
Wnt/β-Catenin-Pathway Alterations and Homologous Recombination Deficiency in Cholangiocarcinoma Cell Lines and Clinical Samples: Towards Specific Vulnerabilities

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and In Vitro Analyses
2.2. Western Blot (WB)
2.3. Immunohistochemistry
2.4. Slide Image Aquisition
2.5. Nucleic Acid Extraction and Quantitative Reverse Transcription Real-Time Polymerase Chain Reaction (qRT-PCR)
2.6. Next-Generation DNA and RNA Sequencing
2.7. HRD Score Analysis
2.8. Nanostring and Statistical Methods
2.9. Patient Samples
3. Results
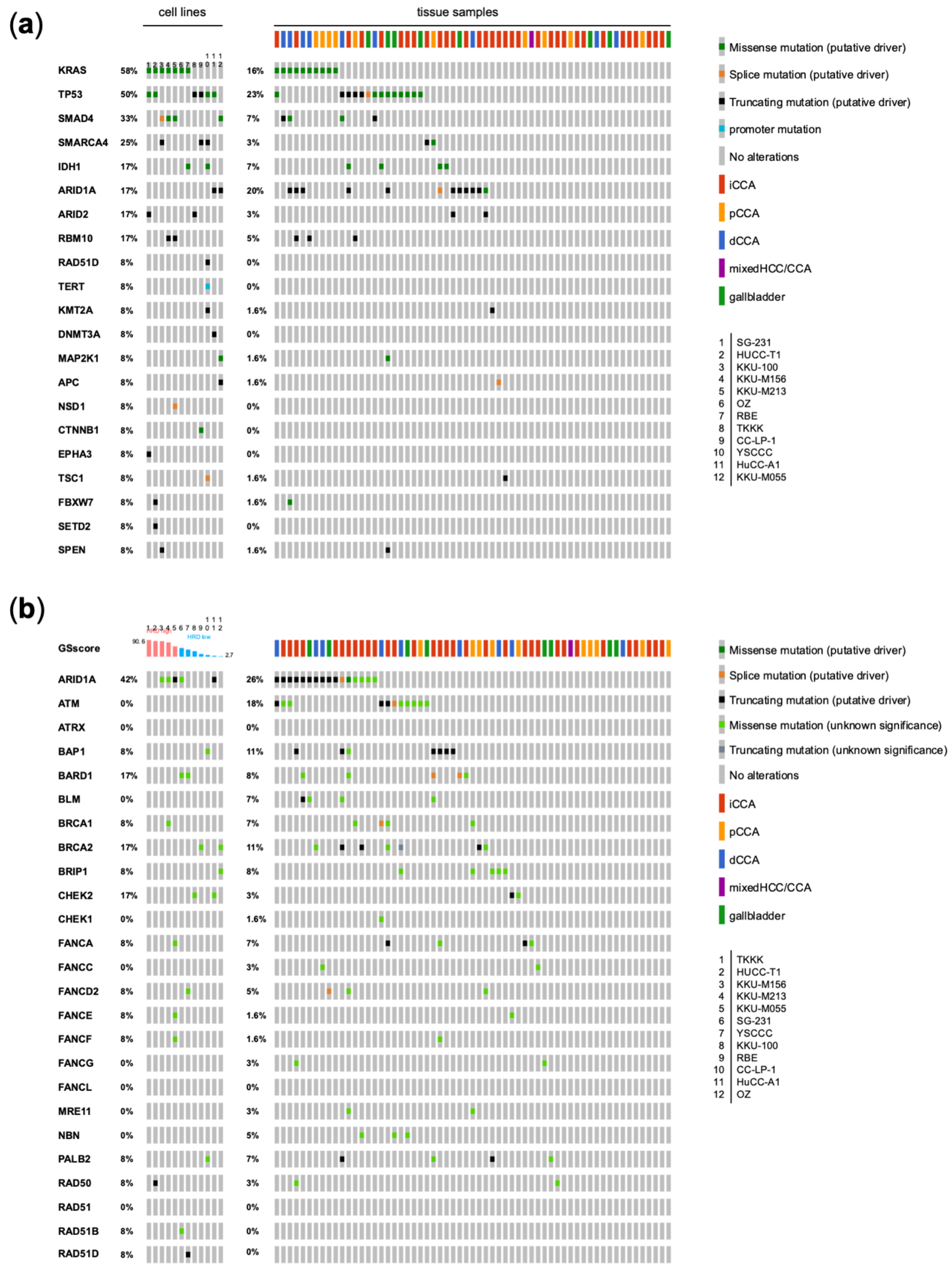
3.1. Genetic Analysis of CCA Cell Lines in Comparison with Human CCA Samples
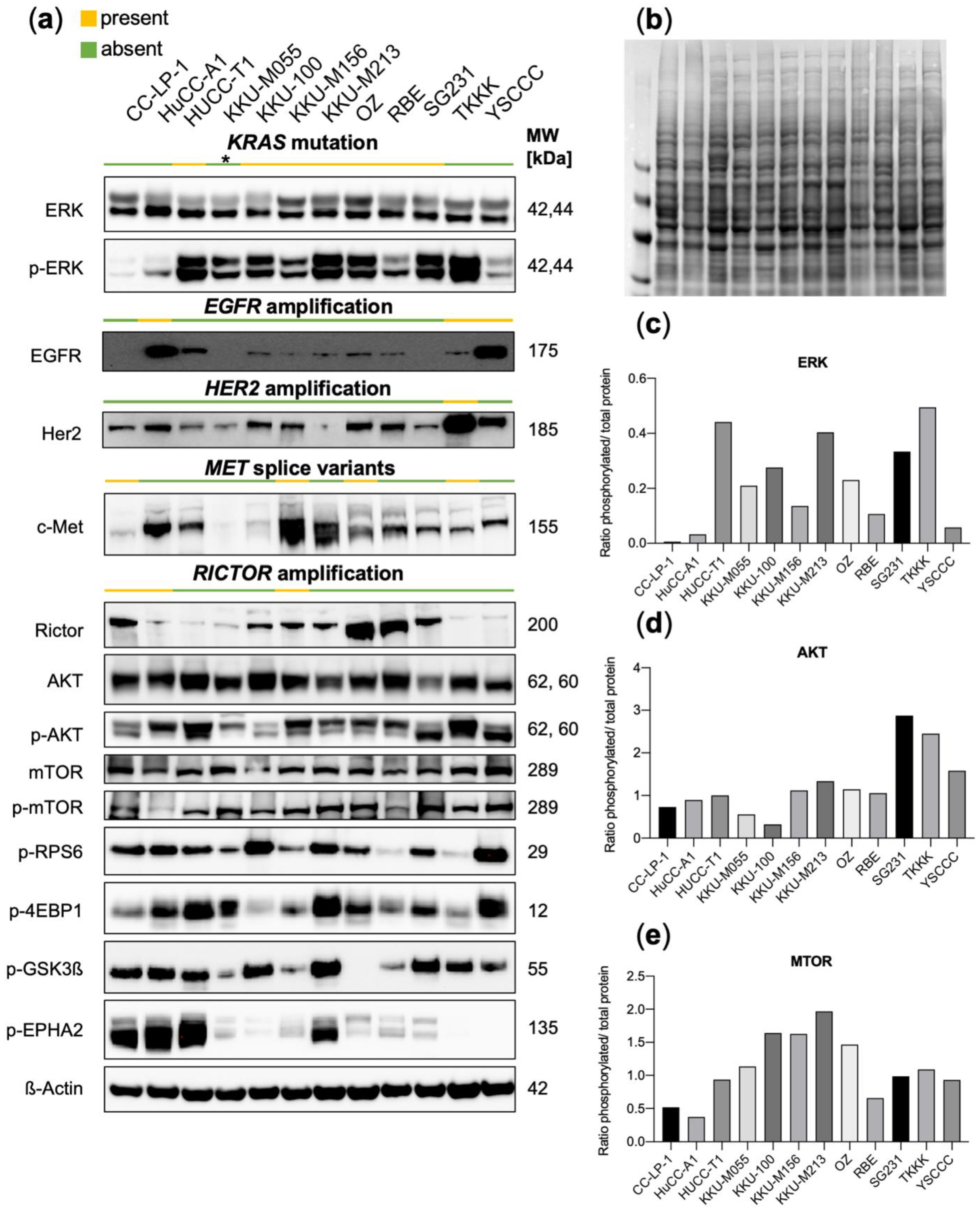
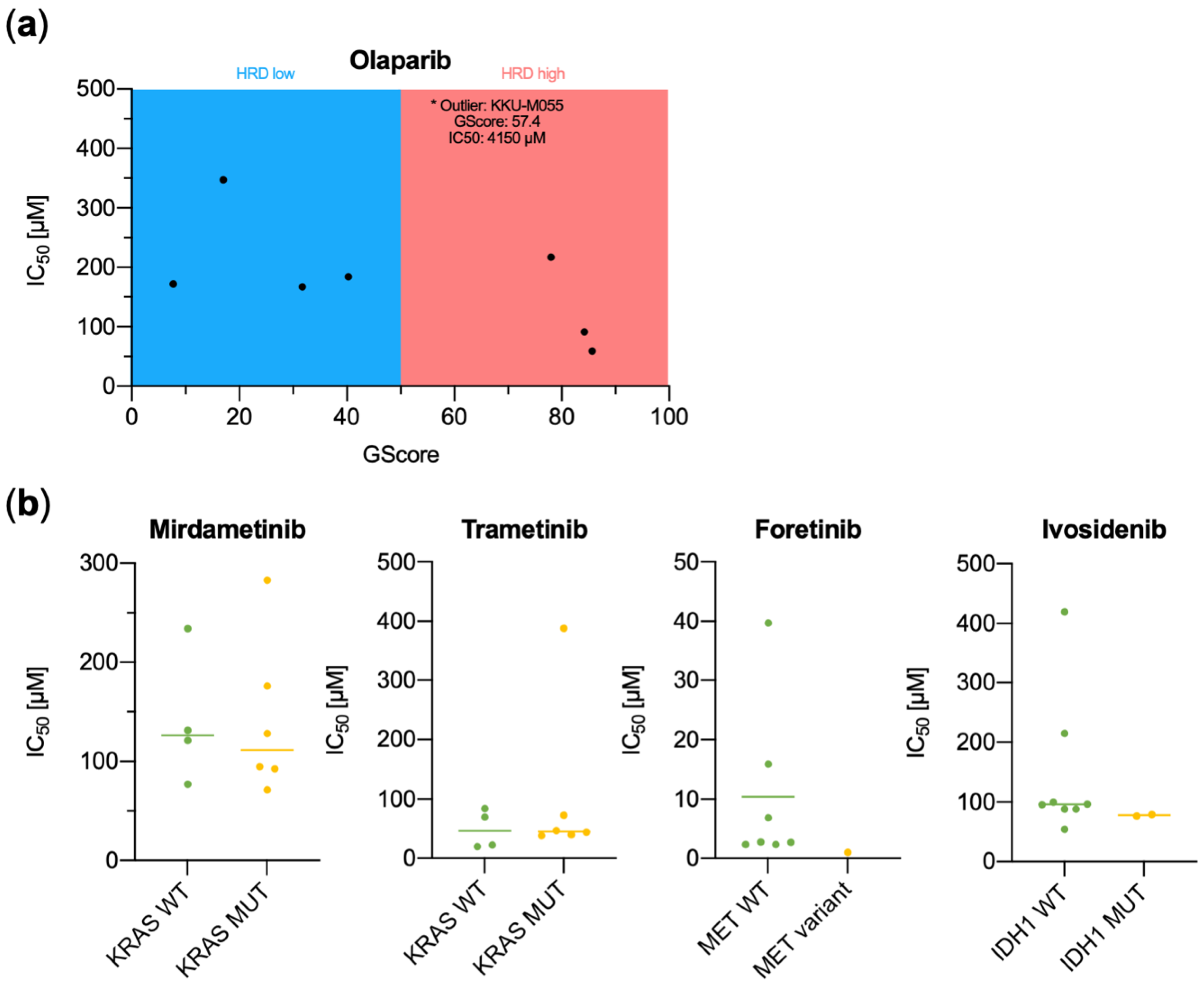
3.2. Functional Characterization of CCA Cell Lines Concerning Genomic Alterations
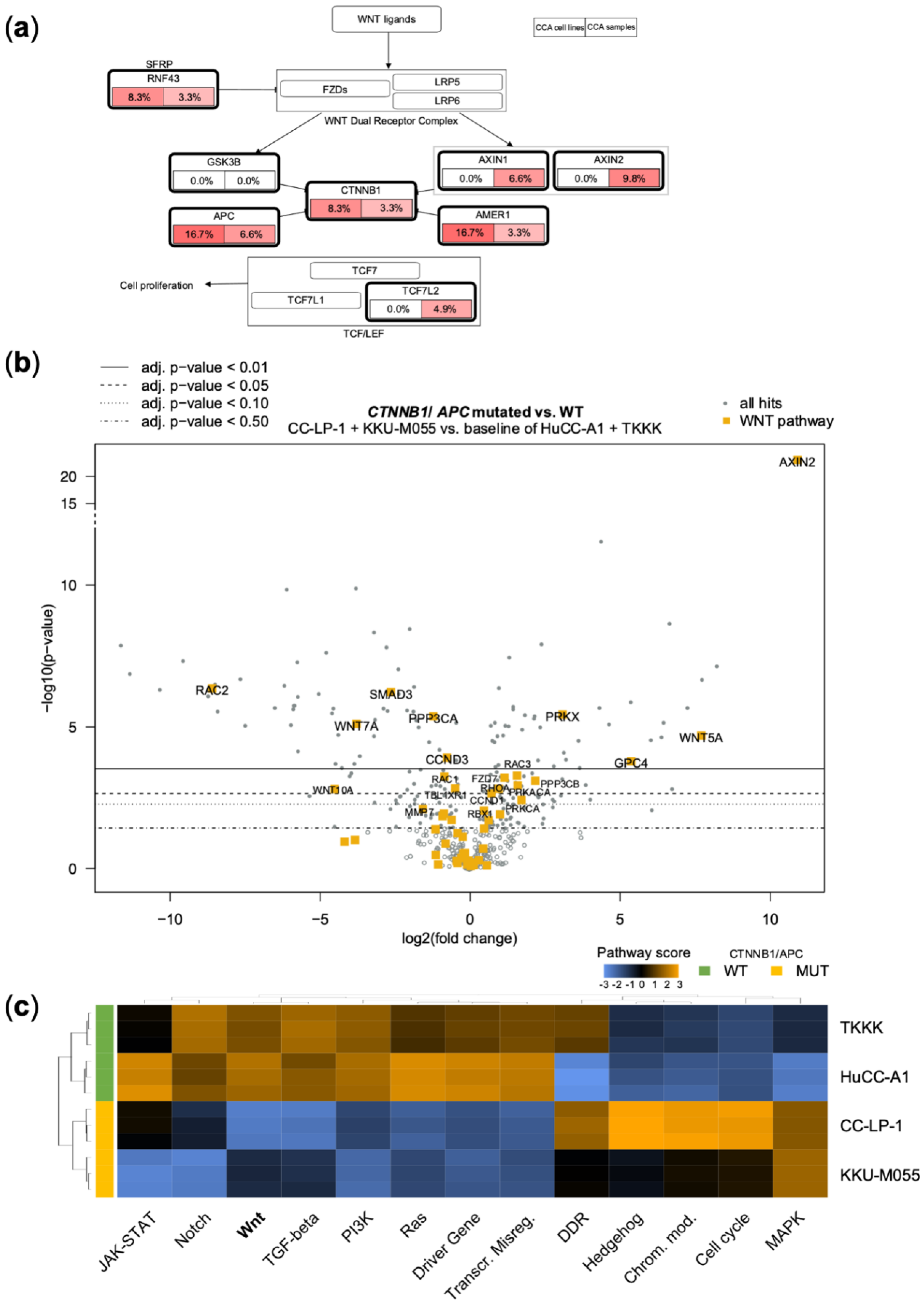
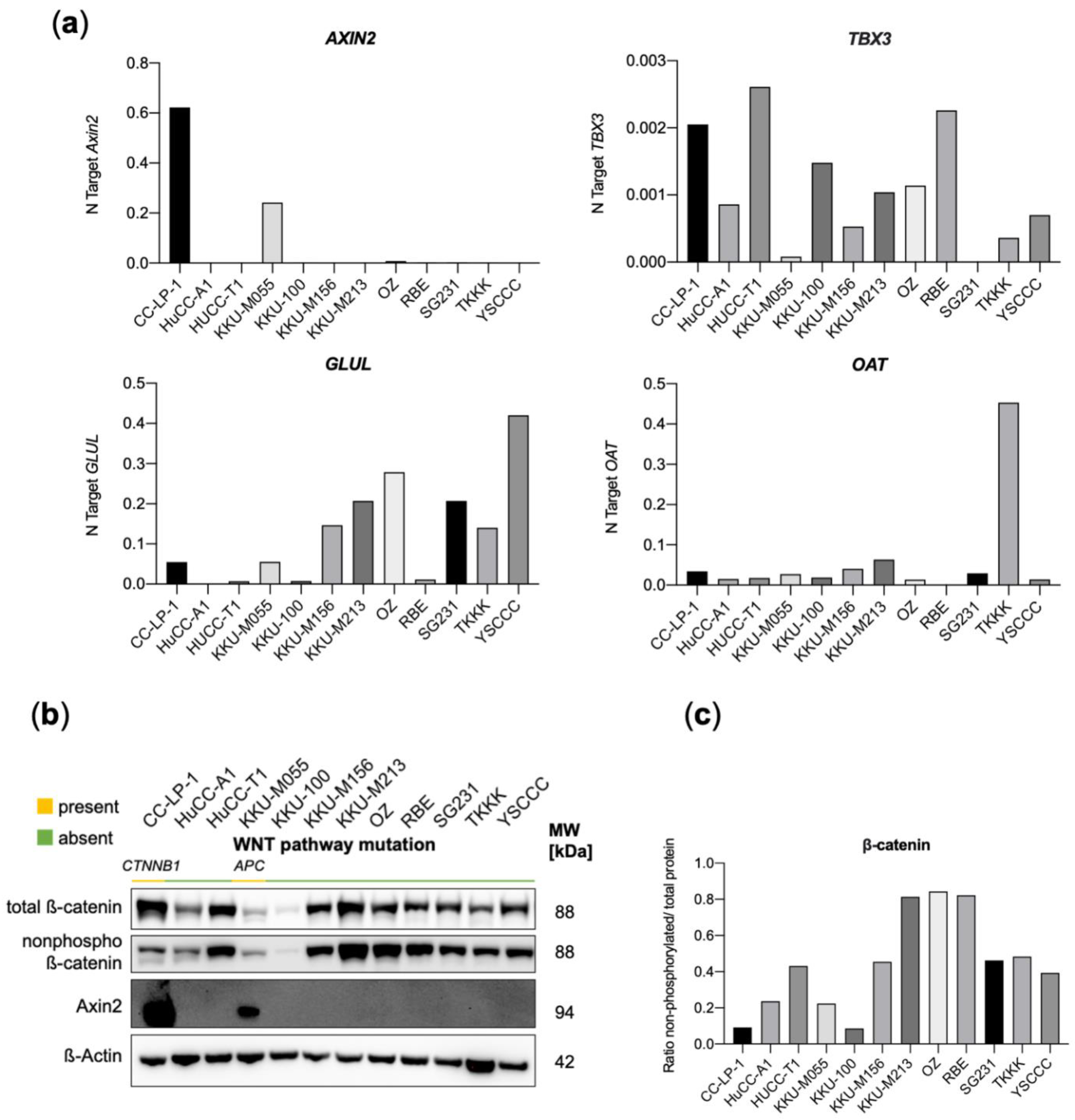
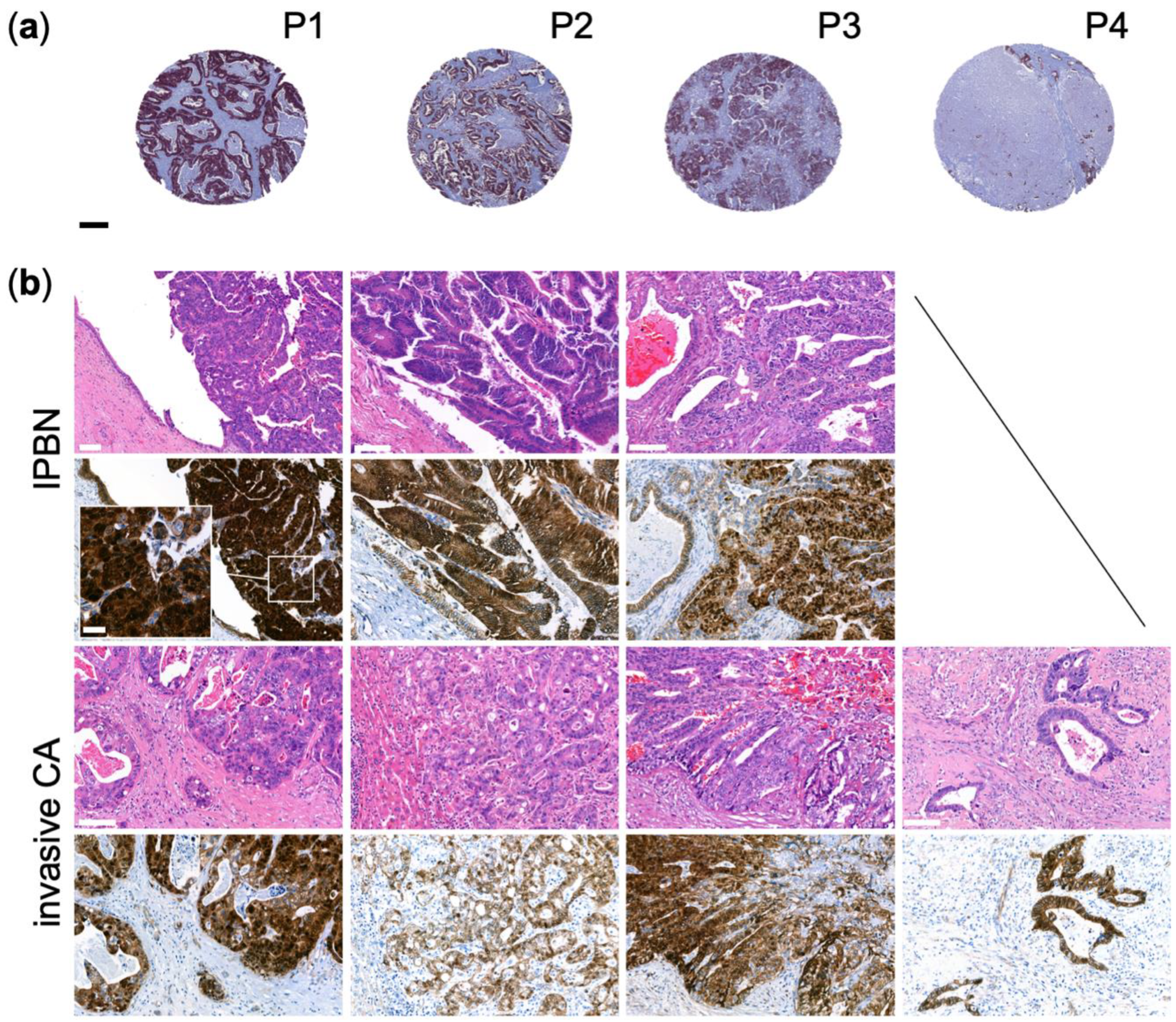
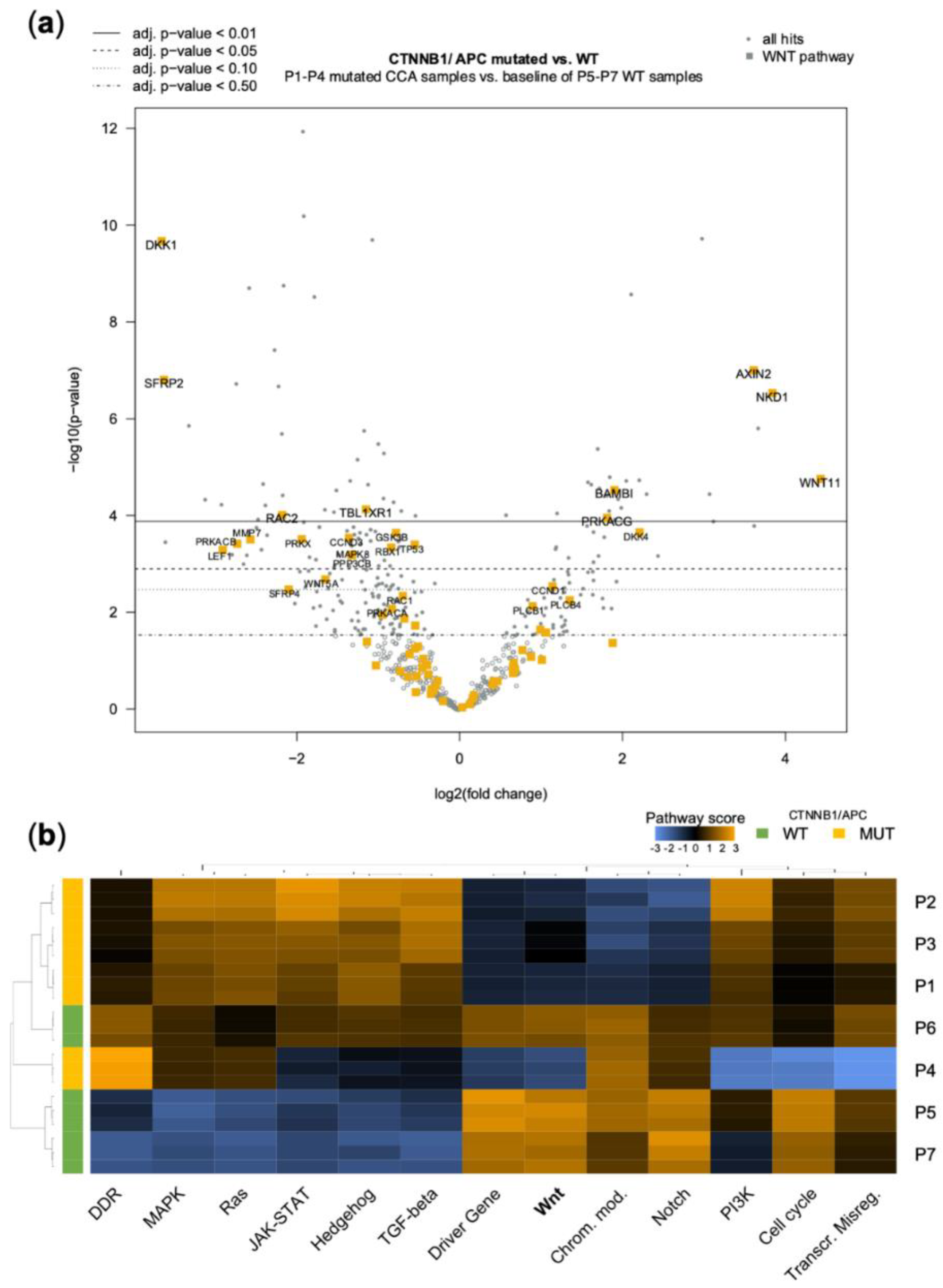
3.3. Genetic Alterations in the WNT-Pathway Are Rare in CCA but Define a Unique Type of Cholangiocarcinogenesis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rizvi, S.; Khan, S.A.; Hallemeier, C.L.; Kelley, R.K.; Gores, G.J. Cholangiocarcinoma—Evolving concepts and therapeutic strategies. Nat. Rev. Clin. Oncol. 2018, 15, 95–111. [Google Scholar] [CrossRef] [Green Version]
- Fan, B.; Malato, Y.; Calvisi, D.F.; Naqvi, S.; Razumilava, N.; Ribback, S.; Gores, G.J.; Dombrowski, F.; Evert, M.; Chen, X.; et al. Cholangiocarcinomas can originate from hepatocytes in mice. J. Clin. Investig. 2012, 122, 2911–2915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izquierdo-Sanchez, L.; Lamarca, A.; La Casta, A.; Buettner, S.; Utpatel, K.; Klümpen, H.-J.; Adeva, J.; Vogel, A.; Lleo, A.; Fabris, L.; et al. Cholangiocarcinoma landscape in Europe: Diagnostic, prognostic and therapeutic insights from the ENSCCA Registry. J. Hepatol. 2022, 76, 1109–1121. [Google Scholar] [CrossRef] [PubMed]
- Abou-Alfa, G.K.; Sahai, V.; Hollebecque, A.; Vaccaro, G.; Melisi, D.; Al-Rajabi, R.; Paulson, A.S.; Borad, M.J.; Gallinson, D.; Murphy, A.G.; et al. Pemigatinib for previously treated, locally advanced or metastatic cholangiocarcinoma: A multicentre, open-label, phase 2 study. Lancet Oncol. 2020, 21, 671–684. [Google Scholar] [CrossRef]
- Hoy, S.M. Pemigatinib: First Approval. Drugs 2020, 80, 923–929. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.X.; Macarulla, T.; Javle, M.M.; Kelley, R.K.; Lubner, S.J.; Adeva, J.; Cleary, J.M.; Catenacci, D.V.T.; Borad, M.J.; Bridgewater, J.A.; et al. Final Overall Survival Efficacy Results of Ivosidenib for Patients With Advanced Cholangiocarcinoma with IDH1 Mutation: The Phase 3 Randomized Clinical ClarIDHy Trial. JAMA Oncol. 2021, 7, 1669–1677. [Google Scholar] [CrossRef] [PubMed]
- Valerie, K.; Povirk, L.F. Regulation and mechanisms of mammalian double-strand break repair. Oncogene 2003, 22, 5792–5812. [Google Scholar] [CrossRef] [Green Version]
- Hartlerode, A.J.; Scully, R. Mechanisms of double-strand break repair in somatic mammalian cells. Biochem. J. 2009, 423, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Heeke, A.L.; Pishvaian, M.J.; Lynce, F.; Xiu, J.; Brody, J.R.; Chen, W.-J.; Baker, T.M.; Marshall, J.L.; Isaacs, C. Prevalence of Homologous Recombination-Related Gene Mutations across Multiple Cancer Types. JCO Precis. Oncol. 2018, 2018, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kan, C.; Zhang, J. BRCA1 Mutation: A Predictive Marker for Radiation Therapy? Int. J. Radiat. Oncol. Biol. Phys. 2015, 93, 281–293. [Google Scholar] [CrossRef] [Green Version]
- Byrski, T.; Huzarski, T.; Dent, R.; Marczyk, E.; Jasiowka, M.; Gronwald, J.; Jakubowicz, J.; Cybulski, C.; Wisniowski, R.; Godlewski, D.; et al. Pathologic complete response to neoadjuvant cisplatin in BRCA1-positive breast cancer patients. Breast Cancer Res. Treat. 2014, 147, 401–405. [Google Scholar] [CrossRef] [PubMed]
- Kaelin, W.G. The Concept of Synthetic Lethality in the Context of Anticancer Therapy. Nat. Rev. Cancer 2005, 5, 689–698. [Google Scholar] [CrossRef] [PubMed]
- Ahn, D.H.; Bekaii-Saab, T. Biliary tract cancer and genomic alterations in homologous recombinant deficiency: Exploiting synthetic lethality with PARP inhibitors. Chinese Clin. Oncol. 2020, 9, 6. [Google Scholar] [CrossRef] [PubMed]
- De Bono, J.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Olaparib for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020, 382, 2091–2102. [Google Scholar] [CrossRef] [PubMed]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.-O.; Hochhauser, D.; Arnold, D.; Oh, D.-Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef]
- Moore, K.; Colombo, N.; Scambia, G.; Kim, B.-G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; Sonke, G.S.; et al. Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2018, 379, 2495–2505. [Google Scholar] [CrossRef]
- Robson, M.E.; Tung, N.; Conte, P.; Im, S.-A.; Senkus, E.; Xu, B.; Masuda, N.; Delaloge, S.; Li, W.; Armstrong, A.; et al. OlympiAD final overall survival and tolerability results: Olaparib versus chemotherapy treatment of physician’s choice in patients with a germline BRCA mutation and HER2-negative metastatic breast cancer. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2019, 30, 558–566. [Google Scholar] [CrossRef]
- Wagle, N.; Grabiner, B.C.; Van Allen, E.M.; Hodis, E.; Jacobus, S.; Supko, J.G.; Stewart, M.; Choueiri, T.K.; Gandhi, L.; Cleary, J.M.; et al. Activating mTOR mutations in a patient with an extraordinary response on a phase I trial of everolimus and pazopanib. Cancer Discov. 2014, 4, 546–553. [Google Scholar] [CrossRef] [Green Version]
- Chae, H.; Kim, D.; Yoo, C.; Kim, K.-P.; Jeong, J.H.; Chang, H.-M.; Lee, S.S.; Park, D.H.; Song, T.J.; Hwang, S.; et al. Therapeutic relevance of targeted sequencing in management of patients with advanced biliary tract cancer: DNA damage repair gene mutations as a predictive biomarker. Eur. J. Cancer 2019, 120, 31–39. [Google Scholar] [CrossRef]
- Xiong, F.; Gong, J.; Wang, Q. Olaparib and Pembrolizumab Treatment for BRCA1-Mutated and PD-L1-Positive Intrahepatic Cholangiocarcinoma Recurrence and Metastasis: A Case Report. OncoTargets Ther. 2020, 13, 6385–6391. [Google Scholar] [CrossRef]
- Cheng, Y.; Zhang, J.; Qin, S.K.; Hua, H.Q. Treatment with olaparib monotherapy for BRCA2-mutated refractory intrahepatic cholangiocarcinoma: A case report. OncoTargets Ther. 2018, 11, 5957–5962. [Google Scholar] [CrossRef]
- Hoppe, M.M.; Sundar, R.; Tan, D.S.P.; Jeyasekharan, A.D. Biomarkers for Homologous Recombination Deficiency in Cancer. J. Natl. Cancer Inst. 2018, 110, 704–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, K.N.; Secord, A.A.; Geller, M.A.; Miller, D.S.; Cloven, N.; Fleming, G.F.; Wahner Hendrickson, A.E.; Azodi, M.; DiSilvestro, P.; Oza, A.M.; et al. Niraparib monotherapy for late-line treatment of ovarian cancer (QUADRA): A multicentre, open-label, single-arm, phase 2 trial. Lancet Oncol. 2019, 20, 636–648. [Google Scholar] [CrossRef]
- Lau, D.K.; Mouradov, D.; Wasenang, W.; Luk, I.Y.; Scott, C.M.; Williams, D.S.; Yeung, Y.H.; Limpaiboon, T.; Iatropoulos, G.F.; Jenkins, L.J.; et al. Genomic Profiling of Biliary Tract Cancer Cell Lines Reveals Molecular Subtypes and Actionable Drug Targets. iScience 2019, 21, 624–637. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.W.; Lee, K.-H.; Kim, J.-W.; Suh, K.J.; Nam, A.-R.; Bang, J.-H.; Bang, Y.-J.; Oh, D.-Y. Enhanced antitumor effect of binimetinib in combination with capecitabine for biliary tract cancer patients with mutations in the RAS/RAF/MEK/ERK pathway: Phase Ib study. Br. J. Cancer 2019, 121, 332–339. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.-S.; Park, J.-I. Wnt signaling in cancer: Therapeutic targeting of Wnt signaling beyond β-catenin and the destruction complex. Exp. Mol. Med. 2020, 52, 183–191. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.-F.; Qiu, L.; Yang, S.-L.; Wu, J.-C.; Liu, T.-J. Wnt/β-catenin signaling as an emerging potential key pharmacological target in cholangiocarcinoma. Biosci. Rep. 2020, 40. [Google Scholar] [CrossRef] [Green Version]
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Wang, W.; Smits, R.; Hao, H.; He, C. Wnt/β-Catenin Signaling in Liver Cancers. Cancers 2019, 11, 926. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Xu, H.; Cui, G.; Liang, B.; Chen, X.; Ko, S.; Affo, S.; Song, X.; Liao, Y.; Feng, J.; et al. β-Catenin sustains and is required for YES-associated protein oncogenic activity in cholangiocarcinoma. Gastroenterology 2022, 163, 481–494. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Weigt, M.; Gao, Q.; Ban, H.; He, H.; Mastrobuoni, G.; Kempa, S.; Chen, W.; Li, F. Rbm10 facilitates heterochromatin assembly via the Clr6 HDAC complex. Epigenet. Chromatin 2021, 14, 8. [Google Scholar] [CrossRef]
- Wanior, M.; Krämer, A.; Knapp, S.; Joerger, A.C. Exploiting vulnerabilities of SWI/SNF chromatin remodelling complexes for cancer therapy. Oncogene 2021, 40, 3637–3654. [Google Scholar] [CrossRef]
- Papillon-Cavanagh, S.; Lu, C.; Gayden, T.; Mikael, L.G.; Bechet, D.; Karamboulas, C.; Ailles, L.; Karamchandani, J.; Marchione, D.M.; Garcia, B.A.; et al. Impaired H3K36 methylation defines a subset of head and neck squamous cell carcinomas. Nat. Genet. 2017, 49, 180–185. [Google Scholar] [CrossRef] [Green Version]
- Russo, M.; Siravegna, G.; Blaszkowsky, L.S.; Corti, G.; Crisafulli, G.; Ahronian, L.G.; Mussolin, B.; Kwak, E.L.; Buscarino, M.; Lazzari, L.; et al. Tumor Heterogeneity and Lesion-Specific Response to Targeted Therapy in Colorectal Cancer. Cancer Discov. 2016, 6, 147–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, D.S.; van Halteren, A.; Quispel, W.T.; van den Bos, C.; Bovée, J.V.M.G.; Patel, B.; Badalian-Very, G.; van Hummelen, P.; Ducar, M.; Lin, L.; et al. MAP2K1 and MAP3K1 mutations in Langerhans cell histiocytosis. Genes. Chromosomes Cancer 2015, 54, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, H.; Peters, M.; Ding, N.; Ribback, S.; Utpatel, K.; Cigliano, A.; Dombrowski, F.; Xu, M.; Chen, X.; et al. Loss of Fbxw7 synergizes with activated Akt signaling to promote c-Myc dependent cholangiocarcinogenesis. J. Hepatol. 2019, 71, 742–752. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Dong, M.; Xu, Z.; Song, X.; Zhang, S.; Qiao, Y.; Che, L.; Gordan, J.; Hu, K.; Liu, Y.; et al. Notch2 controls hepatocyte-derived cholangiocarcinoma formation in mice. Oncogene 2018, 37, 3229–3242. [Google Scholar] [CrossRef]
- Cigliano, A.; Wang, J.; Chen, X.; Calvisi, D.F. Role of the Notch signaling in cholangiocarcinoma. Expert Opin. Ther. Targets 2017, 21, 471–483. [Google Scholar] [CrossRef] [PubMed]
- Krystel-Whittemore, M.; Taylor, M.S.; Rivera, M.; Lennerz, J.K.; Le, L.P.; Dias-Santagata, D.; Iafrate, A.J.; Deshpande, V.; Chebib, I.; Nielsen, G.P.; et al. Novel and established EWSR1 gene fusions and associations identified by next-generation sequencing and fluorescence in-situ hybridization. Hum. Pathol. 2019, 93, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Crispo, F.; Pietrafesa, M.; Condelli, V.; Maddalena, F.; Bruno, G.; Piscazzi, A.; Sgambato, A.; Esposito, F.; Landriscina, M. IDH1 Targeting as a New Potential Option for Intrahepatic Cholangiocarcinoma Treatment-Current State and Future Perspectives. Molecules 2020, 25, 3754. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Hao, H.X.; Growney, J.D.; Woolfenden, S.; Bottiglio, C.; Ng, N.; Cong, F. Inactivating mutations of RNF43 confer Wnt dependency in pancreatic ductal adenocarcinoma. Proc. Natl. Acad. Sci. USA 2013, 110, 12649–12654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eek-hoon, J.; Tong, Z.; Claire, D.; Choun-Ki, J.; Jean-Noel, F.; Frank, C. Wnt/β-Catenin/Tcf Signaling Induces the Transcription of Axin2, a Negative Regulator of the Signaling Pathway. Mol. Cell. Biol. 2002, 22, 1172–1183. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Wang, C.-Y. TBL1-TBLR1 and beta-catenin recruit each other to Wnt target-gene promoter for transcription activation and oncogenesis. Nat. Cell Biol. 2008, 10, 160–169. [Google Scholar] [CrossRef]
- Luo, K. Signaling Cross Talk between TGF-β/Smad and Other Signaling Pathways. Cold Spring Harb. Perspect. Biol. 2017, 9, a022137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujikura, K.; Akita, M.; Ajiki, T.; Fukumoto, T.; Itoh, T.; Zen, Y. Recurrent Mutations in APC and CTNNB1 and Activated Wnt/β-catenin Signaling in Intraductal Papillary Neoplasms of the Bile Duct: A Whole Exome Sequencing Study. Am. J. Surg. Pathol. 2018, 42, 1674–1685. [Google Scholar] [CrossRef] [PubMed]
- Chu, H.Y.; Chen, Z.; Wang, L.; Zhang, Z.-K.; Tan, X.; Liu, S.; Zhang, B.-T.; Lu, A.; Yu, Y.; Zhang, G. Dickkopf-1: A Promising Target for Cancer Immunotherapy. Front. Immunol. 2021, 12, 1850. [Google Scholar] [CrossRef]
- Lin, Z.; Gao, C.; Ning, Y.; He, X.; Wu, W.; Chen, Y.-G. The pseudoreceptor BMP and activin membrane-bound inhibitor positively modulates Wnt/beta-catenin signaling. J. Biol. Chem. 2008, 283, 33053–33058. [Google Scholar] [CrossRef] [Green Version]
- Peraldo-Neia, C.; Cavalloni, G.; Fenocchio, E.; Cagnazzo, C.; Gammaitoni, L.; Cereda, S.; Nasti, G.; Satolli, M.A.; Aprile, G.; Reni, M.; et al. Prognostic and predictive role of EGFR pathway alterations in biliary cancer patients treated with chemotherapy and anti-EGFR. PLoS ONE 2018, 13, e0191593. [Google Scholar] [CrossRef] [Green Version]
- Diamond, E.L.; Durham, B.H.; Ulaner, G.A.; Drill, E.; Buthorn, J.; Ki, M.; Bitner, L.; Cho, H.; Young, R.J.; Francis, J.H.; et al. Efficacy of MEK inhibition in patients with histiocytic neoplasms. Nature 2019, 567, 521–524. [Google Scholar] [CrossRef] [PubMed]
- Fung, A.S.; Graham, D.M.; Chen, E.X.; Stockley, T.L.; Zhang, T.; Le, L.W.; Albaba, H.; Pisters, K.M.; Bradbury, P.A.; Trinkaus, M.; et al. A phase I study of binimetinib (MEK 162), a MEK inhibitor, plus carboplatin and pemetrexed chemotherapy in non-squamous non-small cell lung cancer. Lung Cancer 2021, 157, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Gönül Geyik, Ö.; Anichini, G.; Ulukaya, E.; Marra, F.; Raggi, C. DNA Damage Response Inhibitors in Cholangiocarcinoma: Current Progress and Perspectives. Cells 2022, 11, 1463. [Google Scholar] [CrossRef] [PubMed]
- Sugimachi, K.; Taguchi, K.; Aishima, S.; Tanaka, S.; Shimada, M.; Kajiyama, K.; Sugimachi, K.; Tsuneyoshi, M. Altered Expression of β-Catenin without Genetic Mutation in Intrahepatic Cholangiocarcinoma. Mod. Pathol. 2001, 14, 900–905. [Google Scholar] [CrossRef] [Green Version]
- Schlitter, A.M.; Born, D.; Bettstetter, M.; Specht, K.; Kim-Fuchs, C.; Riener, M.-O.; Jeliazkova, P.; Sipos, B.; Siveke, J.T.; Terris, B.; et al. Intraductal papillary neoplasms of the bile duct: Stepwise progression to carcinoma involves common molecular pathways. Mod. Pathol. 2014, 27, 73–86. [Google Scholar] [CrossRef] [Green Version]
- Boulter, L.; Guest, R.V.; Kendall, T.J.; Wilson, D.H.; Wojtacha, D.; Robson, A.J.; Ridgway, R.A.; Samuel, K.; Van Rooijen, N.; Barry, S.T.; et al. WNT signaling drives cholangiocarcinoma growth and can be pharmacologically inhibited. J. Clin. Investig. 2015, 125, 1269–1285. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antigen [Clone if Monoclonal Antibody] | Dilution | Company | Catalog |
|---|---|---|---|
| Axin2 [H-98] | 1/1000 | Santa Cruz Biotechnology, Inc. | sc-14029 |
| c-Met [EPR19067] | 1/1000 | Abcam plc. | ab216574 |
| EGFR [EP38Y] | 1/1000 | Abcam plc. | ab52894 |
| HER2/ErbB2 | 1/1000 | Cell Signaling Technology, Inc. | 2242 |
| mTOR | 1/1000 | Merck Millipore | 04-385 |
| Non-phospho (Active) β-Catenin (Ser45) [D2U8Y] | 1/1000 | Cell Signaling Technology, Inc. | 19807 |
| Phospho-Akt (S473) | 1/1000 | Cell Signaling Technology, Inc. | 4060 |
| Phospho-mTOR (S2448) [49F9] | 1/1000 | Cell Signaling Technology, Inc. | 2976 |
| Phospho-44/42 MAPK (Erk1/2) [137F5] | 1/1000 | Cell Signaling Technology, Inc. | 4695 |
| Phospho-4EBP1 [53H11] | 1/1000 | Cell Signaling Technology, Inc. | 9644S |
| Pan-AKT [C67E7] | 1/1000 | Cell Signaling Technology, Inc. | 4691 |
| Phospho-EPHA2 | 1/1000 | Santa Cruz Biotechnology, Inc. | sc-924 |
| Phospho-GSK3ß (S9) [D85E12] | 1/1000 | Cell Signaling Technology, Inc. | 5558 |
| Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) [D13.14.4E] | 1/1000 | Cell Signaling Technology, Inc. | 4370 |
| Phospho-RPS6 (S235/236) [2F9] | 1/1000 | Cell Signaling Technology, Inc. | 4856 |
| Rictor | 1/1000 | Bethyl Laboratories | A300-459A |
| ß-catenin (used for IHC) | 1/100 | BD Biosciences | 610154 |
| ß-catenin (used for IHC) | 1/100 | Cell Signaling Technology, Inc. | 9587 |
| ß-catenin (used for IHC) | 1/100 | Roche | 760-4242 |
| β-Actin [8H10D10] | 1/1000 | Cell Signaling Technology, Inc. | 3700 |
| HRP, Goat Anti-Mouse IgG (secondary antibody) | 1/20000 | Abbkine Scientific Co., Ltd. | A21010 |
| HRP, Goat Anti-Rabbit IgG (secondary antibody) | 1/20000 | Abbkine Scientific Co. | A21020 |
| Gene | Assay ID | Fluorescent Dye | Manufacturer |
|---|---|---|---|
| Axin2 | Hs00610344_m1 | FAM | Thermo Fisher Scientific |
| TBX3 | Hs00195612_m1 | FAM | Thermo Fisher Scientific |
| GLUL | Hs00365928_g1 | FAM | Thermo Fisher Scientific |
| OAT | Hs_00236852_m1 | FAM | Thermo Fisher Scientific |
| GAPDH | Hs_02786624_g1 | VIC | Thermo Fisher Scientific |
| OZ | RBE | TKKK | YSCCC | HuCC-A1 | HuCC-T1 | KKU-M055 | KKU-100 | KKU-M156 | KKU-M213 | CC-LP-1 | SG231 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| GScore | 2.7 | 17 | 90.6 | 40.3 | 7.7 | 85.7 | 57.4 | 31.7 | 84.2 | 78 | 9.2 | 47.3 |
| TMB [mutations/MB] | 2.5 | 2.4 | 5.5 | 34.6 | 9.7 | 6.3 | 6.3 | 3.1 | 7.8 | 46.1 | 0.9 | 6.3 |
| Percent unstable MSI Sites | 5.5 | 3.61 | 0.91 | 4.44 | 0 | 6.41 | 0 | 0.91 | 7.08 | 4.35 | 8.33 | 1.69 |
| CNV >2 fold change | none | none | EGFR (2.535) ERBB2 (10.840) | EGFR (13.822) | RICTOR (3.168) FGF10 (2.585) EGFR (2.310) | none | none | ERCC2 (2.133) AR (2.197) | FGF5 (5.452) RICTOR (3.926) ATM (2.617) | NRAS (2.001) FGF5 (3.158) | RICTOR (2.054) | MYC (3.694) |
| Fusions | MET-CAV1 | EWSR1-ADRBK2 | ||||||||||
| Mutations (pathogenic) | KRAS (p.Q61L) GNAS (p.R201H) | IDH1 (p.R132S) KRAS (p.G12V) | TP53 (p.Q104*) | RAD51D (p.K111fs*13) PIK3CA (p.E545K) TP53 (p.V272M) TERT (c.-124C>T) | KRAS (p.G12D) TP53 (p.R175H) | SMAD4 (p.R361H) | KRAS (p.G12D) | KRAS (p.G13C) SMAD4 (p.R361C) | KRAS (p.G13C) SMAD4 (p.R361C) | TP53 (p.R306*) CTNNB1 (p.G34V) | KRAS (p.G12F) TP53 (p.T125P) ARID2 (p.Q1041*) EPHA3 (p.C202*) | |
| MET (p.L130L) | ||||||||||||
| Mutations (likely pathogenic) | ARID2 (p.K569fs*12) | IDH1 (p.R100Q) KMT2A (p.S82*) SMARCA4 (p.E457*) TSC1 (c.107-1G>A) | DNMT3A (p.R143*) TP53 (p.G154V) ARID1A (p.S1138*) | TERT (c.57A>C) FBXW7 (p.S294*) SETD2 (p.Q2285*) | ARID1A (p.P946fs*20) MAP2K1 (p.K57T) APC (p.D1636fs*2) RPS6KA4 (p.Q129*) | SMARCA4 (p.K1412*) SMAD4 (c.1140-1G>T) SPEN (p.Q743*) | RBM10 (p.E670*) | AR (p.L627Q) HRAS (p.V29V) NCOA3 (c.2513-1G>T) NSD1 (c.3115-1G>T) RBM10 (p.E670*) | SMARCA4 (p.E590*) |
| RBE | YSCCC | HuCC-A1 | HuCC-T1 | KKU-M055 | KKU-100 | KKU-M156 | KKU-M213 | CC-LP-1 | SG231 | |
|---|---|---|---|---|---|---|---|---|---|---|
| Mirdametinib | 9.49 × 105 | 2.34 × 104 | 1.21 × 104 | 9.27 × 105 | 7.71 × 105 | 1.76 × 104 | 7.13 × 105 | 2.83 × 104 | 1.31 × 104 | 1.28 × 104 |
| Trametinib | 4.68 × 105 | 6.99 × 105 | 8.38 × 105 | 4.42 × 105 | 2.00 × 105 | 7.26 × 105 | 3.85 × 105 | 3.88 × 104 | 2.27 × 105 | 4.00 × 105 |
| Foretinib | 2.72 × 106 | 2.37 × 106 | 6.84 × 106 | ND | 3.97 × 105 | 1.59 × 105 | ND | 2.77 × 106 | 1.05 × 106 | 2.38 × 106 |
| Olaparib | 3.47 × 104 | 1.84 × 104 | 1.72 × 104 | 5.90 × 105 | 4.15 × 103 | 1.67 × 104 | 9.16 × 105 | 2.17 × 104 | 1.78 × 104 | 1.03 × 104 |
| Ivosidenib | 7.93 × 105 | 7.67 × 105 | 9.68 × 105 | 5.45 × 105 | 8.84 × 105 | 8.80 × 105 | 2.15 × 104 | 9.54 × 105 | 4.19 × 104 | 9.96 × 105 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scheiter, A.; Hierl, F.; Winkel, I.; Keil, F.; Klier-Richter, M.; Coulouarn, C.; Lüke, F.; Kandulski, A.; Evert, M.; Dietmaier, W.; et al. Wnt/β-Catenin-Pathway Alterations and Homologous Recombination Deficiency in Cholangiocarcinoma Cell Lines and Clinical Samples: Towards Specific Vulnerabilities. J. Pers. Med. 2022, 12, 1270. https://doi.org/10.3390/jpm12081270
Scheiter A, Hierl F, Winkel I, Keil F, Klier-Richter M, Coulouarn C, Lüke F, Kandulski A, Evert M, Dietmaier W, et al. Wnt/β-Catenin-Pathway Alterations and Homologous Recombination Deficiency in Cholangiocarcinoma Cell Lines and Clinical Samples: Towards Specific Vulnerabilities. Journal of Personalized Medicine. 2022; 12(8):1270. https://doi.org/10.3390/jpm12081270
Chicago/Turabian StyleScheiter, Alexander, Frederik Hierl, Ingrid Winkel, Felix Keil, Margit Klier-Richter, Cédric Coulouarn, Florian Lüke, Arne Kandulski, Matthias Evert, Wolfgang Dietmaier, and et al. 2022. "Wnt/β-Catenin-Pathway Alterations and Homologous Recombination Deficiency in Cholangiocarcinoma Cell Lines and Clinical Samples: Towards Specific Vulnerabilities" Journal of Personalized Medicine 12, no. 8: 1270. https://doi.org/10.3390/jpm12081270