [CrossRef]
- Reis, J.; Araujo, R. Redescription of Unio tumidiformis Castro, 1885 (Bivalvia, Unionidae), an endemism from the south-western Iberian Peninsula. J. Nat. Hist. 2009, 43, 1929–1945. [Google Scholar] [CrossRef]
- Bolotov, I.N.; Aksenova, O.V.; Bakken, T.; Glasby, C.J.; Gofarov, M.Y.; Kondakov, A.V.; Konopleva, E.S.; Lopes-Lima, M.; Lyubas, A.A.; Wang, Y.; et al. Discovery of a silicate rock-boring organism and macrobioerosion in fresh water. Nat. Commun. 2018, 9, 2882. [Google Scholar] [CrossRef]
- Soroka, M. Characteristics of mitochondrial DNA of unionid bivalves (Mollusca: Bivalvia: Unionidae). I. Detection and characteristics of doubly uniparental inheritance (DUI) of unionid mitochondrial DNA. Folia Malacol. 2010, 18, 147–188. [Google Scholar] [CrossRef] [Green Version]
- Froufe, E.; Sobral, C.; Teixeira, A.; Sousa, R.; Varandas, S.C.; Aldridge, D.; Lopes-Lima, M. Genetic diversity of the pan-European freshwater mussel Anodonta anatina (Bivalvia: Unionoida) based on CO1: New phylogenetic insights and implications for conservation. Aquat. Conserv. Mar. Freshw. Ecosyst. 2014, 24, 561–574. [Google Scholar] [CrossRef] [Green Version]
- Lopes-Lima, M.; Teixeira, A.; Froufe, E.; Lopes, A.; Varandas, S.; Sousa, R. Biology and conservation of freshwater bivalves: Past, present and future perspectives. Hydrobiologia 2014, 735, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Lopes-Lima, M.; Sousa, R.; Geist, J.; Aldridge, D.C.; Araujo, R.; Bergengren, J.; Bespalaya, Y.; Bódis, E.; Burlakova, L.; Van Damme, D.; et al. Conservation status of freshwater mussels in Europe: State of the art and future challenges. Biol. Rev. 2016, 92, 572–607. [Google Scholar] [CrossRef]
- Zając, K.; Florek, J.; Zając, T.; Adamski, P.; Bielański, W.; Ćmiel, A.M.; Klich, M.; Lipińska, A.M. On the Reintroduction of the Endangered Thick-Shelled River Mussel Unio crassus: The Importance of the River’s Longitudinal Profile. Sci. Total Environ. 2018, 624, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Prié, V.; Soler, J.; Araujo, R.; Cucherat, X.; Philippe, L.; Patry, N.; Adam, B.; Legrand, N.; Jugé, P.; Richard, N.; et al. Challenging exploration of troubled waters: A decade of surveys of the giant freshwater pearl mussel Margaritifera auricularia in Europe. Hydrobiologia 2018, 810, 157–175. [Google Scholar] [CrossRef]
- Williams, J.D.; Warren, M.L.J.; Cummings, K.S.; Harris, J.L.; Neves, R.J. Conservation Status of Freshwater Mussels of the United States and Canada. Fisheries 1993, 18, 6–22. [Google Scholar] [CrossRef]
- Strayer, D.L.; Downing, J.A.; Haag, W.R.; King, T.L.; Layzer, J.B.; Newton, T. Changing perspectives on Pearly mussels, North American’s most imperiled animals. Bioscience 2004, 54, 429–439. [Google Scholar] [CrossRef]
- Tedesco, P.A.; Bigorne, R.; Bogan, A.E.; Giam, X.; Jézéquel, C.; Hugueny, B. Estimating how many undescribed species have gone extinct. Conserv. Biol. 2014, 28, 1360–1370. [Google Scholar] [CrossRef]
- King, T.L.; Eackles, M.S.; Gjetvaj, B.; Hoeh, W.R. Intraspecific phylogeography of Lasmigona subviridis (Bivalvia: Unionidae): Conservation implications of range discontinuity. Mol. Ecol. 1999, 8, S65–S78. [Google Scholar] [CrossRef]
- Roe, K.J.; Hartfield, P.D.; Lydeard, C. Phylogeographic analysis of the threatened and endangered superconglutinate-producing mussels of the genus Lampsilis (Bivalvia: Unionidae). Mol. Ecol. 2001, 10, 2225–2234. [Google Scholar] [CrossRef]
- Krebs, R.A. Combining paternally and maternally inherited mitochondrial DNA for analysis of population structure in mussels. Mol. Ecol. 2004, 13, 1701–1705. [Google Scholar] [CrossRef]
- Mock, K.E.; Brim-Box, J.C.; Miller, M.P.; Downing, M.E.; Hoeh, W.R. Genetic diversity and divergence among freshwater mussel (Anodonta) populations in the Bonneville Basin of Utah. Mol. Ecol. 2004, 13, 1085–1098. [Google Scholar] [CrossRef]
- Kelly, M.W.; Rhymer, J.M. Population genetic structure of a rare unionid (Lampsilis cariosa) in a recently glaciated landscape. Conserv. Genet. 2005, 6, 789–802. [Google Scholar] [CrossRef]
- Elderkin, C.L.; Christian, A.D.; Vaughn, C.C.; Metcalfe-Smith, J.L.; Berg, D.J. Population genetics of the freshwater mussel, Amblema plicata (Say 1817) (Bivalvia: Unionidae): Evidence of high dispersal and post-glacial colonization. Conserv. Genet. 2007, 8, 355–372. [Google Scholar] [CrossRef]
- Burlakova, L.E.; Campbell, D.; Karatayev, A.Y.; Barclay, D. Distribution, genetic analysis and conservation priorities for rare Texas freshwater molluscs in the genera Fusconaia and Pleurobema (Bivalvia: Unionidae). Aquat. Biosyst. 2012, 8, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perkins, M.A.; Johnson, N.A.; Gangloff, M.M. Molecular systematics of the critically-endangered North American spinymussels (Unionidae: Elliptio and Pleurobema) and description of Parvaspina gen. nov. Conserv. Genet. 2017, 18, 745–757. [Google Scholar] [CrossRef]
- Pfeiffer, J.M.; Sharpe, A.E.; Johnson, N.A.; Emery, K.F.; Page, L.M. Molecular phylogeny of the Nearctic and Mesoamerican freshwater mussel genus Megalonaias. Hydrobiologia 2018, 811, 139–151. [Google Scholar] [CrossRef] [Green Version]
- Zanatta, D.T.; Stoeckle, B.C.; Inoue, K.; Paquet, A.; Martel, A.L.; Kuehn, R.; Geist, J. High genetic diversity and low differentiation in North American Margaritifera margaritifera (Bivalvia: Unionida: Margaritiferidae). Biol. J. Linn. Soc. 2018, 123, 850–863. [Google Scholar] [CrossRef]
- Zettler, M.L.; Jueg, U. The situation of the freshwater mussel Unio crassus (Philipsson, 1788) in north-east Germany and its monitoring in terms of the EC Habitats Directive. Mollusca 2007, 25, 165–174. [Google Scholar]
- Geist, J. Strategies for the conservation of endangered freshwater pearl mussels (Margaritifera margaritifera L.): A synthesis of Conservation Genetics and Ecology. Hydrobiologia 2010, 644, 69–88. [Google Scholar] [CrossRef] [Green Version]
- Geist, J. Integrative freshwater ecology and biodiversity conservation. Ecol. Indic. 2011, 11, 1507–1516. [Google Scholar] [CrossRef]
- Cuttelod, A.; Seddon, M.; Neubert, E. European Red List of Non-marine Molluscs; Publications Office of the European Union: Luxembourg, 2011; ISBN 9789279201981. [Google Scholar]
- Bolotov, I.N.; Kondakov, A.V.; Konopleva, E.S.; Vikhrev, I.V.; Aksenova, O.V.; Aksenov, A.S.; Bespalaya, Y.V.; Borovskoy, A.V.; Danilov, P.P.; Dvoryankin, G.A.; et al. Integrative taxonomy, biogeography and conservation of freshwater mussels (Unionidae) in Russia. Sci. Rep. 2020, 10, 3072. [Google Scholar] [CrossRef] [Green Version]
- Taeubert, J.-E.; Gum, B.; Geist, J. Host-specificity of the endangered thick-shelled river mussel (Unio crassus, Philipsson 1788) and implications for conservation. Aquat. Conserv. Mar. Freshw. Ecosyst. 2012, 22, 36–46. [Google Scholar] [CrossRef]
- Zając, K. Unio crassus. In Głowaciński Z. Nowicki J. Polska czerwona Księga Zwierząt. Bezkręgowce.; Instytut Ochrony Przyrody PAN w Krakowie, Akademia Rolnicza im. A. Cieszkowskiego w Poznaniu: Kraków, Poland, 2004; p. 448. [Google Scholar]
- Denic, M.; Stoeckl, K.; Gum, B.; Geist, J. Physicochemical assessment of Unio crassus habitat quality in a small upland stream and implications for conservation. Hydrobiologia 2014, 735, 111–122. [Google Scholar] [CrossRef]
- Nagel, K.-O.; Badino, G. Population Genetics and Systematics of European Unionoidea. In Ecology and Evolution of the Freshwater Mussels Unionoida; Springer: Berlin/Heidelberg, Germany, 2001; pp. 51–80. [Google Scholar]
- Prie, V.; Puillandre, N.; Bouchet, P. Bad taxonomy can kill: Molecular reevaluation of Unio mancus Lamarck, 1819 (Bivalvia: Unionidae) and its accepted subspecies. Knowl. Manag. Aquat. Ecosyst. 2012, 405, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Reis, J.; Machordom, A.; Araujo, R. Diversidad morfológica y molecular de los Unionidae (Mollusca, Bivalvia) de Portugal. Graellsia 2013, 69, 17–36. [Google Scholar] [CrossRef]
- Araujo, R.; Buckley, D.; Nagel, K.-O.; García-Jiménez, R.; Machordom, A. Species boundaries, geographic distribution and evolutionary history of the Western Palaearctic freshwater mussels Unio (Bivalvia: Unionidae). Zool. J. Linn. Soc. 2017, 1–25. [Google Scholar] [CrossRef]
- Prié, V.; Puillandre, N. Molecular phylogeny, taxonomy, and distribution of French Unio species (Bivalvia, Unionidae). Hydrobiologia 2014, 735, 95–110. [Google Scholar] [CrossRef]
- Mezhzherin, S.V.; Yanovych, L.M.; Zhalay, Y.I.; Pampura, M.M.; Vasilieva, L.A. Reproductive isolation of two Unio crassus Philipsson, 1788 (Bivalvia, Unionidae) vicarious forms with low genetic differentiation level. Reports Natl. Acad. Sci. Ukr. 2013, 2, 138–143. [Google Scholar]
- Klishko, O.; Lopes-Lima, M.; Froufe, E.; Bogan, A.; Vasiliev, L.; Yanovich, L. Taxonomic reassessment of the freshwater mussel genus Unio (Bivalvia: Unionidae) in Russia and Ukraine based on morphological and molecular data. Zootaxa 2017, 4286, 93–112. [Google Scholar] [CrossRef]
- Gerke, N.; Tiedemann, R. A PCR-basedmolecular identification key to the glochidia of European freshwater mussels (Unionidae). Conserv. Genet. 2001, 2, 287–289. [Google Scholar] [CrossRef]
- Feind, S.; Geist, J.; Kuehn, R. Glacial perturbations shaped the genetic population structure of the endangered thick-shelled river mussel (Unio crassus, Philipsson 1788) in Central and Northern Europe. Hydrobiologia 2018, 810, 177–189. [Google Scholar] [CrossRef]
- Mioduchowska, M.; Kaczmarczyk, A.; Zając, K.; Zając, T.; Sell, J. Gender-Associated Mitochondrial DNA Heteroplasmy in Somatic Tissues of the Endangered Freshwater Mussel Unio crassus (Bivalvia: Unionidae): Implications for Sex Identification and Phylogeographical Studies. J. Exp. Zool. Part A Ecol. Genet. Physiol. 2016, 325, 610–625. [Google Scholar] [CrossRef]
- Geist, J.; Rottmann, O.; Schröder, W.; Kühn, R. Development of microsatellite markers for the endangered freshwater pearl mussel Margaritifera margaritifera L. (Bivalvia: Unionoidea). Mol. Ecol. Notes 2003, 3, 444–446. [Google Scholar] [CrossRef]
- Bouza, C.; Castro, J.; Martínez, P.; Amaro, R.; Fernández, C.; Ondina, P.; Outeiro, A.; San Miguel, E. Threatened freshwater pearl mussel Margaritifera margaritifera L. in NW Spain: Low and very structured genetic variation in southern peripheral populations assessed using microsatellite markers. Conserv. Genet. 2007, 8, 937–948. [Google Scholar] [CrossRef]
- Geist, J.; Wunderlich, H.; Kuehn, R. Use of mollusc shells for DNA-based molecular analyses. J. Molluscan Stud. 2008, 74, 337–343. [Google Scholar] [CrossRef] [Green Version]
- Hutchison, D.W.; Templeton, A.R. Correlation of Pairwise Genetic and Geographic Distance Measures: Inferring the Relative Influences of Gene Flow and Drift on the Distribution of Genetic Variability. Evolution 1999, 53, 1898–1914. [Google Scholar] [CrossRef] [PubMed]
- Hartl, D.L.; Clark, A.G. Principles of population genetics; Sinauer Associates: Sunderland, MA, USA, 1997; ISBN 0878933069. [Google Scholar]
- Bilton, D.T.; Freeland, J.R.; Okamura, B. Dispersal in Freshwater Invertebrates. Annu. Rev. Ecol. Syst. 2001, 32, 159–181. [Google Scholar] [CrossRef] [Green Version]
- Newman, D.; Pilson, D. Increased probability of extinction due to decreased genetic effective population size: Experimantal populations of Clarcia pulchella. Evolution 1997, 51, 354–362. [Google Scholar] [CrossRef]
- Saccheri, I.; Kuussaari, M.; Kankare, M.; Vikman, P.; Hanski, I. Inbreeding and extinction in a butterfly metapopulation. Nature 1998, 392, 491–494. [Google Scholar] [CrossRef]
- Frankham, R. Genetics and conservation biology. C. R. Biol. 2003, 326 (Suppl. 1), S22–S29. [Google Scholar] [CrossRef]
- Reed, D.H.; Frankham, R. Correlation between Fitness and Genetic Diversity. Conserv. Biol. 2003, 17, 230–237. [Google Scholar] [CrossRef]
- Manni, F.; Guérard, E.; Heyer, E. Geographic patterns of (genetic, morphologic, linguistic) variation: How barriers can be detected by using Monmonier’s algorithm. Hum. Biol. 2004, 76, 173–190. [Google Scholar] [CrossRef]
- Article 17 web tool. Available online: https://nature-art17.eionet.europa.eu/article17/reports2012 (accessed on 21 July 2020).
- Piechocki, A.; Dyduch-Falniowska, A. Mięczaki (Mollusca). Małże (Bivalvia); Fauna Słod. Polskie Towarzystwo Hydrobiologiczne. Wydawnictwo Naukowe PWN: Warszawa, Poland, 1993. [Google Scholar]
- Sambrook, J.; Russell, D.W. Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2001; ISBN 0879695773. [Google Scholar]
- White, T.J.; Bruns, S.; Lee, S.; Taylor, J. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In PCR Protocols: A Guide to Methods and Applications; Innis, M.A., Gelfand, D.H., Sninsky, J.J., White, T.J., Eds.; Academic Press: New York, NY, USA, 1990; pp. 315–322. ISBN 008088671X. [Google Scholar]
- Buhay, J.E.; Serb, J.M.; Renee, D.; Parham, Q.; Lydeard, C. Conservation genetics of two endangered unionid bivalve species, Epioblasma florentina walkeri and E. capsaeformis (Unionidae: Lampsilini). J. Molluscan Stud. 2002, 68, 385–391. [Google Scholar] [CrossRef]
- Serb, J.M.; Lydeard, C. Complete mtDNA Sequence of the North American Freshwater Mussel, Lampsilis ornata (Unionidae): An Examination of the Evolution and Phylogenetic Utility of Mitochondrial Genome Organization in Bivalvia (Mollusca). Mol. Biol. Evol. 2003, 20, 1854–1866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Folmer, O.; Black, M.; Hoeh, W.; Lutz, R.; Vrijenhoek, R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol. Mar. Biol. Biotechnol. 1994, 3, 294–299. [Google Scholar] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Hall, T. BioEdit: An important software for molecular biology. GERF Bull. Biosci. 2011, 2, 60–61. [Google Scholar] [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Soding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [Green Version]
- Excoffier, L.; Lischer, H.E.L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef]
- Byers, J.E. The distribution of an introduced mollusc and its role in the long-term demise of a native confamilial species. Biol. Invasions 1999, 1, 339–352. [Google Scholar] [CrossRef]
- Smouse, P.E.; Long, J.C.; Sokal, R.R. Multiple Regression and Correlation Extensions of the Mantel Test of Matrix Correspondence. Syst. Zool. 1986, 35, 627–632. [Google Scholar] [CrossRef]
- Network v. 4.5.1.6 Software. Available online: http://www.fluxus-technology.com (accessed on 21 July 2020).
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasegawa, M.; Kishino, H.; Yano, T. Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J. Mol. Evol. 1985, 22, 160–174. [Google Scholar] [CrossRef] [PubMed]
- Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Voronoi, G. Nouvelles applications des paramètres continus à la théorie des formes quadratiques. J. für die Reine und Angew. Math. 1908, 133, 97–178. [Google Scholar] [CrossRef]
- Brassel, K.E.; Reif, D. A Procedure to Generate Thiessen Polygons. Geogr. Anal. 1979, 11, 289–303. [Google Scholar] [CrossRef]
- Monmonier, M.S. Maximum-Difference Barriers: An Alternative Numerical Regionalization Method*. Geogr. Anal. 1973, 5, 245–261. [Google Scholar] [CrossRef]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian hylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef] [Green Version]
- Newton, M.A.; Raftery, A.E. Approximate Bayesian Inference with the Weighted Likelihood Bootstrap. J. R. Stat. Soc. Ser. B 1994, 56, 3–48. [Google Scholar] [CrossRef]
- Suchard, M.A.; Weiss, R.E.; Sinsheimer, J.S. Bayesian selection of continous-time Markov chain evolutionary models. Mol. Biol. Evol. 2001, 18, 1001–1013. [Google Scholar] [CrossRef] [Green Version]
- Zouros, E.; Ball, A.O.; Saavedra, C.; Freeman, K.R.; Skibinski, D.O.F.; Gallagher, C.; Beynon, C.M. Mitochondrial DNA inheritance. Nature 1994, 368, 818. [Google Scholar] [CrossRef] [PubMed]
- Froufe, E.; Gonçalves, D.V.; Teixeira, A.; Sousa, R.; Varandas, S.; Ghamizi, M.; Zieritz, A.; Lopes-Lima, M. Who lives where? Molecular and morphometric analyses clarify which Unio species (Unionida, Mollusca) inhabit the southwestern Palearctic. Org. Divers. Evol. 2016, 16, 597–611. [Google Scholar] [CrossRef] [Green Version]
- Nagel, K.-O. Testing hypotheses on the dispersal and evolutionary history of freshwater mussels (Mollusca: Bivalvia: Unionidae). J. od Evol. Biol. 2000, 13, 854–865. [Google Scholar] [CrossRef]
- Patton, J.L.; Smith, M.F. Paraphyly, Polyphyly, and the Nature of Species Boundaries in Pocket Gophers (Genus Thomomys). Syst. Biol. 1994, 43, 11–26. [Google Scholar] [CrossRef]
- Sperling, F.A.H.; Harrison, R.G. Mitochondrial DNA variation within and between species of the Papilio machaon group of swallowtail betterflies. Evolution 1994, 48, 408–422. [Google Scholar] [CrossRef] [PubMed]
- Talbot, S.L.; Shields, G.F. Phylogeography of Brown Bears (Ursus arctos) of Alaska and Paraphyly within the Ursidae. Mol. Phylogenet. Evol. 1996, 5, 477–494. [Google Scholar] [CrossRef]
- Avise, J.C.; Wollenberg, K. Phylogenetics and the origin of species. Proc. Natl. Acad. Sci. USA. 1997, 94, 7748–7755. [Google Scholar] [CrossRef] [Green Version]
- Shaw, K.L. Conflict between nuclear and mitochondrial DNA phylogenies of a recent species radiation: What mtDNA reveals and conceals about modes of speciation in Hawaiian crickets. Proc. Natl. Acad. Sci. USA 2002, 99, 16122–16127. [Google Scholar] [CrossRef] [Green Version]
- Griffiths, C.S.; Barrowclough, G.F.; Groth, J.G.; Mertz, L. Phylogeny of the Falconidae (Aves): A comparison of the efficacy of morphological, mitochondrial, and nuclear data. Mol. Phylogenet. Evol. 2004, 32, 101–109. [Google Scholar] [CrossRef]
- Bowen, B.W.; Bass, A.L.; Soares, L.; Toonen, R.J. Conservation implications of complex population structure: Lessons from the loggerhead turtle (Caretta caretta). Mol. Ecol. 2005, 14, 2389–2402. [Google Scholar] [CrossRef]
- Zanatta, D.T.; Fraley, S.J.; Murphy, R.W. Population structure and mantle display polymorphisms in the wavy-rayed lampmussel, Lampsilis fasciola (Bivalvia: Unionidae). Can. J. Zool. 2007, 85, 1169–1181. [Google Scholar] [CrossRef]
- McCauley, D.E. Genetic consequences of local population extinction and recolonization. Trends Ecol. Evol. 1991, 6, 5–8. [Google Scholar] [CrossRef]
- Błońska, D. Geneza słodkowodnej ichtiofauny polski. Kosmos 2012, 2, 261–270. [Google Scholar]
- Ehlers, J.; Gibbard, P.L. Quaternary Glaciations: Extent and Chronology. Part 1, Europe; Elsevier: Amsterdam, The Netherlands, 2004; ISBN 9780080540146. [Google Scholar]
- Witkowski, A. Analiza ichtiofauny baseny Biebrzy. Charakterystyka morfologiczno-systematyczna smoczkoustych i ryb. Acta Univ. Wratislav. 1984, 14, 3–100. [Google Scholar]
- Stoeckl, K.; Taeubert, J.-E.; Geist, J. Fish species composition and host fish density in streams of the thick-shelled river mussel (Unio crassus) - implications for conservation. Aquat. Conserv. Mar. Freshw. Ecosyst. 2015, 25, 276–287. [Google Scholar] [CrossRef]
- Lamand, F.; Roche, K.; Beisel, J.-N. Glochidial infestation by the endangered mollusc Unio crassus in rivers of north-eastern France: Phoxinus phoxinus and Cottus gobio as primary fish hosts. Aquat. Conserv. Mar. Freshw. Ecosyst. 2016, 26, 445–455. [Google Scholar] [CrossRef]
- Lopes-Lima, M.; Froufe, E.; Do, V.T.; Ghamizi, M.; Mock, K.E.; Kebapçı, Ü.; Klishko, O.; Kovitvadhi, S.; Kovitvadhi, U.; Paulo, O.S.; et al. Phylogeny of the most species-rich freshwater bivalve family (Bivalvia: Unionida: Unionidae): Defining modern subfamilies and tribes. Mol. Phylogenet. Evol. 2017, 106, 174–191. [Google Scholar] [CrossRef] [Green Version]
- Witkowski, A.; Kotusz, J.; Przybylski, M.; Marszał, L.; Heese, T.; Amirowicz, A.; Buras, P.; Kukuła, K. Pochodzenie, skład gatunkowy i aktualny stopień zagrożenia ichtiofauny w dorzeczu Wisły i Odry. Arch. Polish Fish. 2004, 12, 7–20. [Google Scholar]
- Konopacka, A. Inwazyjne skorupiaki obunogie (Crustacea, Amphipoda) w wodach Polski. Przegląd Zool. 2004, 48, 141–162. [Google Scholar]
- Durand, J.D.; Persat, H.; Bouvet, Y. Phylogeography and postglacial dispersion of the chub (Leuciscus cephalus) in Europe. Mol. Ecol. 1999, 8, 989–997. [Google Scholar] [CrossRef]
- Nesbø, C.L.; Fossheim, T.; Vollestad, L.A.; Jakobsen, K.S. Genetic divergence and phylogeographic relationships among european perch (Perca fluviatilis) populations reflect glacial refugia and postglacial colonization. Mol. Ecol. 1999, 8, 1387–1404. [Google Scholar] [CrossRef] [PubMed]
- Englbrecht, C.C.; Freyhof, J.; Nolte, A.; Rassmann, K.; Schliewen, U.; Tautz, D. Phylogeography of the bullhead Cottus gobio (Pisces: Teleostei: Cottidae) suggests a pre-Pleistocene origin of the major central European populations. Mol. Ecol. 2000, 9, 709–722. [Google Scholar] [CrossRef] [PubMed]
- Šlechtová, V.; Bohlen, J.; Freyhof, J.; Persat, H.; Delmastro, G.B. The Alps as barrier to dispersal in cold-adapted freshwater fishes? Phylogeographic history and taxonomic status of the bullhead in the Adriatic freshwater drainage. Mol. Phylogenet. Evol. 2004, 33, 225–239. [Google Scholar] [CrossRef] [PubMed]
- Mäkinen, H.S.; Merilä, J. Mitochondrial DNA phylogeography of the three-spined stickleback (Gasterosteus aculeatus) in Europe—Evidence for multiple glacial refugia. Mol. Phylogenet. Evol. 2008, 46, 167–182. [Google Scholar] [CrossRef]
- Hänfling, B.; Brandl, R. Genetic variability, population size and isolation of distinct populations in the freshwater fish Cottus gobio L. Mol. Ecol. 1998, 7, 1625–1632. [Google Scholar] [CrossRef]
- Weiss, S.; Schlötterer, C.; Waidbacher, H.; Jungwirth, M. Haplotype (mtDNA) diversity of brown trout Salmo trutta in tributaries of the Austrian Danube: Massive introgression of Atlantic basin fish-By man or nature? Mol. Ecol. 2001, 10, 1241–1246. [Google Scholar] [CrossRef]
- Berg, D.J.; Cantonwine, E.G.; Hoeh, W.R.; Guttman, S.I. Genetic structure of Quadrula quadrula (Bivalvia: Unionidae): Little variation across large distances. J. Shellfish Res. 1998, 17, 1365–1373. [Google Scholar]
- Hantke, R. Flussgeschichte Mitteleuropas.; Ferdinand Enke Verlag: Stuttgart, Germany, 1993. [Google Scholar]
- Hewitt, G.M. Post-glacial re-colonization of European biota. Biol. J. Linn. Soc. 1999, 68, 87–112. [Google Scholar] [CrossRef]
- Neve, G.; Verlaque, R. Relict species: Phylogeography and conservation biology. In Relict Species: Phylogeography and Conservation Biology; Habel, J.C., Assmann, T., Eds.; Springer: Berlin, Germany, 2010; pp. 277–294. ISBN 9783540921608. [Google Scholar]
- Taberlet, P. Biodiversity at the intraspecific level: The comparative phylogeographic approach. J. Biotechnol. 1998, 64, 91–100. [Google Scholar] [CrossRef]
- Hewitt, G.M. Genetic consequences of climatic oscillations in the Quaternary. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2004, 359, 183–195. [Google Scholar] [CrossRef] [Green Version]
- McDevitt, A.D.; Zub, K.; Kawałko, A.; Oliver, M.K.; Herman, J.S.; Wójcik, J.M. Climate and refugial origin influence the mitochondrial lineage distribution of weasels (Mustela nivalis) in a phylogeographic suture zone. Biol. J. Linn. Soc. 2012, 106, 57–69. [Google Scholar] [CrossRef] [Green Version]
- Haas, F. Superfamilia Unionacea. In Das Tierreich, 88th ed.; de Gruyter: Berlin, Germany, 1969. [Google Scholar]
- Falkner, G. Vorschlag für eine Neufassung der Roten Liste der in Bayern vorkommenden Mollusken (Weichtiere). Mit einem revidierten systematischen Verzeichnis der in Bayern nachgewiesenen Molluskenarten; Schriftenr. Bayer. Landesamt für Umweltschutz: Bayer, Germany, 1990; pp. 61–112. [Google Scholar]
- Nesemann, H. Zoogeographie und Taxonomie der Muschel-Gattungen Unio Philipsson, 1788, Pseudanodonta Bourguignat. 1877 und Pseudunio Haas, 1910 im oberen und mittleren Donausystem (Bivalvia: Unionidae, Margaritiferidae). Nachrichtenblatt der Ersten Vor. Malakol. Gesellschaft 1993, 1, 2–40. [Google Scholar]
- Haas, F. Superfamilia Unionacea. In Das Tierrich.; Mertens, R., Hennig, W., Eds.; Walter de Gruyter: Berlin, Germany, 1969; p. 663. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
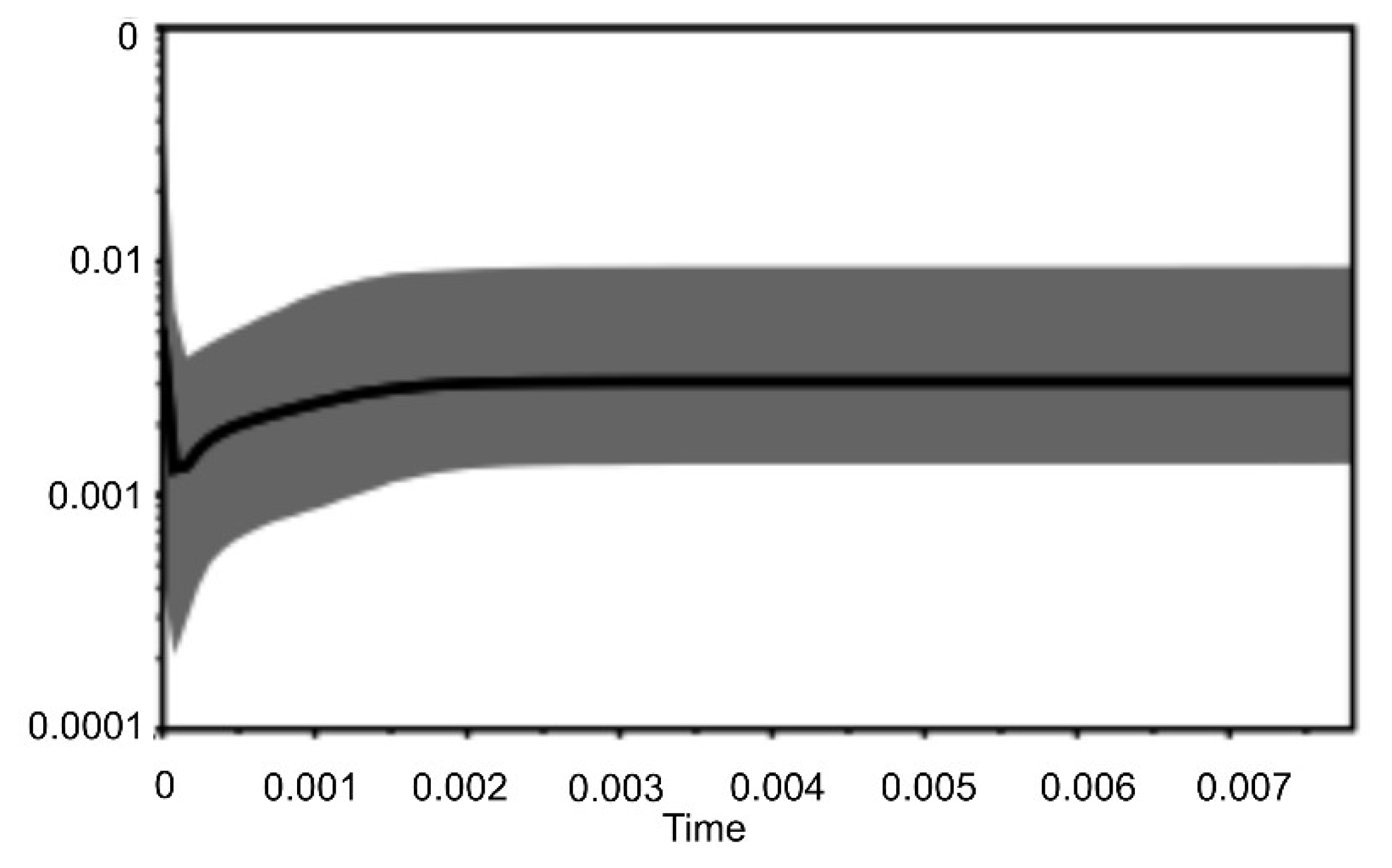{kind=link}
| Haplotype | Vistula Drainage | Neman Drainage | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| cox1 | PIL | CZW | WAR | CED | SKA | JAS | BAB | DUB | LUK | SES | VIR | ZAL |
| C1 | 0.500 | 0.875 | 0.727 | 1.000 | 1.000 | 0.833 | ||||||
| C2 | 0.125 | |||||||||||
| C3 | 0.167 | |||||||||||
| C4 | 0.400 | 0.250 | 1.000 | |||||||||
| C5 | 0.100 | |||||||||||
| C6 | 0.273 | |||||||||||
| C7 | 0.125 | 0.333 | 1.000 | 0.400 | ||||||||
| C8 | 0.500 | 0.667 | 0.600 | |||||||||
| C9 | 0.125 | |||||||||||
| C10 | 0.500 | |||||||||||
| C11 | 0.500 | |||||||||||
| N | 10 | 8 | 11 | 12 | 8 | 12 | 8 | 2 | 1 | 3 | 3 | 5 |
| n | 3 | 2 | 2 | 1 | 1 | 2 | 4 | 2 | 1 | 2 | 1 | 2 |
| HD | 0.644 | 0.250 | 0.436 | 0.000 | 0.000 | 0.303 | 0.750 | - | - | 0.667 | 0.000 | 0.600 |
| (SE) | (0.101) | (0.180) | (0.133) | (0.147) | (0.139) | (0.314) | (0.175) | |||||
| π | 0.016 | 0.008 | 0.012 | 0.000 | 0.000 | 0.001 | 0.003 | - | - | 0.001 | 0.000 | 0.001 |
| (SE) | (0.009) | (0.005) | (0.007) | (0.001) | (0.002) | (0.001) | (0.001) | |||||
| ndh1 | PIL | CZW | WAR | CED | SKA | JAS | BAB | DUB | LUK | SES | VIR | ZAL |
| N1 | 0.500 | 0.750 | 0.727 | 0.833 | 1.000 | 1.000 | 0.200 | |||||
| N2 | 0.083 | |||||||||||
| N3 | 0.125 | 0.083 | ||||||||||
| N4 | 0.200 | 0.125 | 0.272 | 0.500 | 0.667 | 0.571 | 0.200 | 0.250 | ||||
| N5 | 0.100 | |||||||||||
| N6 | 0.100 | |||||||||||
| N7 | 0.125 | 0.200 | 1.000 | 0.250 | ||||||||
| N8 | 0.375 | 0.333 | 0.429 | 0.400 | 0.500 | |||||||
| N | 9 | 8 | 11 | 12 | 9 | 10 | 8 | 3 | 7 | 5 | 5 | 8 |
| n | 4 | 3 | 2 | 3 | 1 | 1 | 3 | 2 | 2 | 4 | 1 | 3 |
| HD | 0.694 | 0.4643 | 0.436 | 0.318 | 0.000 | 0.0000 | 0.679 | 0.667 | 0.571 | 0.900 | 0.000 | 0.714 |
| (SE) | (0.147) | (0.200) | (0.133) | (0.164) | (0.122) | (0.314) | (0.119) | (0.161) | (0.123) | |||
| π | 0.012 | 0.006 | 0.009 | 0.001 | 0.000 | 0.000 | 0.001 | 0.001 | 0.001 | 0.010 | 0.000 | 0.001 |
| (SE) | (0.007) | (0.003) | (0.005) | (0.001) | (0.001) | (0.001) | (0.001) | (0.006) | (0.001) | |||
| cox1+ ndh1 | PIL | CZW | WAR | CED | SKA | JAS | BAB | DUB | LUK | SES | VIR | ZAL |
| CN1 | 0.375 | 0.750 | 0.727 | 0.833 | 1.000 | 0.800 | ||||||
| CN2 | 0.083 | |||||||||||
| CN3 | 0.125 | 0.083 | ||||||||||
| CN4 | 0.125 | |||||||||||
| CN5 | 0.200 | |||||||||||
| CN6 | 0.250 | 0.250 | 1.000 | |||||||||
| CN7 | 0.125 | |||||||||||
| CN8 | 0.125 | |||||||||||
| CN9 | 0.125 | |||||||||||
| CN10 | 0.273 | |||||||||||
| CN11 | 0.125 | 0.333 | 1.000 | 0.200 | ||||||||
| CN12 | 0.375 | 0.667 | 0.600 | |||||||||
| CN13 | 0.125 | |||||||||||
| CN14 | 0.125 | |||||||||||
| CN15 | 0.500 | |||||||||||
| CN16 | 0.500 | |||||||||||
| CN17 | 0.200 | |||||||||||
| N | 8 | 8 | 11 | 12 | 8 | 10 | 8 | 2 | 1 | 3 | 3 | 5 |
| n | 5 | 3 | 2 | 3 | 1 | 2 | 5 | 2 | 1 | 2 | 1 | 2 |
| HD | 0.857 | 0.464 | 0.436 | 0.318 | 0.000 | 0.356 | 0.857 | - | - | 0.667 | 0.000 | 0.700 |
| (SE) | (0.108) | (0.200) | (0.133) | (0.164) | (0.159) | (0.108) | (0.314) | (0.218) | ||||
| π | 0.014 | 0.007 | 0.011 | 0.000 | 0.000 | 0.000 | 0.002 | - | - | 0.001 | 0.000 | 0.001 |
| (SE) | (0.008) | (0.004) | (0.006) | (0.000) | (0.000) | (0.001) | (0.001) | (0.001) | ||||
| Locality Code | CED | CZW | JAS | PIL | SKA | WAR | BAB | DUB | LUK | SES | VIR | ZAL |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CED | 10.726 | 7.635 | 10.726 | inf | 1.889 | 0.018 | 0.010 | 0.005 | 0.008 | 0.003 | 0.009 | |
| CZW | 0.044 | 15.139 | 2.263 | inf | inf | 0.118 | 0.184 | 0.213 | 0.155 | 0.146 | 0.128 | |
| JAS | 0.061 | 0.032 | 0.567 | 5.714 | 2.203 | 0.019 | 0.012 | 0.005 | 0.009 | 0.004 | 0.010 | |
| PIL | 0.502 * | 0.181 | 0.469 * | 0.658 | 31.104 | 0.776 | 2.185 | inf | 1.304 | 1.252 | 0.898 | |
| SKA | −0.038 | 0.000 | 0.080 | 0.432 * | 2.750 | 0.019 | 0.008 | 0.000 | 0.006 | 0.000 | 0.008 | |
| WAR | 0.209 * | −0.047 | 0.185 | 0.016 | 0.154 | 0.291 | 0.486 | 0.682 | 0.400 | 0.389 | 0.330 | |
| BAB | 0.966 * | 0.809 * | 0.962 * | 0.392 * | 0.963 * | 0.632 * | 9.900 | inf | inf | 1.066 | inf | |
| DUB | 0.980 * | 0.730 | 0.977 * | 0.186 | 0.982 * | 0.507 * | 0.048 | inf | inf | 0.536 | 114.931 | |
| LUK | 0.991 | 0.701 | 0.990 | −0.137 | 1.000 | 0.423 | −0.128 | −0.112 | 0.603 | 0.000 | 0.445 | |
| SES | 0.984 * | 0.764 * | 0.982 * | 0.277 * | 0.988 * | 0.555 * | −0.053 | −0.126 | 0.453 | 0.500 | inf | |
| VIR | 0.992 * | 0.774 * | 0.992 * | 0.285 * | 1.000 * | 0.562 * | 0.319 | 0.482 | 1.000 | 0.500 | 0.495 | |
| ZAL | 0.981 * | 0.796 * | 0.979 * | 0.358 * | 0.984 * | 0.602 * | −0.004 | 0.004 | 0.529 | −0.336 | 0.502 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kilikowska, A.; Mioduchowska, M.; Wysocka, A.; Kaczmarczyk-Ziemba, A.; Rychlińska, J.; Zając, K.; Zając, T.; Ivinskis, P.; Sell, J. The Patterns and Puzzles of Genetic Diversity of Endangered Freshwater Mussel Unio crassus Philipsson, 1788 Populations from Vistula and Neman Drainages (Eastern Central Europe). Life 2020, 10, 119. https://doi.org/10.3390/life10070119
Kilikowska A, Mioduchowska M, Wysocka A, Kaczmarczyk-Ziemba A, Rychlińska J, Zając K, Zając T, Ivinskis P, Sell J. The Patterns and Puzzles of Genetic Diversity of Endangered Freshwater Mussel Unio crassus Philipsson, 1788 Populations from Vistula and Neman Drainages (Eastern Central Europe). Life. 2020; 10(7):119. https://doi.org/10.3390/life10070119
Chicago/Turabian StyleKilikowska, Adrianna, Monika Mioduchowska, Anna Wysocka, Agnieszka Kaczmarczyk-Ziemba, Joanna Rychlińska, Katarzyna Zając, Tadeusz Zając, Povilas Ivinskis, and Jerzy Sell. 2020. "The Patterns and Puzzles of Genetic Diversity of Endangered Freshwater Mussel Unio crassus Philipsson, 1788 Populations from Vistula and Neman Drainages (Eastern Central Europe)" Life 10, no. 7: 119. https://doi.org/10.3390/life10070119