
Identification of the Aldo-Keto Reductase Responsible for d-Galacturonic Acid Conversion to l-Galactonate in Saccharomyces cerevisiae



Abstract
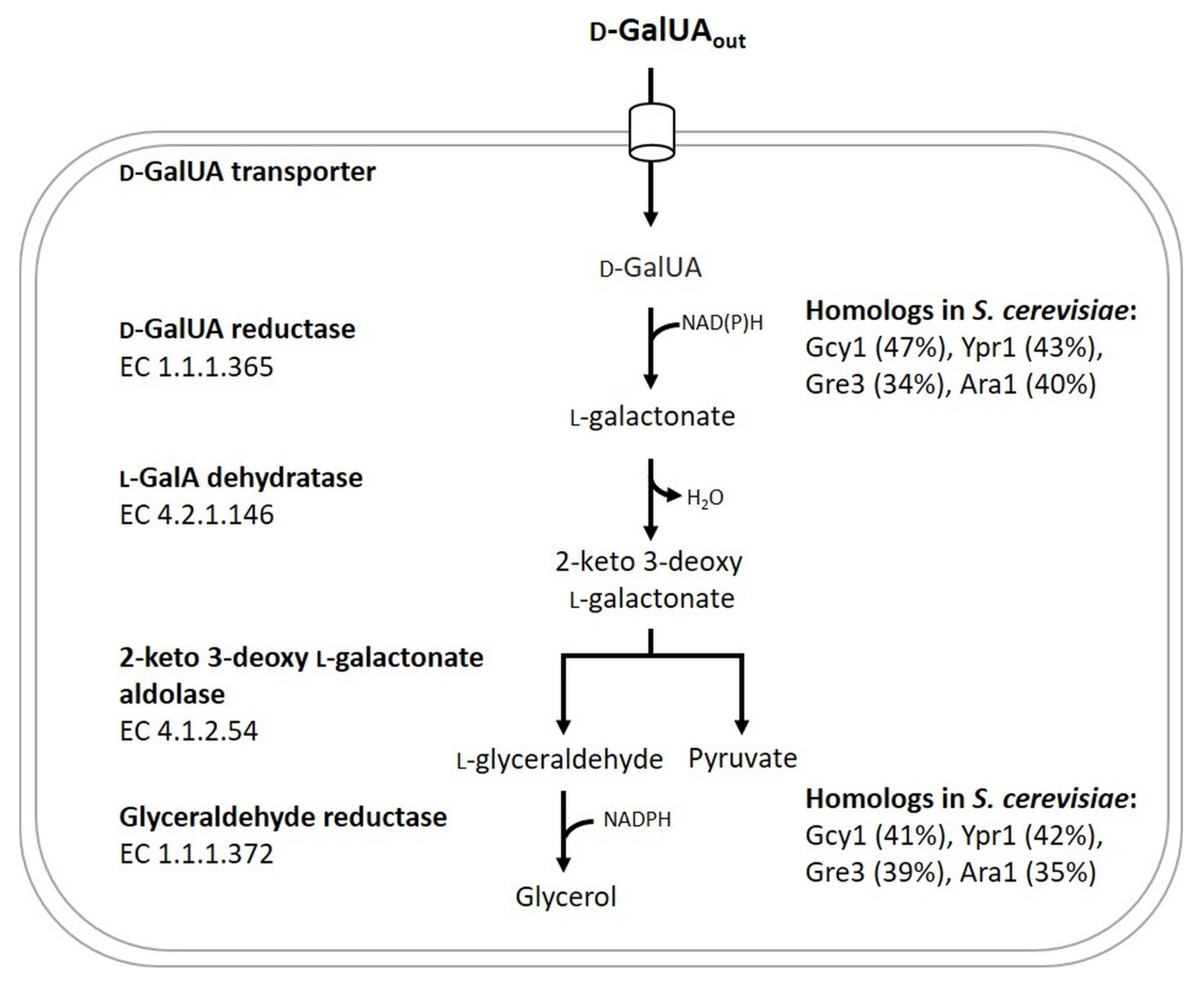
:1. Introduction
2. Materials & Methods
2.1. Strains, Medium Composition and General Cultivation Conditions
2.2. General Molecular Biology Techniques
2.3. Construction and Verification of Deletion Strains
2.4. Construction and Verification of the Reverse-Engineered Strain CEN.PK RE
2.5. Media for Analysing Glycerol and d-GalUA Utilization of Engineered S. cerevisiae Strains
2.6. Characterization of S. cerevisiae in Shake Flask Batch Cultivations
2.7. Metabolite Analysis by HPLC
2.8. Selectable Genetic Marker Replacement in Multicopy Plasmids for GCY1 Overexpression
2.9. In Vitro Measurement of d-GalUA Reductase Activity
3. Results
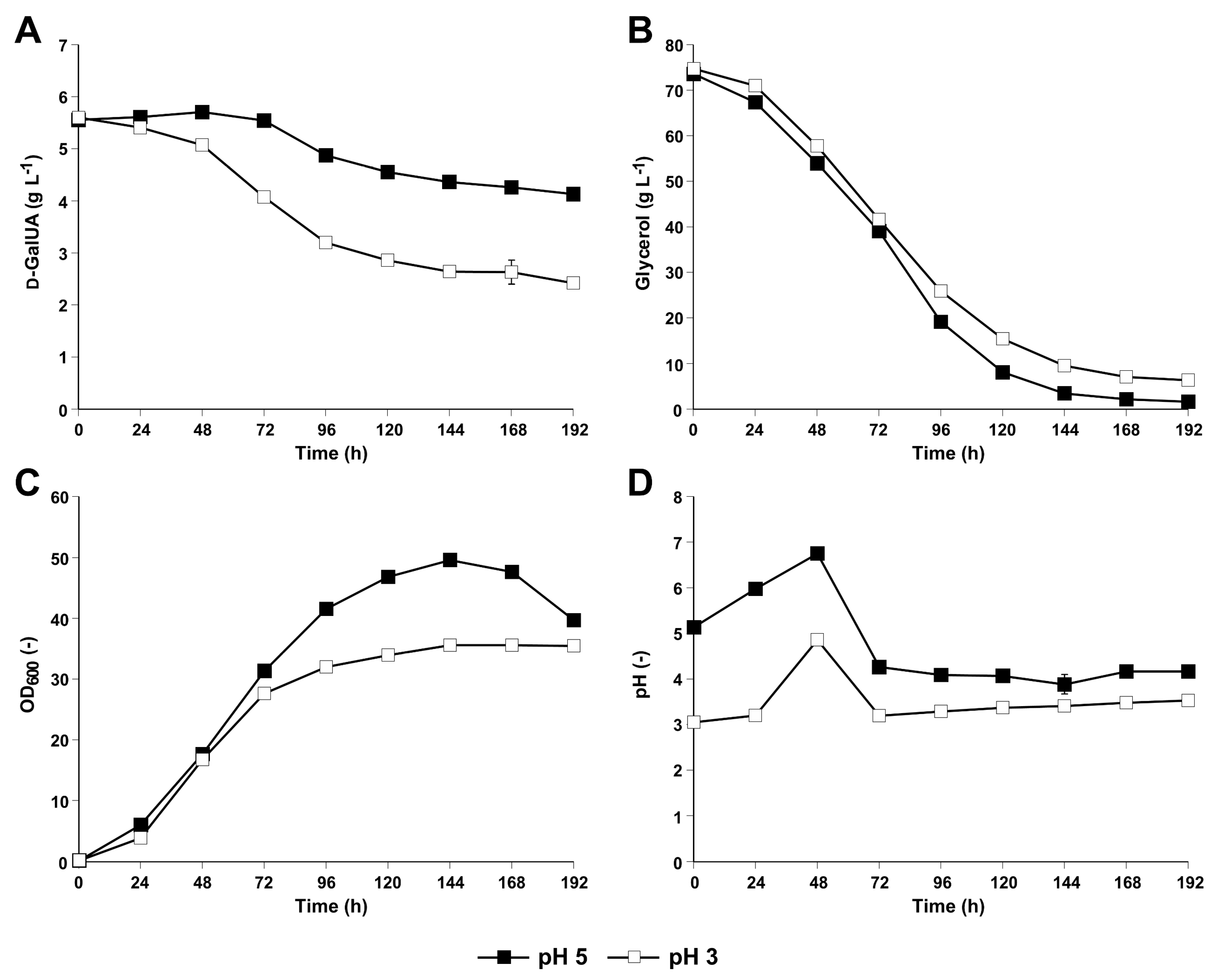
3.1. The S. cerevisiae Derivate CEN.PK113-1A Equipped with the NAD-Dependent DHA Pathway for Glycerol Utilization Was Able to Convert d-GalUA to l-GalA
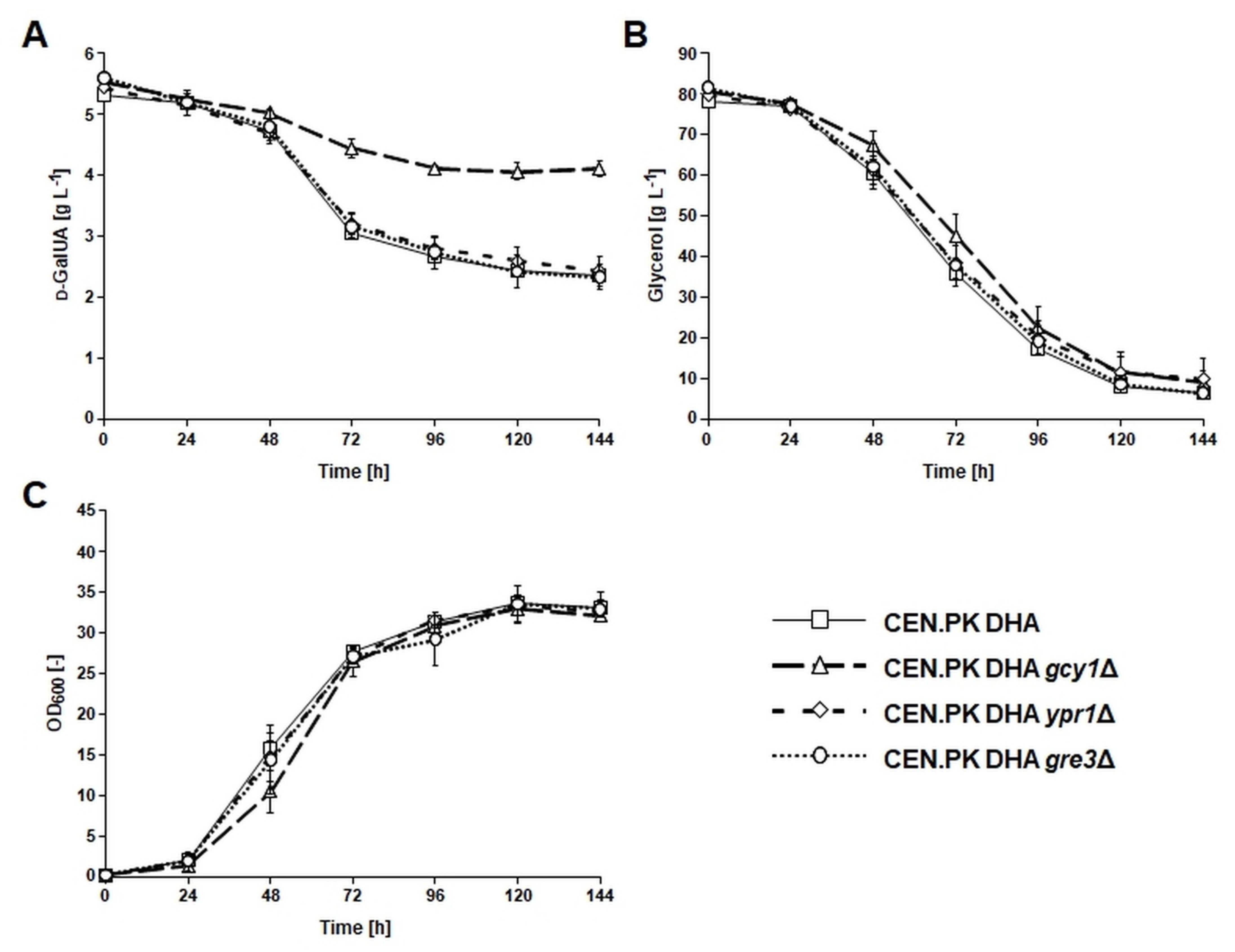
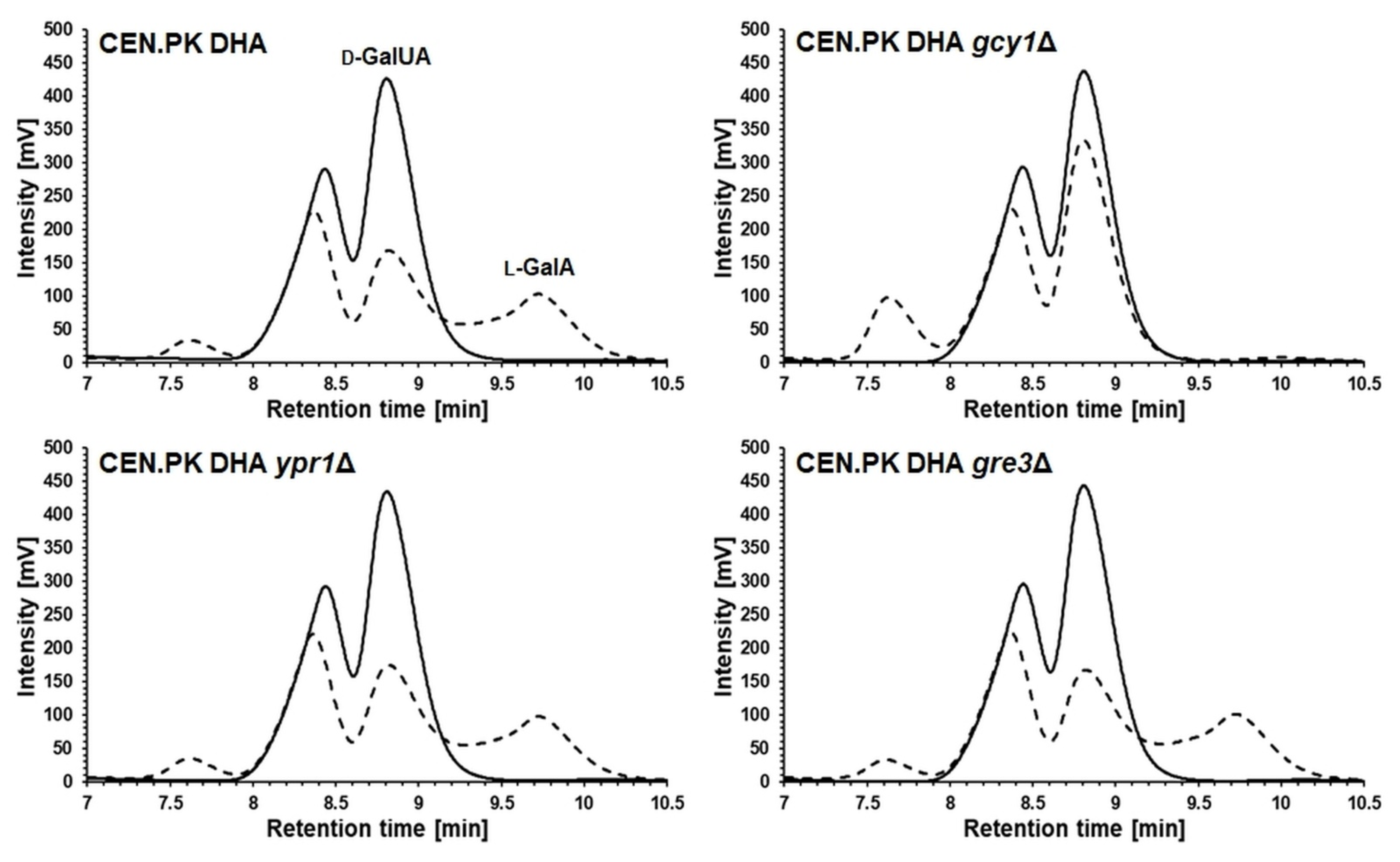
3.2. Deletion of GCY1 Abolished Formation of l-GalA from d-GalUA
3.3. Conversion of d-GalUA to l-GalA Was Not Dependent on the DHA Pathway for Glycerol Catabolism
3.4. Overexpression of GCY1 Resulted in Increased In Vitro Conversion of d-GalUA
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mohnen, D. Pectin structure and biosynthesis. Curr. Opin. Plant Biol. 2008, 11, 266–277. [Google Scholar] [CrossRef] [PubMed]
- Edwards, M.C.; Doran-Peterson, J. Pectin-rich biomass as feedstock for fuel ethanol production. Appl. Microbiol. Biotechnol. 2012, 95, 565–575. [Google Scholar] [CrossRef] [Green Version]
- Berlowska, J.; Binczarski, M.; Dziugan, P.; Wilkowska, A.; Kregiel, D.; Witonska, I. Sugar Beet Pulp as a Source of Valuable Biotechnological Products. In Advances in Biotechnology for Food Industry; Elsevier: Amsterdam, The Netherlands, 2018; Volume 14, pp. 359–392. ISBN 9780128114957. [Google Scholar]
- John, I.; Muthukumar, K.; Arunagiri, A. A review on the potential of citrus waste for D-Limonene, pectin, and bioethanol production. Int. J. Green Energy 2017, 14, 599–612. [Google Scholar] [CrossRef]
- Martins, L.C.; Monteiro, C.C.; Semedo, P.M.; Sá-Correia, I. Valorisation of pectin-rich agro-industrial residues by yeasts: Potential and challenges. Appl. Microbiol. Biotechnol. 2020, 104, 6527–6547. [Google Scholar] [CrossRef]
- Jeong, D.; Park, H.; Jang, B.-K.; Ju, Y.; Shin, M.H.; Oh, E.J.; Lee, E.J.; Kim, S.R. Recent advances in the biological valorization of citrus peel waste into fuels and chemicals. Bioresour. Technol. 2021, 323, 124603. [Google Scholar] [CrossRef]
- Kuivanen, J.; Biz, A.; Richard, P. Microbial hexuronate catabolism in biotechnology. AMB Express 2019, 9, 16. [Google Scholar] [CrossRef]
- Chroumpi, T.; Mäkelä, M.R.; de Vries, R.P. Engineering of primary carbon metabolism in filamentous fungi. Biotechnol. Adv. 2020, 43, 107551. [Google Scholar] [CrossRef] [PubMed]
- Hilditch, S.; Bergha, S.; Kalkkinen, N.; Penttila, M.; Richard, P. The Missing Link in the Fungal D-Galacturonate Pathway. J. Biol. Chem. 2007, 282, 26195–26201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richard, P.; Hilditch, S. d-Galacturonic acid catabolism in microorganisms and its biotechnological relevance. Appl. Microbiol. Biotechnol. 2009, 82, 597–604. [Google Scholar] [CrossRef]
- Biz, A.; Sugai-Guérios, M.H.; Kuivanen, J.; Maaheimo, H.; Krieger, N.; Mitchell, D.A.; Richard, P. The introduction of the fungal d-galacturonate pathway enables the consumption of d-galacturonic acid by Saccharomyces cerevisiae. Microb. Cell Fact. 2016, 15, 144. [Google Scholar] [CrossRef] [Green Version]
- Palma, M.; Guerreiro, J.F.; Sá-Correia, I. Adaptive Response and Tolerance to Acetic Acid in Saccharomyces cerevisiae and Zygosaccharomyces bailii: A Physiological Genomics Perspective. Front. Microbiol. 2018, 9, 1–18. [Google Scholar] [CrossRef]
- Martins, L.C.; Palma, M.; Angelov, A.; Nevoigt, E.; Liebl, W.; Sá-Correia, I. Complete utilization of the major carbon sources present in sugar beet pulp hydrolysates by the oleaginous red yeasts Rhodotorula toruloides and R. mucilaginosa. J. Fungi 2021, 7, 215. [Google Scholar] [CrossRef]
- Huisjes, E.H.; De Hulster, E.; Van Dam, J.C.; Pronk, J.T.; Maris, A.J.A. Van Galacturonic Acid Inhibits the Growth of Saccharomyces cerevisiae on Galactose, Xylose, and Arabinose. Appl. Environ. Microbiol. 2012, 78, 5052–5059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Maris, A.J.A.; Abbott, D.A.; Bellissimi, E.; van den Brink, J.; Kuyper, M.; Luttik, M.A.H.; Wisselink, H.W.; Scheffers, W.A.; van Dijken, J.P.; Pronk, J.T. Alcoholic fermentation of carbon sources in biomass hydrolysates by Saccharomyces cerevisiae: Current status. Antonie Leeuwenhoek 2006, 90, 391–418. [Google Scholar] [CrossRef] [PubMed]
- Rezić, T.; Oros, D.; Marković, I.; Kracher, D.; Ludwig, R.; Santek, B. Integrated hydrolyzation and fermentation of sugar beet pulp to bioethanol. J. Microbiol. Biotechnol. 2013, 23, 1244–1252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lian, J.; Mishra, S.; Zhao, H. Recent advances in metabolic engineering of Saccharomyces cerevisiae: New tools and their applications. Metab. Eng. 2018, 50, 85–108. [Google Scholar] [CrossRef] [PubMed]
- Fraczek, M.G.; Naseeb, S.; Delneri, D. History of genome editing in yeast. Yeast 2018, 35, 361–368. [Google Scholar] [CrossRef] [Green Version]
- Moysés, D.; Reis, V.; Almeida, J.; Moraes, L.; Torres, F. Xylose Fermentation by Saccharomyces cerevisiae: Challenges and Prospects. Int. J. Mol. Sci. 2016, 17, 207. [Google Scholar] [CrossRef] [Green Version]
- Cunha, J.T.; Romaní, A.; Costa, C.E.; Sá-Correia, I.; Domingues, L. Molecular and physiological basis of Saccharomyces cerevisiae tolerance to adverse lignocellulose-based process conditions. Appl. Microbiol. Biotechnol. 2019, 103, 159–175. [Google Scholar] [CrossRef] [Green Version]
- Gonçalves, D.L.; Matsushika, A.; de Sales, B.B.; Goshima, T.; Bon, E.P.S.; Stambuk, B.U. Xylose and xylose/glucose co-fermentation by recombinant Saccharomyces cerevisiae strains expressing individual hexose transporters. Enzyme Microb. Technol. 2014, 63, 13–20. [Google Scholar] [CrossRef]
- Krahulec, S.; Petschacher, B.; Wallner, M.; Longus, K.; Klimacek, M.; Nidetzky, B. Fermentation of mixed glucose-xylose substrates by engineered strains of Saccharomyces cerevisiae: Role of the coenzyme specificity of xylose reductase, and effect of glucose on xylose utilization. Microb. Cell Fact. 2010, 9, 16. [Google Scholar] [CrossRef] [Green Version]
- Zheng, D.Q.; Wu, X.C.; Tao, X.L.; Wang, P.M.; Li, P.; Chi, X.Q.; Li, Y.D.; Yan, Q.F.; Zhao, Y.H. Screening and construction of Saccharomyces cerevisiae strains with improved multi-tolerance and bioethanol fermentation performance. Bioresour. Technol. 2011, 102, 3020–3027. [Google Scholar] [CrossRef]
- Benjaphokee, S.; Hasegawa, D.; Yokota, D.; Asvarak, T.; Auesukaree, C.; Sugiyama, M.; Kaneko, Y.; Boonchird, C.; Harashima, S. Highly efficient bioethanol production by a Saccharomyces cerevisiae strain with multiple stress tolerance to high temperature, acid and ethanol. N. Biotechnol. 2012, 29, 379–386. [Google Scholar] [CrossRef]
- Geng, P.; Zhang, L.; Shi, G.Y. Omics analysis of acetic acid tolerance in Saccharomyces cerevisiae. World J. Microbiol. Biotechnol. 2017, 33, 94. [Google Scholar] [CrossRef] [PubMed]
- Narendranath, N.V.; Power, R. Relationship between pH and medium dissolved solids in terms of growth and metabolism of Lactobacilli and Saccharomyces cerevisiae during ethanol production. Appl. Environ. Microbiol. 2005, 71, 2239–2243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Protzko, R.J.; Latimer, L.N.; Martinho, Z.; de Reus, E.; Seibert, T.; Benz, J.P.; Dueber, J.E. Engineering Saccharomyces cerevisiae for co-utilization of d-galacturonic acid and d-glucose from citrus peel waste. Nat. Commun. 2018, 9, 5059. [Google Scholar] [CrossRef] [Green Version]
- Jeong, D.; Ye, S.; Park, H.; Kim, S.R. Simultaneous fermentation of galacturonic acid and five-carbon sugars by engineered Saccharomyces cerevisiae. Bioresour. Technol. 2020, 295, 122259. [Google Scholar] [CrossRef] [PubMed]
- Perpelea, A.; Wijaya, A.W.; Martins, L.C.; Rippert, D.; Klein, M.; Angelov, A.; Peltonen, K.; Teleki, A.; Liebl, W.; Richard, P.; et al. Towards valorization of pectin-rich agro-industrial residues: Engineering of Saccharomyces cerevisiae for co-fermentation of d-galacturonic acid and glycerol. Metab. Eng. 2021, in press. [Google Scholar] [CrossRef] [PubMed]
- Souffriau, B.; Den Abt, T.; Thevelein, J.M. Evidence for rapid uptake of d-galacturonic acid in the yeast Saccharomyces cerevisiae by a channel-type transport system. FEBS Lett. 2012, 586, 2494–2499. [Google Scholar] [CrossRef] [Green Version]
- Benz, J.P.; Protzko, R.J.; Andrich, J.M.S.; Bauer, S.; Dueber, J.E.; Somerville, C.R. Identification and characterization of a galacturonic acid transporter from Neurospora crassa and its application for Saccharomyces cerevisiae fermentation processes. Biotechnol. Biofuels 2014, 7, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Matsubara, T.; Hamada, S.; Wakabayashi, A.; Kishida, M. Fermentative production of l-galactonate by using recombinant Saccharomyces cerevisiae containing the endogenous galacturonate reductase gene from Cryptococcus diffluens. J. Biosci. Bioeng. 2016, 122, 639–644. [Google Scholar] [CrossRef]
- Swinnen, S.; Ho, P.W.; Klein, M.; Nevoigt, E. Genetic determinants for enhanced glycerol growth of Saccharomyces cerevisiae. Metab. Eng. 2016, 36, 68–79. [Google Scholar] [CrossRef]
- Ho, P.-W.; Klein, M.; Futschik, M.; Nevoigt, E. Glycerol positive promoters for tailored metabolic engineering of the yeast Saccharomyces cerevisiae. FEMS Yeast Res. 2018, 18, foy019. [Google Scholar] [CrossRef]
- Ho, P.-W.; Swinnen, S.; Duitama, J.; Nevoigt, E. The sole introduction of two single-point mutations establishes glycerol utilization in Saccharomyces cerevisiae CEN.PK derivatives. Biotechnol. Biofuels 2017, 10, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Gietz, R.D.; Schiestl, R.H.; Willems, A.R.; Woods, R.A. Studies on the transformation of intact yeast cells by the LiAc/SS-DNA/PEG procedure. Yeast 1995, 11, 355–360. [Google Scholar] [CrossRef]
- Gueldener, U.; Heinisch, J.; Koehler, G.J.; Voss, D.; Hegemann, J.H. A second set of loxP marker cassettes for Cre-mediated multiple gene knockouts in budding yeast. Nucleic Acids Res. 2002, 30, e23. [Google Scholar] [CrossRef] [Green Version]
- Hoffman, C.S.; Winston, F. A ten-minute DNA preparation from yeast efficiently releases autonomous plasmids for transformaion of Escherichia coli. Gene 1987, 57, 267–272. [Google Scholar] [CrossRef]
- Verduyn, C.; Postma, E.; Scheffers, W.A.; Van Dijken, J.P. Effect of benzoic acid on metabolic fluxes in yeasts: A continuous-culture study on the regulation of respiration and alcoholic fermentation. Yeast 1992, 8, 501–517. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, H.T.T.; Nevoigt, E. Engineering of Saccharomyces cerevisiae for the production of dihydroxyacetone (DHA) from sugars: A proof of concept. Metab. Eng. 2009, 11, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Funk, M.; Niedenthal, R.; Mumberg, D.; Brinkmann, K.; Rönicke, V.; Henkel, T. Vector systems for heterologous expression of proteins in Saccharomyces cerevisiae. Methods Enzymol. 2002, 350, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.K.; Krohn, R.I.; Hermanson, G.T.; Mallia, A.K.; Gartner, F.H.; Provenzano, M.D.; Fujimoto, E.K.; Goeke, N.M.; Olson, B.J.; Klenk, D.C. Measurement of protein using bicinchoninic acid. Anal. Biochem. 1985, 150, 76–85. [Google Scholar] [CrossRef]
- Klein, M.; Swinnen, S.; Thevelein, J.M.; Nevoigt, E. Minireview Glycerol metabolism and transport in yeast and fungi: Established knowledge and ambiguities. Environ. Microbiol. 2016, 19, 878–893. [Google Scholar] [CrossRef] [Green Version]
- Klein, M.; Carrillo, M.; Xiberras, J.; Islam, Z.U.; Swinnen, S.; Nevoigt, E. Towards the exploitation of glycerol’s high reducing power in Saccharomyces cerevisiae-based bioprocesses. Metab. Eng. 2016, 38, 464–472. [Google Scholar] [CrossRef]
- Klein, M.; Islam, Z.u.; Knudsen, P.B.; Carrillo, M.; Swinnen, S.; Workman, M.; Nevoigt, E. The expression of glycerol facilitators from various yeast species improves growth on glycerol of Saccharomyces cerevisiae. Metab. Eng. Commun. 2016, 3, 252–257. [Google Scholar] [CrossRef] [Green Version]
- Swinnen, S.; Klein, M.; Carrillo, M.; McInnes, J.; Nguyen, H.T.; Nevoigt, E. Re-evaluation of glycerol utilization in Saccharomyces cerevisiae: Characterization of an isolate that grows on glycerol without supporting supplements. Biotechnol. Biofuels 2013, 6, 157. [Google Scholar] [CrossRef] [Green Version]
- Martens-Uzunova, E.S.; Schaap, P.J. An evolutionary conserved d-galacturonic acid metabolic pathway operates across filamentous fungi capable of pectin degradation. Fungal. Genet. Biol. 2008, 45, 1449–1457. [Google Scholar] [CrossRef]
- Kuorelahti, S.; Kalkkinen, N.; Penttilä, M.; Londesborough, J.; Richard, P. Identification in the mold Hypocrea jecorina of the first fungal d-galacturonic acid reductase. Biochemistry 2005, 44, 11234–11240. [Google Scholar] [CrossRef] [PubMed]
- Petrash, J.M.; Murthy, B.S.N.; Young, M.; Morris, K.; Rikimaru, L.; Griest, T.A.; Harter, T. Functional genomic studies of aldo-keto reductases. Chem. Biol. Interact. 2001, 130, 673–683. [Google Scholar] [CrossRef]
- Chang, Q.; Griest, T.A.; Harter, T.M.; Mark Petrash, J. Functional studies of aldo-keto reductases in Saccharomyces cerevisiae. Biochim. Biophys. Acta Mol. Cell Res. 2007, 1773, 321–329. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.T.; Huh, W.K.; Lee, B.H.; Kang, S.O. D-Arabinose dehydrogenase and its gene from Saccharomyces cerevisiae. Biochim. Biophys. Acta 1998, 1429, 29–39. [Google Scholar] [CrossRef]
- Hur, E.; Wilson, D.K. Crystallization and aldo-keto reductase activity of Gcy1p from Saccharomyces cerevisiae. Acta Crystallogr. Sect. D Biol. Crystallogr. 2000, 56, 763–765. [Google Scholar] [CrossRef] [Green Version]
- Hur, E.; Wilson, D.K. The crystal structure of the GCY1 protein from S. cerevisiae suggests a divergent aldo-keto reductase catalytic mechanism. Chem. Biol. Interact. 2001, 130–132, 527–536. [Google Scholar] [CrossRef]
- Liang, Z.; Wang, X.; Bao, X.; Wei, T.; Hou, J.; Liu, W.; Shen, Y. Newly identified genes contribute to vanillin tolerance in Saccharomyces cerevisiae. Microb. Biotechnol. 2021, 14, 503–516. [Google Scholar] [CrossRef]
- Blank, L.M.; Lehmbeck, F.; Sauer, U. Metabolic-flux and network analysis in fourteen hemiascomycetous yeasts. FEMS Yeast Res. 2005, 5, 545–558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oechsner, U.; Magdolen, V.; Bandlow, W. A nuclear yeast gene (GCY) encodes a polypeptide with high homology to a vertebrate eye lens protein. FEBS Lett. 1988, 238, 123–128. [Google Scholar] [CrossRef] [Green Version]
- Norbeck, J.; Blomberg, A. Metabolic and regulatory changes associated with growth of Saccharomyces cerevisiae in 1.4 M NaCl. J. Biol. Chem. 1997, 272, 5544–5554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blomberg, A. Metabolic surprises in Saccharomyces cerevisiae during adaptation to saline conditions: Questions, some answers and a model. FEMS Microbiol. Lett. 2000, 182, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Roberts, G.G.; Hudson, A.P. Transcriptome profiling of Saccharomyces cerevisiae during a transition from fermentative to glycerol-based respiratory growth reveals extensive metabolic and structural remodeling. Mol. Genet. Genom. 2006, 276, 170–186. [Google Scholar] [CrossRef]
- Zhang, L.; Tang, Y.; Guo, Z.; Shi, G. Engineering of the glycerol decomposition pathway and cofactor regulation in an industrial yeast improves ethanol production. J. Ind. Microbiol. Biotechnol. 2013, 40, 1153–1160. [Google Scholar] [CrossRef] [Green Version]
- Harth, S.; Wagner, J.; Sens, T.; Choe, J.; Benz, J.P.; Weuster-Botz, D.; Oreb, M. Engineering cofactor supply and NADH-dependent d-galacturonic acid reductases for redox-balanced production of l-galactonate in Saccharomyces cerevisiae. Sci. Rep. 2020, 10, 19021. [Google Scholar] [CrossRef]
- Zhang, L.; Thiewes, H.; Kan, J.A.L. Van The d-galacturonic acid catabolic pathway in Botrytis cinerea. Fungal Genet. Biol. 2011, 48, 990–997. [Google Scholar] [CrossRef] [PubMed]
- Liepins, J.; Kuorelahti, S.; Penttilä, M.; Richard, P. Enzymes for the NADPH-dependent reduction of dihydroxyacetone and d-glyceraldehyde and l-glyceraldehyde in the mould Hypocrea jecorina. FEBS J. 2006, 273, 4229–4235. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.; Schäfer, D.; von den Eichen, N.; Haimerl, C.; Harth, S.; Oreb, M.; Benz, J.P.; Weuster-Botz, D. d-Galacturonic acid reduction by S. cerevisiae for l-galactonate production from extracted sugar beet press pulp hydrolysate. Appl. Microbiol. Biotechnol. 2021, 105, 5795–5807. [Google Scholar] [CrossRef] [PubMed]
- Shiramizu, M.; Toste, F.D. Expanding the scope of biomass-derived chemicals through tandem reactions based on oxorhenium-catalyzed deoxydehydration. Angew. Chemie Int. Ed. 2013, 52, 12905–12909. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, T.; Nishikawa, H.; Gao, Y.; Sawa, Y.; Shibata, H.; Yabuta, Y.; Maruta, T.; Shigeoka, S. The pathway via d-galacturonate/l-galactonate is significant for ascorbate biosynthesis in Euglena gracilis: Identification and functional characterization of aldonolactonase. J. Biol. Chem. 2008, 283, 31133–31141. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | Genotype, Description | Reference |
|---|---|---|
| CEN.PK113-1A | MATα (prototrophic) | Euroscarf |
| CEN.PK113-1A UBR2CBS | MATα; ubr2::UBR2CBS 6412-13A | [33] |
| CEN.PK RE | MATα; ubr2::UBR2CBS 6412-13A; gut1::GUT1JL1 | This study * |
| CEN.PK DHA | MATα; ubr2::UBR2CBS 6412-13A; YGLCτ3::PTEF1-CjFPS1-TCYC1; gut1::PTEF1-Opgdh-TCYC1-PADH2-ScDAK1-TTPS1 | [34] |
| CEN.PK DHA gcy1Δ | MATα; ubr2::UBR2CBS 6412-13A; YGLCτ3::PTEF1-CjFPS1-TCYC1; gut1::PTEF1-Opgdh-TCYC1-PADH2-ScDAK1-TTPS1; gcy1::loxP-ble-loxP | This study |
| CEN.PK DHA gre3Δ | MATα; ubr2::UBR2CBS 6412-13A; YGLCτ3::PTEF1-CjFPS1-TCYC1; gut1::PTEF1-Opgdh-TCYC1-PADH2-ScDAK1-TTPS1; gre3::loxP-ble-loxP | This study |
| CEN.PK DHA ypr1Δ | MATα; ubr2::UBR2CBS 6412-13A; YGLCτ3::PTEF1-CjFPS1-TCYC1; gut1::PTEF1-Opgdh-TCYC1-PADH2-ScDAK1-TTPS1; ypr1::loxP-ble-loxP | This study |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rippert, D.; Linguardo, F.; Perpelea, A.; Klein, M.; Nevoigt, E. Identification of the Aldo-Keto Reductase Responsible for d-Galacturonic Acid Conversion to l-Galactonate in Saccharomyces cerevisiae. J. Fungi 2021, 7, 914. https://doi.org/10.3390/jof7110914
Rippert D, Linguardo F, Perpelea A, Klein M, Nevoigt E. Identification of the Aldo-Keto Reductase Responsible for d-Galacturonic Acid Conversion to l-Galactonate in Saccharomyces cerevisiae. Journal of Fungi. 2021; 7(11):914. https://doi.org/10.3390/jof7110914
Chicago/Turabian StyleRippert, Dorthe, Federica Linguardo, Andreea Perpelea, Mathias Klein, and Elke Nevoigt. 2021. "Identification of the Aldo-Keto Reductase Responsible for d-Galacturonic Acid Conversion to l-Galactonate in Saccharomyces cerevisiae" Journal of Fungi 7, no. 11: 914. https://doi.org/10.3390/jof7110914