Current State of Knowledge on Primary Sjögren’s Syndrome, an Autoimmune Exocrinopathy
by

Dorian Parisis
1,2, 
Clara Chivasso
1,
Jason Perret
1, 
Muhammad Shahnawaz Soyfoo
2 and
Christine Delporte
1,* 1
Laboratory of Pathophysiological and Nutritional Biochemistry, Université Libre de Bruxelles, 1070 Brussels, Belgium
2
Department of Rheumatology, Erasme Hospital, Université Libre de Bruxelles, 1070 Brussels, Belgium
*
Author to whom correspondence should be addressed.
J. Clin. Med. 2020, 9(7), 2299; https://doi.org/10.3390/jcm9072299
Submission received: 25 June 2020
/
Revised: 15 July 2020
/
Accepted: 16 July 2020
/
Published: 20 July 2020
(This article belongs to the Special Issue Diseases of the Salivary Glands)
Abstract
:Primary Sjögren’s syndrome (pSS) is a chronic systemic autoimmune rheumatic disease characterized by lymphoplasmacytic infiltration of the salivary and lacrimal glands, whereby sicca syndrome and/or systemic manifestations are the clinical hallmarks, associated with a particular autoantibody profile. pSS is the most frequent connective tissue disease after rheumatoid arthritis, affecting 0.3–3% of the population. Women are more prone to develop pSS than men, with a sex ratio of 9:1. Considered in the past as innocent collateral passive victims of autoimmunity, the epithelial cells of the salivary glands are now known to play an active role in the pathogenesis of the disease. The aetiology of the “autoimmune epithelitis” still remains unknown, but certainly involves genetic, environmental and hormonal factors. Later during the disease evolution, the subsequent chronic activation of B cells can lead to the development of systemic manifestations or non-Hodgkin’s lymphoma. The aim of the present comprehensive review is to provide the current state of knowledge on pSS. The review addresses the clinical manifestations and complications of the disease, the diagnostic workup, the pathogenic mechanisms and the therapeutic approaches.
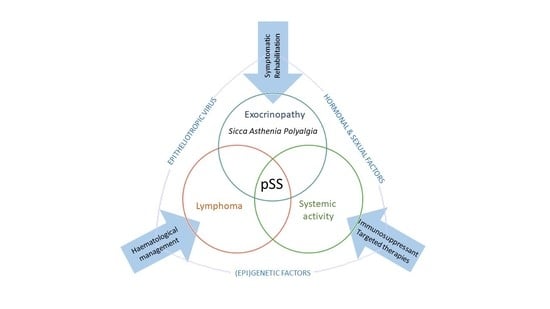
1. Introduction
Sjögren’s syndrome (SS) is a chronic systemic rheumatic disease characterized by lymphoplasmacytic infiltration of the exocrine glands—especially salivary and lachrymal glands—responsible for sicca syndrome and systemic manifestations. The dreaded complication of this dysregulated and unabated lymphocytic activation is the development of lymphoma. SS can be “primary” if it occurs alone (pSS) or “secondary” (sSS) when it is associated with another autoimmune disease [1].
First medical descriptions of SS date back to 1882 when the German Theodor Karl Gustav von Leber (1840–1917) described for the first time a dry inflammation of the ocular surface under the name of “keratitis filamentosa”. Ten years later, the Polish surgeon Jan Mikulicz-Radecki described the case of a man with swelling of the salivary and lacrimal glands, a clinical picture still called Mikulicz syndrome today. At the same time, several cases of patients with ocular and oral dryness were described, whether or not associated with the existence of rheumatism or gout. Dr. W. B. Hadden (1856–1893) described the improvement of xerostomia in one of these patients with the use of an alkaloid called pilocarpine [2]. Despite the involvement of these physicians in the first medical descriptions of SS, only two famous names have remained attached to the disease: Gougerot and Sjögren. Henri Gougerot (1881–1955) was a French dermatologist who described in 1925 three clinical cases characterized by generalized mucous dryness (eyes, mouth, nose, trachea and vagina) associated with atrophy of the salivary glands (SG). He was the first to describe that xerostomia and ocular dryness are part of a larger sicca syndrome resulting from dysfunction of the exocrine glands or their autonomic innervation. In France, the term “Gougerot(-Sjögren) syndrome” is often used to describe pSS. Henrik Samuel Conrad Sjögren (1899–1986) was a Swedish ophthalmologist who was mainly interested in the dryness of the ocular surface. With his wife, Maria Hellgren, daughter of a well-known oculist, he described keratoconjunctivitis sicca (KCS)—distinct from vitamin A deficiency xerophthalmia—using Rose Bengal and methylene blue staining techniques. In 1933, in his PhD thesis, he described the cases of 19 women with KCS and 13 of whom had arthritis. He was therefore, the first to link KCS to a systemic disease beyond the field of ophthalmology. Unfortunately, his thesis was not successful, and he stopped his academic career but not his medical and scientific one. It was only in the years 1935–1943 that Sjögren’s work was recognized and that the term “Sjögren’s syndrome” has been used since. Finally, the autoimmune origin was recognized only in early 1960s [2]. Sjögren was awarded the title of “Doctor” in 1957 by the University of Gothenburg and the honorary title of “Professor” in 1961 by the Swedish Government. Henrik Sjögren died of pneumonia on 17 September 1986, several years after a disabling stroke [3,4,5].
2. Epidemiology
2.1. Prevalence
pSS affects 0.1% to 4.8% of the population with a female to male ratio of 9:1, depending on the cohort studied, classification criteria and methodology used [6,7]. Although pSS is considered a common disorder, its prevalence seems to be overestimated in some studies. Overall, 0.5–1% seems to be a commonly accepted estimate of the prevalence of pSS in the general population [7]. However according to a more recent meta-analysis of 7 studies, prevalence rate is 0.043% with a sex ratio of 10.72. The prevalence of pSS in Europe is higher than in Asia, 0.7122% and 0.045%. Sex ratio does not differ according to the geographic/ethnic origin of the populations studied [8].
2.2. Incidence
There is an overt heterogeneity of SS incidence among several studies. A meta-analysis reported an incidence rate of 6.92 per 100,000 person–years, with an overall average age of 56.2 years at diagnosis and an incidence rate ratio between women and men estimated at 9.29. Six Asian studies reported a relatively higher incidence ratio around 6 per 100,000 person–years. Both Slovenian and American studies reported an incidence ratio of 3.9 per 100,000 person–years. Finally, a Greek study estimated an incidence ratio between the two at 5.3 per 100,000 person–years. Data regarding the incidence of pSS in Africa, Oceania and South America are lacking [8].
3. Physiopathology of Sjögren’s Syndrome
SS is considered as a multifactorial process originating from the interaction between genetic factors and exogenous and endogenous agents able to trigger an abnormal autoimmune response mediated in particular by T and B lymphocytes [9]. The inflammation sustains, perpetuates and amplifies tissue damage and leads to a progressive functional impairment of the affected organs and a chronic inflammatory environment. Three recurrent events are generally associated with SS: (1) a trigger phase induced by environmental factors under specific epigenetic factors, genetic predisposition and hormonal regulation; (2) the dysregulation of normal salivary gland epithelial cell (SGEC) function; (3) a chronic inflammation characterized by SG infiltration made of lymphocytic cells, lymphocytes B hyperactivity and autoantibodies production [10] (Figure 1).
3.1. Trigger Phase
In SS pathogenesis, a trigger phase is induced by environmental factors such as viral infections combined with genetic predisposition, epigenetic factors and sex hormonal regulation (Figure 2).
3.1.1. Environmental Factors
According to the current physiopathogenic model of SS, environmental factors including viral infection lead to SGEC and Toll Like Receptors (TLRs) activation [12,13]. Primary viruses involved in SS induction include Epstein–Barr (EBV) viruses, Human T-lymphotropic virus type I (HTLVI), hepatitis virus C (HCV) and coxsackievirus [13].
EBV is a double stranded DNA virus appertaining to Herpesviridae family, with a strong tropism for B cells. EBV has often been associated with autoimmunity processes and diseases such as Rheumatoid Arthritis (RA), Systemic Lupus Erythematosus (SLE) and Multiple Sclerosis (MS) [14,15]. In addition, the high EBV load found in SG and lacrimal gland biopsies from SS patients as compared to controls [16,17] suggests its role in triggering the activation of the immune system. EBV is able to stimulate the production of proteins that mimic B cell receptor (BCR) and CD40 signalling and induce a strong B cell hyperactivity [18]. Recently, a correlation was established between past EBV infection and the presence of anti-Ro/SSA and anti-La/SSB autoantibodies in SS patients [19]. The RNA encoded by EBV binds TLR3 and induces the secretion of type I IFN and proinflammatory cytokines [20]. Another protein, the latent membrane protein 1 (LMP1) acting as a target for the EBV-induced cytotoxic T lymphocytes response may cause acini atrophy and SG lobule structure destruction observed in SS patients [21].
HTLV-1, a human endemic retrovirus in certain geographical areas such as Japan, has been reported to be present in SGEC [22]. In addition, epidemiologic studies revealed anti-HTLV-1 seropositivity in 23% of SS patients as compared to 3% in controls [23].
Coxsackie virus is a single stranded RNA virus belonging to the Picornaviridae family. A study has identified in SS patients a cross-reactivity between antibodies to the Ro60 epitope and 2B Coxsackie protein sharing 87% sequence homology [24]. However, these data remain controversial [25].
The role of HCV, a single stranded RNA small virus belonging to Flaviviridae family, has been examined in the initial triggering phase of SS. Clinical studies have shown that patients with HCV infection present sicca symptomatology, positive ocular tests, SG lymphocytic infiltration, and autoantibodies [26]. Therefore, HCV-associated SS (patients with HCV fulfilling SS 2002 classification criteria) is indistinguishable from pSS. On this basis, HCV chronic infection should be considered as an exclusion criterion for pSS as HCV infection could participate to SS development in a subset of patients.
Despite possible involvement of viral infection in SS, the most common antiviral drugs do not seem to show real benefit in the treatment of SS [26]. Indeed, as a viral infection may likely trigger onset of the disease, later antiviral treatment may manage a persistent infection but have no effect on the ongoing disease that may no longer be dependent on the presence of the initial viral infection.
3.1.2. Genetic Predisposition
Genetic predisposition to SS plays a role in the trigger phase of the disease. A strong association between human leucocyte antigen (HLA)-DR and HLA-DQ alleles belonging to the group of major histocompatibility genes (MHC) class II genes and SS was observed throughout different populations including Caucasian, Japanese and Chinese populations [27]. All discovered haplotypes are in strong linkage disequilibrium, causing difficulties in establishing which of them contain the locus that confers the risk. SS patients with HLA-DQ1/HLA-DQ2 alleles display more severe autoimmune disease than patients with any other allelic combination at HLA-DQ [28]. In addition to the HLA system, most recent studies have focused their attention on polymorphic genes that code for molecules physiologically involved in apoptosis such as Fas and Fas ligand (FasL). Using MRL/lpr-murine model, a retrotransposon inserted in Fas gene was identified as playing a role in cell apoptosis and induction of progressive sialadenitis [29,30]. Fas/FasL gene polymorphisms have also been found in SS patients [31] but have not clearly been identified as disease-determining factors. Ro52 gene encoding the 52-kd Ro autoantigen display single nucleotide polymorphism (SNP) located 13bp upstream of exon 4 identified as significantly associated with the presence of anti-Ro 52kD autoantibodies in SS patients [32]. Numerous additional genes including IL-10 [33], TNF alpha [34], alpha chain of the IL-4 receptor [35], IRF5, STAT4 [36] and CXCL13 [37] also display a gene polymorphism possibly associated with SS as well. Recent studies carried out in several SS cohorts of different ethnicity have revealed additional candidate genes probably associated with the risk to develop the lymphoma in SS patients. The presence of a polymorphism in the tumour necrosis factor alpha induced protein 3 (TNFAIP3) gene is associated with the risk to develop the non-Hodkin’s lymphoma in a SS Caucasian cohort [38,39,40]. In addition, two polymorphisms of methylene-tetrapholate reductase (MTHFR) gene are considered risk factors for lymphoma in SS patients [41]. While gene polymorphism plays an indisputable role in the triggering phase of SS, the individual contribution of each genetic factor remains to be assessed [42].
3.1.3. Epigenetic Factors
Several studies have analysed the contribution of epigenetics to SS and auto-antibodies production [43]. The epigenetic processes more closely linked to the disease are DNA methylation, miRNA, circular mRNA and long non-coding RNA function.
DNA methylation is a mechanism that consists in the addition of a methyl group from a methyl donor S-adenosylmethionine (SAM) to cytosine residues in the context of the CpG dinucleotide catalysed by DNA methyltransferases (DNMTs). In general, the addition of a methyl group onto DNA is associated with gene silencing due to a structural modification of chromatin. DNA methylation is one of most important mechanisms used by different type of cells to change their genetic expression such as the transition from naïve steady to effector B- and T-cells. An epigenome-wide analysis has identified several genes and epigenetic modification probably associated with SS [44]. The most frequent modification observed is the demethylation of several sites in SS patients’ genome. Labial SG DNA methylation is significantly reduced in SS patients as compared to the control subjects. This defect was conserved when the SGEC were primarily cultured. Apparently, the SGEC from SS patients were associated with a 7-fold decrease in DNMT1 and a 2-fold increase in demethylating partner Gadd45-alpha expression. This demethylation process was also associated in part with the infiltration of SG by B cells and the pathology severity [45]. Different studies have also reported a link between demethylating drugs and SS. In fact, mice receiving an oral administration of hydralazine or isoniazid (demethylating agents) for several weeks develop a pathology similar to SS in terms of immunological features and autoantibodies production. The signs of SS pathology disappeared after discontinuation of the drug [46]. A recent study conducted in CD19 + B cells and minor SG of SS patients has also identified a hypomethylation site on interferon (IFN)-regulated genes which induces an increase of IFN response activation normally observed in SS disease [47]. In addition, DNA demethylation of the pro-apoptotic death associated protein kinase (DAP-kinase) gene [48] and the runt-related transcription factor (RUNX1) gene in CD4 + T cells [49] have been associated with non-Hodgkin B cell lymphoma predisposition in SS. In conclusion, the genome methylation analysis represents a useful tool to identify links between epigenetic modifications in various cell types related to SS.
miRNAs are small endogenous non-coding RNAs that regulate gene-expression transcriptionally and post-transcriptionally. Interestingly, miR-17-92 cluster, is downregulated [50] and associated with a lymphoproliferative disease and autoimmunity [51,52] in SG of SS patients. Another study has shown increased levels of miR-146a that regulates the inflammatory response, inducing the repression of IRAK1 and the increase of TRAF6 expression which, in turn, promote NF-κB expression in the peripheral mononuclear cells of SS patients [53]. Aberrations in microRNA expression are often observed in various autoimmune diseases and for this reason they could be used as a potential diagnostic or prognostic biomarkers. Furthermore, the small size of mature miRNA offers a high level of stability that renders them useful in disease follow-up using paraffin embedded samples stored for long periods of time [54,55].
Circular RNA (circRNA) consist in a class of RNA generated after an alternative splicing process of pre-mRNA named “backsplicing”, in which a downstream 5′ donor links an upstream 3′ acceptor throughout a 3′ → 5′ phosphodiester bond. circRNa are divided in three subgroups: exonic circRNAs (ecircRNAs), intronic circRNAs (ciRNAs) and exon-intron circRNAs (EIciRNAs) [56]. Recent studies have observed that circRNA could be involved in development of autoimmune diseases such as RA, MS, SLE and SS [57]. A microarray analysis has identified 234 differentially expressed circRNAs between SS patients and healthy controls, whereby 2 are significantly upregulated and 3 downregulated in SS. Functional analysis has also shown that these circRNAs are related to arthritis and the presence of autoantibodies [58]. All this data taken into account, we can conclude that circRNAs could be used as biomarkers for a potentially valuable diagnostic tool for SS disease, but supplementary investigations assessing which of them is the most specific of pathology are necessary.
Long non-coding RNAs (lncRNA) are a novel class of functional non-translated RNAs with a length of over 200 nucleotides. Several studies revealed a strong link between lncRNAs and the immune responses [59]. The expression analysis of lncRNAs in SS patients has shown lncRNAs LINC00657, LINC00511 and CTD-2020K17.1 potentially associated with the disease. These 3 lncRNAs target different genes involved in B cell physiology and malignancy, including IL15, WDR5, GNAI2, LTßR, CBX8, BAK1, BAX ext [60]. IL15 and WDR5 play an important role in B cell proliferation and differentiation; GNAI2 regulates B cell trafficking to the lymph nodes [61]; LTßR and CBX8 are involved in GC formation in inflamed tissues [62,63], and BAK1 and BAX are overexpressed in B cell lymphoma [64]. These results illustrate an important role of lncRNAs in multiple processes and the understanding of their modulation and function could provide deeper insight into the pathogenesis of SS and facilitate the identification of novel therapeutic strategies.
3.1.4. Sex Hormones Deregulation and X-Chromosome Linked Factors
Nine out of ten SS patients are women and generally during menopause [65]. The strong predisposition of women to develop SS clearly demonstrates the role of sex hormones as a risk factor of the disease. In a recent case-control study, pSS in women was associated with lower oestrogen exposure and lower cumulative menstrual cycling time compared to sicca controls. Conversely, an increasing oestrogen exposure was negatively associated with development of pSS [66]. Finally, an effect of X chromosome per se is also evoked since men with Klinefelter’s syndrome have a higher risk of developing pSS—20 times higher—compared to healthy men, despite normal sex hormone levels [67,68]. Similarly, the association between pSS and mixed connective tissue disease has been reported in a 16-year-old Japanese patient with trisomy X [69].
Androgens suppress the inflammation and enhance the function of lacrimal glands in female SS mouse models (MRL/MpJ-Tnfrsf6lpr[MRL/lpr]) [70]. The androgens could help maintaining acini structure in healthy SG, while their reduction observed in SS patients could cause a decrease in integrin expression and probably a dysregulation of acini architecture [71]. SS patients present low levels of androgen hormones both in the bloodstream and in SG [72]. In Klinefelter’s syndrome associated SS and SLE, correction of hypogonadism by testosterone therapy for 60 days leads to remission in one case-series report [73].
Healthy ovariectomized C57BL/6 mice display an exocrinopathy with autoimmune characteristics similar to SS including SG focal adenitis, lacrimal glands lesions, Ro/SSA, La/SSB and α-fodrin autoantibodies [74]. Similarly to ovariectomized mice, both mice rendered deficient in aromatase, an enzyme important in the biosynthesis of oestrogens, as well as mice that received an aromatase inhibitor develop a lymphoproliferative autoimmune disease resembling SS [75,76]. How oestrogen deficiency promotes autoimmune lesions remains unclear. However, one putative explanation could be that oestrogen deficiency stimulates SGEC to secrete IFN-α and IL-8, and to express MHC class II, enabling them to act as antigen-presenting cells. Oestrogen deficiency is responsible for RbAp48 overexpression, which induces p53-mediated apoptosis in exocrine glands [77]. In another study, transgenic mice overexpressing RbAp48 develop SS-like exocrinopathy characterized by an increased propensity to apoptosis and the acquisition of an active immunocompetent role by epithelial cells, producing IFN-γ and IL-18 [78]. In primary cultures of human SG cells, pre-treatment with 7β-estradiol impede IFNγ-induced upregulation of ICAM-1 in control group but not in pSS group. These data suggest a protective role of oestrogens on epithelial activation and the existence of a deficient estrogenic responsiveness in pSS [79]. Not surprisingly, the use of aromatase inhibitors in the treatment of breast cancer is associated with arthralgia or even authentic SS [80,81,82].
Humans and other primates, secrete large amount of sex steroid precursors, such as dehydroepiandrosterone (DHEA) and DHEA-sulphate precursors, metabolic intermediates in the biosynthesis of androgens and oestrogens. According to tissue needs, the prohormones are directly processed within tissues. DHEA is present in low concentrations in patients with SS as compared to age-matched healthy controls [83]. Several studies have shown that human MSG possess an organized intracrine machinery capable to convert DHEA(-sulphate) pro-hormone to its active metabolites, dihydrotestosterone (DHT) and 17β-oestradiol [84] (Figure 3). However, the non- functionality of this enzymatic machinery in MSG from SS patients could account for the diminished local concentrations of DHT and androgen-regulated biomarker Cysteine-Rich Secretory Protein 3 (CRISP-3) in SS patients [85].
Taken together, these data suggest that women affected by SS at menopause, when the levels of testosterone produced by the ovaries has already declined, may be particularly vulnerable to androgen deficiency because the only source of DHT in SG is dependent on local conversion of DHEA. Whereas in men, the level of systemic androgens produces by gonads may satisfy the specific needs of SG, not requiring the intermediate metabolite.
3.2. SGEC Deregulation
3.2.1. Upregulation of Adhesion Molecules
According to recent observations, several SS pathogenic models could explain the role of SGEC in glandular damage. The current SS pathogenic model is the “autoimmune epithelitis”. This model considers SGEC as a crucial player in the initial triggering phase of the disease [86]. SGEC from SS patients express significantly higher levels of TLRs mRNA levels, including TLR-1, TLR-2, TLR-3 and TLR-4 as compared to control SGEC [87]. Under physiological conditions, TLRs are activated by the recognition of pathogen-associated molecular patterns (PAMPs) derived from microorganisms and endogenous mediators of inflammation known as danger-associated molecular patterns (DAMPs) [88]. TLR signalling pathway acts as link between innate and adaptive immunity in autoimmune diseases. Indeed, upon activation, TLRs recruit adapter proteins in order to propagate the intracellular signal that results in the transcription of genes involved in inflammation, immune regulation, cell survival and proliferation and subsequent activation of the immune system. TLR signalling in SGEC upregulates several molecules such as MHC class I and class II, costimulatory molecules such as B7.1 (CD80) and B7.2 (CD86) and adhesion molecules 1 (ICAM-1) [89].
3.2.2. Antigen-Presenting Cell Properties
The expression of MHC class I, MHC class II, costimulatory molecules and adhesion molecules on SGECs empower them to present antigen to T cells (acting as non-professional antigen presenting cells).
3.2.3. Chemokines Production
The activation of Interferon Regulatory Factor (IRF) and nuclear factor kappa-light-chain-enhancer of activated B cells (NFkB) pathways increases the production of inflammatory cytokines, including type I IFN, tumour necrosis factor-α (TNF-α), interleukin(IL)-1, IL-6 and BAFF [90].
3.2.4. Apoptosis and Expression of Self-Antigens
In addition to chemokines production, the ribonucleoproteins, normally hidden from the immune system, are exposed on the cell surface. In particular, the expression of antigen Ro/SSA and La/SSB proteins on apoptotic SGEC promotes the initiation of autoimmunity.
3.2.5. Alteration of Proteins Involved in Saliva Secretion
Apoptosis of the acinar epithelial cells and altered expression and distribution of proteins involved in saliva secretion has been proposed as possible mechanisms responsible for the impairment of secretory function of SS SG. For example, an increase in AQP3 expression was observed at the apical membrane of acinar cell of SG from SS [91], while AQP1 [92] and AQP4 [93] expression was decreased in myoepithelial cells. Rituximab treatment, used in SS patients to deplete B cells, increases AQP1 protein expression in myoepithelial cell and induces an improvement of saliva flow [94]. These data could suggest a crucial role to AQP1 in saliva secretion. However, AQP1-null mice model has shown that this protein is not essential for saliva production [95]. Nevertheless, one cannot exclude a compensatory effect in such mouse models, whereby other AQPs could be alternatively used. In contrast, AQP5 is today considered the most important protein involved in saliva secretion [96]. Under physiological conditions, AQP5 translocates from the intracellular vesicular compartments to the apical membrane of SG acinar cells after activation of muscarinic and adrenergic receptors [97]. In SS patients and SS mice models, aberrant localization of AQP5 has been observed [98], which is predominately basolateral instead of apical [99,100,101]. The reason why the AQP5 localization is altered is still unknown but several hypotheses have been proposed.
The presence of autoantibodies against M3 receptor could impair its activation and block the translocation signal normally sent to AQP5 [102]. Another possible mechanism could be the alteration of protein–protein interactions between AQP5 and its partner proteins [103]. Prolactin inducible protein (PIP) is a known AQP5 protein partner in lacrimal glands in mice models. Aberrant binding of PIP to the c-terminal domain of AQP5 impairs AQP5 trafficking to the apical membrane of epithelial cells [104]. Lastly, the inflammatory environment that characterizes SS disease could also directly or indirectly be involved in these modifications [105,106]. IFN-γ for example, contributes to SS pathogenesis inducing SG apoptosis and expression of several chemoattractant cytokines and enhancing the antigen presenting function of epithelial cells [107,108,109]. IFN-γ administration leads to increased production of anti-M3R antibody, which affect the SG secretory function in response to an adequate stimulus [110]. Neutralization of IFN-γ in anti-programmed death ligand 1 (PDL1)-treated non-obese diabetic (NOD)/ShiLtJ mice improves AQP5 expression and saliva secretion [111]. TNF-α is another pro-inflammatory cytokine that is increased in SS [112]. Elevated TNF-α levels in both serum and SG has been observed in SS patients compared to controls [113]. In human SG acinar cells, TNF-α treatment down-regulates the expression of AQP5 [114]. The injection of antibodies against TNF-α in NOD mice reduces SG inflammatory foci and increases AQP5 protein expression [115]. It seems clear that correct expression, trafficking and localization of AQP5 are essential to overcome the impaired salivary secretion process and the combination of inflammation, antibodies production, protein–protein interaction and salivary epithelial cells deregulation are probably involved in the hypofunction of SG of SS patients.
3.3. Chronic Inflammation
3.3.1. T-Cell Infiltration
In the early stages of SS, the lymphocytic infiltrates, present in SG from SS patients, are constituted by a vast majority (>75%) of T lymphocytes being mostly CD4 T cells [116]. However, saliva from SS patients contains greater Th1 cytokines than saliva from controls [109,117], including IL-1β, IL-6, tumour necrosis factor (TNF)-α, and IFN-γ [118]. Th2-derived cytokines, such as IL-10 and IL-4, were also found in greater quantity in SG tissue from SS patients than in controls [119]. The two T cell responses are in a dynamic balance with a predominance of Th1 activity in patients suffering from SS [120]. In patients with SS, the activated T cells respond to an intense antigenic stimulus, such as the recognition of Ro and La autoantigens expressed on blebs of apoptotic cells [121], which induces a proliferative response [122]. Therefore, T-cell recognition of self-antigens and their subsequent activation are crucial for the cascade of events leading to the development of SS pathology. T cells may proliferate locally in SG or be re-directed by chemokines from the circulation to the glands. Two chemokines involved in the attraction of T-cells in SS SG are CXCL9 and CXCL10 [123]. In SS SG, T cells are likely to be involved in the disruption of the glandular architecture throughout the apoptosis mechanism mediated by FasL pathway [124], by a direct cytotoxic activity involving the release of perforin and/or secretion of cytokines and by the activation of B cells [125]. Th17 cells represent another subpopulation of T-cells strongly activated in SS patients [126]. In general, Th17 plays an important physiological role in mucosal defence in healthy individuals. In SS patients, the activated Th17 cells promote inflammation by secreting IL-6, IL-17, IL-21, IL-22 and IL-23 [127,128,129]. Follicular helper T cells have been shown to play an important role in lymphoid follicle formation and ectopic germinal centre formation in SS SG [130]. During pathology, SGEC induce activation and differentiation of T helper to T follicular helper by the release of IL6 and ICOS ligand expression. The activated follicular cells in turn secrete IL-21 cytokine which mediates B cell maturation and proliferation [131]. In conclusion, the combined activation of T-cell subtypes creates an optimal environment for detrimental B cell activation and the breakdown of tolerance.
3.3.2. Breakdown of B Cells Tolerance
Under physiological condition, B cells originate in the bone marrow from haematopoietic stem cells and during their development undergo several stages of selection because of a large portion of self-reactive and polyreactive B cell are normally generated [132]. The first checkpoint removes the polyreactive B cells in the bone marrow (central tolerance checkpoint), the second in the periphery ensures that only a small amount of self-reactive, and polyreactive mature naïve B cells survive. Finally, a third tolerance checkpoint called pre-germinal centre checkpoint, excludes self-reactive naïve B cells from entering B cell follicles [133].
A recent study has revealed the existence of deficiencies in both early and late B cell tolerance checkpoints in patients with SS. Indeed, the accumulation of circulating autoreactive naïve B cells in SS suggests an impairment of the autoreactive B cell clearance during the early peripheral tolerance checkpoints and an increased frequency of autoreactive unswitched and switched memory B cells reveals a possible impairment also in pre- and/or post-germinal centre tolerance checkpoints [134]. These observations have also been made in patients with SLE, RA and type 1 diabetes [135,136]. B cell depletion using anti-CD20 antibodies in Id3 knockout mice model leads to a significant histological improvement associated with a recovery of saliva secretory function and corroborate the hypothesis that B cells could play an important role in SS disease [137].
B cell hyperactivity is an important hallmark of SS. Two cytokines have been shown to be fundamental in B cell survival and proliferation: B cell Activating Factor of the TNF Family (BAFF) and APRIL (A proliferating ligand) [138]. Once SG tissue infiltration is established, a large number of cells such as dendritic cells, monocytes and macrophages but also SGEC and T lymphocytes can secrete BAFF. BAFF overexpression has indeed been documented in SS as well as in other systemic autoimmune diseases and has been correlated with autoantibodies [139].
3.3.3. Formation of Germinal-Like Structures
Germinal centres (GCs) were described for the first time by Walther Flemming in 1884 [140]. GCs are specific region in secondary lymphoid tissues such lymph nodes and spleen. GCs provide the environment for proliferation of mature B cells, differentiation and mutation of their immunoglobulin variable-region gene segments during a process called somatic hypermutation, which generates a diversity of clones. Following this process, the cells migrate from the dark zone to the lighter zone of the lymphoid tissues, where the affinity of immunoglobulins is tested on follicular dendritic cells (FDC) and follicular helper T cells (TFH) cells presenting the antigens. The non-selected cells undergo apoptosis while the selected cells are stimulated by T cells to undergo class switch recombination and differentiation into antibody-producing plasma cells or memory B cells [141,142]. SG from SS patients can contain similar GC structures made of T, B, and plasma cells, macrophages, and follicular dendritic cells [143]. Given the strong similarity of SG GC with the lymphoid organ GC, the SG GC observed in SS patients were defined as ectopic GC-like structures, also known as “tertiary lymphoid organs” [144]. Several studies have reported the association between GCs and the immunopathological features of SS [145]. Other important studies have observed a 6.5- to 15.6-fold increased risk to develop non-Hodgkin lymphomas in SS with an elevated presence of GCs [146,147].
3.3.4. Local Production of Autoantibodies
The most common and studied antibodies in SS patients are those directed against the autoantigens Ro/SSA and La/SSB [148]. Anti-Ro, Anti-La, anti-SSA and anti-SSB were originally described as four antibodies directed against antigens expressed by salivary and lacrimal glands tissues from SS patients. Later, anti-Ro and anti-La were shown to be the same antibodies as anti-SSA and anti-SSB, respectively [149,150].
Ro antigen is constituted of two distinct Ro proteins of 52 and 60 kDa, with the latter binding to small cytoplasmic RNAs known as hY RNAs. The Ro52 protein, also known as TRIM21, is frequently targeted by SS antibodies, which makes it a useful diagnostic marker, but its function and why it becomes a target protein in a lot of rheumatic diseases is not completely understood. Ro52 is a member of the tripartite motif (TRIM) protein family, and it plays an important role in the ubiquitination of proteins. Several targets have been suggested as substrate of Ro52 activity, including various members of the IFN-regulatory factor (IRF) transcription factor family. The most speculated hypothesis attributes to Ro52 a role of IFN negative regulator. Indeed, in a Ro52-null mouse, the lack of ubiquitination mediated by Ro52 leads to an aberrant expression of type I IFNs and proinflammatory cytokines, such as IL-6, IL-12, IL-23, and TNF-α [151]. La/SSB antigen is a 48 kDa phosphorylated protein located in the nucleus and the cytoplasm. La/SSB binds to many RNA molecules newly synthesized by RNA polymerase III [152]. These two antibodies are detected in 50% to 70% of primary SS patients, but the anti-La/SSB alone is observed in only 2% of patients [153,154].
In most cases, anti-Ro/SSA and anti-La/SSB are correlated with severe dysfunction of the exocrine glands, associated with parotid gland enlargement and large number of lymphocytic infiltrates in the MSG [155,156].
Other antibodies believed to be pathogenic in SS are anti-centromere antibodies (ACA), anti-citrullinated protein antibodies (ACPA), anti-carbonic anhydrase II antibodies, anti-aquaporin-5, anti-muscarinic receptor 3 (anti-M3R) and anti-fodrin antibodies. ACA are directed against six antigens associated with the centromere (complex of kinetochore proteins). The incidence of ACA antibody ranges from 3.7% to 4% [157,158]. ACPA are directed against fibrin and fibrinogen, vimentine and alpha-enolase (CEP-1). In general, ACPA antibodies are the marker most observed in rheumatoid arthritis but are usually present in low concentrations in pSS as well, in about 3–22% of cases [159]. Anti-carbonic anhydrase II antibodies have been detected in 12.5–20.8% of SS patients and also play a pathogenic role in renal tubular acidosis (RTA) [160,161]. In fact, immunization of mice with human carbonic anhydrase II resulted in autoimmune sialadenitis, production of anti-carbonic-anhydrase-II antibodies and urinary acidification defect [162,163]. Anti-AQP5 antibodies were observed to be associated with serologic and histopathological features of SS [164]. Anti-M3R antibodies are present in serum of up to 90% of subjects with SS [165]. Antibodies against alpha-fodrin are detected in serum samples from patients with primary or secondary SS, especially in patients with sicca symptoms. However, anti-alpha-fodrin antibodies do not represent a sensitive nor a specific serological marker of SS [166]. Other novel tissue-specific autoantibodies are currently under investigation: autoantibodies against salivary protein 1 (SP-1), parotid secretory protein (PSP) and carbonic anhydrase 6 have been described in pSS and non-pSS patients with chronic pain, which may help to understand and diagnose early pSS and pSS-associated widespread pain syndrome in the future [167]. Anti-cofilin-1, anti-alpha-enolase and anti-RGI2 antibodies are associated with pSS MALT lymphoma [168]. Other autoantibodies have also been described to be more frequently found in pSS patients and variously associated with the clinical and biological characteristics of the disease [168]. Table 1 summarizes the novel autoantibodies that have been detected in pSS patients.
3.3.5. Damage of Salivary Acini Architecture
One of the pathomorphological characteristics of SG from SS patients is the presence of focal infiltration made of lymphocytic cells. The focus infiltrate is defined as the “focus score” and “focus score = 1” is a group of 50 or more lymphocytes per 4 mm2 of tissue [196]. SG infiltration is normally associated with destruction and fragmentation of the glandular tissue, acinar hyperplasia and replacement of acinar cells with fatty or fibrotic infiltrations [197]. These events lead to a deep modification and impaired function of the glandular tissue. An architectural disorganization of the epithelial cells has been described in the pSS: detachment of the basement membrane, alterations of the apical microvilli and disorganization of the tight junctions separating the apical and basolateral poles [198]. Several studies have shown that SS labial SG (LSG) display significant increase in proteolytic activity of matrix metalloproteinases (MMPs) and higher expression of MMP-3 and MMP-9 exclusively in acinar and ductal cells [199]. Some of the cytokines synthesized by the inflammatory cells, acinar and ductal cells of SS LSG can induce increased MMPs expression [108,200]. In turn, high MMPs expression triggers a high level of remodelling activity in the basal lamina that enhances the vulnerability of SGEC to direct contact with cytotoxic inflammatory cells [201]. The disorganisation of the basal lamina of acini and ducts of LSG from patients with SS is the most frequent modification observed that positively correlates with the number of inflammatory cells within the gland.
4. Clinical Manifestations
Although often reduced to its sicca syndrome due to its tropism for glandular tissue, pSS remains a systemic disease that can affect virtually all organs. These clinical manifestations can be due to various mechanisms: dryness secondary to exocrinopathy, autoimmune epithelitis with periepithelial lymphocytic infiltration of target organs, associated organ-specific autoimmunity with specific autoantibodies, systemic manifestations linked to the presence of immune complexes or cryoglobulinemia and clonal lymphocytic expansion. Three-quarters of pSS patients will have at least one extraglandular manifestation, ranging from mild inflammatory arthralgia to life-threatening manifestations. The clinical manifestations can occur at diagnosis or during follow-up, even after more than 10 years, which must justify careful monitoring of patients. In general, the manifestations due to lymphocytic infiltration around an epithelium of a target organ have a stable and indolent course (e.g., sicca syndrome, renal tubular acidosis, pulmonary involvement) while the autoimmune disorders linked to immune complexes or autoantibodies have a more unpredictable course, with flares and remissions.
4.1. General Manifestations
More than half of pSS patients report disabling fatigue and non-restful sleep [202], partly related to poor sleep quality due to dryness, night pain and an increased prevalence of obstructive sleep apnoea [203]. Low-grade fever is found in 6% to 41% of pSS patients [204], while periodic fever is found more anecdotally [204]. Weight loss and night sweats may also be due to the systemic activity of the disease, autonomic involvement or lymphoma development. B symptoms—the triad of fever, night sweating and weight loss classically described in lymphomas—are found only in 15% of low-grade lymphomas associated with pSS [205].
4.2. Ocular Manifestations
Dry eye is a classic manifestation of pSS, part of the sicca syndrome affecting more than 95% of pSS patients. Patients can report inability to tear, foreign-body sensation, conjunctival inflammation, eye fatigue and decreased visual acuity. Ocular dryness can be complicated by keratoconjunctivitis sicca, blepharitis, bacterial keratitis or corneal ulcer [206]. Uveitis, episcleritis and orbital pseudotumor are rare but possible systemic manifestations [207].
4.3. Stomatologic Manifestations
Lymphocytic infiltration of SG generates exocrinopathy with hyposialia responsible for soreness, adherence of food to the mucosa, dysphagia, difficulties in speaking or eating, dental caries, tooth loss, periodontal involvement, lip dryness and nonspecific ulcerations and aphthae [206,208]. Oral candidiasis and angular cheilitis are mycotic complications related to the loss of antimicrobial action of saliva [209]. Parenchymal involvement can be complicated by recurrent parotid enlargement of infectious, lithiasic, inflammatory or lymphomatous origin [210]. SG may be the site of bilateral multicystic parotid masses and lymphoma.
4.4. Musculoskeletal Manifestations
Joint inflammatory manifestations are, after sicca syndrome, the most frequent manifestations of pSS (50% of patients) [211]. Patients may have arthralgia with inflammatory characteristics (morning stiffness > 30 min) or less frequently true symmetric polysynovitis mimicking rheumatoid arthritis (RA). Joint involvement of the pSS is generally moderate (<5 affected joints) and preferentially affects the small joints of the hands and upper limbs [211,212]. Joint involvement is conventionally non-erosive—except in case of an overlap with RA—but can be deforming (Jaccoud arthropathy) [211]. More rarely, pSS can be responsible for myositis. Finally, widespread pain is frequent—nearly 50% of pSS patients—resembling primary fibromyalgia [213,214].
4.5. Neurological Manifestations
Neurological manifestations of pSS are relatively frequent (18–45% of patients) and affect both the central and peripheral (sensitivomotor and autonomic included) nervous systems, with a higher prevalence of peripheral manifestations [215].
The peripheral manifestations are polymorphic and can be differentiated according to electromyographic examinations in mixed polyneuropathy, axon sensory polyneuropathy, sensory ataxic neuronopathy, axon sensorimotor polyneuropathy, pure sensory neuronopathy, mononeuritis multiplex or rarely chronic demyelinating polyradiculoneuropathy. The mechanisms mentioned are mainly lymphocytic infiltration of the dorsal root ganglia (for sensory ganglioneuronopathy), vasculitic lesions of the vasa nervorum and/or the presence of axon-specific autoantibodies. The cranial nerves can also be involved, essentially the trigeminal nerve by involvement of the Gasser ganglion (associated or not with a more extensive ganglionopathy) and the facial nerve (uni- or bilateral paralysis). The other cranial nerves are affected anecdotally. Finally, damage to non-myelinated fibres can be responsible of autonomic neuropathy or small-fibre neuropathy.
In the central nervous system, pSS may be responsible for encephalic or spinal manifestations, with stroke-like or Multiple Sclerosis-like damage secondary to cerebral vasculitis. Some demyelinating manifestations combining myelitis and optic neuritis are part of an associated neuromyelitis optica spectrum disorder (NMOSD), a condition linked to the presence of anti-aquaporin 4 autoantibodies. Neuro-pSS can also manifest as a recurrent aseptic lymphocytic meningitis. Rarely, the association of upper and lower motor neuron diseases resulting in an amyotrophic lateral sclerosis-like syndrome has been described during pSS.
Finally, cognitive dysfunction (“brain frog”), restless leg syndrome and psychiatric abnormalities are classically linked to pSS, but it is not clear whether these manifestations are reactive or directly linked to the pathophysiology of the disease.
4.6. Pulmonary Manifestations
The prevalence of clinically significant lung disease in pSS is 9–20% although subclinical manifestations can be found in more than 50% of patients by CT-scan or bronchoalveolar lavage findings. pSS exocrinopathy also affects the lower airways causing coughing, tracheobronchitis sicca, bronchial hyperresponsiveness (mimicking late-onset asthma), cylindrical bronchiectasis and bronchiolitis (mainly follicular bronchiolitis). This involvement of the small airway epithelium is rarely responsible for an obstructive ventilatory syndrome (11–14%) but can be complicated by recurrent pulmonary infections or atelectasis [216,217].
Nonspecific interstitial pneumonia (NSIP) and usual interstitial pneumonia (UIP) are the most frequent interstitial lung diseases (ILD) patterns during pSS, corresponding to 45% and 16% of cases respectively. Lymphocytic interstitial pneumonitis (LIP) arrives in 3rd position (15% of ILD cases) and can be considered as a more specific benign diffuse lymphoproliferative disorder of pSS, probably starting from the follicular bronchiolitis. It must be differentiated from pulmonary lymphoma, which is found in 2% of pSS-ILD. Other patterns such as organizing pneumonitis are less frequent (11%) or even rare such as pulmonary amyloidosis, alveolar haemorrhage, Langerhans’ histiocytosis, cavitary lung disease and/or combined pulmonary fibrosis and emphysema syndrome. However, presence of multifocal cysts on CT-scan should raise clinical suspicion for pSS-ILD [211,216,217].
4.7. Dermatological Manifestations
Cutaneous involvement in pSS is relatively common and multiple manifestations are described such as xeroderma, eyelid dermatitis, annular erythema/subacute cutaneous lupus-like lesions and vascular purpura (caused by cutaneous vasculitis, urticarial vasculitis, cryoglobulinemia or hypergammaglobulinemic purpura of Waldenström) [211]. More rarely pSS can be responsible for cutaneous ulcer, livedo, erythema nodosum, panniculitis, amyloidosis or granuloma annulare [209].
4.8. Cardiovascular Manifestations
Raynaud phenomenon is the most frequent vascular manifestation, affecting 15% of patients [207]. Fortunately, cardiac manifestations such as pericarditis, pulmonary hypertension and cardiomyopathy are very rare, affecting <1% of pSS patients, respectively [207]. Cardiac rhythm disturbances have been described, secondary to ionic disorders, dysautonomia or direct impairment of the electrical conduction system of the heart [224,225].
4.9. Oeso-Gastrointestinal Manifestations
Dysphagia is a frequent complaint in pSS patients generally related to inadequate lubrication of the upper aerodigestive tract and food bolus resulting from hyposalivation. Oesophageal dysmobility is also mentioned in certain cases, explaining the lack of correlation between xerostomia and dysphagia [226,227]. Dyspepsia is frequent, occurring in 23% of pSS patients, and often linked to chronic atrophic gastritis where inflammatory infiltrates similar to those of the SG are found following tissue histological examination. Antibodies against parietal cells or intrinsic factor can be found, but pernicious anaemia remains rare [226]. Manifestations such as diffuse abdominal pain, diarrhoea or malabsorption can occur as part of a protein losing enteropathy or in case of overlap with Celiac disease [226,227]. Interestingly, pSS patients with Primary Biliary Cirrhosis overlap (PBC) are at higher risk of developing duodenal ulcers (85% of cases) [226]. The digestive tract can be the site of acute and serious complications in the context of cryoglobulinaemic vasculitis.
4.10. Pancreatic and Hepatobiliary Manifestations
The pancreas being an exocrine gland, it is not surprising to find cases of acute pancreatitis, chronic pancreatitis or pancreatic insufficiency in 0–7% of pSS patients. Moreover, 25% to 33% prevalence of chronic pancreatitis-like morphologic changes suggest that there are many asymptomatic cases [226]. Hepatomegaly is found in 10–20% of patients. Liver tests are disrupted in 10–50% of patients, usually mildly and with no particular clinical significance. pSS can be associated with Primary Biliary Cirrhosis (PBC)—another autoimmune epithelitis—or with autoimmune hepatitis (AH). Pseudolymphoma has been described to occur in liver like it may occur in salivary or lacrimal glands [226,227].
4.11. Uronephrologic Manifestations
Schematically, renal involvement linked to pSS can be divided into 3 groups: (1) tubulointerstitial nephritis linked to autoimmune epithelitis characterized by peritubular lymphocyte infiltration, (2) glomerulonephritis associated with immune complexes and (3) disorders linked to the presence of specific autoantibodies. According to different cohorts, about 5% of pSS patients have a renal involvement. However, this figure seems clearly underestimated if occult tubular involvement is systematically assessed [211,228].
Tubular involvement can be associated with dysfunction of any part of the renal tubule and can be responsible for polyuropolydypsic syndrome, low molecular weight proteinuria, aminoaciduria, euglycemic glycosuria, acidosis with normal anion gap, hypokalaemia that may be complicated by paralysis or disturbed heart rhythm, hypophosphoremia linked to increased phosphate excretion that may be complicated by osteomalacia, nephrocalcinosis or the formation of recurrent kidney stones [228,229]. More anecdotally, acquired Gitelman or Bartter syndrome has been described, possibly linked to the presence of specific autoantibodies targeting transporters (ie NaCl co-transporter in Gitelman syndrome) [228,230]. Glomerular disease occurs later in the history of the disease and most often corresponds to a mesangioproliferative glomerulonephritis (MPGN) caused by the deposition of immune complexes, usually cryoglobulinemia, which should be looked for [211,228].
Interstitial cystitis is a chronic inflammatory disease of the bladder that can be found in pSS patients. This rare manifestation is characterized by complaints such as pollakiuria, lower abdominal pain, urinary urgency, painful micturition, haematuria and dysuria [231]. Interstitial cystitis can be complicated by bilateral hydronephrosis and obstructive renal failure [231].
4.12. Haematological Manifestations
Anaemia is present in 20% of pSS cases, usually normochromic normocytic, of various mechanisms: anaemia of chronic disease or haemolytic, more rarely secondary to aplastic or pernicious anaemia or myelodysplastic syndrome [232,233]. Leukopenia is found in 15% of patients and most often corresponds to lymphocytopenia. Agranulocytosis is rare. Thrombocytopenia is found in 15% of patients, of peripheral origin, whether or not involved in Evans syndrome [232,233]. Rare cases of Thrombotic Thrombocytopenic Purpura (TTP) [234,235,236] and Hemophagocytic lymphohistiocytosis (HLH) [237] have been described.
Reactive multiple lymphadenopathy is possible, statistically associated with the presence of synovitis [212]. The intense stimulation of B cells explains the occurrence of hypergammaglobulinemia, hyperviscosity syndrome, monoclonal gammapathy, cryoglobulinemia and amyloidosis [232,238]. The formation of immune complexes leads to complement fraction consumption.
CD4-Lymphocytopenia is mainly found in anti-Ro-SSA positive patients and is associated with an increased risk of non-Hodgkin’s lymphoma (NHL) [232]. NHL has a prevalence of 4.3% in pSS patients [205]. Schematically, pSS-associated NHL can be divided into two main categories: the first has an indolent course and is dominated by the extranodal marginal zone (MZ) B cell lymphomas of MALT-type, and the second corresponds to the high-grade lymphomas such as de novo or secondary diffuse large B cell lymphoma (DLBCL). In pSS patients, MALT lymphomas are indolent diseases characterized by a good performance status, small tumour burden and infrequent B symptoms. They are preferably located in one or more extranodal sites such as SG, stomach, nasopharynx, lung, liver, kidney, orbit and skin [205]. It is interesting to note that almost all of these sites are organs involved in autoimmune epithelitis. Locoregional nodal involvement can be observed while bone marrow infiltration is rare. DLBCL are aggressive and have a poor prognosis. A certain proportion of them probably come from a transformation from a low-grade lymphoma. NHL mainly occurs in pSS patients with cryoglobulinemia, palpable purpura and C4 fraction consumption [205].
4.13. Ear–Nose–Throat (ENT) Manifestations
ENT complaints are common (40–50%) in pSS patients but objective fibroscopic abnormalities are less frequent (20%) [239]. Exocrinopathy can generate rhinitis sicca—reported by about 40% of pSS patients—which is a source of discomfort, nasal crusting, sinusitis, epistaxis or smell and taste disorders [240]. pSS patients are more likely to develop laryngopharyngeal reflux (LPR) because oesophageal involvement impairs anti-reflux mechanisms. LPR—in addition to pharyngitis sicca—manifests itself through various ENT complaints such as dysphonia, throat pain, chronic throat clearing or Eustachian tube dysfunction [241].
As with other systemic vasculitides, pSS may be responsible for sensorineural hearing loss or chondritis [242], responding to corticosteroid treatments. In an appealing way, pSS is associated with a sensorineural hearing loss in a significant proportion of patients, mainly affecting high frequencies, but whose clinical impact is not obvious [243].
4.14. Gynaecological and Obstetrical Manifestations
pSS does not have a negative impact on fertility, but chronic pain and vaginal dryness can be the cause of dyspareunia having a negative impact on the sexuality of female patients [244]. During pregnancy, pSS can be responsible for two rare but classic manifestations: autoimmune congenital heart block and neonatal lupus [245,246,247]. These two manifestations are linked to the transplacental passage of anti-Ro/SSA autoantibodies. Congenital heart block occurs in 2% of anti-Ro/SSA positive pregnancies but with a 10 to 20% risk of recurrence in subsequent pregnancies. More rarely, neonatal lupus can be associated with endocardial fibroelastosis, valvular malformations or septal defects. Neonatal lupus—affecting one fifth of anti-Ro/SSA positive pregnancies—is characterized by an erythematous rash and photosensitivity that can be associated with hepatic, haematological and neurological involvement. Compared with healthy pregnancy, patients with pSS had significantly higher chance of pregnancy loss or neonatal death. However, there were no significant associations between pSS and premature birth, spontaneous or artificial abortion or stillbirth [248]. These data should be taken with caution because they are based on a limited number of heterogeneous—and not necessarily recent—studies.
5. Diagnosis Workup
5.1. Diagnosis Versus Classification Criteria
Faced with one or more compatible manifestations, the diagnosis of pSS must be evoked and investigated. Making a diagnosis is the basis of medical care. For the patient, it represents the end of questioning and diagnostic wandering. For the physician, the diagnosis makes it possible to clarify the management. Finally, for the researcher, the diagnosis makes it possible to create homogeneous groups around a consensus definition. Unfortunately, there is no single diagnostic test to confirm the diagnosis of pSS. Due to its protean and willingly insidious presentation, pSS is sometimes difficult to recognize and may delay diagnosis by more than 10 years. Sicca syndrome, fatigue and unspecific musculoskeletal pain can be wrongly taken for manifestations of age, anxio-depression or perimenopause in people with pSS. Systemic manifestations can sometimes precede sicca syndrome, resulting in an “occult pSS” [249]. For these various reasons, the gold standard for individual diagnosis of pSS remains the opinion of an expert clinician. To allow the study of the disease in groups of pSS patients, several consensuses have defined classification criteria allowing a common definition of what pSS is. The 3 most recent sets of classification criteria are presented in Table 2. By definition, classification criteria are specific but may lack sensitivity and should not be used blindly as diagnostic criteria but as a guide in clinical practice.
5.2. Sicca Syndrome and Glandular Assessment
The investigation for objective dysfunction of the salivary and lacrimal glands is useful for the diagnosis and symptomatic management of the patient. Anatomical or functional imaging can be used to assess changes in the major SG during pSS.
The evaluation of dry eyes requires a simple ophthalmological examination. The Schirmer test consists of positioning a small strip of filter paper inside the inferior fornix of each eye. The eyes are then closed for 5 min. After this time, the strips are removed, and the amount of tears absorbed by capillarity is measured in millimetres from the edge of the strip in contact with the ocular surface. Dryness is significant if ≤5 mm/5 min. The evaluation then continues with the evaluation of the stability of the tear film by the Break-up Time (BUT) and the search for conjunctival or corneal lesions linked to dryness (keratoconjunctivitis sicca). These various tests use the slit lamp and the ocular instillation of dyes. BUT is measured by placing a drop of fluorescein in each eye and measuring the time during which the coloured tear film uniformly covers the ocular surface, before the appearance of dry spots. A tear BUT test of less than 10 s (averaged over 3 testings’) is considered pathological but is not specific of pSS manifestations. Finally, damage to the conjunctiva and cornea is highlighted by ocular surface staining techniques (fluorescein and lissamine green) [253]. The anomalies are scored using standardized scores: van Bijsterveld scale or the SICCA Ocular Staining Score (OSS). Respective cut-offs of ≥4 and ≥5 correspond to pathological situations suggestive of pSS. Those tests are more specific of pSS than Schirmer and Break-up time tests. Rose Bengal dye is no longer used because of its poor tolerance and local toxicity.
The evaluation of hyposalivation can be easily performed by sialometry. In its simplest form, sialometry consists of measuring the Unstimulated Whole Salivary Flow rate (UWSF) and the Stimulated Whole Salivary Flow rate (SWSF). UWSF is performed by asking the patient—fasted for minimum 2 h—to passively drain all the saliva produced in a tared jar for 15 min. The jar is then weighed and the saliva volume estimated. UWSF less than 0.1 mL/min is considered pathological (normal range 0.3–0.4 mL/min). UWSF represents a minor classification criterion. SWSF is measured in the presence of mechanical stimulation. SWSF can be measured using the Saxon test or Gum test protocols. Saxon test is performed by asking the patient to chew for 2 min a tared compress which will then be weighed. Gum test is performed as USWF, but in this case, the patient chews chewing gum and then spits saliva in a container. A diagnosis of hyposalivation is made if SWSF is ≤0.5–0.7 mL/min (normal range 1.5–2.0 mL/min). It is also possible to measure the salivary flow specific to each major SG by aspiration or cannulation. However, these techniques are of little use to the rheumatologist and especially uncomfortable for the patient.
Radiosialography is an X-ray imaging technique requiring the retrograde injection of a contrast solution into the excretory ducts of the major SG. This technique indirectly highlights glandular damage by studying changes in the “tree structure” of the excretory ducts [254]. Given the invasive nature and the complications of this technique, it has been abandoned in favour of other non-invasive techniques.
SG scintigraphy (SGS) studies the uptake, the concentration and the basal or stimulated secretion of a radioactive tracer by the parotid and submandibular glands following an infusion of Technethium-99 pertechnetate. SGS interpretation is mainly based on Schall’s classification [255], a qualitative score classifying anomalies in 4 grades—from grade 1 (normal) to grade 4 (the total absence of uptake and mouth activity). With ≥3 as cut-off, sensitivity and specificity are 54–87% and 78–98%, respectively [256]. Salivary scintigraphy is one of the classification criteria of 2002 for pSS but has disappeared from the most recent classification criteria of 2016. An abnormal scintigraphy makes it possible to objectify a dysfunction of the SG but does not allow etiological diagnosis as no image is specific of pSS. However, it may be of interest for treatment: if the examination shows SG with normal uptake but with a major dysfunction of excretion (possibly due to an autonomic disorder), the patient could benefit from a sialagogue treatment. In case of a scintigraphy demonstrating no uptake of the tracer, the parenchyma is probably totally destroyed and a sialagogue treatment will be useless.
Ultrasound is a simple, non-invasive way to assess the parenchyma of parotid and submandibular glands for diagnostic and prognostic evidence for pSS. Mode-B ultrasound using a high frequency linear probe allows characterization of size, homogeneity, presence of hypo-/anechoic areas, hyperechoic bands and clearness of SG borders. These different items were included in several diagnostic scores [257]. The OMERACT group, in an attempt to standardize, developed in 2019 a semi-quantitative scoring (0–3) based on the presence of hypoechoic/anechoic zones within the parenchyma of the parotid and submandibular glands [258]. A score ≥ 2 is abnormal and suggestive of pSS. At present, SG ultrasound (SGUS) is not part of classification criteria but may well be in the future [259]. Unfortunately, correlations between histological abnormalities (lymphocytic infiltration, diseased parenchyma or ductal ectasia/cysts) and SGUS lesions have not been corroborated [254]. SGUS scores improvement after treatment with Rituximab prove that part of the abnormalities are correlated with the disease activity and not only damage accrual [260,261]. To date, there is currently insufficient evidence to use SGUS as a prognostic or treatment response factor. Thanks to its high spatial and contrast resolution, low cost and accessibility, SGUS has replaced MRI in the diagnosis of the pSS patient.
5.3. Labial Minor SG Biopsy
The minor SG biopsy (MSGB) is a simple procedure that can be performed with little equipment. Several biopsy techniques have been described in the literature [262,263]. After disinfection, the reappearance of small drops of saliva makes it possible to identify the accessory SG at the level of the lateral third of the lower lip. The mucosa above these glands is anesthetized with an injection of lidocaine. The mucosa is then opened with a scalpel over 5–10 mm and the glands removed with forceps. The individualization and extraction of the glands is made easier by the hydrodissection that occurs during local anaesthesia and by the eversion of the lip. Lobules are herniated towards the surface of the wound by the application of pressure—digital or instrumental—on the external part of the lip. For quality concerns, the removal of 4–6 glands—allowing the study of minimum 8 mm2 of glands—is recommended [264]. A parotid biopsy is only exceptionally performed because technically more complex with a theoretical risk of damage to the facial nerve, for a diagnostic contribution identical to MSGB based on focus-score. On the other hand, the detection of lymphoepithelial lesions and early stage lymphomas—having a prognostic value—is more frequent/easier to detect on parotid biopsies [263].
The central element of MSGB pathology is the presence of clusters of more than 50 mononuclear cells (mainly lymphocytes) called foci. These foci in periductal or perivascular areas adjacent to normal acini are counted, reported to the area investigated and expressed as a Chisholm–Mason score [265] or a Focus-score [266]. Compared to the initial descriptions of those scores, some experts recommend counting all foci, including those associated with areas of fibrosis or atrophy, for fear of changing the Focus-score [264]. The Focus-score corresponds to the average number of foci per 4 mm2 of gland. It goes from 0 to 12, 12 corresponding by convention to the coalescence of the foci. The Chisholm score ranks chronic sialadenitis from 0 to 4. Grade 0 corresponds in the absence of infiltration; grade 1 corresponds to a slight infiltration of mononuclear cells, however not forming a focus; grade 2 corresponds to the presence of an infiltrate of mononuclear cells organizing in foci but whose density is <1 focus per 4 mm2; grades 3 and 4 correspond to the presence of 1 or > 1 focus per 4 mm2, respectively. The presence of focal sialadenitis characterized by a Focus-score ≥ 1 (Chisholm grade ≥ 3) is a major diagnostic argument for pSS and is included in the different classification criteria. Due to its sensitivity and specificity >80% and its significant positive predictive value [267], the presence of a chronic focal sialadenitis (Focus-score ≥ 1) is particularly useful in the diagnosis of early pSS, even with specific manifestations and autoantibodies negativity [249].
Although not part of the classification criteria, other anomalies can be described: fibrosis, acinar atrophy, ectasia or metaplasia of the excretory ducts, histiocytic granulomas, presence of germinal centre-like structures, lymphoepithelial or myoepithelial sialadenitis (LESA/MESA) [268,269]. LESA/MESA are characterized by lymphocytic infiltration of ducts and basal cell hyperplasia, resulting in a multilayered epithelium. In addition, pathology allows differential diagnosis with sarcoidosis, IgG4-related disease, amyloidosis and lymphoma. Finally, MSGB provides information on the patient’s prognosis: a Focus-score ≥3 and the presence of germinal centre-like structures or LESA/MESA are associated with more severe disease and an increased frequency of local and systemic manifestations, including lymphoma. For this reason, we recommend doing MSGB even if the diagnosis can be made based on anti-Ro/SSA positivity with objective sicca syndrome.
The parotid biopsy has fallen somewhat into disuse due to the ease of performing a minor SG biopsy with equivalent diagnostic performance. On the other hand, the possible discrepancies with MSGB [270,271], the possibility of early detection of lesions associated with a poor prognosis, the possibility of biopsying the same gland again to monitor the disease and the possibility of correlating it with SGUS semiology make parotid biopsy a tool that would need to be reassessed in the future [263].
5.4. Antinuclear Antibodies (ANA) Profile
The other major element in the diagnosis of pSS is the presence of anti-Ro/SSA and/or anti-La/SSB autoantibodies. The Ro/La system is a heterogeneous antigenic complex, composed by three different proteins (52kDa Ro, 60kDa Ro and La) and four small RNAs particles [272]. The search for antinuclear antibodies (ANA) by Immunofluorescence (IF) on HEp-2/HeLa cells is therefore an important element in the diagnosis of pSS. ANA is positive in 70% of pSS patients, usually with a fine speckled fluorescence [273]. Anti-Ro/SSA and/or anti-La/SSB autoantibodies are identified in 50–90% and 25–60% of patients, respectively [274]. It should be borne in mind that the Hep-2 cells do not sufficiently express Ro/SSA antigen, explaining the fact that 10% of patients anti-Ro/SSA-positive in ELISA have negative ANA in IF on HEp-2 cells [274]. Therefore, in case of suspicion of pSS, it is necessary to request the anti-Ro/SSA antibodies identification by ELISA, even in the presence of a negative ANA IF screening. Two types of anti-Ro/SSA autoantibodies can be differentiated: anti-Ro52 and anti-Ro60 [272]. Anti-Ro52/SSA have no specific ANA fluorescence staining pattern (might even exhibit a cytoplasmic pattern [274]), is precipitin negative and is not detected by ELISAs based on natural SSA/Ro. Ro52+ Ro60+ patients are likely to have pSS while Ro52+ Ro60- patients are not [275]. Isolated anti-Ro52/SSA positivity is statistically linked to primary myositis and systemic sclerosis. On the other hand, anti-Ro52/SSA and anti-La/SSB have the highest relative risks of congenital heart block in offspring from anti-Ro/SSA positive patients because these two antigens are expressed in foetal cardiac tissue from the 18th to 24th week [272]. Anti-La/SSB is mainly found in the presence of an anti-Ro/SSA, evoking a mechanism of epitope spreading. In only 2–3% of cases, pSS patients present with an isolated Anti-La/SSB antibody [276,277]. The presence of another ANA pattern or the identification of “atypical” ANAs can allow the identification of a secondary SS, an overlap with another systemic disease or a specific pSS subgroup [159]. The prognostic implication of these antibodies is discussed in the prognosis section.
5.5. Blood Workup
In addition to ANA testing, the initial blood workup for suspected autoimmune systemic disease includes a complete blood count; a coagulation profile with antiphospholipid panel; urea/creatinine dosage and urine sediment and 24-h urine protein or urine protein/creatinine levels; Na+/K+/HCO3−/Cl−/Uric Acid levels to investigate renal tubulopathy; hepatic enzymes levels; creatine phosphokinase (CPK) to investigate myositis; C3/C4/CH50 levels, Rheumatoid Factor (RF), Cyclic Citrullinated Peptide (CCP) antibodies, Coombs test; serum protein electrophoresis and total IgG, IgM and IgA levels to investigate presence of polyclonal hypergammaglobulinemia and/or monoclonal gammapathy; HCV serology; VDRL/TPHA; free T4 levels, TSH, anti-thyroid peroxidase, anti-thyroglobulin, anti-mitochondrial, anti-smooth muscle, anti-gastric parietal cell antibodies in case of associated auto-immune diseases. Hypergammaglobulinemia and lymphopenia are classically described during pSS. Their presence may be an additional argument, but their diagnostic performance is not known.
5.6. Sjögren’s Syndrome Differential Diagnosis
5.7. Primary versus Secondary Sjögren’s Syndrome
It is classic in medical nosology to describe the isolated and idiopathic form of a disorder as “primary” and to qualify as “secondary” the forms associated with specific causes or entities. SS is no exception. Historically, this dichotomy differentiated pSS patients from patients suffering from RA complicated by sicca syndrome. Subsequently, “secondary SS” (sSS) extended to other connective tissue diseases (e.g., SLE and Systemic Sclerosis (SScl)) and autoimmune diseases (e.g., primary biliary cirrhosis, thyroiditis and vasculitis) [279]. This nomenclature has also been indirectly “ratified” in AECG Classification Criteria from 2002 [250], classifying as “sSS” patients with another well-defined major connective tissue disease and at least one dry symptom (ocular or buccal) and 2 out of 3 signs of exocrine dysfunction (MSGB, SG signs or ocular signs in Table 2).
In light of current data, this dichotomy seems obsolete and should be reviewed. While polyautoimmunity and overlap syndromes are currently recognized, one can wonder why SS is still considered a second-class disorder.
Based on the examination of salivary gland biopsies of 34 RA patients with sicca symptoms, two phenotypes can be differentiated [280]. One group of patients presented a phenotype characterized by mild salivary gland lesions and negative autoantibody. Histologically, minor SG biopsies display increased prevalence of antigen-presenting cells and CD8+ T cells, decreased presence of B cells, and “non-activated” epithelial cells (based on the expression of HLA-DR and co-stimulation proteins D80/B7.1). A second group of patients presented a phenotype characterized by glandular manifestations and/or auto-antibodies positivity. Their minor SG biopsies demonstrated CD80/B7.1 overexpression and low frequency of S100+ cells, correlated with the positivity of anti-Ro/SSA autoantibodies and/or focus score ≥ 1. Both groups had an historical RA-sSS and an RA-pSS overlap, respectively. In this study, compared to RA patients without sicca symptoms, RA-sicca patients statistically present more Raynaud’s phenomenon, SG enlargement, palpable purpura and renal, lung and liver involvement. They displayed more frequent ANA, anti-Ro/SSA autoantibodies and RF positivity. The published data do not allow us to know if these manifestations are over-represented in the second group.
From a serohistological point of view, there is no difference in terms of anti-Ro/SSA positivity, anti-La/SSB positivity and SG infiltration between a pSS alone and an sSS associated with a SLE [281] or SScl [282]. It therefore seems more like an overlap than a so-called sSS. On the other hand, as for RA patients, SS overlap modifies the associated clinical phenotype. Compared with SLE-alone patients, patients with SLE-SS overlap are older and had a higher frequency of Raynaud’s phenomenon, anti-Ro/SSA positivity, anti-La/SSB positivity and rheumatoid factor. They also had a significantly lower frequency of renal involvement, lymphadenopathy and thrombocytopenia [281]. Compared with SScl-alone patients, patients with SScl-SS overlap seem less at risk of serious complications from SScl namely lung fibrosis, pulmonary artery hypertension and scleroderma renal crisis [282].
To summarize, “secondary SS” is to be banned from our vocabulary [283] or—at a pinch—redefined very restrictively for some exocrine involvement occurring in rheumatoid arthritis not corresponding to a real SS, if such an entity exists. Moreover, “secondary SS” has disappeared from the classification criteria of 2012 and 2016. The patient has or does not have (p)SS, which may or not be associated with other autoimmune diseases, reflecting common etiopathogenic pathways. In this way, the clinician avoids three pitfalls: (1) minimizing the SS-related symptoms, which decrease the quality of life of the patients; (2) forgetting that overlap may change the clinical phenotype and (3) forgetting the risk of lymphoma. Unfortunately, pSS overlap syndromes had been under-recognized, under-researched and possibly under-treated in the past because of the historical label of “secondary SS” and their exclusions from the majority of clinical trials [284]. Their management is therefore based on the clinician’s expertise, patient choices, best evidence and practice for the management of all associated diseases. To better individualize pSS in the future, it would be necessary to be able to move from a clinical definition to a molecular or even epigenetic signature.
6. Prognosis
Once the pSS diagnosis is made, treatment and medical decisions will be based on the expected course of the disease and its impact on the patient’s life. This burden can be summarized in “5D”: Death (mortality), Disease activity, Damage accrual, Discomfort (pain and sicca symptoms) and Disability. To assess the effect of therapeutic interventions on the natural history and functional repercussions of the disease, scores that can be used as clinical outcomes in trials have been developed.
6.1. Death
Although overall pSS mortality is low and similar to the general population [285], a subgroup of patients will have a poorer vital prognosis. The excess mortality observed in such subgroup of patients is generally attributed to the development of lymphoma or to uncommon but severe visceral involvement. The leading causes of mortality in pSS patients are cardiovascular events, followed by solid-organ and lymphoid malignancies and infections [285]. Risk factors associated with increased mortality are advanced age at diagnosis, male sex, parotid enlargement, abnormal parotid scintigraphy, extraglandular involvement, vasculitis, anti-SSB positivity, low C3 and C4 and cryoglobulinaemia [285].
pSS is associated with increased risks of overall cancer (pooled RR 1.17 to 1.88), non-Hodgkin lymphoma (NHL) (pooled RR 8.53 to 18.99) and thyroid cancer (pooled RR 1.14 to 4.03) [286,287]. Biomarkers associated with the development of lymphoma are mainly signs associated with exuberant B cell proliferation and immune-complex production [288,289,290]: parotid swelling, Focus-Score ≥3, germinal centre-like lesions, skin vasculitis or palpable purpura, complement consumption (Low C3, C4 or CH50), presence of cryoglobulinemia or monoclonal paraproteinemia, rheumatoid factor, increased β-2 microglobulin, lymphocytopenia, hypoglobulinemia, lymphadenopathy or splenomegaly and head and neck irradiation.
6.2. Disease Activity
Disease activity may be defined as the functional or structural changes in an organ related to inflammatory burden of the disease and are reversible under treatment. As in other inflammatory diseases, disease activity can fluctuate over time and progress between relapses and remissions. A significant proportion of pSS patients—nearly 50–70%—display a systemic manifestation at the time of glandular onset or within 6 months, mainly lymphadenopathy/splenomegaly, non-erosive arthritis and neurologic involvement [291]. The long-term study of the Antonius Nieuwegein Sjögren (ANS) cohort revealed that, within 10 years of diagnosis, 30.7% of the 140 patients included in this study developed an associated extraglandular or autoimmune manifestation such as polyneuropathy, interstitial lung disease, arthritis, discoid or subacute cutaneous lupus erythematosus (LE) and Hashimoto’s disease [292]. The presence of cryoglobulinemia is associated with an increased risk of developing a systemic manifestation [211,292]. On the other hand, presenting widespread pain seems to be a “protective phenotype” [292].
Currently the European League Against Rheumatism (EULAR) SS disease activity index (ESSDAI) score has been used to quantify the inflammatory systemic activity of the disease. Within ESSDAI, clinical or biological manifestations are classified as “low” (1 point), “moderate” (2 points) or “high activity” (3 points) in 12 domains. To calculate the ESSDAI score, the value of the highest level of activity for each domain is multiplied by the domain weight (1 to 6) and then added together. The maximum theoretical ESSDAI score is 123. Minimal clinically important improvement was defined as an improvement of at least three points. More recently, ClinESSDAI score, a variant of the ESSDAI score without the biological domain, has also been used [293] (Table 4).
However, it should be borne in mind that (clin) ESSDAI score does not investigate all of the possible events related to pSS. Out of 6331 patients included in the international register “The Big Data Sjögren Project Consortium” [207], 1641 patients (26%) had at least one non-ESSDAI systemic manifestation on a predefined list of 26 organ-specific features not currently included in the ESSDAI classification. Patients with non-ESSDAI manifestations are patients with higher systemic activity than patients without non-ESSDAI manifestations (mean ESSDAI 10.3 vs. 5.5, p < 0.001).
Patients with significant systemic activity are generally patients with early onset disease, antinuclear antibodies (ANA) positivity with a higher frequency of anti-Ro/SSA (with or without anti-La/SSB), low C3, low C4 and cryoglobulinemia [154,276,277,298]. Children of anti-Ro/SSA positive mothers are at risk of specific neonatal complications such as neonatal lupus and congenital heart block [277]. Paradoxically, patients with higher disease activity are less disabled by sicca syndrome or widespread pain [276,277]. Conversely, patients with late-onset seronegative disease will mainly present a more disabling sicca syndrome but fewer systemic manifestations linked to the activity of the disease [277]. Finally, isolated anti-La/SSB positivity occurs in only 3% of pSS patients and is associated with an intermediate phenotype between Ro/SSA positive- and seronegative patients [277]. Thus, systemic complications could appear many years after initial pSS diagnosis and justify long-term surveillance, especially in cryoglobulinemia or “high risk” phenotype patients.
The immunological profile of pSS highlights the presence of atypical ANA—12% of cases [299]—or other specific autoantibodies. A subset of pSS patients with anti-centromere positivity develops a clinical phenotype overlapping between SS and systemic sclerosis with a higher age, more frequent Raynaud’s phenomenon and keratoconjonctivitis sicca and a lower proportion of anti-Ro/SSA and anti-La/SSB, rheumatoid factor, leukocytopenia and hypergammaglobulinemia [159,299]. In most cases, a minority of these patients appear to progress to an authentic systemic sclerosis. Anti-Cyclic Citrullinated Peptides (anti-CCP) positivity—present in 3–10% of patients—is associated with a greater frequency of joint manifestations or with overlap with rheumatoid arthritis (RA) [159,277]. The presence of anti-mitochondrial antibodies (1.7–13%) and anti-smooth muscle/anti-liver kidney microsomal antibodies (30–62%) is associated with overlap with primary biliary cirrhosis and autoimmune hepatitis [159].
6.3. Damage Accrual
Disease damage may be defined as the addition over time of irreversible functional or structural changes resulting from disease activity, iatrogenic treatments or co-morbidities.
Two scores exist to quantify damage related to pSS: SS Disease Damage Index (SSDDI) [296] and SS Damage Index (SSDI) [297]. SSDDI is composed of a list of 18 irreversible damages affecting 6 organ-domains (oral, ocular, neurologic, pleuropulmonary, renal and lymphoproliferative), divided into 9 items weighted for severity. SSDI is an unweighted checklist of 27 items divided into 3 lists: ocular damage, oral damage and systemic damage. Systemic damage is further subclassified into 7 areas: neurological, renal, pulmonary, cardiovascular, gastrointestinal, musculoskeletal and malignancy (Table 4).
In a retrospective study using 148 pSS patients attending the UCLH Sjögren’s clinic followed for 10 years, Krylova et al. revealed that 28.3%, 36.7% and 45% of patients displayed SSDI damage (excluding oral damage that was not assessed in the study) after 1, 5 and 10 years of disease, respectively [300]. Items most involved are in the ocular domain, parotid swelling and malignancy. These results suggested that pSS patients accumulate less damage—calculated on different scores—over time than lupus patients, who have a greater inflammatory burden and use of immunosuppressive treatments [300].
Another retrospective study using 155 pSS patients showed that the total increase of patients with damage was 28% after 1 year, 44% after 3 years, 74% after 5 years and 83% at 10 years, with a good correlation between SSDDI and SSDI [301]. More specifically, teeth loss and/or caries, salivary flow impairment, corneal ulcers and tear flow impairment were reported in 49.5%, 34%, 22.6% and 11% of patients, respectively. Unsurprisingly, systemic damage—observed in 13.5% of patients—was correlated with basal ESSDAI, low C4 and lymphopenia. In the same way, persistent SG swelling—detected in 14% of patients—was associated with (bio)markers of systemic activity and B cell proliferation (lower age at diagnosis, anti-Ro/SSA positivity, cryoglobulinemia, low C4, hypergammaglobulinemia and lymphopenia). Lymphoproliferative disorders were detected in 4.5% and malignancy in 9% of cases at 10 years post-diagnosis [301].
6.4. Discomfort and Disability
SS can be disabling and associated with significant functional status impairment related to oral and/or ocular dryness, systemic activity, pain, fatigue and daytime somnolence, anxiety and depression symptoms [302,303,304]. Objective assessments of sicca syndrome correlated poorly with symptoms and remain generally stable over time [305]. Besides the associated symptoms, sicca syndrome also has a negative impact on smell, taste, pruritus, voice, swallowing and sexual function [306,307]. Fatigue and pain are both correlated with reduced quality of life and psychological distress [307]. Patients with widespread pain—34.9% of the cohort—were more frequently negative for anti-La/SSB, more frequently seronegative for all autoantibodies (ANA/SSA/SSB/RF) and had statistically fewer extraglandular manifestations in a Dutch study including 83 patients [308]. Another Italian study on 100 pSS patients demonstrated a prevalence of widespread pain of 22%, a phenotype statistically associated with fewer systemic and immunological manifestations (hypergammaglobulinemia, rheumatoid factor, focus-score ≥ 1) [309]. A subset of pSS patients therefore seem to develop a clinical phenotype with lower visceral involvement but with significant morbidity linked to glandular manifestations and a significant psychosomatic burden [302,310], bringing them closer to the notion of “Sicca Asthenia Polyalgia (SAP) Syndrome” [311,312,313]. At diagnosis, one in 4 patients is unable to work. This figure increases to more than 1 in 3 at 1 year. Work disability at 2 years is 40% and is related to fibromyalgia pattern, age and incapacity for work at diagnosis [314]. pSS has a high individual and societal cost, especially due to dental cost, symptomatic therapies and disease compensation [307].
EULAR SS Patient Reported Index (ESSPRI) is a consensus index calculated as the mean of 3 visual analogue scales (VAS)—self-assessment of dryness, (limb) pain and fatigue—allowing easy measurement of patients’ symptoms in pSS [295]. By convention, patient-acceptable symptom state was defined by an ESSPRI <5/10 and the minimal clinically important improvement by a decrease of at least one point or 15%. The ESSPRI score is correlated with the Patient Global Assessment [PGA] [295] and with more complex and time-consuming scores such as the Profile of Fatigue and Discomfort [PROFAD] [295], Sicca Symptoms Inventory [SSI] [295], Health Assessment Questionnaire [HAQ] [315], Short Form 36 health survey [SF-36] [302], time trade-off values [TTO] and EuroQol5D VAS [316,317]. Very interestingly, a study using baseline data from 120 patients included in the TEARS study revealed that—even if there is a small correlation between ESSPRI and ESSDAI—ESSPRI is the only determinant associated with the quality of life score SF-36 in a multivariate model [318]. The ESSPRI score is therefore a good clinical screening and monitoring tool as well as a good surrogate endpoint to study the effectiveness of therapeutic interventions on pSS associated “Sicca Asthenia Polyalgia” Syndrome (Table 4).
It is therefore important, a fortiori in mild cases with low activity score but disabling sicca syndrome, to focus on improving the quality of life of patients through attentive and multimodal symptomatic management and to offer a multidisciplinary management program for the most disabled.
7. Therapeutic
Despite a better understanding of its pathophysiology, treatment of SS remains disappointing and essentially palliative. Systemic activity is treated by immunosuppressant drugs, based on scarce evidence. Manifestations linked to damage caused by local or systemic activity of pSS should be identified because they are by definition irreversible and cannot therefore be improved by immunosuppressive treatments. In the last 5 years, pSS management has been addressed by guidelines from EULAR [210], British Society of Rheumatology and National Institute for Health and Care Excellence (NICE) [319], Brazilian Society of Rheumatology [320], Research Team for Autoimmune Diseases [321] and Sjögren’s Syndrome Foundation [322]. The main principles for care are summarized below.
7.1. Sicca Syndrome and Non-Visceral Manifestations
Despite the dysimmune origin of the disease, no immunosuppressive treatment has demonstrated sufficient efficacy associated with a satisfactory risk–benefit balance in the treatment of sicca syndrome and non-visceral aspecific manifestations (non-inflammatory widespread chronic pain, fatigue). Treatment is mainly focused on symptom management and prevention or treatment of complications resulting from exocrinopathy (Table 5).
Therapeutic approach to oral dryness must be driven by baseline objective and subjective severity of hyposialia and xerostomia. To this end, current guidelines recommend evaluating baseline SG function by measuring unstimulated (UWSF) and stimulated salivary flow (SWSF) or using salivary scintigraphy. Subjective xerostomia impact is captured by a simple Visual Analogue Scale, as part of the ESSPRI score. EULAR guidelines propose an algorithmic approach to the management of dry mouth: patients with an UWSF < 0.1 mL/min are categorized based on their SWSF as mild (>0.7 mL/min), moderate (0.1–0.7 mL/min) or severe dysfunction (<0.1 mL/min). Self-care advice and non-pharmacological stimulation are proposed to mild cases as first line therapy [210]. Pharmacological stimulation (pilocarpine per os or as a mouthwash, cevimeline per os) is the treatment of choice in moderate cases (with residual SG function) or in mild dysfunction patients who failed to respond to basic recommendations, in addition to first line therapy. Saliva substitutes are reserved for patients with no residual function or as a third line treatment in non-responding patients.
The stomatological complications of exocrinopathy affecting the SG are cavities formation, periodontal disease, candida infections and glandular swellings linked to abscess or to a lithiasic disease. It is therefore strongly recommended that patient adopts impeccable dental hygiene and be evaluated at least 2 times per year by a dental professional. Local fluoride-based treatments can be administrated. Candida simple infection (visible white plaques) are treated with Nystatin mouthwash for 7 days. One-week prophylactic treatment may be repeated every 8 weeks in the event of recurrence. Erythematous infection of tongue or oral cavity is treated with Fluconazole 50 mg for 10 days. Angular cheilitis is treated with Miconazole topically on each side of the mouth for 2 weeks. Presence of abscess or lithiasic involvement can be treated with antibiotic treatment and stomatologist involvement is indicated. If no infectious or mechanical cause is found in case of gland swelling, a distinction must be made between primary neoplasia, systemic activity of the disease (as scored in ESSDAI, treated by glucocorticoid in loco by sialendoscopy, per os or intra-muscular) and the appearance of a lymphomatous complication.
The management of dry eyes must also be guided by the objective and subjective severity of keratoconjunctivitis sicca (KCS), resulting from damage to corneal and conjunctival epithelium secondary to accelerated tear-film break-up and hyperosmolar tear composition. EULAR guidelines propose an algorithmic approach based on Ocular Staining Score (OSS) score and Ocular Surface Disease Index (OSDI) questionnaire to classify patients as non-severe or severe KCS [210]. The British Society of Rheumatology recommended a classification into 3 categories (mild, moderate and severe dry eyes) based on the Schirmer’s test, Break Up Time (BUT) and ocular staining [319]. First line therapy for all patients with dry eyes is the instillation of preservative-free artificial tears containing methylcellulose or hyaluronate, and ointment at night. In DREAM studies, use of supplements of n-3 fatty acids for 12 month and beyond does not improve OSDI, staining scores, BUT or Schirmer test compared to olive oil in dry eyes patients [323,324]. Although these treatments are not associated with an improvement in objective parameters, substantial subjective improvement in both groups suggests that daily olive oil teaspoon should be used in dry eye management [325]. Although the origin of the dryness is the decrease in the production of tears, a dysfunction of the Meibomian glands can also be associated and must be treated by daily eyelid massage with hot pad or liposomal spray to reconstitute the lipid layer preventing the evaporation of the tear film. In patients with persistent Meibomian inflammation and blepharitis, doxycycline 50 mg once daily for a minimum of 3 months is effective as a metallomatrix proteinase inhibitor. In case of refractory case of severe KCS, local treatment using NSAID-, glucocorticoid- or cyclosporin-containing eyedrops can be used under the strict supervision of an ophthalmologist. Rescue therapies by serum eye drops, oral or topical muscarinic agonists, lifitegrast-containing eyedrops or lacrimal plugs insertion must be evaluated in specialized settings.
Only two Disease Modifying Anti-Rheumatic Drug (DMARDs) have demonstrated a significant effect on sicca syndrome: Methotrexate in a small uncontrolled trial [326], and Mizoribine (a Japanese DMARD) in 2 cohort studies [327,328]. With regard to biological therapies, infliximab, etanercept, belimumab and tocilizumab have failed to demonstrate a favourable effect on exocrine glandular function in their respective RCTs. “Abatacept Sjögren Active Patients” (ASAP) proof-of-concept trial on abatacept showed a significant improvement in ESSPRI and BUT, but not on SWSF while another trial showed no effect on ESSPRI and SWSF. Some randomized trials, but not all, find an improvement in exocrine function and dryness with rituximab. In TEARS study, a study using 120 patients, aims for a >30% improvement in at least 2 VAS in 4 (fatigue, pain, dryness and PGA) at 6–16–24 weeks, primary endpoint is only reached at week 6, and this effect is no longer found thereafter. Dryness VAS is statistically different from the placebo group from week 6 to 24, but no group achieved a clinically significant decrease. The other large trial, TRACTISS, studying the effect of rituximab on 133 patients with a primary endpoint of >30% improvement oral dryness and fatigue VAS at 48w, did not show significant improvements in any outcome measure, except unstimulated salivary flow. However, this intervention does not seem cost-effective. The clinical significance of those differences remains to be determined and is interpreted according to the various guidelines. Only the Sjögren’s Syndrome Foundation proposes to use rituximab as rescue therapy for sicca syndrome [322].
In pSS patients, complaints regarding general non-specific symptoms (non-inflammatory musculoskeletal pain and fatigue) mimicking a fibromyalgia picture are common and can be challenging for the clinician. In this context, differential diagnosis is important. Non-specific manifestation of another condition (e.g., hypothyroidism, hypocortisolism, osteoarthritis, depression, neoplasia) or resulting from a misleading manifestation linked to the systemic activity of the disease (e.g., myositis, inflammatory arthralgia or arthritis, hypokalaemia or osteomalacia due to tubular involvement, small fibre neuropathy or lymphoma) must be ruled out. When no secondary cause is identified, this fibromyalgia-like presentation can be treated as such [329]. These can be quantified and monitored using the ESSPRI score or standardized scores such as the Profile of Fatigue and the Brief Pain Inventory. Education and management according to the biopsychosocial model of chronic pain, lifestyle adaptation, sleep management strategies and the practice of moderate physical activity are the cornerstones of the management of fatigue and pain. Many patients report benefit from joining a SS support group. If drug treatment is necessary, it will consist of the prescription of conventional painkillers (short-term acetaminophen or NSAID). Antidepressants and anticonvulsants may be considered as co-analgesic medications in chronic musculoskeletal or neuropathic pain, keeping in mind the anticholinergic effect of these drugs, which can worsen sicca syndrome. Opioids are not suitable treatments for chronic pain patients. DHEA supplementation is not recommended.
As a rule of thumb, systemic immunomodulatory drugs should not be used to treat non-specific systemic manifestations because evidence is scarce. In currently available biotherapies, abatacept and belimumab failed to demonstrate an effect on fatigue and pain VAS. Data on rituximab are conflicting: 3 RCTs showed an improvement in fatigue VAS, results not found in the large TRACTISS trial. A phase 2 RCT on a total of 17 patients failed to demonstrate >20% improvement of fatigue VAS at 24 weeks, fatigue VAS improvement at 24w or >30% improvement of fatigue VAS at 24w. The authors only report a statistically significant improvement in fatigue VAS in treated group compared to baseline, while the placebo group did not reach a statistically significant difference [330]. In two other studies, patients with early pSS and active disease treated with RTX displayed a significant improvement in fatigue VAS compared to placebo from different time points post-treatment [331,332]. All RCTs have shown that rituximab is not associated with an improvement in pain VAS. An RCT investigating the effect of anakinra on fatigue, although not reaching its primary endpoint, shows a significant improvement in VAS fatigue [333]. Off-label use of DMARD or biological treatments, even as a rescue therapy, is currently not mainstream recommendation in this indication. However, some guidelines suggest a trial of hydroxychloroquine in patients with recurrent musculoskeletal complaints or fatigue, mainly based on “experience-based medicine”. In its 2015 guidelines, Sjögren’s Syndrome Committee of Brazilian Society of Rheumatology highlighted the possibility of using rituximab as rescue therapy for fatigue (but not sicca syndrome) management [320].
7.2. Systemic Manifestations
Management of visceral manifestations linked to disease systemic activity is currently based only on rare randomized controlled trials, cohort studies or case-reports [334]. Treatment regimens are often borrowed from systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), mixed cryoglobulinemia or idiopathic organ-specific autoimmune disease management.
Therapeutic regimen must be tailored to organ specific involvement and severity of the disorder. This approach requires organ-by-organ examination of disease activity and pre-existing damage. To this end, ESSDAI score may be used as a guide but does not take into count all the systemic manifestations of pSS [210]. As a rule of thumb, systemic immunosuppressive therapy will only be offered to patients with moderate or severe organ activity (as define in ESSDAI score) or moderate overall systemic activity (ESSDAI ≥5) [210]. Organ manifestation classified as mild usually requires only self-care advice, local treatment or pain relief medication (NSAID for inflammatory arthralgia or co-analgesic for neuropathic pain). In case of treatment failure, low-dose corticosteroid treatment and/or conventional DMARD may be considered depending on clinical manifestation.
In cases requiring immunosuppressive therapy, an induction/remission biphasic regimen is recommended for the rapid control of organ damage and the preservation of its function [210]. Corticosteroid therapy is an almost essential treatment for moderate to severe systemic manifestations. To date, no steroid-free regimen has been studied in pSS and 95% of the published regimens include corticosteroid therapy, alone or in combination with an immunosuppressant [210]. When immunosuppressive therapy is prescribed, it is usually a conventional broad-spectrum immunosuppressant used as a cortisone-sparing or as a remission-inducing agent: hydroxychloroquine, methotrexate, other conventional DMARDs (leflunomide, salazopirine), mycophenolate mofetil or cyclosporine. As there are no head-to-head comparisons, the choice of immunosuppressant is mainly based on the clinician’s experience and on the therapeutic regimens used in idiopathic or lupus-related disorders (HCQ and MTX in skin and articular involvement, AZA, CyA or MMF in pulmonary or renal involvement). Severe life- or organ-threatening manifestations (central nervous system involvement, glomerulonephritis), generally require an aggressive regimen including methylprednisolone pulse-therapy combined with an alkylating agent (usually cyclophosphamide IV or PO, more rarely chlorambucil) as remission-inducing agents. IVIG at immunomodulatory doses are used in neuropathies or myositis. Biological therapies (mainly rituximab) generally come only in the third line as rescue therapies. The exception to this rule concerns the manifestations associated with cryoglobulinemia where rituximab is proposed as an immunosuppressant of choice, in combination with corticosteroid therapy or even plasmapheresis in life-threatening cases. As with other autoimmune diseases, corticosteroid therapy should be reasoned with a tapering regimen guaranteeing the shortest possible exposure to supraphysiological doses while maintaining remission. Complications of chronic corticosteroid therapy must be addressed proactively.
Hydroxychloroquine is commonly used as first line DMARD for moderate systemic manifestations mainly affecting the skin and joints. Its use is mainly based on the similarities between pSS and SLE, as pSS is sometimes considered as “lupus of mucous membranes”. As opposed to SLE, the evidence for its use in systemic manifestations of pSS does not actually exist, and its use is completely empirical. The first—JOQUER trial—attempting to demonstrate the effect of hydroxychloroquine over 24 weeks failed to reach the primary endpoint (30% or greater reduction between weeks 0 and 24 in scores on 2 of 3 VAS (dryness, pain, and fatigue)) [335]. In a more recent RCT performed over 2 weeks, no effect of hydroxychloroquine was seen on BUT test, Schirmer test, corneal staining score or OSDI score [336]. While those RCT have not been designed to investigate the effect of the drug on systemic manifestations of the disease, and the number of patients was small, hypergammaglobulinemia statistically improved significantly [335,336].
7.3. pSS-Associated Lymphoma
The occurrence of lymphoma is a complication that must be screened clinically, especially in patients at risk (see above). Any appearance of a firm, painless glandular swelling must be investigated if it does not disappear spontaneously. The exams of choice to detect lymphoma are an MRI of the major SG and a CT of chest, abdomen and pelvis for staging or a PET scan to investigate the entire body in a single examination. pSS patients with lymphoma require personalized treatment provided by an oncohematologist according to the histological type, the extent of the involvement and the systemic manifestations.
7.4. Obstetrical Considerations
Ideally, pSS patients of childbearing age should benefit from a preconception consultation aimed at reviewing their treatment and their serological profile (anti-Ro/SSA, anti-La/SSB and antiphospholipid panel). Low-dose aspirin can be considered to promote placental implantation [319]. Anti-Ro/SSA positive mothers should be followed regularly by foetal ultrasound in a specialized centre [210,319]. Prophylactic treatment of neonatal atrioventricular block with hydroxychloroquine may be offered, since this drug is compatible with pregnancy [210]. If a conduction disorder appears on a follow-up ultrasound, rescue therapy with glucocorticoid with or without IVIG may be attempted [210]. In the event of atrioventricular block at birth, a pacemaker must be quickly implanted.
7.5. Targeted Therapies: Revolution or Disillusion?
Targeted therapies have revolutionized Rheumatology in recent years, especially in chronic inflammatory rheumatism—such as in RA—and, to a lesser extent, systemic diseases such as SLE and vasculitis. In terms of pSS, many targeted therapies have been tested or are currently in the pipeline. Unfortunately, a revolution like the one known in the field of RA has not yet occurred. These targeted drugs are shown in Figure 4 and summarized in Table 6, Table 7, Table 8, Table 9, Table 10, Table 11 and Table 12.
Given their predominant role in the production of autoantibodies, germinal centres and the evolution towards lymphoma, B cell depletion is one of the therapeutic mechanisms studied in pSS (Table 6, Table 7 and Table 8). In addition to the mixed results of the anti-CD20 Rituximab RCTs, other targeted drugs have been studied. Epratuzumab, an anti-CD22 B cell depleting therapy studied in SLE patients had a positive effect on the systemic activity of SLE patients with Sjögren syndrome in a post-hoc analysis of EMBODY trial [337]. However, an RCT should be designed to assess the effect of the therapy on both ESSDAI and ESSPRI in pSS patients. Other B cell depletion strategies aiming at blocking the BAFF pathway showed a positive effect on the ESSDAI and ESSPRI scores at 28–52 weeks [338,339]. However, the confirmation of these promising results against a placebo is necessary. Other strategies targeting BAFF pathway are also under investigation: a TACI-antibody fusion protein called RC18, rituximab + belimumab combo therapy, Tibulizumab—a dual anti-BAFF (belimumab) and anti-IL-17 antibody (Ixekizumab)—and Ianalumab (anti-BAFF receptor). The results of these different studies are expected during 2020. B cell targeting drugs by Bruton tyrosine kinase inhibitor (4 molecules), LTßR fusion protein, PI3Kδ inhibitor (3 molecules) and Cathepsin S inhibitor are currently being evaluated with inconclusive results to date. Bortezomib, a proteasome inhibitor used for the treatment of multiple myeloma, has been successfully used in 2 cases of refractory pSS reports but has never been studied on a larger scale [236,340].
T-cells play a central role in the modulation and polarization of the local autoimmune reaction within lymphocyte infiltrates in the exocrine glands. They are also used as therapeutic target by biotherapies interfering with the T-cell co-stimulation (Table 9). To date, there is no convincing result to recommend these treatments in pSS, but most studies targeting the CD40-ligand (CD154)/CD40 pathway are in progress. Therapies targeting T-cell trafficking, such as Fingolimod or Natalizumab, have not been studied in pSS.
With regard to anti-cytokine targeted therapies, RCTs using anti-TNF (infliximab and etanercept) and anti-IL6 receptor (tocilizumab) are negative (Table 11). Anakinra demonstrated a statistically significant decrease in fatigue VAS, without however reaching its primary clinical endpoint. The development of GSK2618960—an anti-IL-7Rα biotherapy—was stopped by the company due to the prioritization of their portfolio. So far, only one RCT studying the effect of Ustekinumab—an anti-IL-12/IL-23 antibody—on ESSDAI score at week 24 as primary endpoint is expected to give results in 2022 [341].
In a phase II trial, Filgotinib—a Jak1 inhibitor—and Lanraplenib—a SIK inhibitor—failed to demonstrate a significant effect on the ESSDAI and ESSPRI scores [342]. Finally, innovative therapies targeting plasmacytoid dendritic cells, immune complexes by RNase1-Fc fusion protein or the induction of T-reg cells by low-dose IL-2 injections are being evaluated. These various therapies are reviewed in Table 12.
8. Conclusions
pSS is a multifaceted disease combining pleiomorphic systemic autoimmune manifestations, glandular manifestations, a frequently added psychosomatic component and the possible progression to non-Hodgkin lymphoma. Its management has two complementary facets: improving the quality of life of patients by tackling dryness, fatigue and chronic pain symptomatically in a multidisciplinary way and treating systemic manifestations to prevent damage, which will worsen the vital and functional prognosis. Although we understand more and more its pathophysiology, many questions remain unanswered, and its treatment remains disappointing compared to other autoimmune diseases. pSS therefore remains a vast field of investigation where much fundamental and clinical research remains to be done. Ten take-home messages:
- SS is characterized by lymphoplasmacytic infiltration of exocrine glands. The cause of SS is complex and influenced by a combination of genetic, epigenetic, hormonal and environmental factors.
- The pathogenic mechanisms remain unclear. However, the immune system-mediated loss of glands function, specifically of salivary and lacrimal glands, certainly explains the common symptoms of dry mouth and dry eyes. In this inflammatory environment, T-cells mediate a direct destruction of glandular tissue and B-cell activation, leading to the production of autoantibodies. More than 20 autoantibodies could be involved in SS, but the most commonly used for SS diagnosis are anti-Ro/SSA and anti-La/SSB.
- Although often reduced to its sicca syndrome due to its tropism for glandular tissue, pSS remains a systemic disease that can affect virtually all organs. These clinical manifestations can be due to various mechanisms: dryness secondary to exocrinopathy, autoimmune epithelitis with periepithelial lymphocytic infiltration of target organs, autoimmunity and clonal lymphocytic expansion.
- Due to its protean and willingly insidious presentation, pSS is sometimes difficult to recognize and may delay diagnosis by more than 10 years. Classification criteria are used to create cohorts for study purposes and should not be used blindly as diagnostic criteria but as a guide in clinical practice. For these various reasons, the gold standard for individual diagnosis of pSS remains the opinion of an expert clinician.
- From a serohistological point of view, so-called “secondary Sjögren’s syndrome” in SLE and SScl patients does not differ from pSS. It is therefore preferable to forget this historical dichotomy. In this way, the clinician avoids three pitfalls: (1) minimizing the SS-related symptoms, which decrease the quality of life of the patients; (2) forget that overlap may change the clinical phenotype and (3) forget the risk of lymphoma.
- Although overall pSS mortality is low and similar to the general population, a subgroup of patients will have a poorer vital prognosis linked to cardiovascular events, solid-organ and lymphoid malignancies and infections. Biomarkers associated with the development of MALT lymphoma are mainly signs associated with exuberant B cell proliferation and immune-complex production.
- The impact of pSS can be assessed according to three clinical dimensions: “sicca asthenia polyalgia” complex, inflammatory disease activity and structural damage. They are assessed by the ESSPRI, ESSDAI and SSD(D)I scores, respectively. Even in the absence of florid systemic manifestations, pSS can be disabling and associated with significant functional status impairment related to oral and/or ocular dryness, systemic activity, pain, fatigue and daytime somnolence, anxiety and depression symptoms.
- The treatment of manifestations linked to the “sicca asthenia polyalgia” complex mainly involves symptomatic measures and rehabilitation. To date, no immunosuppressant has demonstrated a favourable risk–benefit balance in this indication.
- The treatment of manifestations related to inflammatory disease activity is currently based on scarce evidence. Therapeutic regimen must be tailored to organ specific involvement and severity of the disorder. Mild manifestations will be treated with hydroxychloroquine or local corticosteroids while moderate to severe systemic involvement will require the use of systemic corticosteroid therapy, combined or not with a broad-spectrum immunosuppressant. Rituximab will only be used as a third line, except in cases of cryoglobulinemia where it is the treatment of choice.
- Despite targeted therapies having revolutionized rheumatology in recent years and the impressive number of molecules tested so far in pSS, a revolution like the one known in the field of RA has not yet occurred.
Author Contributions
D.P., C.C., J.P., M.S.S. and C.D. contributed to the writing of the review. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded the EU H2020 contract HarmonicSS (H2020-SC1-2016-RTD/731944), Fonds Erasme, Fonds de la Recherche Scientifique—FNRS (grant J.0053.20). DP is a Research Fellow of the Fonds de la Recherche Scientifique—FNRS.
Acknowledgments
The authors thank Bahija Jellouli for her secretarial help.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
| ACA | Anti-centromere antibodies |
| ACPA | Anti-citrullinated protein antibodies |
| ACR | American College of Rheumatology |
| AECG | American European Consensus Group |
| AH | Autoimmune Hepatitis |
| ANA | Antinuclear antibodies |
| anti-M3R | Anti-muscarinic receptor 3 |
| APRIL | A proliferation-inducing ligand |
| ASAP | “Abatacept Sjögren Active Patients” study |
| AZA | Azathioprine |
| BAFF | B cell Activating Factor |
| BCR | B cell receptor |
| BUT | Break-up Time |
| CCP | Cyclic Citrullinated Peptide |
| circRNA | Circular RNA |
| ciRNAs | Intronic circRNAs |
| ClinESSDAI | Clinical ESSDAI variant |
| CPK | Creatine phosphokinase |
| CRISP-3 | Cysteine-Rich Secretory Protein 3 () |
| CT-scan | Computerized tomography |
| CyA | Ciclosporin A |
| DAMPS | Danger-associated molecular patterns |
| DAP-kinase | Pro-apoptotic death associated protein kinase |
| DHEA | Dehydroepiandrosterone |
| DHT | Dihydrotestosterone |
| DLBCL | Diffuse large B cell lymphoma |
| DMARD | Disease Modifying Anti-Rheumatic Drug |
| DNMTs | DNA methyltransferases |
| DREAM | “Dry Eye Assessment and Management” study |
| EBV | Epstein-Barr virus |
| ecircRNAs | Exonic circRNAs |
| EIciRNAs | Exon-intron circRNAs |
| ELISA | Enzyme-linked immunosorbent assay |
| ENT | Ear-Nose-Throat |
| ESSDAI | EULAR Sjögren’s syndrome disease activity index |
| ESSPRI | EULAR Sjögren’s Syndrome Patient Reported Index |
| EULAR | European League Against Rheumatism |
| FASl | Fas ligand |
| FDC | Follicular dendritic cells |
| GCs | Germinal centres |
| HCQ | Hydroxychloroquine |
| HCV | Hepatitis C virus |
| HLH | Hemophagocytic lymphohistiocytosis |
| HTLV1 | Human T-lymphotropic virus type I |
| ICAM-1 | InterCellular Adhesion Molecule 1 |
| IF | Immunofluorescence |
| IFN | Interferon |
| IgG,A,M | Immunoglobulin G, A and M |
| IL- | Interleukin |
| ILD | Interstitial lung disease(s) |
| IRF | Interferon Regulatory Factor |
| IV | Intravenous therapy |
| IVIG | Intravenous Immunoglobulin |
| KCS | Keratoconjunctivitis sicca |
| LEMA | Myoepithelial sialadenitis |
| LESA | lymphoepithelial sialadenitis |
| LIP | Lymphocytic interstitial pneumonitis |
| LMP1 | Latent membrane protein 1 |
| lncRNA | Long non-coding RNAs |
| LPR | Laryngopharyngeal reflux |
| LSG | Labial SG |
| MALT | mucosa-associated lymphoid tissue |
| MHC | Major histocompatibility genes |
| MMF | Mycophenolate mofetil |
| MMP | Matrix metalloproteinases |
| MPGN | Mesangioproliferative glomerulonephritis |
| MRI | Magnetic Resonance Imaging |
| MS | Multiple Sclerosis |
| MSGB | Minor salivary gland biopsy |
| MTX | Methotrexate |
| NAC | N-acetylcystein |
| NFkB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NHL | Non-Hodgkin’s lymphoma |
| NICE | National Institute for Health and Care Excellence |
| NMOSD | Neuromyelitis optica spectrum disorder |
| NOD | Non-obese diabetic |
| NSAID | Nonsteroidal anti-inflammatory drugs |
| NSIP | Nonspecific interstitial pneumonia |
| OMERACT | Outcome Measures in Rheumatology group |
| OSDI | Ocular Surface Disease Index |
| OSS | Ocular Staining Score |
| PAMPs | Pathogen-associated molecular patterns |
| PBC | Primary Biliary Cirrhosis |
| PDC | Plasmacytoid dendritic cells |
| PDL1 | Programmed death ligand 1 |
| PET scan | Positron emission tomography |
| PGA | Patient Global Assessment |
| PIP | Prolactin inducible protein |
| PO | per os |
| PSP | Parotid secretory protein |
| pSS | Primary Sjögren’s Syndrome |
| pSS-ILD | pSS-related interstitial lung disease |
| q6h, q8h | Every 6 h, every 8 h |
| RA | Rheumatoid Arthritis |
| RCT | Randomized controlled trial |
| RF | Rheumatoid Factor |
| RTA | Renal tubular acidosis |
| RTX | Rituximab |
| RX1 | Runt-related transcription factor |
| SAM | Methyl donor S-adenosylmethionine |
| SAP | Sicca Asthenia Polyalgia |
| SF-36 | Short Form 36 health survey |
| SG | Salivary Gland |
| SGS | Salivary glands scintigraphy |
| SGUS | Salivary glands ultrasound |
| SICCA | Sjögren’s International Collaborative Clinical Alliance |
| SLE | Systemic lupus erythematosus |
| SNP | Single nucleotide polymorphism |
| SP-1 | Salivary protein 1 |
| SSDDI | Sjögren’s Syndrome Disease Damage Index |
| SSDI | Sjögren’s Syndrome Damage Index |
| sSS | Secondary Sjögren’s Syndrome |
| SWSF | Stimulated Whole Salivary Flow rate |
| TACI | Transmembrane Activator and CAML Interactor |
| TEARS | “Tolerance and efficacy of rituximab in primary Sjögren syndrome” trial |
| Tfh | Follicular helper T cells |
| TLRs | Toll Like Receptors |
| TNF-α | Tumour necrosis factor-α |
| TPHA | Treponema Pallidum Hemagglutinations Assay |
| TRACTISS | “TRial of Anti-B-Cell Therapy In patients with primary Sjögren’s Syndrome” trial |
| TSH | Thyroid-stimulating hormone |
| TTP | Thrombotic Thrombocytopenic Purpura |
| UCLH | University College London Hospitals |
| UIP | Usual interstitial pneumonia |
| UWSF | Unstimulated Whole Saliva Flow rate |
| VAS | Visual analogue scales |
| VDRL | Venereal Disease Research Laboratory |
References
- Fox, R.I. Sjögren’s syndrome. Lancet 2005, 366, 321–331. [Google Scholar] [CrossRef]
- Gerli, R.; Bartoloni, E.; Alunno, A. (Eds.) Sjögren’s Syndrome: Novel Insights in Pathogenic, Clinical, and Therapeutic Aspects; Elsevier: Amsterdam, The Netherlands; Academic Press: Cambridge, MA, USA, 2016; ISBN 978-0-12-803604-4. [Google Scholar]
- Ghafoor, M. Sjögren’s Before Sjögren: Did Henrik Sjögren (1899–1986) Really Discover Sjögren’s Disease? J. Maxillofac. Oral Surg. 2012, 11, 373–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murube, J. Henrik Sjögren, 1899–1986. Ocul. Surf. 2010, 8, 2–7. [Google Scholar] [CrossRef]
- Wollheim, F.A. Henrik Sjögren and Sjögren’s syndrome. Scand. J. Rheumatol. Suppl. 1986, 61, 11–16. [Google Scholar]
- Binard, A.; Devauchelle-Pensec, V.; Fautrel, B.; Jousse, S.; Youinou, P.; Saraux, A. Epidemiology of Sjögren’s syndrome: Where are we now? Clin. Exp. Rheumatol. 2007, 25, 1–4. [Google Scholar]
- Mavragani, C.P.; Moutsopoulos, H.M. The geoepidemiology of Sjögren’s syndrome. Autoimmun. Rev. 2010, 9, A305–A310. [Google Scholar] [CrossRef]
- Qin, B.; Wang, J.; Yang, Z.; Yang, M.; Ma, N.; Huang, F.; Zhong, R. Epidemiology of primary Sjögren’s syndrome: A systematic review and meta-analysis. Ann. Rheum. Dis. 2015, 74, 1983–1989. [Google Scholar] [CrossRef]
- Delaleu, N.; Jonsson, M.V.; Appel, S.; Jonsson, R. New concepts in the pathogenesis of Sjögren’s syndrome. Rheum. Dis. Clin. N. Am. 2008, 34, 833–845. [Google Scholar] [CrossRef]
- Konttinen, Y.T.; Käsnä-Ronkainen, L. Sjögren’s syndrome: Viewpoint on pathogenesis. One of the reasons I was never asked to write a textbook chapter on it. Scand. J. Rheumatol. Suppl. 2002, 116, 15–22. [Google Scholar] [CrossRef]
- Mitsias, D.I.; Kapsogeorgou, E.K.; Moutsopoulos, H.M. Sjögren’s syndrome: Why autoimmune epithelitis? Oral Dis. 2006, 12, 523–532. [Google Scholar] [CrossRef]
- Igoe, A.; Scofield, R.H. Autoimmunity and infection in Sjögren’s syndrome. Curr. Opin. Rheumatol. 2013, 25, 480–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Björk, A.; Mofors, J.; Wahren-Herlenius, M. Environmental factors in the pathogenesis of primary Sjögren’s syndrome. J. Intern. Med. 2020, 287, 475–492. [Google Scholar] [CrossRef] [PubMed]
- Ascherio, A.; Munger, K.L. Epstein-barr virus infection and multiple sclerosis: A review. J. Neuroimmune Pharmacol. 2010, 5, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Toussirot, E.; Roudier, J. Epstein-Barr virus in autoimmune diseases. Best Pract. Res. Clin. Rheumatol. 2008, 22, 883–896. [Google Scholar] [CrossRef] [PubMed]
- Saito, I.; Servenius, B.; Compton, T.; Fox, R.I. Detection of Epstein-Barr virus DNA by polymerase chain reaction in blood and tissue biopsies from patients with Sjogren’s syndrome. J. Exp. Med. 1989, 169, 2191–2198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mariette, X.; Gozlan, J.; Clerc, D.; Bisson, M.; Morinet, F. Detection of Epstein-Barr virus DNA by in situ hybridization and polymerase chain reaction in salivary gland biopsy specimens from patients with Sjögren’s syndrome. Am. J. Med. 1991, 90, 286–294. [Google Scholar] [CrossRef]
- Dimitriou, I.; Xanthou, G.; Kapsogeorgou, E.; Abu-Helu, R.; Moutsopoulos, H.; Manoussakis, M. High spontaneous CD40 expression by salivary gland epithelial cells in Sjogren’s syndrome: Possible evidence for intrinsic activation of epithelial cells. Arthritis Res. 2001, 3, P018. [Google Scholar] [CrossRef] [Green Version]
- Kivity, S.; Arango, M.T.; Ehrenfeld, M.; Tehori, O.; Shoenfeld, Y.; Anaya, J.-M.; Agmon-Levin, N. Infection and autoimmunity in Sjogren’s syndrome: A clinical study and comprehensive review. J. Autoimmun. 2014, 51, 17–22. [Google Scholar] [CrossRef]
- Iwakiri, D.; Zhou, L.; Samanta, M.; Matsumoto, M.; Ebihara, T.; Seya, T.; Imai, S.; Fujieda, M.; Kawa, K.; Takada, K. Epstein-Barr virus (EBV)-encoded small RNA is released from EBV-infected cells and activates signaling from Toll-like receptor 3. J. Exp. Med. 2009, 206, 2091–2099. [Google Scholar] [CrossRef] [Green Version]
- Murray, R.J.; Wang, D.; Young, L.S.; Wang, F.; Rowe, M.; Kieff, E.; Rickinson, A.B. Epstein-Barr virus-specific cytotoxic T-cell recognition of transfectants expressing the virus-coded latent membrane protein LMP. J. Virol. 1988, 62, 3747–3755. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, H.; Takahashi, Y.; Yamamoto-Fukuda, T.; Horai, Y.; Nakashima, Y.; Arima, K.; Nakamura, T.; Koji, T.; Kawakami, A. Direct Infection of Primary Salivary Gland Epithelial Cells by Human T Lymphotropic Virus Type I in Patients With Sjögren’s Syndrome. Arthritis Rheumatol. 2015, 67, 1096–1106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terada, K.; Katamine, S.; Eguchi, K.; Moriuchi, R.; Kita, M.; Shimada, H.; Yamashita, I.; Iwata, K.; Tsuji, Y.; Nagataki, S. Prevalence of serum and salivary antibodies to HTLV-1 in Sjögren’s syndrome. Lancet 1994, 344, 1116–1119. [Google Scholar] [CrossRef]
- Stathopoulou, E.A.; Routsias, J.G.; Stea, E.A.; Moutsopoulos, H.M.; Tzioufas, A.G. Cross-reaction between antibodies to the major epitope of Ro60 kD autoantigen and a homologous peptide of Coxsackie virus 2B protein. Clin. Exp. Immunol. 2005, 141, 148–154. [Google Scholar] [CrossRef]
- Gottenberg, J.-E.; Pallier, C.; Ittah, M.; Lavie, F.; Miceli-Richard, C.; Sellam, J.; Nordmann, P.; Cagnard, N.; Sibilia, J.; Mariette, X. Failure to confirm coxsackievirus infection in primary Sjögren’s syndrome. Arthritis Rheum. 2006, 54, 2026–2028. [Google Scholar] [CrossRef] [PubMed]
- Flores-Chávez, A.; Carrion, J.A.; Forns, X.; Ramos-Casals, M. Extrahepatic manifestations associated with Chronic Hepatitis C Virus Infection. Rev. Espanola Sanid. Penit. 2017, 19, 87–97. [Google Scholar] [CrossRef]
- Kang, H.I.; Fei, H.M.; Saito, I.; Sawada, S.; Chen, S.L.; Yi, D.; Chan, E.; Peebles, C.; Bugawan, T.L.; Erlich, H.A. Comparison of HLA class II genes in Caucasoid, Chinese, and Japanese patients with primary Sjögren’s syndrome. J. Immunol. 1950 1993, 150, 3615–3623. [Google Scholar]
- Kerttula, T.O.; Collin, P.; Polvi, A.; Korpela, M.; Partanen, J.; Mäki, M. Distinct immunologic features of Finnish Sjögren’s syndrome patients with HLA alleles DRB1*0301, DQA1*0501, and DQB1*0201. Alterations in circulating T cell receptor gamma/delta subsets. Arthritis Rheum. 1996, 39, 1733–1739. [Google Scholar] [CrossRef]
- Mountz, J.D.; Zhou, T.; Su, X.; Wu, J.; Cheng, J. The role of programmed cell death as an emerging new concept for the pathogenesis of autoimmune diseases. Clin. Immunol. Immunopathol. 1996, 80, S2–S14. [Google Scholar] [CrossRef]
- Adachi, M.; Watanabe-Fukunaga, R.; Nagata, S. Aberrant transcription caused by the insertion of an early transposable element in an intron of the Fas antigen gene of lpr mice. Proc. Natl. Acad. Sci. USA 1993, 90, 1756–1760. [Google Scholar] [CrossRef] [Green Version]
- Bolstad, A.I.; Wargelius, A.; Nakken, B.; Haga, H.J.; Jonsson, R. Fas and Fas ligand gene polymorphisms in primary Sjögren’s syndrome. J. Rheumatol. 2000, 27, 2397–2405. [Google Scholar]
- Nakken, B.; Jonsson, R.; Bolstad, A.I. Polymorphisms of the Ro52 gene associated with anti-Ro 52-kd autoantibodies in patients with primary Sjögren’s syndrome. Arthritis Rheum. 2001, 44, 638–646. [Google Scholar] [CrossRef]
- Hulkkonen, J.; Pertovaara, M.; Antonen, J.; Lahdenpohja, N.; Pasternack, A.; Hurme, M. Genetic association between interleukin-10 promoter region polymorphisms and primary Sjögren’s syndrome. Arthritis Rheum. 2001, 44, 176–179. [Google Scholar] [CrossRef]
- Qin, B.; Wang, J.; Liang, Y.; Yang, Z.; Zhong, R. The association between TNF-α, IL-10 gene polymorphisms and primary Sjögren’s syndrome: A meta-analysis and systemic review. PLoS ONE 2013, 8, e63401. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Casals, M.; Font, J.; Brito-Zeron, P.; Trejo, O.; García-Carrasco, M.; Lozano, F. Interleukin-4 receptor alpha polymorphisms in primary Sjögren’s syndrome. Clin. Exp. Rheumatol. 2004, 22, 374. [Google Scholar]
- Imgenberg-Kreuz, J.; Rasmussen, A.; Sivils, K.; Nordmark, G. Genetics and epigenetics in primary Sjögren’s syndrome. Rheumatology 2019. [Google Scholar] [CrossRef] [Green Version]
- Traianos, E.Y.; Locke, J.; Lendrem, D.; Bowman, S.; Hargreaves, B.; Macrae, V. Serum CXCL13 levels are associated with lymphoma risk and lymphoma occurrence in primary Sjögren’s syndrome. Rheumatol. Int. 2020, 40, 541–548. [Google Scholar] [CrossRef] [Green Version]
- Ben-Eli, H.; Gomel, N.; Aframian, D.J.; Abu-Seir, R.; Perlman, R.; Ben-Chetrit, E.; Mevorach, D.; Kleinstern, G.; Paltiel, O.; Solomon, A. SNP variations in IL10, TNFα and TNFAIP3 genes in patients with dry eye syndrome and Sjogren’s syndrome. J. Inflamm. 2019, 16, 6. [Google Scholar] [CrossRef] [Green Version]
- Nocturne, G.; Tarn, J.; Boudaoud, S.; Locke, J.; Miceli-Richard, C.; Hachulla, E.; Dubost, J.J.; Bowman, S.; Gottenberg, J.E.; Criswell, L.A.; et al. Germline variation of TNFAIP3 in primary Sjögren’s syndrome-associated lymphoma. Ann. Rheum. Dis. 2016, 75, 780–783. [Google Scholar] [CrossRef]
- Nezos, A.; Gkioka, E.; Koutsilieris, M.; Voulgarelis, M.; Tzioufas, A.G.; Mavragani, C.P. TNFAIP3 F127C Coding Variation in Greek Primary Sjogren’s Syndrome Patients. J. Immunol. Res. 2018, 2018, 6923213. [Google Scholar] [CrossRef] [Green Version]
- Fragkioudaki, S.; Nezos, A.; Souliotis, V.L.; Chatziandreou, I.; Saetta, A.A.; Drakoulis, N.; Tzioufas, A.G.; Voulgarelis, M.; Sfikakis, P.P.; Koutsilieris, M.; et al. MTHFR gene variants and non-MALT lymphoma development in primary Sjogren’s syndrome. Sci. Rep. 2017, 7, 7354. [Google Scholar] [CrossRef] [Green Version]
- Nezos, A.; Mavragani, C.P. Contribution of Genetic Factors to Sjögren’s Syndrome and Sjögren’s Syndrome Related Lymphomagenesis. J. Immunol. Res. 2015, 2015, 754825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arvaniti, P.; Le Dantec, C.; Charras, A.; Arleevskaya, M.A.; Hedrich, C.M.; Zachou, K.; Dalekos, G.N.; Renaudineau, Y. Linking genetic variation with epigenetic profiles in Sjögren’s syndrome. Clin. Immunol. 2020, 210, 108314. [Google Scholar] [CrossRef]
- Konsta, O.D.; Thabet, Y.; Le Dantec, C.; Brooks, W.H.; Tzioufas, A.G.; Pers, J.-O.; Renaudineau, Y. The contribution of epigenetics in Sjögren’s Syndrome. Front. Genet. 2014, 5, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thabet, Y.; Le Dantec, C.; Ghedira, I.; Devauchelle, V.; Cornec, D.; Pers, J.-O.; Renaudineau, Y. Epigenetic dysregulation in salivary glands from patients with primary Sjögren’s syndrome may be ascribed to infiltrating B cells. J. Autoimmun. 2013, 41, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Cannat, A.; Seligmann, M. Induction by isoniazid and hydrallazine of antinuclear factors in mice. Clin. Exp. Immunol. 1968, 3, 99–105. [Google Scholar]
- Imgenberg-Kreuz, J.; Sandling, J.K.; Almlöf, J.C.; Nordlund, J.; Signér, L.; Norheim, K.B.; Omdal, R.; Rönnblom, L.; Eloranta, M.-L.; Syvänen, A.-C.; et al. Genome-wide DNA methylation analysis in multiple tissues in primary Sjögren’s syndrome reveals regulatory effects at interferon-induced genes. Ann. Rheum. Dis. 2016, 75, 2029–2036. [Google Scholar] [CrossRef] [Green Version]
- Toso, A.; Aluffi, P.; Capello, D.; Conconi, A.; Gaidano, G.; Pia, F. Clinical and molecular features of mucosa-associated lymphoid tissue (MALT) lymphomas of salivary glands. Head Neck 2009, 31, 1181–1187. [Google Scholar] [CrossRef]
- Altorok, N.; Coit, P.; Hughes, T.; Koelsch, K.A.; Stone, D.U.; Rasmussen, A.; Radfar, L.; Scofield, R.H.; Sivils, K.L.; Farris, A.D.; et al. Genome-wide DNA methylation patterns in naive CD4+ T cells from patients with primary Sjögren’s syndrome. Arthritis Rheumatol. 2014, 66, 731–739. [Google Scholar] [CrossRef] [Green Version]
- Alevizos, I.; Illei, G.G. MicroRNAs in Sjögren’s syndrome as a prototypic autoimmune disease. Autoimmun. Rev. 2010, 9, 618–621. [Google Scholar] [CrossRef] [Green Version]
- Mendell, J.T. miRiad roles for the miR-17-92 cluster in development and disease. Cell 2008, 133, 217–222. [Google Scholar] [CrossRef] [Green Version]
- Xiao, C.; Srinivasan, L.; Calado, D.P.; Patterson, H.C.; Zhang, B.; Wang, J.; Henderson, J.M.; Kutok, J.L.; Rajewsky, K. Lymphoproliferative disease and autoimmunity in mice with increased miR-17-92 expression in lymphocytes. Nat. Immunol. 2008, 9, 405–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zilahi, E.; Tarr, T.; Papp, G.; Griger, Z.; Sipka, S.; Zeher, M. Increased microRNA-146a/b, TRAF6 gene and decreased IRAK1 gene expressions in the peripheral mononuclear cells of patients with Sjögren’s syndrome. Immunol. Lett. 2012, 141, 165–168. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.; Tetzlaff, M.T.; Vanbelle, P.; Elder, D.; Feldman, M.; Tobias, J.W.; Sepulveda, A.R.; Xu, X. MicroRNA expression profiling outperforms mRNA expression profiling in formalin-fixed paraffin-embedded tissues. Int. J. Clin. Exp. Pathol. 2009, 2, 519–527. [Google Scholar] [PubMed]
- Howe, K. Extraction of miRNAs from Formalin-Fixed Paraffin-Embedded (FFPE) Tissues. Methods Mol. Biol. 2017, 1509, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Sun, B.; Huang, S.; Zhao, L. Roles of circular RNAs in immune regulation and autoimmune diseases. Cell Death Dis. 2019, 10, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Xia, X.; Tang, X.; Wang, S. Roles of CircRNAs in Autoimmune Diseases. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef]
- Su, L.-C.; Xu, W.-D.; Liu, X.-Y.; Fu, L.; Huang, A.-F. Altered expression of circular RNA in primary Sjögren’s syndrome. Clin. Rheumatol. 2019, 38, 3425–3433. [Google Scholar] [CrossRef]
- Roy, S.; Awasthi, A. Emerging roles of noncoding RNAs in T cell differentiation and functions in autoimmune diseases. Int. Rev. Immunol. 2019, 38, 232–245. [Google Scholar] [CrossRef]
- Dolcino, M.; Tinazzi, E.; Vitali, C.; Del Papa, N.; Puccetti, A.; Lunardi, C. Long Non-Coding RNAs Modulate Sjögren’s Syndrome Associated Gene Expression and Are Involved in the Pathogenesis of the Disease. J. Clin. Med. 2019, 8, 1349. [Google Scholar] [CrossRef] [Green Version]
- Han, S.-B.; Moratz, C.; Huang, N.-N.; Kelsall, B.; Cho, H.; Shi, C.-S.; Schwartz, O.; Kehrl, J.H. Rgs1 and Gnai2 regulate the entrance of B lymphocytes into lymph nodes and B cell motility within lymph node follicles. Immunity 2005, 22, 343–354. [Google Scholar] [CrossRef] [Green Version]
- Coca, A.; Sanz, I. Updates on B-cell immunotherapies for systemic lupus erythematosus and Sjogren’s syndrome. Curr. Opin. Rheumatol. 2012, 24, 451–456. [Google Scholar] [CrossRef] [PubMed]
- Béguelin, W.; Teater, M.; Gearhart, M.D.; Calvo Fernández, M.T.; Goldstein, R.L.; Cárdenas, M.G.; Hatzi, K.; Rosen, M.; Shen, H.; Corcoran, C.M.; et al. EZH2 and BCL6 Cooperate to Assemble CBX8-BCOR Complex to Repress Bivalent Promoters, Mediate Germinal Center Formation and Lymphomagenesis. Cancer Cell 2016, 30, 197–213. [Google Scholar] [CrossRef] [PubMed]
- Bai, M.; Skyrlas, A.; Agnantis, N.J.; Kamina, S.; Tsanou, E.; Grepi, C.; Galani, V.; Kanavaros, P. Diffuse large B-cell lymphomas with germinal center B-cell-like differentiation immunophenotypic profile are associated with high apoptotic index, high expression of the proapoptotic proteins bax, bak and bid and low expression of the antiapoptotic protein bcl-xl. Mod. Pathol. 2004, 17, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Wei, W.; He, X.; Xie, Y.; Kamal, M.A.; Li, J. Influence of Hormones on Sjögren’s Syndrome. Curr. Pharm. Des. 2018, 24, 4167–4176. [Google Scholar] [CrossRef] [PubMed]
- McCoy, S.S.; Sampene, E.; Baer, A.N. Sjögren’s Syndrome is Associated With Reduced Lifetime Sex Hormone Exposure: A Case-Control Study. Arthritis Care Res. 2019, acr.24014. [Google Scholar] [CrossRef] [PubMed]
- Harris, V.M.; Sharma, R.; Cavett, J.; Kurien, B.T.; Liu, K.; Koelsch, K.A.; Rasmussen, A.; Radfar, L.; Lewis, D.; Stone, D.U.; et al. Klinefelter’s syndrome (47,XXY) is in excess among men with Sjögren’s syndrome. Clin. Immunol. 2016, 168, 25–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seminog, O.O.; Seminog, A.B.; Yeates, D.; Goldacre, M.J. Associations between Klinefelter’s syndrome and autoimmune diseases: English national record linkage studies. Autoimmunity 2015, 48, 125–128. [Google Scholar] [CrossRef]
- Fujimoto, M.; Ikeda, K.; Nakamura, T.; Iwamoto, T.; Furuta, S.; Nakajima, H. Development of mixed connective tissue disease and Sjögren’s syndrome in a patient with trisomy X. Lupus 2015, 24, 1217–1220. [Google Scholar] [CrossRef]
- Morthen, M.K.; Tellefsen, S.; Richards, S.M.; Lieberman, S.M.; Rahimi Darabad, R.; Kam, W.R.; Sullivan, D.A. Testosterone Influence on Gene Expression in Lacrimal Glands of Mouse Models of Sjögren Syndrome. Investig. Ophthalmol. Vis. Sci. 2019, 60, 2181–2197. [Google Scholar] [CrossRef]
- Porola, P.; Laine, M.; Virtanen, I.; Pöllänen, R.; Przybyla, B.D.; Konttinen, Y.T. Androgens and Integrins in Salivary Glands in Sjögren’s Syndrome. J. Rheumatol. 2010, 37, 1181–1187. [Google Scholar] [CrossRef]
- Taiym, S.; Haghighat, N.; Al-Hashimi, I. A comparison of the hormone levels in patients with Sjogren’s syndrome and healthy controls. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod. 2004, 97, 579–583. [Google Scholar] [CrossRef] [PubMed]
- Bizzarro, A.; Valentini, G.; Martino, G.D.; Daponte, A.; De Bellis, A.; Iacono, G. Influence of Testosterone Therapy on Clinical and Immunological Features of Autoimmune Diseases Associated with Klinefelter’s Syndrome. J. Clin. Endocrinol. Metab. 1987, 64, 32–36. [Google Scholar] [CrossRef] [PubMed]
- Ishimaru, N.; Arakaki, R.; Watanabe, M.; Kobayashi, M.; Miyazaki, K.; Hayashi, Y. Development of autoimmune exocrinopathy resembling Sjögren’s syndrome in estrogen-deficient mice of healthy background. Am. J. Pathol. 2003, 163, 1481–1490. [Google Scholar] [CrossRef]
- Iwasa, A.; Arakaki, R.; Honma, N.; Ushio, A.; Yamada, A.; Kondo, T.; Kurosawa, E.; Kujiraoka, S.; Tsunematsu, T.; Kudo, Y.; et al. Aromatase Controls Sjögren Syndrome–Like Lesions through Monocyte Chemotactic Protein-1 in Target Organ and Adipose Tissue–Associated Macrophages. Am. J. Pathol. 2015, 185, 151–161. [Google Scholar] [CrossRef]
- Shim, G.-J.; Warner, M.; Kim, H.-J.; Andersson, S.; Liu, L.; Ekman, J.; Imamov, O.; Jones, M.E.; Simpson, E.R.; Gustafsson, J.-A. Aromatase-deficient mice spontaneously develop a lymphoproliferative autoimmune disease resembling Sjogren’s syndrome. Proc. Natl. Acad. Sci. USA 2004, 101, 12628–12633. [Google Scholar] [CrossRef] [Green Version]
- Ishimaru, N.; Arakaki, R.; Omotehara, F.; Yamada, K.; Mishima, K.; Saito, I.; Hayashi, Y. Novel Role for RbAp48 in Tissue-Specific, Estrogen Deficiency-Dependent Apoptosis in the Exocrine Glands. Mol. Cell. Biol. 2006, 26, 2924–2935. [Google Scholar] [CrossRef] [Green Version]
- Ishimaru, N.; Arakaki, R.; Yoshida, S.; Yamada, A.; Noji, S.; Hayashi, Y. Expression of the retinoblastoma protein RbAp48 in exocrine glands leads to Sjögren’s syndrome–like autoimmune exocrinopathy. J. Exp. Med. 2008, 205, 2915–2927. [Google Scholar] [CrossRef] [Green Version]
- Manoussakis, M.N.; Tsinti, M.; Kapsogeorgou, E.K.; Moutsopoulos, H.M. The salivary gland epithelial cells of patients with primary Sjögren’s syndrome manifest significantly reduced responsiveness to 17β-estradiol. J. Autoimmun. 2012, 39, 64–68. [Google Scholar] [CrossRef]
- Laroche, M.; Borg, S.; Lassoued, S.; De Lafontan, B.; Roché, H. Joint pain with aromatase inhibitors: Abnormal frequency of Sjögren’s syndrome. J. Rheumatol. 2007, 34, 2259–2263. [Google Scholar]
- Shanmugam, V.K.; McCloskey, J.; Elston, B.; Allison, S.J.; Eng-Wong, J. The CIRAS study: A case control study to define the clinical, immunologic, and radiographic features of aromatase inhibitor-induced musculoskeletal symptoms. Breast Cancer Res. Treat. 2012, 131, 699–708. [Google Scholar] [CrossRef] [Green Version]
- Guidelli, G.M.; Martellucci, I.; Galeazzi, M.; Francini, G.; Fioravanti, A. Sjögren’s syndrome and aromatase inhibitors treatment: Is there a link? Clin. Exp. Rheumatol. 2013, 31, 653–654. [Google Scholar] [PubMed]
- Laine, M.; Porola, P.; Udby, L.; Kjeldsen, L.; Cowland, J.B.; Borregaard, N.; Hietanen, J.; Ståhle, M.; Pihakari, A.; Konttinen, Y.T. Low salivary dehydroepiandrosterone and androgen-regulated cysteine-rich secretory protein 3 levels in Sjögren’s syndrome. Arthritis Rheum. 2007, 56, 2575–2584. [Google Scholar] [CrossRef] [PubMed]
- Konttinen, Y.T.; Fuellen, G.; Bing, Y.; Porola, P.; Stegaev, V.; Trokovic, N.; Falk, S.S.I.; Liu, Y.; Szodoray, P.; Takakubo, Y. Sex steroids in Sjögren’s syndrome. J. Autoimmun. 2012, 39, 49–56. [Google Scholar] [CrossRef]
- Spaan, M.; Porola, P.; Laine, M.; Rozman, B.; Azuma, M.; Konttinen, Y.T. Healthy human salivary glands contain a DHEA-sulphate processing intracrine machinery, which is deranged in primary Sjögren’s syndrome. J. Cell. Mol. Med. 2009, 13, 1261–1270. [Google Scholar] [CrossRef] [PubMed]
- Moutsopoulos, H.M.; Kordossis, T. Sjögren’s syndrome revisited: Autoimmune epithelitis. Br. J. Rheumatol. 1996, 35, 204–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spachidou, M.P.; Bourazopoulou, E.; Maratheftis, C.I.; Kapsogeorgou, E.K.; Moutsopoulos, H.M.; Tzioufas, A.G.; Manoussakis, M.N. Expression of functional Toll-like receptors by salivary gland epithelial cells: Increased mRNA expression in cells derived from patients with primary Sjögren’s syndrome. Clin. Exp. Immunol. 2007, 147, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-Q.; Szodoray, P.; Zeher, M. Toll-Like Receptor Pathways in Autoimmune Diseases. Clin. Rev. Allergy Immunol. 2016, 50, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spachidou, M.; Kapsogeorgou, E.; Bourazopoulou, E.; Moutsopoulos, H.; Manoussakis, M. Cultured salivary gland epithelial cells from patients with primary Sjögren’s syndrome and disease controls are sensitive to signaling via Toll-like receptors 2 and 3: Upregulation of intercellular adhesion molecule-1 expression. Arthritis Res. Ther. 2005, 7, P154. [Google Scholar] [CrossRef] [Green Version]
- Iwanaszko, M.; Kimmel, M. NF-κB and IRF pathways: Cross-regulation on target genes promoter level. BMC Genom. 2015, 16, 307. [Google Scholar] [CrossRef] [Green Version]
- Ichiyama, T.; Nakatani, E.; Tatsumi, K.; Hideshima, K.; Urano, T.; Nariai, Y.; Sekine, J. Expression of aquaporin 3 and 5 as a potential marker for distinguishing dry mouth from Sjögren’s syndrome. J. Oral Sci. 2018, 60, 212–220. [Google Scholar] [CrossRef] [Green Version]
- Beroukas, D.; Hiscock, J.; Gannon, B.J.; Jonsson, R.; Gordon, T.P.; Waterman, S.A. Selective down-regulation of aquaporin-1 in salivary glands in primary Sjögren’s syndrome. Lab. Investig. J. Tech. Methods Pathol. 2002, 82, 1547–1552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sisto, M.; Lorusso, L.; Ingravallo, G.; Nico, B.; Ribatti, D.; Ruggieri, S.; Lofrumento, D.D.; Lisi, S. Abnormal distribution of AQP4 in minor salivary glands of primary Sjögren’s syndrome patients. Autoimmunity 2017, 50, 202–210. [Google Scholar] [CrossRef] [PubMed]
- Ring, T.; Kallenbach, M.; Praetorius, J.; Nielsen, S.; Melgaard, B. Successful treatment of a patient with primary Sjögren’s syndrome with Rituximab. Clin. Rheumatol. 2006, 25, 891–894. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Ying, X.; Qian, Y.; Liu, H.; Lan, Y.; Xie, A.; Zhu, X. Physiological and pathological impact of AQP1 knockout in mice. Biosci. Rep. 2019, 39. [Google Scholar] [CrossRef]
- Verkman, A.S.; Yang, B.; Song, Y.; Manley, G.T.; Ma, T. Role of water channels in fluid transport studied by phenotype analysis of aquaporin knockout mice. Exp. Physiol. 2000, 85, 233S–241S. [Google Scholar] [CrossRef]
- Hosoi, K.; Yao, C.; Hasegawa, T.; Yoshimura, H.; Akamatsu, T. Dynamics of Salivary Gland AQP5 under Normal and Pathologic Conditions. Int. J. Mol. Sci. 2020, 21, 1182. [Google Scholar] [CrossRef] [Green Version]
- Delporte, C.; Bryla, A.; Perret, J. Aquaporins in Salivary Glands: From Basic Research to Clinical Applications. Int. J. Mol. Sci. 2016, 17, 166. [Google Scholar] [CrossRef] [Green Version]
- Steinfeld, S.; Cogan, E.; King, L.S.; Agre, P.; Kiss, R.; Delporte, C. Abnormal distribution of aquaporin-5 water channel protein in salivary glands from Sjögren’s syndrome patients. Lab. Investig. J. Tech. Methods Pathol. 2001, 81, 143–148. [Google Scholar] [CrossRef] [Green Version]
- Soyfoo, M.S.; De Vriese, C.; Debaix, H.; Martin-Martinez, M.D.; Mathieu, C.; Devuyst, O.; Steinfeld, S.D.; Delporte, C. Modified aquaporin 5 expression and distribution in submandibular glands from NOD mice displaying autoimmune exocrinopathy. Arthritis Rheum. 2007, 56, 2566–2574. [Google Scholar] [CrossRef] [Green Version]
- Yoshimura, S.; Nakamura, H.; Horai, Y.; Nakajima, H.; Shiraishi, H.; Hayashi, T.; Takahashi, T.; Kawakami, A. Abnormal distribution of AQP5 in labial salivary glands is associated with poor saliva secretion in patients with Sjögren’s syndrome including neuromyelitis optica complicated patients. Mod. Rheumatol. 2016, 26, 384–390. [Google Scholar] [CrossRef]
- Lee, B.H.; Gauna, A.E.; Perez, G.; Park, Y.; Pauley, K.M.; Kawai, T.; Cha, S. Autoantibodies against Muscarinic Type 3 Receptor in Sjögren’s Syndrome Inhibit Aquaporin 5 Trafficking. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Roche, J.V.; Törnroth-Horsefield, S. Aquaporin Protein-Protein Interactions. Int. J. Mol. Sci. 2017, 18, 2255. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, Y.; Tsuzaka, K.; Takeuchi, T.; Sasaki, Y.; Tsubota, K. Altered distribution of aquaporin 5 and its C-terminal binding protein in the lacrimal glands of a mouse model for Sjögren’s syndrome. Curr. Eye Res. 2008, 33, 621–629. [Google Scholar] [CrossRef]
- Soyfoo, M.S.; Konno, A.; Bolaky, N.; Oak, J.S.; Fruman, D.; Nicaise, C.; Takiguchi, M.; Delporte, C. Link between inflammation and aquaporin-5 distribution in submandibular gland in Sjögren’s syndrome? Oral Dis. 2012, 18, 568–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soyfoo, M.S.; Bolaky, N.; Depoortere, I.; Delporte, C. Relationship between aquaporin-5 expression and saliva flow in streptozotocin-induced diabetic mice? Oral Dis. 2012, 18, 501–505. [Google Scholar] [CrossRef]
- Jin, J.-O.; Yu, Q. T Cell-Associated Cytokines in the Pathogenesis of Sjögren’s Syndrome. J. Clin. Cell. Immunol. 2013, S1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, R.I.; Kang, H.I.; Ando, D.; Abrams, J.; Pisa, E. Cytokine mRNA expression in salivary gland biopsies of Sjögren’s syndrome. J. Immunol. 1950 1994, 152, 5532–5539. [Google Scholar]
- Boumba, D.; Skopouli, F.N.; Moutsopoulos, H.M. Cytokine mRNA expression in the labial salivary gland tissues from patients with primary Sjögren’s syndrome. Br. J. Rheumatol. 1995, 34, 326–333. [Google Scholar] [CrossRef]
- Sumida, T.; Tsuboi, H.; Iizuka, M.; Hirota, T.; Asashima, H.; Matsumoto, I. The role of M3 muscarinic acetylcholine receptor reactive T cells in Sjögren’s syndrome: A critical review. J. Autoimmun. 2014, 51, 44–50. [Google Scholar] [CrossRef]
- Zhou, J.; Jin, J.-O.; Kawai, T.; Yu, Q. Endogenous programmed death ligand-1 restrains the development and onset of Sjögren’s syndrome in non-obese diabetic mice. Sci. Rep. 2016, 6, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Arce-Franco, M.; Dominguez-Luis, M.; Pec, M.K.; Martínez-Gimeno, C.; Miranda, P.; Alvarez de la Rosa, D.; Giraldez, T.; García-Verdugo, J.M.; Machado, J.D.; Díaz-González, F. Functional effects of proinflammatory factors present in Sjögren’s syndrome salivary microenvironment in an in vitro model of human salivary gland. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, E.H.; Lee, Y.J.; Hyon, J.Y.; Yun, P.Y.; Song, Y.W. Salivary cytokine profiles in primary Sjögren’s syndrome differ from those in non-Sjögren sicca in terms of TNF-α levels and Th-1/Th-2 ratios. Clin. Exp. Rheumatol. 2011, 29, 970–976. [Google Scholar] [PubMed]
- Yamamura, Y.; Motegi, K.; Kani, K.; Takano, H.; Momota, Y.; Aota, K.; Yamanoi, T.; Azuma, M. TNF-α inhibits aquaporin 5 expression in human salivary gland acinar cells via suppression of histone H4 acetylation. J. Cell. Mol. Med. 2012, 16, 1766–1775. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Kawai, T.; Yu, Q. Pathogenic role of endogenous TNF-α in the development of Sjögren’s-like sialadenitis and secretory dysfunction in non-obese diabetic mice. Lab. Investig. 2017, 97, 458–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, R.I.; Adamson, T.C.; Fong, S.; Young, C.; Howell, F.V. Characterization of the phenotype and function of lymphocytes infiltrating the salivary gland in patients with primary Sjogren syndrome. Diagn. Immunol. 1983, 1, 233–239. [Google Scholar] [PubMed]
- Roescher, N.; Tak, P.P.; Illei, G.G. Cytokines in Sjögren’s syndrome. Oral Dis. 2009, 15, 519–526. [Google Scholar] [CrossRef] [Green Version]
- Fox, R.I.; Kang, H.I. Pathogenesis of Sjögren’s syndrome. Rheum. Dis. Clin. N. Am. 1992, 18, 517–538. [Google Scholar]
- Bertorello, R.; Cordone, M.P.; Contini, P.; Rossi, P.; Indiveri, F.; Puppo, F.; Cordone, G. Increased levels of interleukin-10 in saliva of Sjögren’s syndrome patients. Correlation with disease activity. Clin. Exp. Med. 2004, 4, 148–151. [Google Scholar] [CrossRef]
- Youinou, P.; Pers, J.-O. Disturbance of cytokine networks in Sjögren’s syndrome. Arthritis Res. Ther. 2011, 13, 227. [Google Scholar] [CrossRef] [Green Version]
- Ohlsson, M.; Jonsson, R.; Brokstad, K.A. Subcellular redistribution and surface exposure of the Ro52, Ro60 and La48 autoantigens during apoptosis in human ductal epithelial cells: A possible mechanism in the pathogenesis of Sjögren’s syndrome. Scand. J. Immunol. 2002, 56, 456–469. [Google Scholar] [CrossRef]
- Davies, M.L.; Taylor, E.J.; Gordon, C.; Young, S.P.; Welsh, K.; Bunce, M.; Wordsworth, B.P.; Davidson, B.; Bowman, S.J. Candidate T cell epitopes of the human La/SSB autoantigen. Arthritis Rheum. 2002, 46, 209–214. [Google Scholar] [CrossRef]
- Hasegawa, H.; Inoue, A.; Kohno, M.; Muraoka, M.; Miyazaki, T.; Terada, M.; Nakayama, T.; Yoshie, O.; Nose, M.; Yasukawa, M. Antagonist of interferon-inducible protein 10/CXCL10 ameliorates the progression of autoimmune sialadenitis in MRL/lpr mice. Arthritis Rheum. 2006, 54, 1174–1183. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Ogawa, N.; Nakabayashi, T.; Liu, G.T.; D’Souza, E.; McGuff, H.S.; Guerrero, D.; Talal, N.; Dang, H. Fas and Fas ligand expression in the salivary glands of patients with primary Sjögren’s syndrome. Arthritis Rheum. 1997, 40, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Ibrahem, H.M. B cell dysregulation in primary Sjögren’s syndrome: A review. Jpn. Dent. Sci. Rev. 2019, 55, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Verstappen, G.M.; Corneth, O.B.J.; Bootsma, H.; Kroese, F.G.M. Th17 cells in primary Sjögren’s syndrome: Pathogenicity and plasticity. J. Autoimmun. 2018, 87, 16–25. [Google Scholar] [CrossRef]
- Katsifis, G.E.; Rekka, S.; Moutsopoulos, N.M.; Pillemer, S.; Wahl, S.M. Systemic and local interleukin-17 and linked cytokines associated with Sjögren’s syndrome immunopathogenesis. Am. J. Pathol. 2009, 175, 1167–1177. [Google Scholar] [CrossRef] [Green Version]
- Sonnenberg, G.F.; Nair, M.G.; Kirn, T.J.; Zaph, C.; Fouser, L.A.; Artis, D. Pathological versus protective functions of IL-22 in airway inflammation are regulated by IL-17A. J. Exp. Med. 2010, 207, 1293–1305. [Google Scholar] [CrossRef] [Green Version]
- Lavoie, T.N.; Stewart, C.M.; Berg, K.M.; Li, Y.; Nguyen, C.Q. Expression of interleukin-22 in Sjögren’s syndrome: Significant correlation with disease parameters. Scand. J. Immunol. 2011, 74, 377–382. [Google Scholar] [CrossRef]
- Monteiro, R.; Martins, C.; Barcelos, F.; Nunes, G.; Lopes, T.; Borrego, L.-M. Follicular helper and follicular cytotoxic T cells in primary Sjögren’s Syndrome: Clues for an abnormal antiviral response as a pathogenic mechanism. Ann. Med. 2019, 51, 42. [Google Scholar] [CrossRef] [Green Version]
- Saito, M.; Otsuka, K.; Ushio, A.; Yamada, A.; Arakaki, R.; Kudo, Y.; Ishimaru, N. Unique Phenotypes and Functions of Follicular Helper T Cells and Regulatory T Cells in Sjögren’s Syndrome. Curr. Rheumatol. Rev. 2018, 14, 239–245. [Google Scholar] [CrossRef]
- Scheid, J.F.; Mouquet, H.; Kofer, J.; Yurasov, S.; Nussenzweig, M.C.; Wardemann, H. Differential regulation of self-reactivity discriminates between IgG+ human circulating memory B cells and bone marrow plasma cells. Proc. Natl. Acad. Sci. USA 2011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mouquet, H.; Nussenzweig, M.C. Polyreactive antibodies in adaptive immune responses to viruses. Cell. Mol. Life Sci. 2012, 69, 1435–1445. [Google Scholar] [CrossRef] [PubMed]
- Corsiero, E.; Sutcliffe, N.; Pitzalis, C.; Bombardieri, M. Accumulation of self-reactive naïve and memory B cell reveals sequential defects in B cell tolerance checkpoints in Sjögren’s syndrome. PLoS ONE 2014, 9, e114575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samuels, J.; Ng, Y.-S.; Coupillaud, C.; Paget, D.; Meffre, E. Impaired early B cell tolerance in patients with rheumatoid arthritis. J. Exp. Med. 2005, 201, 1659–1667. [Google Scholar] [CrossRef]
- Mietzner, B.; Tsuiji, M.; Scheid, J.; Velinzon, K.; Tiller, T.; Abraham, K.; Gonzalez, J.B.; Pascual, V.; Stichweh, D.; Wardemann, H.; et al. Autoreactive IgG memory antibodies in patients with systemic lupus erythematosus arise from nonreactive and polyreactive precursors. Proc. Natl. Acad. Sci. USA 2008, 105, 9727–9732. [Google Scholar] [CrossRef] [Green Version]
- Hayakawa, I.; Tedder, T.F.; Zhuang, Y. B-lymphocyte depletion ameliorates Sjögren’s syndrome in Id3 knockout mice. Immunology 2007, 122, 73–79. [Google Scholar] [CrossRef]
- Baff and April: A Tutorial on B Cell Survival. PubMed NCBI. Available online: https://www.ncbi.nlm.nih.gov/pubmed/12427767 (accessed on 2 April 2020).
- Pers, J.-O.; Daridon, C.; Devauchelle, V.; Jousse, S.; Saraux, A.; Jamin, C.; Youinou, P. BAFF overexpression is associated with autoantibody production in autoimmune diseases. Ann. N. Y. Acad. Sci. 2005, 1050, 34–39. [Google Scholar] [CrossRef]
- Nieuwenhuis, P.; Opstelten, D. Functional anatomy of germinal centers. Am. J. Anat. 1984, 170, 421–435. [Google Scholar] [CrossRef]
- Maeda, T.; Wakasawa, T.; Shima, Y.; Tsuboi, I.; Aizawa, S.; Tamai, I. Role of polyamines derived from arginine in differentiation and proliferation of human blood cells. Biol. Pharm. Bull. 2006, 29, 234–239. [Google Scholar] [CrossRef] [Green Version]
- Meyer-Hermann, M. A mathematical model for the germinal center morphology and affinity maturation. J. Theor. Biol. 2002, 216, 273–300. [Google Scholar] [CrossRef] [Green Version]
- Jonsson, M.V.; Skarstein, K. Follicular dendritic cells confirm lymphoid organization in the minor salivary glands of primary Sjögren’s syndrome. J. Oral Pathol. Med. 2008, 37, 515–521. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, M.V.; Skarstein, K.; Jonsson, R.; Brun, J.G. Serological implications of germinal center-like structures in primary Sjögren’s syndrome. J. Rheumatol. 2007, 34, 2044–2049. [Google Scholar]
- Salomonsson, S.; Jonsson, M.V.; Skarstein, K.; Brokstad, K.A.; Hjelmström, P.; Wahren-Herlenius, M.; Jonsson, R. Cellular basis of ectopic germinal center formation and autoantibody production in the target organ of patients with Sjögren’s syndrome. Arthritis Rheum. 2003, 48, 3187–3201. [Google Scholar] [CrossRef] [PubMed]
- Johnsen, S.J.; Brun, J.G.; Gøransson, L.G.; Småstuen, M.C.; Johannesen, T.B.; Haldorsen, K.; Harboe, E.; Jonsson, R.; Meyer, P.A.; Omdal, R. Risk of non-Hodgkin’s lymphoma in primary Sjögren’s syndrome: A population-based study. Arthritis Care Res. 2013, 65, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Theander, E.; Henriksson, G.; Ljungberg, O.; Mandl, T.; Manthorpe, R.; Jacobsson, L.T.H. Lymphoma and other malignancies in primary Sjögren’s syndrome: A cohort study on cancer incidence and lymphoma predictors. Ann. Rheum. Dis. 2006, 65, 796–803. [Google Scholar] [CrossRef] [Green Version]
- Nardi, N.; Brito-Zerón, P.; Ramos-Casals, M.; Aguiló, S.; Cervera, R.; Ingelmo, M.; Font, J. Circulating auto-antibodies against nuclear and non-nuclear antigens in primary Sjögren’s syndrome: Prevalence and clinical significance in 335 patients. Clin. Rheumatol. 2006, 25, 341–346. [Google Scholar] [CrossRef]
- Jones, B. Lacrimal and salivary precipitating antibodies in Sjögren’s syndrome. Lancet 1958, 272, 773–776. [Google Scholar] [CrossRef]
- Anderson, J.R.; Gray, K.; Beck, J.S.; Kinnear, W.F. Precipitating autoantibodies in Sjögren’s syndrome. Lancet 1961, 278, 456–460. [Google Scholar] [CrossRef]
- Espinosa, A.; Dardalhon, V.; Brauner, S.; Ambrosi, A.; Higgs, R.; Quintana, F.J.; Sjöstrand, M.; Eloranta, M.L.; Ní Gabhann, J.; Winqvist, O.; et al. Loss of the lupus autoantigen Ro52/Trim21 induces tissue inflammation and systemic autoimmunity by disregulating the IL-23-Th17 pathway. J. Exp. Med. 2009, 206, 1661–1671. [Google Scholar] [CrossRef]
- Keene, J.D. Molecular structure of the La and Ro autoantigens and their use in autoimmune diagnostics. J. Autoimmun. 1989, 2, 329–334. [Google Scholar] [CrossRef]
- Elkon, K.B.; Gharavi, A.E.; Hughes, G.R.; Moutsoupoulos, H.M. Autoantibodies in the sicca syndrome (primary Sjögren’s syndrome). Ann. Rheum. Dis. 1984, 43, 243–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baer, A.N.; McAdams DeMarco, M.; Shiboski, S.C.; Lam, M.Y.; Challacombe, S.; Daniels, T.E.; Dong, Y.; Greenspan, J.S.; Kirkham, B.W.; Lanfranchi, H.E.; et al. The SSB-positive/SSA-negative antibody profile is not associated with key phenotypic features of Sjögren’s syndrome. Ann. Rheum. Dis. 2015, 74, 1557–1561. [Google Scholar] [CrossRef] [PubMed]
- Manoussakis, M.N.; Pange, P.J.; Moutsopulos, H.M. The autoantibody profile in Sjögren’s syndrome. Ter. Arkh. 1988, 60, 17–20. [Google Scholar] [PubMed]
- Mavragani, C.P.; Tzioufas, A.G.; Moutsopoulos, H.M. Sjögren’s syndrome: Autoantibodies to cellular antigens. Clinical and molecular aspects. Int. Arch. Allergy Immunol. 2000, 123, 46–57. [Google Scholar] [CrossRef]
- Bournia, V.-K.K.; Diamanti, K.D.; Vlachoyiannopoulos, P.G.; Moutsopoulos, H.M. Anticentromere antibody positive Sjögren’s Syndrome: A retrospective descriptive analysis. Arthritis Res. Ther. 2010, 12, R47. [Google Scholar] [CrossRef] [Green Version]
- Salliot, C.; Gottenberg, J.-E.; Bengoufa, D.; Desmoulins, F.; Miceli-Richard, C.; Mariette, X. Anticentromere antibodies identify patients with Sjögren’s syndrome and autoimmune overlap syndrome. J. Rheumatol. 2007, 34, 2253–2258. [Google Scholar]
- Bournia, V.-K.; Vlachoyiannopoulos, P.G. Subgroups of Sjögren syndrome patients according to serological profiles. J. Autoimmun. 2012, 39, 15–26. [Google Scholar] [CrossRef]
- Kyriakidis, N.C.; Kapsogeorgou, E.K.; Tzioufas, A.G. A comprehensive review of autoantibodies in primary Sjögren’s syndrome: Clinical phenotypes and regulatory mechanisms. J. Autoimmun. 2014, 51, 67–74. [Google Scholar] [CrossRef]
- Takemoto, F.; Hoshino, J.; Sawa, N.; Tamura, Y.; Tagami, T.; Yokota, M.; Katori, H.; Yokoyama, K.; Ubara, Y.; Hara, S.; et al. Autoantibodies against carbonic anhydrase II are increased in renal tubular acidosis associated with Sjogren syndrome. Am. J. Med. 2005, 118, 181–184. [Google Scholar] [CrossRef]
- Nishimori, I.; Bratanova, T.; Toshkov, I.; Caffrey, T.; Mogaki, M.; Shibata, Y.; Hollingsworth, M.A. Induction of experimental autoimmune sialoadenitis by immunization of PL/J mice with carbonic anhydrase II. J. Immunol. 1950 1995, 154, 4865–4873. [Google Scholar]
- Takemoto, F.; Katori, H.; Sawa, N.; Hoshino, J.; Suwabe, T.; Sogawa, Y.; Nomura, K.; Nakanishi, S.; Higa, Y.; Kanbayashi, H.; et al. Induction of anti-carbonic-anhydrase-II antibody causes renal tubular acidosis in a mouse model of Sjogren’s syndrome. Nephron Physiol. 2007, 106, p63–p68. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.; Lee, J.; Park, S.-H.; Kim, H.-D.; Choi, Y. Associations of Anti-Aquaporin 5 Autoantibodies with Serologic and Histopathological Features of Sjögren’s Syndrome. J. Clin. Med. 2019, 8, 1863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clinical Associations of Autoantibodies to Human Muscarinic Acetylcholine Receptor 3 (213–228) in Primary Sjogren’s Syndrome. PubMed NCBI. Available online: https://www.ncbi.nlm.nih.gov/pubmed/?term=10.1093%2Frheumatology%2Fkeh672 (accessed on 2 April 2020).
- Sordet, C.; Gottenberg, J.E.; Goetz, J.; Bengoufa, D.; Humbel, R.-L.; Mariette, X.; Sibilia, J. Anti-α-fodrin autoantibodies are not useful diagnostic markers of primary Sjögren’s syndrome. Ann. Rheum. Dis. 2005, 64, 1244–1245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Applbaum, E.; Lichtbroun, A. Novel Sjögren’s autoantibodies found in fibromyalgia patients with sicca and/or xerostomia. Autoimmun. Rev. 2019, 18, 199–202. [Google Scholar] [CrossRef] [PubMed]
- Martín-Nares, E.; Hernández-Molina, G. Novel autoantibodies in Sjögren’s syndrome: A comprehensive review. Autoimmun. Rev. 2019, 18, 192–198. [Google Scholar] [CrossRef] [PubMed]
- De Langhe, E.; Bossuyt, X.; Shen, L.; Malyavantham, K.; Ambrus, J.L.; Suresh, L. Evaluation of Autoantibodies in Patients with Primary and Secondary Sjogren’s Syndrome. Open Rheumatol. J. 2017, 11, 10–15. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.; Li, J.; Chen, J.; Shao, M.; Zhang, R.; Liang, Y.; Zhang, X.; Zhang, X.; Zhang, Q.; Li, F.; et al. Tissue-Specific Autoantibodies Improve Diagnosis of Primary Sjögren’s Syndrome in the Early Stage and Indicate Localized Salivary Injury. J. Immunol. Res. 2019, 2019, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Suresh, L.; Malyavantham, K.; Shen, L.; Ambrus, J.L. Investigation of novel autoantibodies in Sjogren’s syndrome utilizing Sera from the Sjogren’s international collaborative clinical alliance cohort. BMC Ophthalmol. 2015, 15, 38. [Google Scholar] [CrossRef] [Green Version]
- Shen, L.; Suresh, L.; Lindemann, M.; Xuan, J.; Kowal, P.; Malyavantham, K.; Ambrus, J.L. Novel autoantibodies in Sjogren’s syndrome. Clin. Immunol. 2012, 145, 251–255. [Google Scholar] [CrossRef]
- Xuan, J.; Wang, Y.; Xiong, Y.; Qian, H.; He, Y.; Shi, G. Investigation of autoantibodies to SP-1 in Chinese patients with primary Sjögren’s syndrome. Clin. Immunol. 2018, 188, 58–63. [Google Scholar] [CrossRef]
- Everett, S.; Vishwanath, S.; Cavero, V.; Shen, L.; Suresh, L.; Malyavantham, K.; Lincoff-Cohen, N.; Ambrus, J.L. Analysis of novel Sjogren’s syndrome autoantibodies in patients with dry eyes. BMC Ophthalmol. 2017, 17, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubschman, S.; Rojas, M.; Kalavar, M.; Kloosterboer, A.; Sabater, A.L.; Galor, A. Association Between Early Sjögren Markers and Symptoms and Signs of Dry Eye. Cornea 2020, 39, 311–315. [Google Scholar] [CrossRef] [PubMed]
- Uchida, K.; Akita, Y.; Matsuo, K.; Fujiwara, S.; Nakagawa, A.; Kazaoka, Y.; Hachiya, H.; Naganawa, Y.; Oh-Iwa, I.; Ohura, K.; et al. Identification of specific autoantigens in Sjögren’s syndrome by SEREX. Immunology 2005, 116, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liao, X.; Wang, Y.; Chen, S.; Sun, Y.; Lin, Q.; Shi, G. Autoantibody to MDM2: A potential serological marker of primary Sjogren’s syndrome. Oncotarget 2017, 8, 14306–14313. [Google Scholar] [CrossRef] [PubMed]
- Nozawa, K.; Ikeda, K.; Satoh, M.; Reeves, W.H.; Stewart, C.M.; Li, Y.-C.; Yen, T.J.; Rios, R.M.; Takamori, K.; Ogawa, H.; et al. Autoantibody to NA14 is an independent marker primarily for Sjogren’s syndrome. Front. Biosci. Landmark Ed. 2009, 14, 3733–3739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uomori, K.; Nozawa, K.; Ikeda, K.; Doe, K.; Yamada, Y.; Yamaguchi, A.; Fujishiro, M.; Kawasaki, M.; Morimoto, S.; Takamori, K.; et al. A re-evaluation of anti-NA-14 antibodies in patients with primary Sjögren’s syndrome: Significant role of interferon-γ in the production of autoantibodies against NA-14. Autoimmunity 2016, 49, 347–356. [Google Scholar] [CrossRef]
- Duda, S.; Witte, T.; Stangel, M.; Adams, J.; Schmidt, R.E.; Baerlecken, N.T. Autoantibodies binding to stathmin-4: New marker for polyneuropathy in primary Sjögren’s syndrome. Immunol. Res. 2017, 65, 1099–1102. [Google Scholar] [CrossRef]
- Fiorentino, D.F.; Presby, M.; Baer, A.N.; Petri, M.; Rieger, K.E.; Soloski, M.; Rosen, A.; Mammen, A.L.; Christopher-Stine, L.; Casciola-Rosen, L. PUF60: A prominent new target of the autoimmune response in dermatomyositis and Sjögren’s syndrome. Ann. Rheum. Dis. 2016, 75, 1145–1151. [Google Scholar] [CrossRef]
- Tay, S.H.; Fairhurst, A.-M.; Mak, A. Clinical utility of circulating anti-N-methyl-d-aspartate receptor subunits NR2A/B antibody for the diagnosis of neuropsychiatric syndromes in systemic lupus erythematosus and Sjögren’s syndrome: An updated meta-analysis. Autoimmun. Rev. 2017, 16, 114–122. [Google Scholar] [CrossRef]
- Lauvsnes, M.B.; Beyer, M.K.; Kvaløy, J.T.; Greve, O.J.; Appenzeller, S.; Kvivik, I.; Harboe, E.; Tjensvoll, A.B.; Gøransson, L.G.; Omdal, R. Association of hippocampal atrophy with cerebrospinal fluid antibodies against the NR2 subtype of the N-methyl-D-aspartate receptor in patients with systemic lupus erythematosus and patients with primary Sjögren’s syndrome. Arthritis Rheumatol. 2014, 66, 3387–3394. [Google Scholar] [CrossRef]
- Wolska, N.; Rybakowska, P.; Rasmussen, A.; Brown, M.; Montgomery, C.; Klopocki, A.; Grundahl, K.; Scofield, R.H.; Radfar, L.; Stone, D.U.; et al. Brief Report: Patients With Primary Sjögren’s Syndrome Who Are Positive for Autoantibodies to Tripartite Motif-Containing Protein 38 Show Greater Disease Severity. Arthritis Rheumatol. 2016, 68, 724–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alunno, A.; Bistoni, O.; Carubbi, F.; Valentini, V.; Cafaro, G.; Bartoloni, E.; Giacomelli, R.; Gerli, R. Prevalence and significance of anti-saccharomyces cerevisiae antibodies in primary Sjögren’s syndrome. Clin. Exp. Rheumatol. 2018, 36, 73–79. [Google Scholar] [PubMed]
- Birnbaum, J.; Hoke, A.; Lalji, A.; Calabresi, P.; Bhargava, P.; Casciola-Rosen, L. Brief Report: Anti-Calponin 3 Autoantibodies: A Newly Identified Specificity in Patients With Sjögren’s Syndrome. Arthritis Rheumatol. 2018, 70, 1610–1616. [Google Scholar] [CrossRef] [Green Version]
- Mukaino, A.; Nakane, S.; Higuchi, O.; Nakamura, H.; Miyagi, T.; Shiroma, K.; Tokashiki, T.; Fuseya, Y.; Ochi, K.; Umeda, M.; et al. Insights from the ganglionic acetylcholine receptor autoantibodies in patients with Sjögren’s syndrome. Mod. Rheumatol. 2016, 26, 708–715. [Google Scholar] [CrossRef] [PubMed]
- Birnbaum, J.; Atri, N.M.; Baer, A.N.; Cimbro, R.; Montagne, J.; Casciola-Rosen, L. Relationship Between Neuromyelitis Optica Spectrum Disorder and Sjögren’s Syndrome: Central Nervous System Extraglandular Disease or Unrelated, Co-Occurring Autoimmunity?: Relationship Between Sjögren’s Syndrome and NMOSD. Arthritis Care Res. 2017, 69, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Tzartos, J.S.; Stergiou, C.; Daoussis, D.; Zisimopoulou, P.; Andonopoulos, A.P.; Zolota, V.; Tzartos, S.J. Antibodies to aquaporins are frequent in patients with primary Sjögren’s syndrome. Rheumatology 2017, 56, 2114–2122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.-H.; Zhou, P.-F.; Long, G.-F.; Tian, X.; Guo, Y.-F.; Pang, A.-M.; Di, R.; Shen, Y.-N.; Liu, Y.-D.; Cui, Y.-J. Elevated Plasma P-Selectin Autoantibodies in Primary Sjögren Syndrome Patients with Thrombocytopenia. Med. Sci. Monit. 2015, 21, 3690–3695. [Google Scholar] [CrossRef] [Green Version]
- Bergum, B.; Koro, C.; Delaleu, N.; Solheim, M.; Hellvard, A.; Binder, V.; Jonsson, R.; Valim, V.; Hammenfors, D.S.; Jonsson, M.V.; et al. Antibodies against carbamylated proteins are present in primary Sjögren’s syndrome and are associated with disease severity. Ann. Rheum. Dis. 2016, 75, 1494–1500. [Google Scholar] [CrossRef] [Green Version]
- Pecani, A.; Alessandri, C.; Spinelli, F.R.; Priori, R.; Riccieri, V.; Di Franco, M.; Ceccarelli, F.; Colasanti, T.; Pendolino, M.; Mancini, R.; et al. Prevalence, sensitivity and specificity of antibodies against carbamylated proteins in a monocentric cohort of patients with rheumatoid arthritis and other autoimmune rheumatic diseases. Arthritis Res. Ther. 2016, 18, 276. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Hussain, M.; Yang, X.; Chen, P.; Yang, C.; Xun, Y.; Tian, Y.; Du, H. Identification of Moesin as a Novel Autoantigen in Patients with Sjögren’s Syndrome. Protein Pept. Lett. 2018, 25, 350–355. [Google Scholar] [CrossRef]
- Cui, L.; Elzakra, N.; Xu, S.; Xiao, G.G.; Yang, Y.; Hu, S. Investigation of three potential autoantibodies in Sjogren’s syndrome and associated MALT lymphoma. Oncotarget 2017, 8, 30039–30049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nezos, A.; Cinoku, I.; Mavragani, C.P.; Moutsopoulos, H.M. Antibodies against citrullinated alpha enolase peptides in primary Sjogren’s syndrome. Clin. Immunol. 2017, 183, 300–303. [Google Scholar] [CrossRef] [PubMed]
- Segerberg-Konttinen, M.; Konttinen, Y.T.; Bergroth, V. Focus score in the diagnosis of Sjögren’s syndrome. Scand. J. Rheumatol. Suppl. 1986, 61, 47–51. [Google Scholar] [PubMed]
- Bodeutsch, C.; de Wilde, P.C.; Kater, L.; van Houwelingen, J.C.; van den Hoogen, F.H.; Kruize, A.A.; Hené, R.J.; van de Putte, L.B.; Vooijs, G.P. Quantitative immunohistologic criteria are superior to the lymphocytic focus score criterion for the diagnosis of Sjögren’s syndrome. Arthritis Rheum. 1992, 35, 1075–1087. [Google Scholar] [CrossRef]
- Barrera, M.J.; Bahamondes, V.; Sepúlveda, D.; Quest, A.F.G.; Castro, I.; Cortés, J.; Aguilera, S.; Urzúa, U.; Molina, C.; Pérez, P.; et al. Sjögren’s syndrome and the epithelial target: A comprehensive review. J. Autoimmun. 2013, 42, 7–18. [Google Scholar] [CrossRef]
- Pérez, P.; Goicovich, E.; Alliende, C.; Aguilera, S.; Leyton, C.; Molina, C.; Pinto, R.; Romo, R.; Martinez, B.; González, M.J. Differential expression of matrix metalloproteinases in labial salivary glands of patients with primary Sjögren’s syndrome. Arthritis Rheum. 2000, 43, 2807–2817. [Google Scholar] [CrossRef]
- Sun, D.; Emmert-Buck, M.R.; Fox, P.C. Differential cytokine mRNA expression in human labial minor salivary glands in primary Sjögren’s syndrome. Autoimmunity 1998, 28, 125–137. [Google Scholar] [CrossRef]
- Molina, C.; Alliende, C.; Aguilera, S.; Kwon, Y.-J.; Leyton, L.; Martínez, B.; Leyton, C.; Pérez, P.; González, M.-J. Basal lamina disorganisation of the acini and ducts of labial salivary glands from patients with Sjogren’s syndrome: Association with mononuclear cell infiltration. Ann. Rheum. Dis. 2006, 65, 178–183. [Google Scholar] [CrossRef] [Green Version]
- Ng, W.-F.; Bowman, S.J. Primary Sjogren’s syndrome: Too dry and too tired. Rheumatology 2010, 49, 844–853. [Google Scholar] [CrossRef] [Green Version]
- Hackett, K.L.; Gotts, Z.M.; Ellis, J.; Deary, V.; Rapley, T.; Ng, W.-F.; Newton, J.L.; Deane, K.H.O. An investigation into the prevalence of sleep disturbances in primary Sjögren’s syndrome: A systematic review of the literature. Rheumatology 2017, 56, 570–580. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.-C.; Chang, K.; Lin, C.-Y.; Chen, Y.-H.; Lu, P.-L. Periodic fever as the manifestation of primary Sjogren’s syndrome: A case report and literature review. Clin. Rheumatol. 2012, 31, 1517–1519. [Google Scholar] [CrossRef] [PubMed]
- Voulgarelis, M.; Moutsopoulos, H.M. Mucosa-associated lymphoid tissue lymphoma in Sjögren’s syndrome: Risks, management, and prognosis. Rheum. Dis. Clin. N. Am. 2008, 34, 921–933. [Google Scholar] [CrossRef] [PubMed]
- Kassan, S.S.; Moutsopoulos, H.M. Clinical manifestations and early diagnosis of Sjögren syndrome. Arch. Intern. Med. 2004, 164, 1275–1284. [Google Scholar] [CrossRef]
- Retamozo, S.; Acar-Denizli, N.; Rasmussen, A.; Horváth, I.F.; Baldini, C.; Priori, R.; Sandhya, P.; Hernandez-Molina, G.; Armagan, B.; Praprotnik, S.; et al. Systemic manifestations of primary Sjögren’s syndrome out of the ESSDAI classification: Prevalence and clinical relevance in a large international, multi-ethnic cohort of patients. Clin. Exp. Rheumatol. 2019, 37, 97–106. [Google Scholar] [PubMed]
- López-Pintor, R.M.; Fernández Castro, M.; Hernández, G. Oral involvement in patients with primary Sjögren’s syndrome. Multidisciplinary care by dentists and rheumatologists. Reumatol. Clin. 2015, 11, 387–394. [Google Scholar] [CrossRef] [PubMed]
- Generali, E.; Costanzo, A.; Mainetti, C.; Selmi, C. Cutaneous and Mucosal Manifestations of Sjögren’s Syndrome. Clin. Rev. Allergy Immunol. 2017, 53, 357–370. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Casals, M.; Brito-Zerón, P.; Bombardieri, S.; Bootsma, H.; De Vita, S.; Dörner, T.; Fisher, B.A.; Gottenberg, J.-E.; Hernandez-Molina, G.; Kocher, A.; et al. EULAR recommendations for the management of Sjögren’s syndrome with topical and systemic therapies. Ann. Rheum. Dis. 2020, 79, 3–18. [Google Scholar] [CrossRef] [Green Version]
- Ramos-Casals, M.; Brito-Zerón, P.; Seror, R.; Bootsma, H.; Bowman, S.J.; Dörner, T.; Gottenberg, J.-E.; Mariette, X.; Theander, E.; Bombardieri, S.; et al. Characterization of systemic disease in primary Sjögren’s syndrome: EULAR-SS Task Force recommendations for articular, cutaneous, pulmonary and renal involvements. Rheumatology 2015, 54, 2230–2238. [Google Scholar] [CrossRef] [Green Version]
- Mirouse, A.; Seror, R.; Vicaut, E.; Mariette, X.; Dougados, M.; Fauchais, A.-L.; Deroux, A.; Dellal, A.; Costedoat-Chalumeau, N.; Denis, G.; et al. Arthritis in primary Sjögren’s syndrome: Characteristics, outcome and treatment from French multicenter retrospective study. Autoimmun. Rev. 2019, 18, 9–14. [Google Scholar] [CrossRef]
- Vitali, C.; Del Papa, N. Pain in primary Sjögren’s syndrome. Best Pract. Res. Clin. Rheumatol. 2015, 29, 63–70. [Google Scholar] [CrossRef]
- Atzeni, F.; Cazzola, M.; Benucci, M.; Di Franco, M.; Salaffi, F.; Sarzi-Puttini, P. Chronic widespread pain in the spectrum of rheumatological diseases. Best Pract. Res. Clin. Rheumatol. 2011, 25, 165–171. [Google Scholar] [CrossRef]
- Alunno, A.; Carubbi, F.; Bartoloni, E.; Cipriani, P.; Giacomelli, R.; Gerli, R. The kaleidoscope of neurological manifestations in primary Sjögren’s syndrome. Clin. Exp. Rheumatol. 2019, 37, 192–198. [Google Scholar]
- Flament, T.; Bigot, A.; Chaigne, B.; Henique, H.; Diot, E.; Marchand-Adam, S. Pulmonary manifestations of Sjögren’s syndrome. Eur. Respir. Rev. 2016, 25, 110–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatron, P.-Y.; Tillie-Leblond, I.; Launay, D.; Hachulla, E.; Fauchais, A.L.; Wallaert, B. Pulmonary manifestations of Sjögren’s syndrome. Presse Med. 1983 2011, 40, e49–e64. [Google Scholar] [CrossRef] [PubMed]
- Tavoni, A.; Vitali, C.; Cirigliano, G.; Frigelli, S.; Stampacchia, G.; Bombardieri, S. Shrinking lung in primary Sjögren’s syndrome. Arthritis Rheum. 1999, 42, 2249–2250. [Google Scholar] [CrossRef]
- Singh, R.; Huang, W.; Menon, Y.; Espinoza, L.R. Shrinking lung syndrome in systemic lupus erythematosus and Sjogren’s syndrome. J. Clin. Rheumatol. 2002, 8, 340–345. [Google Scholar] [CrossRef] [PubMed]
- Langenskiöld, E.; Bonetti, A.; Fitting, J.W.; Heinzer, R.; Dudler, J.; Spertini, F.; Lazor, R. Shrinking lung syndrome successfully treated with rituximab and cyclophosphamide. Respiration 2012, 84, 144–149. [Google Scholar] [CrossRef]
- Blanco Pérez, J.J.; Pérez González, A.; Guerra Vales, J.L.; Melero Gonzalez, R.; Pego Reigosa, J.M. Shrinking Lung in Primary Sjogrën Syndrome Successfully Treated with Rituximab. Arch. Bronconeumol. 2015, 51, 475–476. [Google Scholar] [CrossRef] [PubMed]
- Baenas, D.F.; Retamozo, S.; Pirola, J.P.; Caeiro, F. Shrinking lung syndrome and pleural effusion as an initial manifestation of primary Sjögren’s syndrome. Síndrome de pulmón encogido y derrame pleural como manifestación inicial de síndrome de Sjögren primario. Rheumatol. Clin. 2020, 16, 65–68. [Google Scholar] [CrossRef]
- Uslu, S.; Köken Avşar, A.; Erez, Y.; Sarı, İ. Shrinking Lung Syndrome in Primary Sjögren Syndrome. Balk. Med. J. 2020. [Google Scholar] [CrossRef]
- Liang, M.; Bao, L.; Xiong, N.; Jin, B.; Ni, H.; Zhang, J.; Zou, H.; Luo, X.; Li, J. Cardiac arrhythmias as the initial manifestation of adult primary Sjögren’s syndrome: A case report and literature review. Int. J. Rheum. Dis. 2015, 18, 800–806. [Google Scholar] [CrossRef] [PubMed]
- Sung, M.J.; Park, S.-H.; Kim, S.-K.; Lee, Y.-S.; Park, C.-Y.; Choe, J.-Y. Complete atrioventricular block in adult Sjögren’s syndrome with anti-Ro autoantibody. Kor. J. Intern. Med. 2011, 26, 213–215. [Google Scholar] [CrossRef] [PubMed]
- Popov, Y.; Salomon-Escoto, K. Gastrointestinal and Hepatic Disease in Sjogren Syndrome. Rheum. Dis. Clin. N. Am. 2018, 44, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Ebert, E.C. Gastrointestinal and hepatic manifestations of Sjogren syndrome. J. Clin. Gastroenterol. 2012, 46, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Evans, R.; Zdebik, A.; Ciurtin, C.; Walsh, S.B. Renal involvement in primary Sjögren’s syndrome. Rheumatology 2015, 54, 1541–1548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geng, Y.; Zhao, Y.; Zhang, Z. Tubulointerstitial nephritis-induced hypophosphatemic osteomalacia in Sjögren’s syndrome: A case report and review of the literature. Clin. Rheumatol. 2018, 37, 257–263. [Google Scholar] [CrossRef]
- Gu, X.; Su, Z.; Chen, M.; Xu, Y.; Wang, Y. Acquired Gitelman syndrome in a primary Sjögren syndrome patient with a SLC12A3 heterozygous mutation: A case report and literature review. Nephrology 2017, 22, 652–655. [Google Scholar] [CrossRef] [Green Version]
- Darrieutort-Laffite, C.; André, V.; Hayem, G.; Saraux, A.; Le Guern, V.; Le Jeunne, C.; Puéchal, X. Sjögren’s syndrome complicated by interstitial cystitis: A case series and literature review. Joint Bone Spine 2015, 82, 245–250. [Google Scholar] [CrossRef]
- Manganelli, P.; Fietta, P.; Quaini, F. Hematologic manifestations of primary Sjögren’s syndrome. Clin. Exp. Rheumatol. 2006, 24, 438–448. [Google Scholar]
- Ramos-Casals, M.; Font, J.; Garcia-Carrasco, M.; Brito, M.-P.; Rosas, J.; Calvo-Alen, J.; Pallares, L.; Cervera, R.; Ingelmo, M. Primary Sjögren syndrome: Hematologic patterns of disease expression. Medicine 2002, 81, 281–292. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, H.; Takahashi, Y.; Kaneko, H.; Kano, T.; Mimori, A. Thrombotic thrombocytopenic purpura with an autoantibody to ADAMTS13 complicating Sjögren’s syndrome: Two cases and a literature review. Mod. Rheumatol. 2013, 23, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Zhu, T.; Wu, D.; Zhang, L. Sjögren’s syndrome initially presented as thrombotic thrombocytopenic purpura in a male patient: A case report and literature review. Clin. Rheumatol. 2018, 37, 1421–1426. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Gu, W.; Ma, Y.; Wang, J.; Wu, M. Relapsed/refractory acquired thrombotic thrombocytopenic purpura in a patient with Sjögren syndrome: Case report and review of the literature. Medicine 2018, 97, e12989. [Google Scholar] [CrossRef]
- García-Montoya, L.; Sáenz-Tenorio, C.N.; Janta, I.; Menárguez, J.; López-Longo, F.J.; Monteagudo, I.; Naredo, E. Hemophagocytic lymphohistiocytosis in a patient with Sjögren’s syndrome: Case report and review. Rheumatol. Int. 2017, 37, 663–669. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Molina, G.; Faz-Munoz, D.; Astudillo-Angel, M.; Iturralde-Chavez, A.; Reyes, E. Coexistance of Amyloidosis and Primary Sjögren’s Syndrome: An Overview. Curr. Rheumatol. Rev. 2018, 14, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Freeman, S.R.M.; Sheehan, P.Z.; Thorpe, M.A.; Rutka, J.A. Ear, Nose, and Throat Manifestations of Sjögren’s Syndrome: Retrospective Review of a Multidisciplinary Clinic. J. Otolaryngol. 2005, 34, 20. [Google Scholar] [CrossRef] [PubMed]
- Midilli, R.; Gode, S.; Oder, G.; Kabasakal, Y.; Karci, B. Nasal and paranasal involvement in primary Sjogren‘s syndrome. Rhinol. J. 2013, 51, 265–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belafsky, P.C.; Postma, G.N. The laryngeal and esophageal manifestations of Sjögren’s syndrome. Curr. Rheumatol. Rep. 2003, 5, 297–303. [Google Scholar] [CrossRef]
- Rodriguez, M.A.; Tapanes, F.J.; Stekman, I.L.; Pinto, J.A.; Camejo, O.; Abadi, I. Auricular chondritis and diffuse proliferative glomerulonephritis in primary Sjogren’s syndrome. Ann. Rheum. Dis. 1989, 48, 683–685. [Google Scholar] [CrossRef] [Green Version]
- Tumiati, B. Hearing Loss in the Sjogren Syndrome. Ann. Intern. Med. 1997, 126, 450. [Google Scholar] [CrossRef]
- Isik, H.; Isik, M.; Aynioglu, O.; Karcaaltincaba, D.; Sahbaz, A.; Beyazcicek, T.; Harma, M.I.; Demircan, N. Are the women with Sjögren’s Syndrome satisfied with their sexual activity? Rev. Bras. Reumatol. Engl. Ed. 2017, 57, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Capone, C.; Buyon, J.P.; Friedman, D.M.; Frishman, W.H. Cardiac Manifestations of Neonatal Lupus: A Review of Autoantibody-associated Congenital Heart Block and its Impact in an Adult Population. Cardiol. Rev. 2012, 20, 72–76. [Google Scholar] [CrossRef] [Green Version]
- Picone, O.; Alby, C.; Frydman, R.; Mariette, X. Sjögren syndrome in Obstetric and Gynecology: Literature review. J. Gynecol. Obstet. Biol. Reprod. 2006, 35, 169–175. [Google Scholar] [CrossRef]
- Costedoat-Chalumeau, N.; Amoura, Z.; Villain, E.; Cohen, L.; Fermont, L.; Le Thi Huong, D.; Vauthier, D.; Georgin-Lavialle, S.; Wechsler, B.; Dommergues, M.; et al. Prise en charge obstétricale des patientes à risque de « lupus néonatal ». J. Gynécologie Obstétrique Biol. Reprod. 2006, 35, 146–156. [Google Scholar] [CrossRef]
- Upala, S.; Yong, W.C.; Sanguankeo, A. Association between primary Sjögren’s syndrome and pregnancy complications: A systematic review and meta-analysis. Clin. Rheumatol. 2016, 35, 1949–1955. [Google Scholar] [CrossRef]
- Brito-Zerón, P.; Theander, E.; Baldini, C.; Seror, R.; Retamozo, S.; Quartuccio, L.; Bootsma, H.; Bowman, S.J.; Dörner, T.; Gottenberg, J.-E.; et al. Early diagnosis of primary Sjögren’s syndrome: EULAR-SS task force clinical recommendations. Expert Rev. Clin. Immunol. 2016, 12, 137–156. [Google Scholar] [CrossRef]
- Vitali, C.; Bombardieri, S.; Jonsson, R.; Moutsopoulos, H.M.; Alexander, E.L.; Carsons, S.E.; Daniels, T.E.; Fox, P.C.; Fox, R.I.; Kassan, S.S.; et al. Classification criteria for Sjögren’s syndrome: A revised version of the European criteria proposed by the American-European Consensus Group. Ann. Rheum. Dis. 2002, 61, 554–558. [Google Scholar] [CrossRef] [Green Version]
- Shiboski, S.C.; Shiboski, C.H.; Criswell, L.A.; Baer, A.N.; Challacombe, S.; Lanfranchi, H.; Schiødt, M.; Umehara, H.; Vivino, F.; Zhao, Y.; et al. American College of Rheumatology classification criteria for Sjögren’s syndrome: A data-driven, expert consensus approach in the Sjögren’s International Collaborative Clinical Alliance cohort. Arthritis Care Res. 2012, 64, 475–487. [Google Scholar] [CrossRef]
- Shiboski, C.H.; Shiboski, S.C.; Seror, R.; Criswell, L.A.; Labetoulle, M.; Lietman, T.M.; Rasmussen, A.; Scofield, H.; Vitali, C.; Bowman, S.J.; et al. 2016 American College of Rheumatology/European League Against Rheumatism Classification Criteria for Primary Sjögren’s Syndrome: A Consensus and Data-Driven Methodology Involving Three International Patient Cohorts. Arthritis Rheumatol. 2017, 69, 35–45. [Google Scholar] [CrossRef]
- Begley, C.; Caffery, B.; Chalmers, R.; Situ, P.; Simpson, T.; Nelson, J.D. Review and analysis of grading scales for ocular surface staining. Ocul. Surf. 2019, 7, 208–220. [Google Scholar] [CrossRef]
- Baldini, C.; Zabotti, A.; Filipovic, N.; Vukicevic, A.; Luciano, N.; Ferro, F.; Lorenzon, M.; De Vita, S. Imaging in primary Sjögren’s syndrome: The “obsolete and the new”. Clin. Exp. Rheumatol. 2018, 36, 215–221. [Google Scholar] [PubMed]
- Schall, G.L.; Anderson, L.G.; Wolf, R.O.; Herdt, J.R.; Tarpley, T.M.; Cummings, N.A.; Zeiger, L.S.; Talal, N. Xerostomia in Sjögren’s syndrome. Evaluation by sequential salivary scintigraphy. JAMA 1971, 216, 2109–2116. [Google Scholar] [CrossRef]
- Vinagre, F.; Santos, M.J.; Prata, A.; da Silva, J.C.; Santos, A.I. Assessment of salivary gland function in Sjögren’s syndrome: The role of salivary gland scintigraphy. Autoimmun. Rev. 2009, 8, 672–676. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Song, S.; Wu, S.; Duan, T.; Chen, L.; Ye, J.; Xiao, J. Diagnostic accuracy of salivary gland ultrasonography with different scoring systems in Sjögren’s syndrome: A systematic review and meta-analysis. Sci. Rep. 2018, 8, 17128. [Google Scholar] [CrossRef] [PubMed]
- Jousse-Joulin, S.; Milic, V.; Jonsson, M.V.; Plagou, A.; Theander, E.; Luciano, N.; Rachele, P.; Baldini, C.; Bootsma, H.; Vissink, A.; et al. Is salivary gland ultrasonography a useful tool in Sjögren’s syndrome? A systematic review. Rheumatology 2016, 55, 789–800. [Google Scholar] [CrossRef] [Green Version]
- Nimwegen, J.F.; Mossel, E.; Delli, K.; Ginkel, M.S.; Stel, A.J.; Kroese, F.G.M.; Spijkervet, F.K.L.; Vissink, A.; Arends, S.; Bootsma, H. Incorporation of Salivary Gland Ultrasonography Into the American College of Rheumatology/European League Against Rheumatism Criteria for Primary Sjögren’s Syndrome. Arthritis Care Res. 2020, 72, 583–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, B.A.; Everett, C.C.; Rout, J.; O’Dwyer, J.L.; Emery, P.; Pitzalis, C.; Ng, W.-F.; Carr, A.; Pease, C.T.; Price, E.J.; et al. Effect of rituximab on a salivary gland ultrasound score in primary Sjögren’s syndrome: Results of the TRACTISS randomised double-blind multicentre substudy. Ann. Rheum. Dis. 2018, 77, 412–416. [Google Scholar] [CrossRef] [PubMed]
- Jousse-Joulin, S.; Devauchelle-Pensec, V.; Cornec, D.; Marhadour, T.; Bressollette, L.; Gestin, S.; Pers, J.O.; Nowak, E.; Saraux, A. Brief Report: Ultrasonographic Assessment of Salivary Gland Response to Rituximab in Primary Sjögren’s Syndrome: Ultrasonographic response to Rituximab in primary SS. Arthritis Rheumatol. 2015, 67, 1623–1628. [Google Scholar] [CrossRef]
- Varela-Centelles, P.; Seoane-Romero, J.-M.; Sánchez-Sánchez, M.; González-Mosquera, A.; Diz-Dios, P.; Seoane, J. Minor salivary gland biopsy in Sjögren’s syndrome: A review and introduction of a new tool to ease the procedure. Med. Oral Patol. Oral Cirugia Bucal 2014, 19, e20–e23. [Google Scholar] [CrossRef]
- Spijkervet, F.K.L.; Haacke, E.; Kroese, F.G.M.; Bootsma, H.; Vissink, A. Parotid Gland Biopsy, the Alternative Way to Diagnose Sjögren Syndrome. Rheum. Dis. Clin. N. Am. 2016, 42, 485–499. [Google Scholar] [CrossRef]
- Fisher, B.A.; Jonsson, R.; Daniels, T.; Bombardieri, M.; Brown, R.M.; Morgan, P.; Bombardieri, S.; Ng, W.-F.; Tzioufas, A.G.; Vitali, C.; et al. Standardisation of labial salivary gland histopathology in clinical trials in primary Sjögren’s syndrome. Ann. Rheum. Dis. 2017, 76, 1161–1168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chisholm, D.M.; Mason, D.K. Labial salivary gland biopsy in Sjogren’s disease. J. Clin. Pathol. 1968, 21, 656–660. [Google Scholar] [CrossRef] [PubMed]
- Greenspan, J.S.; Daniels, T.E.; Talal, N.; Sylvester, R.A. The histopathology of Sjögren’s syndrome in labial salivary gland biopsies. Oral Surg. Oral Med. Oral Pathol. 1974, 37, 217–229. [Google Scholar] [CrossRef]
- Guellec, D.; Cornec, D.; Jousse-Joulin, S.; Marhadour, T.; Marcorelles, P.; Pers, J.-O.; Saraux, A.; Devauchelle-Pensec, V. Diagnostic value of labial minor salivary gland biopsy for Sjögren’s syndrome: A systematic review. Autoimmun. Rev. 2013, 12, 416–420. [Google Scholar] [CrossRef]
- Campos, J.; Hillen, M.R.; Barone, F. Salivary Gland Pathology in Sjögren’s Syndrome. Rheum. Dis. Clin. N. Am. 2016, 42, 473–483. [Google Scholar] [CrossRef]
- Barone, F.; Campos, J.; Bowman, S.; Fisher, B.A. The value of histopathological examination of salivary gland biopsies in diagnosis, prognosis and treatment of Sjögren’s Syndrome. Swiss Med. Wkly. 2015, 145, w14168. [Google Scholar] [CrossRef]
- Pijpe, J.; Kalk, W.W.I.; van der Wal, J.E.; Vissink, A.; Kluin, P.M.; Roodenburg, J.L.N.; Bootsma, H.; Kallenberg, C.G.M.; Spijkervet, F.K.L. Parotid gland biopsy compared with labial biopsy in the diagnosis of patients with primary Sjogren’s syndrome. Rheumatology 2006, 46, 335–341. [Google Scholar] [CrossRef] [Green Version]
- Marx, R.E.; Hartman, K.S.; Rethman, K.V. A prospective study comparing incisional labial to incisional parotid biopsies in the detection and confirmation of sarcoidosis, Sjögren’s disease, sialosis and lymphoma. J. Rheumatol. 1988, 15, 621–629. [Google Scholar]
- Franceschini, F.; Cavazzana, I. Anti-Ro/SSA and La/SSB antibodies. Autoimmunity 2005, 38, 55–63. [Google Scholar] [CrossRef]
- Tzioufas, A.G.; Tatouli, I.P.; Moutsopoulos, H.M. Autoantibodies in Sjögren’s syndrome: Clinical presentation and regulatory mechanisms. Presse Med. 1983 2012, 41, e451–e460. [Google Scholar] [CrossRef]
- Trevisani, V.F.M.; Pasoto, S.G.; Fernandes, M.L.M.S.; Lopes, M.L.L.; de Magalhães Souza Fialho, S.C.; Pinheiro, A.C.; dos Santos, L.C.; Appenzeller, S.; Fidelix, T.; Ribeiro, S.L.E.; et al. Recommendations from the Brazilian society of rheumatology for the diagnosis of Sjögren’s syndrome (Part I): Glandular manifestations (systematic review). Adv. Rheumatol. 2019, 59, 58. [Google Scholar] [CrossRef] [Green Version]
- Robbins, A.; Hentzien, M.; Toquet, S.; Didier, K.; Servettaz, A.; Pham, B.-N.; Giusti, D. Diagnostic Utility of Separate Anti-Ro60 and Anti-Ro52/TRIM21 Antibody Detection in Autoimmune Diseases. Front. Immunol. 2019, 10, 444. [Google Scholar] [CrossRef] [Green Version]
- Kontny, E.; Lewandowska-Poluch, A.; Chmielińska, M.; Olesińska, M. Subgroups of Sjögren’s syndrome patients categorised by serological profiles: Clinical and immunological characteristics. Reumatologia 2018, 56, 346–353. [Google Scholar] [CrossRef] [PubMed]
- Brito-Zerón, P.; Retamozo, S.; Ramos-Casals, M. Phenotyping Sjögren’s syndrome: Towards a personalised management of the disease. Clin. Exp. Rheumatol. 2018, 36, 198–209. [Google Scholar]
- Cornec, D.; Saraux, A.; Jousse-Joulin, S.; Pers, J.O.; Boisramé-Gastrin, S.; Renaudineau, Y.; Gauvin, Y.; Roguedas-Contios, A.M.; Genestet, S.; Chastaing, M.; et al. The Differential Diagnosis of Dry Eyes, Dry Mouth, and Parotidomegaly: A Comprehensive Review. Clin. Rev. Allergy. Immunol. 2015, 49, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Moutsopoulos, H.M.; Chused, T.M.; Mann, D.L.; Klippel, J.H.; Fauci, A.S.; Frank, M.M.; Lawley, T.J.; Hamburger, M.I. Sjögren’s syndrome (Sicca syndrome): Current issues. Ann. Intern. Med. 1980, 92, 212–226. [Google Scholar] [CrossRef] [PubMed]
- Fragoulis, G.E.; Fragkioudaki, S.; Reilly, J.H.; Kerr, S.C.; McInnes, I.B.; Moutsopoulos, H.M. Analysis of the cell populations composing the mononuclear cell infiltrates in the labial minor salivary glands from patients with rheumatoid arthritis and sicca syndrome. J. Autoimmun. 2016, 73, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Manoussakis, M.N.; Georgopoulou, C.; Zintzaras, E.; Spyropoulou, M.; Stavropoulou, A.; Skopouli, F.N.; Moutsopoulos, H.M. Sjögren’s syndrome associated with systemic lupus erythematosus: Clinical and laboratory profiles and comparison with primary Sjögren’s syndrome. Arthritis Rheum. 2004, 50, 882–891. [Google Scholar] [CrossRef]
- Salliot, C.; Mouthon, L.; Ardizzone, M.; Sibilia, J.; Guillevin, L.; Gottenberg, J.E.; Mariette, X. Sjogren’s syndrome is associated with and not secondary to systemic sclerosis. Rheumatology 2007, 46, 321–326. [Google Scholar] [CrossRef] [Green Version]
- Rojas-Villarraga, A.; Amaya-Amaya, J.; Rodriguez-Rodriguez, A.; Mantilla, R.D.; Anaya, J.M. Introducing polyautoimmunity: Secondary autoimmune diseases no longer exist. Autoimmune Dis. 2012, 2012, 254319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kollert, F.; Fisher, B.A. Equal rights in autoimmunity: Is Sjögren’s syndrome ever ’secondary’? Rheumatology 2020, 59, 1218–1225. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.G.; Singh, S.; Matteson, E.L. Rate, risk factors and causes of mortality in patients with Sjögren’s syndrome: A systematic review and meta-analysis of cohort studies. Rheumatology 2016, 55, 450–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, Y.; Yang, Z.; Qin, B.; Zhong, R. Primary Sjogren’s syndrome and malignancy risk: A systematic review and meta-analysis. Ann. Rheum. Dis. 2014, 73, 1151–1156. [Google Scholar] [CrossRef] [PubMed]
- Zintzaras, E.; Voulgarelis, M.; Moutsopoulos, H.M. The risk of lymphoma development in autoimmune diseases: A meta-analysis. Arch. Intern. Med. 2005, 165, 2337–2344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jonsson, M.V.; Theander, E.; Jonsson, R. Predictors for the development of non-Hodgkin lymphoma in primary Sjögren’s syndrome. Presse Med. 1983 2012, 41, e511–e516. [Google Scholar] [CrossRef] [PubMed]
- Nishishinya, M.B.; Pereda, C.A.; Muñoz-Fernández, S.; Pego-Reigosa, J.M.; Rúa-Figueroa, I.; Andreu, J.-L.; Fernández-Castro, M.; Rosas, J.; Loza Santamaría, E. Identification of lymphoma predictors in patients with primary Sjögren’s syndrome: A systematic literature review and meta-analysis. Rheumatol. Int. 2015, 35, 17–26. [Google Scholar] [CrossRef]
- Papageorgiou, A.; Voulgarelis, M.; Tzioufas, A.G. Clinical picture, outcome and predictive factors of lymphoma in Sjögren syndrome. Autoimmun. Rev. 2015, 14, 641–649. [Google Scholar] [CrossRef]
- Hernandez-Molina, G.; Michel-Peregrina, M.; Bermúdez-Bermejo, P.; Sánchez-Guerrero, J. Early and late extraglandular manifestations in primary Sjögren’s syndrome. Clin. Exp. Rheumatol. 2012, 30, 455. [Google Scholar]
- Ter Borg, E.J.; Kelder, J.C. Development of new extra-glandular manifestations or associated auto-immune diseases after establishing the diagnosis of primary Sjögren’s syndrome: A long-term study of the Antonius Nieuwegein Sjögren (ANS) cohort. Rheumatol. Int. 2017, 37, 1153–1158. [Google Scholar] [CrossRef]
- Seror, R.; Meiners, P.; Baron, G.; Bootsma, H.; Bowman, S.J.; Vitali, C.; Gottenberg, J.-E.; Theander, E.; Tzioufas, A.; De Vita, S.; et al. Development of the ClinESSDAI: A clinical score without biological domain. A tool for biological studies. Ann. Rheum. Dis. 2016, 75, 1945–1950. [Google Scholar] [CrossRef]
- Seror, R.; Ravaud, P.; Bowman, S.J.; Baron, G.; Tzioufas, A.; Theander, E.; Gottenberg, J.-E.; Bootsma, H.; Mariette, X.; Vitali, C. EULAR Sjögren’s syndrome disease activity index: Development of a consensus systemic disease activity index for primary Sjögren’s syndrome. Ann. Rheum. Dis. 2010, 69, 1103–1109. [Google Scholar] [CrossRef] [PubMed]
- Seror, R.; Ravaud, P.; Mariette, X.; Bootsma, H.; Theander, E.; Hansen, A.; Ramos-Casals, M.; Dörner, T.; Bombardieri, S.; Hachulla, E.; et al. EULAR Sjögren’s Syndrome Patient Reported Index (ESSPRI): Development of a consensus patient index for primary Sjögren’s syndrome. Ann. Rheum. Dis. 2011, 70, 968–972. [Google Scholar] [CrossRef] [PubMed]
- Vitali, C.; Palombi, G.; Baldini, C.; Benucci, M.; Bombardieri, S.; Covelli, M.; Del Papa, N.; De Vita, S.; Epis, O.; Franceschini, F.; et al. Sjögren’s syndrome disease damage index and disease activity index: Scoring systems for the assessment of disease damage and disease activity in Sjögren’s syndrome, derived from an analysis of a cohort of Italian patients. Arthritis Rheum. 2007, 56, 2223–2231. [Google Scholar] [CrossRef] [PubMed]
- Barry, R.J.; Sutcliffe, N.; Isenberg, D.A.; Price, E.; Goldblatt, F.; Adler, M.; Canavan, A.; Hamburger, J.; Richards, A.; Regan, M.; et al. The Sjogren’s Syndrome Damage Index—A damage index for use in clinical trials and observational studies in primary Sjogren’s syndrome. Rheumatology 2008, 47, 1193–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quartuccio, L.; Baldini, C.; Bartoloni, E.; Priori, R.; Carubbi, F.; Corazza, L.; Alunno, A.; Colafrancesco, S.; Luciano, N.; Giacomelli, R.; et al. Anti-SSA/SSB-negative Sjögren’s syndrome shows a lower prevalence of lymphoproliferative manifestations, and a lower risk of lymphoma evolution. Autoimmun. Rev. 2015, 14, 1019–1022. [Google Scholar] [CrossRef] [PubMed]
- Fauchais, A.L.; Martel, C.; Gondran, G.; Lambert, M.; Launay, D.; Jauberteau, M.O.; Hachulla, E.; Vidal, E.; Hatron, P.Y. Immunological profile in primary Sjögren syndrome: Clinical significance, prognosis and long-term evolution to other auto-immune disease. Autoimmun. Rev. 2010, 9, 595–599. [Google Scholar] [CrossRef] [PubMed]
- Krylova, L.; Isenberg, D. Assessment of patients with primary Sjogren’s syndrome--outcome over 10 years using the Sjogren’s Syndrome Damage Index. Rheumatology 2010, 49, 1559–1562. [Google Scholar] [CrossRef] [Green Version]
- Baldini, C.; Ferro, F.; Pepe, P.; Luciano, N.; Sernissi, F.; Cacciatore, C.; Martini, D.; Tavoni, A.; Mosca, M.; Bombardieri, S. Damage Accrual In a Single Centre Cohort Of Patients With Primary Sjögren’s Syndrome Followed Up For Over 10 Years. In Sjögren’s Syndrome: Clinical Aspects, Proceedings of the 2013 ACR/ARHP Annual Meeting, San Diego, CA, USA, 25–30 October 2013; WILEY: Hoboken, NJ, USA, 2013. [Google Scholar]
- Cho, H.J.; Yoo, J.J.; Yun, C.Y.; Kang, E.H.; Lee, H.-J.; Hyon, J.Y.; Song, Y.W.; Lee, Y.J. The EULAR Sjogren’s syndrome patient reported index as an independent determinant of health-related quality of life in primary Sjogren’s syndrome patients: In comparison with non-Sjogren’s sicca patients. Rheumatology 2013, 52, 2208–2217. [Google Scholar] [CrossRef] [Green Version]
- Hackett, K.L.; Newton, J.L.; Frith, J.; Elliott, C.; Lendrem, D.; Foggo, H.; Edgar, S.; Mitchell, S.; Ng, W.-F. Impaired functional status in primary Sjögren’s syndrome. Arthritis Care Res. 2012, 64, 1760–1764. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, X.; Chen, H.; Shen, B. Sjögren’s syndrome is associated with negatively variable impacts on domains of health-related quality of life: Evidence from Short Form 36 questionnaire and a meta-analysis. Patient Prefer. Adherence 2017, 11, 905–911. [Google Scholar] [CrossRef] [Green Version]
- Haldorsen, K.; Moen, K.; Jacobsen, H.; Jonsson, R.; Brun, J.G. Exocrine function in primary Sjögren syndrome: Natural course and prognostic factors. Ann. Rheum. Dis. 2008, 67, 949–954. [Google Scholar] [CrossRef]
- Al-Ezzi, M.Y.; Pathak, N.; Tappuni, A.R.; Khan, K.S. Primary Sjögren’s syndrome impact on smell, taste, sexuality and quality of life in female patients: A systematic review and meta-analysis. Mod. Rheumatol. 2017, 27, 623–629. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, S.T.; Valim, V.; Fisher, B.A. Health-related quality of life and costs in Sjögren’s syndrome. Rheumatology 2019. [Google Scholar] [CrossRef] [PubMed]
- Ter Borg, E.J.; Kelder, J.C. Lower prevalence of extra-glandular manifestations and anti-SSB antibodies in patients with primary Sjögren’s syndrome and widespread pain: Evidence for a relatively benign subset. Clin. Exp. Rheumatol. 2014, 32, 349–353. [Google Scholar] [PubMed]
- Ostuni, P.; Botsios, C.; Sfriso, P.; Punzi, L.; Chieco-Bianchi, F.; Semerano, L.; Grava, C.; Todesco, S. Fibromyalgia in Italian patients with primary Sjögren’s syndrome. Joint Bone Spine 2002, 69, 51–57. [Google Scholar] [CrossRef]
- Champey, J.; Corruble, E.; Gottenberg, J.; Buhl, C.; Meyer, T.; Caudmont, C.; Bergé, E.; Pellet, J.; Hardy, P.; Mariette, X. Quality of life and psychological status in patients with primary Sjögren’s syndrome and sicca symptoms without autoimmune features. Arthritis Rheum. 2006, 55, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Mariette, X. Dry eyes and mouth syndrome or sicca, asthenia and polyalgia syndrome? Rheumatology 2003, 42, 914–915. [Google Scholar] [CrossRef] [Green Version]
- Mavragani, C.P.; Skopouli, F.N.; Moutsopoulos, H.M. Increased Prevalence of Antibodies to Thyroid Peroxidase in Dry Eyes and Mouth Syndrome or Sicca Asthenia Polyalgia Syndrome. J. Rheumatol. 2009, 36, 1626–1630. [Google Scholar] [CrossRef]
- Price, E.J. Dry eyes and mouth syndrome—A subgroup of patients presenting with sicca symptoms. Rheumatology 2002, 41, 416–422. [Google Scholar] [CrossRef] [Green Version]
- Mandl, T.; Jørgensen, T.S.; Skougaard, M.; Olsson, P.; Kristensen, L.-E. Work Disability in Newly Diagnosed Patients with Primary Sjögren Syndrome. J. Rheumatol. 2017, 44, 209–215. [Google Scholar] [CrossRef] [Green Version]
- Pertovaara, M.; Korpela, M. ESSPRI and other patient-reported indices in patients with primary Sjogren’s syndrome during 100 consecutive outpatient visits at one rheumatological clinic. Rheumatology 2014, 53, 927–931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lendrem, D.; Mitchell, S.; McMeekin, P.; Gompels, L.; Hackett, K.; Bowman, S.; Price, E.; Pease, C.T.; Emery, P.; Andrews, J.; et al. Do the EULAR Sjogren’s syndrome outcome measures correlate with health status in primary Sjogren’s syndrome? Rheumatology 2015, 54, 655–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koh, J.; Kwok, S.; Lee, J.; Son, C.; Kim, J.-M.; Kim, H.; Park, S.; Sung, Y.; Choe, J.; Lee, S.; et al. Pain, xerostomia, and younger age are major determinants of fatigue in Korean patients with primary Sjögren’s syndrome: A cohort study. Scand. J. Rheumatol. 2017, 46, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Cornec, D.; Devauchelle-Pensec, V.; Mariette, X.; Jousse-Joulin, S.; Berthelot, J.; Perdriger, A.; Puéchal, X.; Le Guern, V.; Sibilia, J.; Gottenberg, J.; et al. Severe Health-Related Quality of Life Impairment in Active Primary Sjögren’s Syndrome and Patient-Reported Outcomes: Data From a Large Therapeutic Trial. Arthritis Care Res. 2017, 69, 528–535. [Google Scholar] [CrossRef]
- Price, E.J.; Rauz, S.; Tappuni, A.R.; Sutcliffe, N.; Hackett, K.L.; Barone, F.; Granata, G.; Ng, W.-F.; Fisher, B.A.; Bombardieri, M.; et al. The British Society for Rheumatology guideline for the management of adults with primary Sjögren’s Syndrome. Rheumatology 2017. [Google Scholar] [CrossRef] [Green Version]
- Valim, V.; Trevisani, V.F.M.; Pasoto, S.G.; Serrano, E.V.; Ribeiro, S.L.E.; de Fidelix, T.S.A.; Vilela, V.S.; do Prado, L.L.; Tanure, L.A.; Libório-Kimura, T.N.; et al. Recommendations for the treatment of Sjögren’s syndrome. Rev. Bras. Reumatol. 2015, 55, 446–457. [Google Scholar] [CrossRef]
- Sumida, T.; Azuma, N.; Moriyama, M.; Takahashi, H.; Asashima, H.; Honda, F.; Abe, S.; Ono, Y.; Hirota, T.; Hirata, S.; et al. Clinical practice guideline for Sjögren’s syndrome 2017. Mod. Rheumatol. 2018, 28, 383–408. [Google Scholar] [CrossRef] [Green Version]
- Vivino, F.B.; Carsons, S.E.; Foulks, G.; Daniels, T.E.; Parke, A.; Brennan, M.T.; Forstot, S.L.; Scofield, R.H.; Hammitt, K.M. New Treatment Guidelines for Sjögren’s Disease. Rheum. Dis. Clin. N. Am. 2016, 42, 531–551. [Google Scholar] [CrossRef] [Green Version]
- The Dry Eye Assessment and Management Study Research Group. n−3 Fatty Acid Supplementation for the Treatment of Dry Eye Disease. N. Engl. J. Med. 2018, 378, 1681–1690. [Google Scholar] [CrossRef]
- Hussain, M.; Shtein, R.M.; Pistilli, M.; Maguire, M.G.; Oydanich, M.; Asbell, P.A. The Dry Eye Assessment and Management (DREAM) extension study—A randomized clinical trial of withdrawal of supplementation with omega-3 fatty acid in patients with dry eye disease. Ocul. Surf. 2020, 18, 47–55. [Google Scholar] [CrossRef]
- Asbell, P.A.; Maguire, M.G. Why DREAM should make you think twice about recommending Omega-3 supplements. Ocul. Surf. 2019, 17, 617–618. [Google Scholar] [CrossRef]
- Skopouli, F.N.; Jagiello, P.; Tsifetaki, N.; Moutsopoulos, H.M. Methotrexate in primary Sjögren’s syndrome. Clin. Exp. Rheumatol. 1996, 14, 555–558. [Google Scholar] [PubMed]
- Nakayamada, S.; Saito, K.; Umehara, H.; Ogawa, N.; Sumida, T.; Ito, S.; Minota, S.; Nara, H.; Kondo, H.; Okada, J.; et al. Efficacy and safety of mizoribine for the treatment of Sjögren’s syndrome: A multicenter open-label clinical trial. Mod. Rheumatol. 2007, 17, 464–469. [Google Scholar] [CrossRef] [PubMed]
- Nakayamada, S.; Fujimoto, T.; Nonomura, A.; Saito, K.; Nakamura, S.; Tanaka, Y. Usefulness of initial histological features for stratifying Sjogren’s syndrome responders to mizoribine therapy. Rheumatology 2009, 48, 1279–1282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacFarlane, G.J.; Kronisch, C.; Dean, L.E.; Atzeni, F.; Häuser, W.; Fluß, E.; Choy, E.; Kosek, E.; Amris, K.; Branco, J.; et al. EULAR revised recommendations for the management of fibromyalgia. Ann. Rheum. Dis. 2017, 76, 318–328. [Google Scholar] [CrossRef] [PubMed]
- Dass, S.; Bowman, S.J.; Vital, E.M.; Ikeda, K.; Pease, C.T.; Hamburger, J.; Richards, A.; Rauz, S.; Emery, P. Reduction of fatigue in Sjogren syndrome with rituximab: Results of a randomised, double-blind, placebo-controlled pilot study. Ann. Rheum. Dis. 2008, 67, 1541–1544. [Google Scholar] [CrossRef] [PubMed]
- Devauchelle-Pensec, V.; Mariette, X.; Jousse-Joulin, S.; Berthelot, J.-M.; Perdriger, A.; Puéchal, X.; Le Guern, V.; Sibilia, J.; Gottenberg, J.-E.; Chiche, L.; et al. Treatment of Primary Sjögren Syndrome With Rituximab: A Randomized Trial. Ann. Intern. Med. 2014, 160, 233–242. [Google Scholar] [CrossRef]
- Carubbi, F.; Cipriani, P.; Marrelli, A.; Benedetto, P.; Ruscitti, P.; Berardicurti, O.; Pantano, I.; Liakouli, V.; Alvaro, S.; Alunno, A.; et al. Efficacy and safety of rituximab treatment in early primary Sjögren’s syndrome: A prospective, multi-center, follow-up study. Arthritis Res. Ther. 2013, 15, R172. [Google Scholar] [CrossRef] [Green Version]
- Norheim, K.B.; Harboe, E.; Gøransson, L.G.; Omdal, R. Interleukin-1 Inhibition and Fatigue in Primary Sjögren’s Syndrome—A Double Blind, Randomised Clinical Trial. PLoS ONE 2012, 7, e30123. [Google Scholar] [CrossRef] [Green Version]
- Van der Heijden, E.H.M.; Kruize, A.A.; Radstake, T.R.D.J.; van Roon, J.A.G. Optimizing conventional DMARD therapy for Sjögren’s syndrome. Autoimmun. Rev. 2018, 17, 480–492. [Google Scholar] [CrossRef]
- Gottenberg, J.-E.; Ravaud, P.; Puéchal, X.; Le Guern, V.; Sibilia, J.; Goeb, V.; Larroche, C.; Dubost, J.-J.; Rist, S.; Saraux, A.; et al. Effects of Hydroxychloroquine on Symptomatic Improvement in Primary Sjögren Syndrome: The JOQUER Randomized Clinical Trial. JAMA 2014, 312, 249. [Google Scholar] [CrossRef] [PubMed]
- Yoon, C.H.; Lee, H.J.; Lee, E.Y.; Lee, E.B.; Lee, W.-W.; Kim, M.K.; Wee, W.R. Effect of Hydroxychloroquine Treatment on Dry Eyes in Subjects with Primary Sjögren’s Syndrome: A Double-Blind Randomized Control Study. J. Kor. Med. Sci. 2016, 31, 1127. [Google Scholar] [CrossRef] [PubMed]
- Gottenberg, J.-E.; Dörner, T.; Bootsma, H.; Devauchelle-Pensec, V.; Bowman, S.J.; Mariette, X.; Bartz, H.; Oortgiesen, M.; Shock, A.; Koetse, W.; et al. Efficacy of Epratuzumab, an Anti-CD22 Monoclonal IgG Antibody, in Systemic Lupus Erythematosus Patients with Associated Sjögren’s Syndrome: Post Hoc Analyses From the EMBODY Trials. Arthritis Rheumatol. 2018, 70, 763–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mariette, X.; Seror, R.; Quartuccio, L.; Baron, G.; Salvin, S.; Fabris, M.; Desmoulins, F.; Nocturne, G.; Ravaud, P.; De Vita, S. Efficacy and safety of belimumab in primary Sjögren’s syndrome: Results of the BELISS open-label phase II study. Ann. Rheum. Dis. 2015, 74, 526–531. [Google Scholar] [CrossRef]
- De Vita, S.; Quartuccio, L.; Seror, R.; Salvin, S.; Ravaud, P.; Fabris, M.; Nocturne, G.; Gandolfo, S.; Isola, M.; Mariette, X. Efficacy and safety of belimumab given for 12 months in primary Sjögren’s syndrome: The BELISS open-label phase II study. Rheumatology 2015. [Google Scholar] [CrossRef] [Green Version]
- Jakez-Ocampo, J.; Atisha-Fregoso, Y.; Llorente, L. Refractory Primary Sjögren Syndrome Successfully Treated With Bortezomib. JCR J. Clin. Rheumatol. 2015, 21, 31–32. [Google Scholar] [CrossRef]
- Shah, U. Pilot Trial of Ustekinumab for Primary Sjögren’s Syndrome. 2020. Available online: clinicaltrials.gov (accessed on 9 July 2020).
- Gilead Sciences. Study to Assess Safety and Efficacy of Filgotinib, Lanraplenib and Tirabrutinib in Adults With Active Sjogren’s Syndrome; Clinical Trial Registration; Gilead Sciences, Inc.: Foster City, CA, USA, 2020. [Google Scholar]
- Meijer, J.M.; Meiners, P.M.; Vissink, A.; Spijkervet, F.K.L.; Abdulahad, W.; Kamminga, N.; Brouwer, E.; Kallenberg, C.G.M.; Bootsma, H. Effectiveness of rituximab treatment in primary Sjögren’s syndrome: A randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2010, 62, 960–968. [Google Scholar] [CrossRef]
- Bowman, S.J.; Everett, C.C.; O’Dwyer, J.L.; Emery, P.; Pitzalis, C.; Ng, W.-F.; Pease, C.T.; Price, E.J.; Sutcliffe, N.; Gendi, N.S.T.; et al. Randomized Controlled Trial of Rituximab and Cost-Effectiveness Analysis in Treating Fatigue and Oral Dryness in Primary Sjögren’s Syndrome: Rituximab for symptomatic fatigue and oral dryness in primary SS. Arthritis Rheumatol. 2017, 69, 1440–1450. [Google Scholar] [CrossRef] [Green Version]
- RemeGen A Phase II Study of RC18, a Recombinant Human B Lymphocyte Stimulator Receptor: Immunoglobulin G (IgG) Fc Fusion Protein for Injection for the Treatment of Subjects with Primary Sjögren’s Syndrome. 2019. Available online: clinicaltrials.gov (accessed on 20 May 2020).
- GlaxoSmithKline A Randomized, Double Blind (Sponsor Open), Comparative, Multicenter Study to Evaluate the Safety and Efficacy of Subcutaneous Belimumab (GSK1550188) and Intravenous Rituximab Co-administration in Subjects With Primary Sjögren’s Syndrome. 2020. Available online: clinicaltrials.gov (accessed on 9 July 2020).
- Eli Lilly and Company A Multiple Ascending Dose Study to Evaluate the Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of LY3090106 in Subjects With Sjögren’s Syndrome. 2018. Available online: clinicaltrials.gov (accessed on 20 May 2020).
- Dörner, T.; Posch, M.G.; Li, Y.; Petricoul, O.; Cabanski, M.; Milojevic, J.M.; Kamphausen, E.; Valentin, M.-A.; Simonett, C.; Mooney, L.; et al. Treatment of primary Sjögren’s syndrome with ianalumab (VAY736) targeting B cells by BAFF receptor blockade coupled with enhanced, antibody-dependent cellular cytotoxicity. Ann. Rheum. Dis. 2019, 78, 641–647. [Google Scholar] [CrossRef]
- Novartis Pharmaceuticals Study of Safety and Efficacy of Multiple VAY736 Doses in Patients with Moderate to Severe Primary Sjogren’s Syndrome (pSS). 2020. Available online: clinicaltrials.gov (accessed on 9 July 2020).
- Novartis Pharmaceuticals An Adaptive Phase 2 Randomized Double-blind, Placebo-controlled Multi-center Study to Evaluate the Safety and Efficacy of Multiple LOU064 Doses in Patients With Moderate to Severe Sjögren’s Syndrome (LOUiSSe). 2020. Available online: clinicaltrials.gov (accessed on 9 July 2020).
- Bristol-Myers Squibb A Phase II, Randomized, Multi-Center, Double-Blind, Placebo Controlled Study to Evaluate the Efficacy and Safety of BMS-931699 (Lulizumab) or BMS-986142 in Subjects With Moderate to Severe Primary Sjögren’s Syndrome. 2018. Available online: clinicaltrials.gov (accessed on 20 May 2020).
- Bristol-Myers Squibb A Randomized, Placebo-Controlled, Double-Blind, Multicenter Study to Assess the Efficacy and Safety of Branebrutinib Treatment in Subjects With Active Systemic Lupus Erythematosus or Primary Sjögren’s Syndrome, or Branebrutinib Treatment Followed by Open-label Abatacept Treatment in Subjects With Active Rheumatoid Arthritis. 2020. Available online: clinicaltrials.gov (accessed on 9 July 2020).
- St.Clair, E.W.; Baer, A.N.; Wei, C.; Noaiseh, G.; Parke, A.; Coca, A.; Utset, T.O.; Genovese, M.C.; Wallace, D.J.; McNamara, J.; et al. Clinical Efficacy and Safety of Baminercept, a Lymphotoxin β Receptor Fusion Protein, in Primary Sjögren’s Syndrome: Results From a Phase II Randomized, Double-Blind, Placebo-Controlled Trial. Arthritis Rheumatol. 2018, 70, 1470–1480. [Google Scholar] [CrossRef]
- Incyte Corporation An Open-Label Phase 2 Study of INCB050465 in Participants With Primary Sjögren’s Syndrome. 2020. Available online: clinicaltrials.gov (accessed on 20 May 2020).
- Juarez, M.; Diaz, N.; Johnston, G.I.; Nayar, S.; Payne, A.; Helmer, E.; Cain, D.; Williams, P.; Ng, W.F.; Fisher, B.; et al. AB0458 a phase II randomised double-blind, placebo-controlled, proof of concept study of oral Seletalisib in patients with priary Sjögren’s syndrome (PSS). Ann. Rheum. Dis. 2019, 78, 1692–1693. [Google Scholar]
- Dörner, T.; Zeher, M.; Laessing, U.; Chaperon, F.; De Buck, S.; Hasselberg, A.; Valentin, M.-A.; Ma, S.; Cabanski, M.; Kalis, C.; et al. OP0250 A randomised, double-blind study to assess the safety, tolerability and preliminary efficacy of leniolisib (CDZ173) in patients with primary sjÖgren’s syndrome. Ann. Rheum. Dis. 2018, 77, 174. [Google Scholar]
- Baer, A.; Gottenberg, J.-E.; St.Clair, W.E.; Sumida, T.; Takeuchi, T.; Seror, R.; Foulks, G.; Nys, M.; Johnsen, A.; Wong, R.; et al. OP0039 Efficacy and safety of Abatacept in active primary Sjögren’s syndrome: Results of a randomised placebo-controlled phase III trial. Ann. Rheum. Dis. 2019, 78, 89–90. [Google Scholar]
- Meiners, P.M.; Vissink, A.; Kroese, F.G.M.; Spijkervet, F.K.L.; Smitt-Kamminga, N.S.; Abdulahad, W.H.; Bulthuis-Kuiper, J.; Brouwer, E.; Arends, S.; Bootsma, H. Abatacept treatment reduces disease activity in early primary Sjogren’s syndrome (open-label proof of concept ASAP study). Ann. Rheum. Dis. 2014, 73, 1393–1396. [Google Scholar] [CrossRef] [PubMed]
- Van Nimwegen, J.F.; Mossel, E.; van Zuiden, G.S.; Wijnsma, R.F.; Delli, K.; Stel, A.J.; van der Vegt, B.; Haacke, E.A.; Olie, L.; Los, L.I.; et al. Abatacept treatment for patients with early active primary Sjögren’s syndrome: A single-centre, randomised, double-blind, placebo-controlled, phase 3 trial (ASAP-III study). Lancet Rheumatol. 2020, 2, e153–e163. [Google Scholar] [CrossRef]
- Fisher, B.A.; Szanto, A.; Ng, W.-F.; Bombardieri, M.; Posch, M.G.; Papas, A.S.; Gergely, P. Assessment of the anti-CD40 antibody iscalimab in patients with primary Sjögren’s syndrome: A multicentre, randomised, double-blind, placebo-controlled, proof-of-concept study. Lancet Rheumatol. 2020, 2. [Google Scholar] [CrossRef]
- Novartis Pharmaceuticals A 48-week, 6-arm, Randomized, Double-blind, Placebo-controlled Multicenter Trial to Assess the Safety and Efficacy of Multiple CFZ533 Doses Administered Subcutaneously in Two Distinct Populations of Patients with Sjögren’s Syndrome (TWINSS). 2020. Available online: clinicaltrials.gov (accessed on 9 July 2020).
- Viela Bio A Phase 2 Randomized, Double-blind, Placebo-controlled, Proof of Concept Study to Evaluate the Efficacy and Safety of VIB4920 in Subjects With Sjögren’s Syndrome (SS). 2020. Available online: clinicaltrials.gov (accessed on 9 July 2020).
- Mariette, X.; Bombardieri, M.; Alevizos, I.; Moate, R.; Sullivan, B.; Noaiseh, G.; Kvarnström, M.; Rees, W.; Wang, L.; Illei, G. A Phase 2a Study of MEDI5872 (AMG557), a Fully Human Anti-ICOS Ligand Monoclonal Antibody in Patients with Primary Sjögren’s Syndrome. In Sjögrenʼs Syndrome—Basic & Clinical Science Poster I, Proceedings of the 2019 ACR/ARP Annual Meeting, Atlanta, GA, USA, 8–13 November 2019; WILEY: Hoboken, NJ, USA, 2019. [Google Scholar]
- Hoffmann-La Roche A Multi-Center, Randomized, Double-Blind, Placebo-Controlled, Parallel Group Phase 2A Study to Assess the Efficacy of RO5459072 in Patients With Primary Sjogren’s Syndrome. 2018. Available online: clinicaltrials.gov (accessed on 20 May 2020).
- Gabor Illei, M. D A Randomized, Placebo Controlled, Proof of Concept, Study of Raptiva, a Humanized Anti-CD-11a Monoclonal Antibody, in Patients With Sjogren’s Syndrome. 2015. Available online: clinicaltrials.gov (accessed on 20 May 2020).
- Cacoub, P.; Felten, R.; Devauchelle-Pensec, V.; Duffau, P.; Hachulla, E.; Hatron, P.Y.; Salliot, C.; Perdriger, A.; Morel, J.; Mekinian, A.; et al. Inhibition du récepteur de l’interleukine-6 au cours du syndrome de Gougerot-Sjögren primaire: Essai randomisé multicentrique académique en double aveugle tocilizumab versus placebo (ETAP study). Rev. Médecine Interne 2019, 40, A33. [Google Scholar] [CrossRef]
- Mariette, X.; Ravaud, P.; Steinfeld, S.; Baron, G.; Goetz, J.; Hachulla, E.; Combe, B.; Puéchal, X.; Pennec, Y.; Sauvezie, B.; et al. Inefficacy of infliximab in primary Sjögren’s syndrome: Results of the randomized, controlled trial of remicade in primary Sjögren’s syndrome (TRIPSS): Infliximab in Primary Sjögren’s Syndrome. Arthritis Rheum. 2004, 50, 1270–1276. [Google Scholar] [CrossRef]
- Sankar, V.; Brennan, M.T.; Kok, M.R.; Leakan, R.A.; Smith, J.A.; Manny, J.; Baum, B.J.; Pillemer, S.R. Etanercept in Sjögren’s syndrome: A twelve-week randomized, double-blind, placebo-controlled pilot clinical trial: Randomized Controlled Pilot Study of Etanercept in SS. Arthritis Rheum. 2004, 50, 2240–2245. [Google Scholar] [CrossRef]
- GlaxoSmithKline A Two Part Phase IIa Study, to Evaluate the Safety and Tolerability, Pharmacokinetics, Proof of Mechanism and Potential for Efficacy of an Anti-IL-7 Receptor-α Monoclonal Antibody (GSK2618960) in the Treatment of Primary Sjögren’s Syndrome. 2018. Available online: clinicaltrials.gov (accessed on 20 May 2020).
- Viela Bio A Phase 1 Randomized, Placebo-Controlled, Blinded, Multiple Ascending Dose Study to Evaluate VIB7734 in Systemic Lupus Erythematosus, Cutaneous Lupus Erythematosus, Sjogren’s Syndrome, Systemic Sclerosis, Polymyositis, and Dermatomyositis. 2020. Available online: clinicaltrials.gov (accessed on 20 May 2020).
- Fisher, B.; Barone, F.; Jobling, K.; Gallagher, P.; Macrae, V.; Filby, A.; Hulmes, G.; Milne, P.; Traianos, E.; Iannizzotto, V.; et al. OP0202 Effects of RSLV-132 on fatigue in patients with primary Sjögren’s syndrome—Results of a phase II randomised double-blind, placebo-controlled proof of concept study. Ann. Rheum. Dis. 2019, 78, 177. [Google Scholar] [CrossRef] [Green Version]
- Assistance Publique—Hôpitaux de Paris Induction of Regulatory t Cells by Low Dose il2 in Autoimmune and Inflammatory Diseases. 2020. Available online: clinicaltrials.gov (accessed on 9 July 2020).
Figure 1.
Overview of physiopathological mechanism underlying Sjögren’s syndrome (SS). Environmental triggers, such as viral infections, genetic predispositions, epigenetics and sex hormone deregulation, cause the disruption of salivary gland epithelial cell (SGEC), the production of type I interferon (IFN) and other cytokines such as B cell Activating Factor of the tumour necrosis factor (TNF) Family (BAFF) [11] and the alteration of proteins involved in saliva secretion. Dendritic cells, as well as SGEC acquire the characteristics of antigen-presenting cells capable of processing viral and self-antigens, leading to the activation of autoreactive T and B cells. Autoreactive T cells induce tissue damage through the release of cytotoxic granules and cause the exposure of autoantigens on the surface of SGEC. In addition, activated B cells produce autoantibodies that induce SGEC apoptosis and create an inflammatory microenvironment. This complex mechanism triggers a self-perpetuating cycle of autoimmunity.
Figure 1.
Overview of physiopathological mechanism underlying Sjögren’s syndrome (SS). Environmental triggers, such as viral infections, genetic predispositions, epigenetics and sex hormone deregulation, cause the disruption of salivary gland epithelial cell (SGEC), the production of type I interferon (IFN) and other cytokines such as B cell Activating Factor of the tumour necrosis factor (TNF) Family (BAFF) [11] and the alteration of proteins involved in saliva secretion. Dendritic cells, as well as SGEC acquire the characteristics of antigen-presenting cells capable of processing viral and self-antigens, leading to the activation of autoreactive T and B cells. Autoreactive T cells induce tissue damage through the release of cytotoxic granules and cause the exposure of autoantigens on the surface of SGEC. In addition, activated B cells produce autoantibodies that induce SGEC apoptosis and create an inflammatory microenvironment. This complex mechanism triggers a self-perpetuating cycle of autoimmunity.

Figure 2.
Factors involved in SS trigger phase.

Figure 3.
Intracrine steroidogenic machinery in healthy acinar cells. The figure shows the conversion of dehydroepiandrosterone (DHEA) to active sex steroids. STS: steroid sulphatase, SULT2B1: sulfotransferase 2B1, HSD: hydroxy steroid dehydrogenase, 5-α-R: 5α-reductase, TEST: testosterone, DHT: dihydrotestosterone. DHEA-S: DHEA-sulphate.
Figure 3.
Intracrine steroidogenic machinery in healthy acinar cells. The figure shows the conversion of dehydroepiandrosterone (DHEA) to active sex steroids. STS: steroid sulphatase, SULT2B1: sulfotransferase 2B1, HSD: hydroxy steroid dehydrogenase, 5-α-R: 5α-reductase, TEST: testosterone, DHT: dihydrotestosterone. DHEA-S: DHEA-sulphate.

Figure 4.
Synoptic view of targeted drugs (being) studied in pSS. Therapeutic classes are in bold. Biotherapies and small molecules are in black if they have been the subject of one or more trials in pSS or in grey if they exist but have not been tested in pSS. Names in strikethrough are drugs whose development has been stopped because of unacceptable side effects or because of portfolio prioritization.
Figure 4.
Synoptic view of targeted drugs (being) studied in pSS. Therapeutic classes are in bold. Biotherapies and small molecules are in black if they have been the subject of one or more trials in pSS or in grey if they exist but have not been tested in pSS. Names in strikethrough are drugs whose development has been stopped because of unacceptable side effects or because of portfolio prioritization.


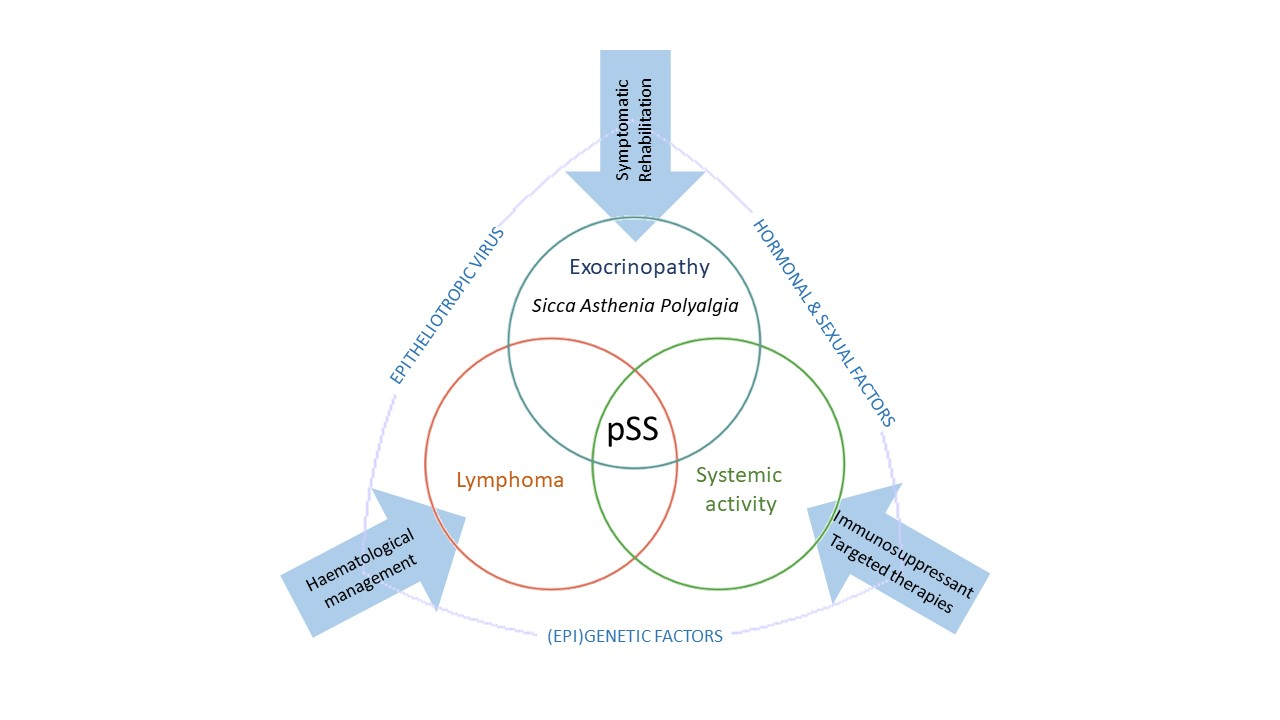{kind=link}
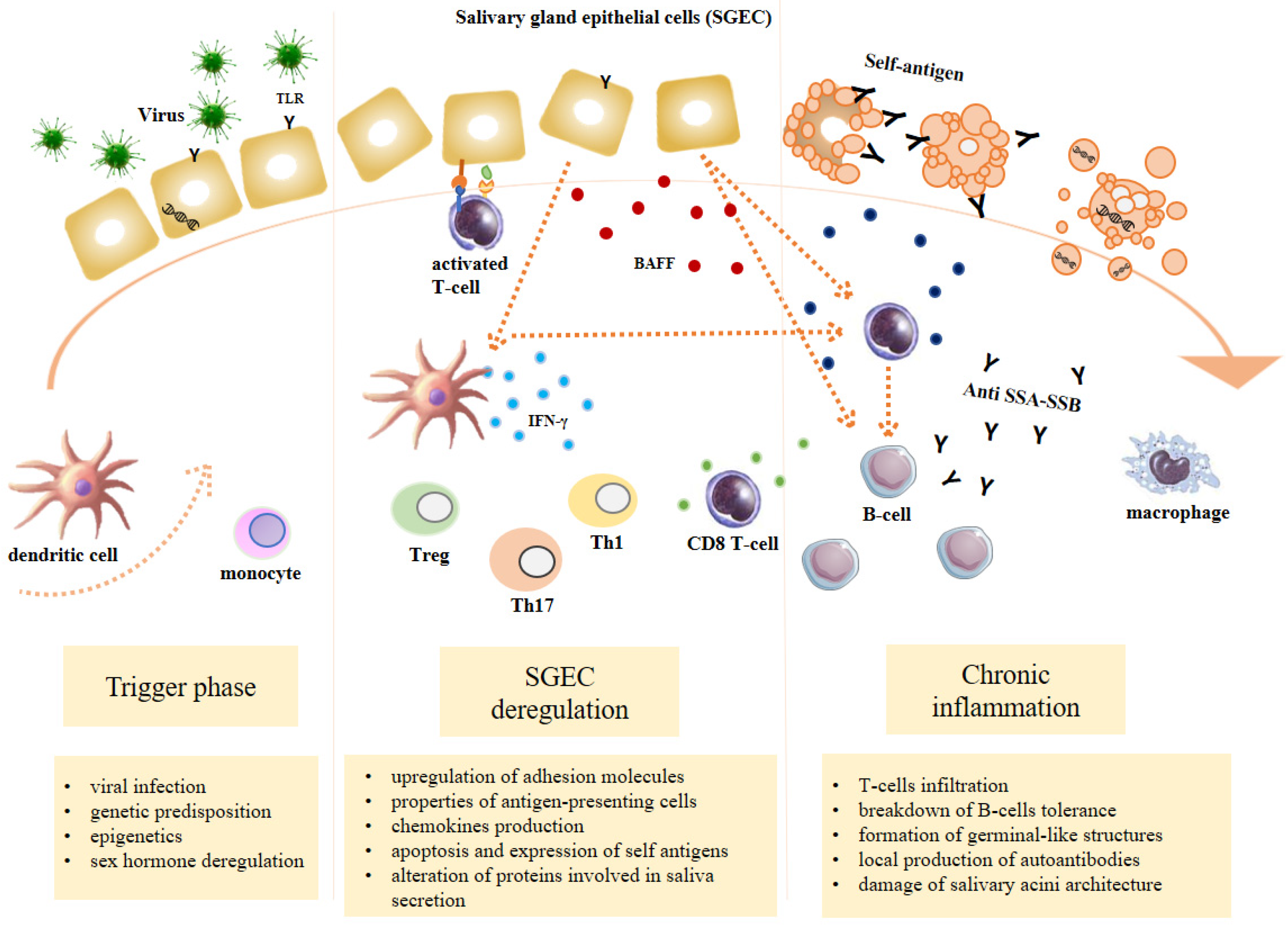{kind=link}
{kind=link}
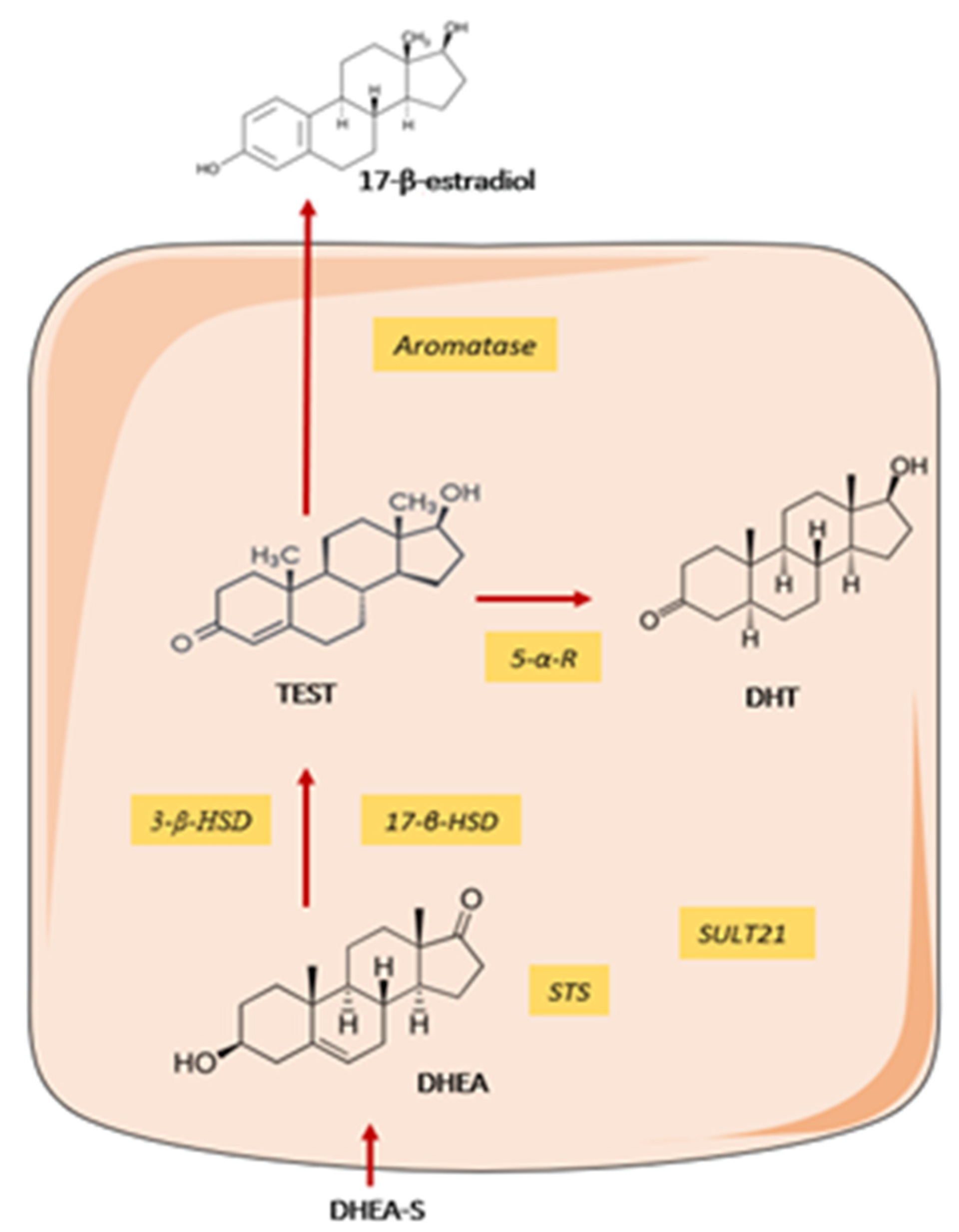{kind=link}
{kind=link}
Table 1.
Rapid overview of original publications describing novel autoantibodies in pSS.
| Autoantigen Targeted by Autoantibody | Number of Patients (N Total/Pooled) | Autoantibody Prevalence (% of Total) | Clinical Associations | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| pSS | pSS MALT | Sicca | FM Sicca | Crtl | pSS | pSS MALT | Sicca | FM Sicca | Crtl | ||
| Salivary protein 1 (SP1) | 270 | _ | 29 | 151 | 148 | 46.3 | _ | 75.9 | 45.7 | 27 | Early disease, low focus-score, SSA−/SSB− [169,170,171,172,173] Found in non-pSS dry eye and fibromyalgia with sicca syndrome [167,174,175] |
| Carbonic anhydrase 6 (CA6) | 13 | _ | _ | 151 | 23 | 53.8 | _ | _ | 7.3 | 4.3 | |
| Parotid secretory protein (PSP) | 13 | _ | _ | 151 | 23 | 15.4 | _ | _ | 11.3 | 4.3 | |
| Interferon-inducible protein-16 | 250 | _ | _ | _ | 255 | 37.2 | _ | _ | _ | 2.7 | High focus-score and GC, hyperγ, ANA > 1:320 [176] |
| Mouse double minute 2 (MDM2) | 100 | _ | _ | _ | 74 | 21 | _ | _ | _ | 5.4 | ⇧ disease duration, ESSDAI, ⇧ focus-score, anaemia, thrombocytopenia, SSB+ [177] |
| Nuclear autoantigen 14 kDa (NA-14) | 204 | _ | _ | _ | 144 | 12.7 | _ | _ | _ | 0 | ⇧ IgA level, ANA < 1:320, ANA−, shorter disease duration [178,179] |
| Stathmin-4 | 72 | _ | _ | _ | 128 | 15 | _ | _ | _ | 5 | Polyneuropathy, vasculitis [180] |
| Poly(U)-binding splicing factor 60 kDa | 84 | _ | _ | _ | 38 | 30 | _ | _ | _ | 5.3 | Asian or African descent, ANA+, RF+, hyperγ, SSA+, SSB+ [181] |
| NR2 | 66 | _ | _ | _ | 99 | 20 | _ | _ | _ | 7.6 | ⇩ memory function, ⇧ depression rate [182] ⇩ hippocampal grey matter [183] |
| 50 | _ | _ | _ | _ | 12 * | _ | _ | _ | _ | ||
| TRIM38 | 235 | _ | _ | _ | 50 | 10 | _ | _ | _ | 4 | ⇧ ocular stain scores, ⇩ Schirmer’s test, focus-score ≥ 3, SSA+, RF+, hyperγ [184] |
| Saccharomyces cerevisiae | 104 | _ | _ | _ | _ | 5 | _ | _ | _ | _ | Triple Ro52+/Ro60+/La+, hypocomplementemia, cutaneous involvement [185] |
| Calponin-3 | 209 | _ | _ | _ | 46 | 11 | _ | _ | _ | 2.2 | Peripheral neuropathy [186] |
| Ganglionic acetylcholine receptor | 39 | _ | _ | _ | 39 | 23 | _ | _ | _ | 0 | Autonomic neuropathy [187] |
| Aquaporin-4 | 109 | _ | _ | _ | _ | 10 | _ | _ | _ | _ | NMOSD overlap [188] |
| Aquaporin-5 | 112 | _ | _ | _ | 53 | 73 | _ | _ | _ | 32 | Low resting salivary flow [164] |
| Other aquaporins (1, 3, 8, 9) | 34 | _ | _ | _ | _ | 38 | _ | _ | _ | _ | ⇧ ocular stain scores [189] |
| P-selectin | 70 | _ | _ | _ | 35 | 21 | _ | _ | 0 | Low platelet count [190] | |
| Carbamylated proteins | 123 | _ | _ | _ | 172 | 28.5 | _ | _ | _ | 3.5 | ⇧ total IgG, IgM, RF+, β2-microglobulin, ⇧ focus-score and GC [191,192] |
| Moesin | 50 | _ | _ | _ | 50 | 42 | _ | _ | _ | 4 | [193] |
| Cofilin-1 | 50 | 20 | _ | _ | 50 | 76 | 80 | _ | _ | 18 | Association with pSS lymphoma [194] IgA isotype of anti-Ro/SSA ACPA+ and high urine pH for anti-alpha-enolase [195] |
| Alpha-enolase | 50 | 20 | _ | _ | 50 | 82 | 90 | _ | _ | 26 | |
| Rho GDP-dissociation inhibitor 2 | 50 | 20 | _ | _ | 50 | 86 | 90 | _ | _ | 26 | |
* = antibody positivity in cerebrospinal fluid; Sicca = non-pSS Sicca syndrome; FM = fibromyalgia with non-pSS sicca syndrome similar to “Sicca Asthenia Polyalgia” syndrome; Crtl = healthy controls; hyperγ = hypergammaglobulinemia; ANA = antinuclear antibodies; GC = germinal centre; SSA and SSB = anti-Ro/SSA (Ro52 and/or Ro60) and anti-La/SSB; ESSDAI = Eular Sjögren Syndrome Disease Activity Index; RF+ = rheumatoid factor positivity; NMOSD = Neuromyelitis Optica Spectrum Disorder; ACPA+ = anti-citrullinated protein antibodies positivity; ⇧ = increase(d)/higher; ⇩ = decrease(d)/lower; “−” = negativity.
Table 2.
Modern pSS Classification Criteria—comparisons of items, definitions and diagnosis performance compared to experts’ opinions.
Table 2.
Modern pSS Classification Criteria—comparisons of items, definitions and diagnosis performance compared to experts’ opinions.
| AECG Classification Criteria (2002) [250] | SICCA Classification Criteria (2012) [251] | ACR-EULAR Classification Criteria (2016) [252] | ||||
|---|---|---|---|---|---|---|
| Domain | Item Definition | Value | Item Definition | Value | Item Definition | Value |
| Subjective eye dryness | ≥1/3 specific questions | minor | / | _ | / | _ |
| Subjective oral dryness | ≥1/3 specific questions | minor | / | _ | / | _ |
| Ocular signs | Schirmer (≤5 mm/5 min) OR Van Bijsterveld ≥ 4 | minor | OSS ≥3 | 1 | Schirmer (<5 mm/5 min) | 1 |
| OSS ≥ 5 OR Van Bijsterveld ≥ 4 | 1 | |||||
| SG dysfunction | UWSF (≤1.5 mL/15 min) OR Compatible parotid sialography OR Anormal salivary scintigraphy | minor | / | _ | UWSF (≤0.1 mL/min) | 1 |
| MSGB | Focus-score ≥ 1 | Major | Focus-score ≥ 1 | 1 | Focus-score ≥ 1 | 3 |
| Auto antibodies | Anti-Ro/SSA or Anti-La/SSB | Major | Anti-Ro/SSA or Anti-La/SSB OR RF(+) with ANA(+) ≥1:320 | 1 | Anti-Ro/SSA | 3 |
| pSS definition | 4 out of 6 with ≥ 1 Major (or 3 out of 4 objectives findings) | pSS signs and/or symptoms with ≥2/3 criteria | Sicca or ESSDAI manifestation with a total score ≥ 4 | |||
| Exclusions criteria |
|
|
| |||
| Sensitivity | 93.5% | 92.5% | 96% | |||
| Specificity | 94.0% | 95.4% | 95% | |||
AECG = American European Consensus Group, SICCA = Sjögren’s International Collaborative Clinical Alliance, ACR-EULAR = American College of Rheumatology—European League Against Rheumatism, UWSF = unstimulated whole saliva flow, RF = rheumatoid factor, ANA = antinuclear antibodies, ESSDAI = EULAR Sjögren’s syndrome disease activity index.
Table 3.
Differential diagnosis of Sjögren’s syndrome (non-exhaustive list).
| Sicca Symptoms Complex | Glandular Involvement | Articular Involvement | Systemic Involvement | |
|---|---|---|---|---|
| Xerogenic medications | X | _ | _ | _ |
| Aromatase inhibitors | (X) | _ | X | (X) pSS-like |
| Age-related dryness | X | _ | _ | _ |
| Metabolic sialadenosis | _ | X | _ | _ |
| Non-SS dry eye diseases | X | _ | _ | _ |
| Head and neck irradiation | X | _ | _ | _ |
| Sarcoïdosis | X | X | X | X |
| Hyperlipoproteinemia (II, IV, V type) | X | X | (X) | _ |
| Chronic Graft vs. Host disease | X | X | X | X |
| Primary lymphoma | X | X | _ | (X) |
| Amyloïdosis | X | X | (X) | (X) Renal, purpura |
| Viral chronic sialadenitis (HCV, HIV, HTLV-1) | X | (X) | X | X |
| Other chronic Non-specific sialadenitis | X | X Usually unilateral | _ | _ |
| Diabetes Mellitus | X | (X) Sialadenosis | (X) Cheiroarthropathy | (X) Neuropathy |
| Haemochromatosis | X | (X) | X CPPD | (X) |
| Other connective tissue disease | X | _ | X | X |
| Rheumatoid arthritis | (X) | _ | X | (X) |
| Granulomatosis with polyangiitis | X | (X) | X | X |
| IgG4-related disease (Mikulicz syndrome) | X | X | (X) | (X) |
| Anxiety, fibromyalgia | X | _ | (X) | _ |
| Checkpoint inhibitors | X | (X) | X | X |
Table 4.
Common damage, burden and activity scores for clinical monitoring of pSS patients.
| EULAR Sjögren’s Syndrome Disease Activity Index | EULAR Sjögren’s Syndrome Patient Reported Index | Sjögren’s Syndrome Disease Damage Index | Sjögren’s Syndrome Damage Index | |
|---|---|---|---|---|
| Abbreviation | ESSDAI | ESSPRI | SSDDI | SSDI |
| First description | Seror et al. [294] | Seror et al. [295] | Vitali et al. [296] | Barry et al. [297] |
| Year | 2010 | 2011 | 2007 | 2008 |
| Type | Activity index | PRO | Damage index | Damage index |
| Domains (n) | 12 | 1 | 6 | 9 |
| Items (n) | 44 | 3 | 9 | 27 |
| Items scoring | 0 to 3 | VAS (0–10) | 1, 2 or 5 | 1 |
| Domain weight | 1 to 6 | 1 | 1 | 1 |
| Calculation | Sum | Mean | Sum | Sum |
| Score range | 0–123 | 0–10 | 0–16 | 0–27 |
| Clinically significant threshold | <5 Low ≥5, ≤13 moderate ≥14 high | ≥5/10 is an unsatisfactory symptom state | - | - |
| Minimal clinically important difference | ≥3 points improvement | ≥1 point or ≥15% improvement | - | - |
VAS = visual analogue scale, PRO = patient reported outcome.
Table 5.
Current treatment for sicca-related manifestations.
| Salivary Gland Involvement | Lachrymal Gland Involvement | Skin and Vaginal Mucosa Involvement | |
|---|---|---|---|
| Self-Care |
|
| |
| Conserve |
| ||
| Replace |
|
|
|
| Stimulate |
|
|
|
| Complications Prevention and Management |
|
|
Table 6.
B cell targeted drugs in pSS part 1: monoclonal antibodies directed against B cell specific Cluster of Differentiation (CD).
Table 6.
B cell targeted drugs in pSS part 1: monoclonal antibodies directed against B cell specific Cluster of Differentiation (CD).
| DRUG | TRIAL (Reference) | Inclusion Criteria | Number of Subjects | Age (Years) | Disease Duration (Years) | Mean ESSDAI | Primary Outcome | Results | Effects (Statistically Significant) | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Drg | Ctrl | Sicca Syndrome | Fibro-Like | Systemic | ||||||||
| Rituximab Anti-CD20 | NCT00363350 Phase I/II [343] | AECG criteria RF+ and SSa and/or SSb+ SWS >0.15 mL/min | 20 | 10 | 43 ± 11 | 5.25 ± 4.17 | 8 (4–13) | SWS ⇧ at 48w | met | SWS/UWS ⇧ LG test ⇧ Schirmer = BUT = | SF36 ⇧ MFI ⇩ | Vasculitis ⇩ |
| Phase III [330] | AECG criteria SSa and/or SSb+ F-VAS ≥5/10 | 8 | 9 | 51 (22–64) | 7.25 (1–18) | na | ⇩ > 20% of F-VAS at 24w; ⇩ F-VAS at 24w; ⇩ >30% of F-VAS at 24w | not met not met not met | UWS = Schirmer = | F-VAS ⇩ PROFAD ⇩ P-VAS = Soc-SF36 ⇧ | Glandular ⇩ | |
| Phase III [332] | AECG criteria SSa and/or SSb+ Disease duration ≤ 2y 2/5 of [PhGA >50 mm or ESSDAI ≥ 6 or subESSPRI ≥ 5] | 19 | 22 | 40 (27–53) | 1 (1–2) | 20 (6–41) | ΔESSDAI until 120W | met from 24w to 120w | D-VAS ⇩ Schirmer ⇧ UWS ⇧ | P-VAS = F-VAS ⇩ | ESSDAI ⇩ | |
| NCT00740948 Phase III TEARS [331] | AECG criteria with 2/4 VAS ≥ 5/10 for PhGA, pain, fatigue and dryness AND biologically active OR 1 extra-glandular manifestation or parotid gland enlargement. | 63 | 57 | 52.9 ± 13.3 | 4.6 ± 4.8 | 10 ± 6.9 | ⇩ 30% of at least 2/4 VAS at 6-16-24w | met at 6w not met at 16-24w | D-VAS ⇩ Schirmer = | P-VAS = F-VAS ⇩ | ESSDAI = Glandular = Articular = | |
| Phase III TRACTISS [344] | pSS with SSa+ UWS >0 mL/min F-VAS and D-VAS >5/10 | 67 | 66 | 54 ± 11.5 | 5.7 ± 5.4 | 5.7 ± 4.5 | ⇩ 30% D-VAS and F-VAS at 48w | not met | UWS ⇧ ESSPRI = D-VAS = | F-VAS = SF36 = PROFAD = | ESSDAI ⇩ | |
| Epratuzumab Anti-CD22 | Post-hoc Phase I/II EMBODY [337] | SLE with SSa+ and SS diagnosis | 31 + 41 | 40 | 46.4 ± 12.3 | 5.1 (0–34) | na | BICLA at 48w ΔBILAG at 48w ΔSLEDAI at 48w ΔPhGA at 48w | met met not met not met | na | na | BILAG ⇩ |
AECG = American European Consensus Group, Drg = drug/treatment group, Ctrl = control group, Fibro-like = fibromyalgia-like symptoms such as fatigue and widespread pain, FR+ = presence of rheumatoid factor, SSa/SSb = anti-Ro/SSa and anti-La/SSb, SWS = stimulated whole saliva flow, UWS = unstimulated whole saliva flow, LG test = lissamine green test, BUT = break-up time, SF36 = Short Form 36 health survey score, Soc-SF36 = social component of SF36 score, Phys-SF36 = physical component of SF36 score, MFI = Multidimensional Fatigue Inventory score, F-VAS = fatigue visual analogue scale, Schirmer = Schirmer test, P-VAS = Pain visual analogue scale, PROFAD = Profile of Fatigue and Discomfort, DSST = Digit Symbol Substitution Test, ESSDAI = EULAR SS disease activity index, D-VAS = dryness visual analogue scale, PhGA = physician global activity visual analogue scale, subESSPRI = P-VAS, D-VAS or F-VAS, BILAG = British Isles Lupus Assessment Group index, BICLA = BILAG-based Combined Lupus Assessment, ESSPRI = EULAR SS Patient Reported Index, SAEs = serious adverse effects, SGUS = salivary gland ultrasound, Ig = immunoglobulin, ⇩⇧ = decrease/increase, Δ = difference.
Table 7.
B cell targeted drugs in pSS part 2: BAFF/APRIL system targeted therapies.
| DRUG | TRIAL (References) | Inclusion Criteria | Number of Subjects | Age (Years) | Disease Duration (Years) | Mean ESSDAI | Primary Outcome | Results | Effects (Statistically Significant) | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Drg | Ctrl | Sicca Syndrome | Fibro-Like | Systemic | ||||||||
| Belimumab Anti-BAFF | NCT01160666 NCT01008982 Phase II BELISS [338,339] | AECG criteria SSa and/or SSb+ AND systemic complication OR B cell activation OR early disease (≤5 years) | 30 | - | 49.5 ± 6.5 | 5.7 ± 5.6 | 8.8 ± 7.4 | ⇩ of 2/5 VAS at 28w
| 60% response | ESSPRI ⇩ D-VAS ⇩ UWS = Schirmer = | ESSPRI ⇩ P-VAS = F-VAS = SF36 = | ESSDAI ⇩ Glandular ⇩ |
| Follow-up of previous study | 15 | - | 40.2 ± 11.8 | 5.9 ± 5.7 | 3.8 ± 3.1 | Idem between 28–52w | 86.7% Stable response | ESSPRI ⇩ D-VAS ⇩ UWS = Schirmer = | ESSPRI ⇩ P-VAS = F-VAS = Phys-SF36⇧ | ESSDAI ⇩ Glandular ⇩ Articular ⇩ Biologic ⇩ | ||
| RC18 TACI-Igfusion protein | NCT04078386 Phase II [345] | AECG criteria SSa+ ESSDAI ≥ 5 | 30 | ? | ? | ? | ΔESSDAI at 24w | December 2020 | Secondary endpoint | Secondary endpoint | Primary endpoint | |
| Rituximab Anti-CD20 + Belimumab Anti-BAFF | NCT02631538 Phase II [346] | AECG criteria SSa and/or SSb+ ESSDAI ≥ 5 UWS >0 mL/min D-VAS ≥ 5/10 | 70 | ? | ? | ? | SAEs at 104w AESIs at 104w | Study completed on June 2020 | Secondary endpoint | na | Secondary endpoint | |
| Tibulizumab (LY3090106) Anti-BAFF + Anti-IL-17 | NCT02614716 Phase I [347] | AECG criteria SSa and/or SSb+ | 32 | ? | ? | ? | SAEs at 197d | Not published | na | na | na | |
| Ianalumab (VAY736) Anti-BAFFR | NCT02149420 Phase II [348] | AECG criteria ANA ≥1:160 SSa and/or SSb+ ESSDAI ≥ 6 UWS >0 mL/min | 6+12 | 9 | 50.5 ± 12.16 | ? | 12.5 (6, 31) | ΔESSDAI at 12w | not met | D-VAS ⇩ | SF-36 = MFI ⇩ F-VAS ⇩ | ESSDAI = Articular ⇩ |
| NCT02962895 Phase II [349] | AECG criteria SSa+ ESSDAI ≥ 6 (from 7 domains only) | 195 | ? | ? | ? | Change in multi-dimensional disease activity at 24w | Study completed on June 2020 | Secondary endpoint | Secondary endpoint | Primary endpoint | ||
Table 8.
B cell targeted drugs in pSS part 3: drugs targeting other B cells survival and function pathways.
Table 8.
B cell targeted drugs in pSS part 3: drugs targeting other B cells survival and function pathways.
| DRUG | TRIAL (Reference) | Inclusion Criteria | Number of Subjects | Age (Years) | Disease Duration (Years) | Mean ESSDAI | Primary Outcome | Results | Effects (Statistically Significant) | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Drg | Ctrl | Sicca Syndrome | Fibro-Like | Systemic | ||||||||
| LOU064 BTK inhibitor | NCT04035668 Phase II LOUISSe [350] | 2016 ACR/EULAR criteria SSa and/or SSb+ ESSDAI ≥ 6 UWS >0 mL/min | 252 | ? | ? | ? | ΔESSDAI at 24w | Estimated Study Completion on January 2023 | Secondary endpoint | Secondary endpoint | Secondary endpoint | |
| Tirabrutinib (GS-4059) BTK inhibitor | NCT03100942 Phase II [342] | AECG criteria SSa and/or SSb+ ESSDAI ≥ 4 | 38 | 37 | 55.8 ± 10.06 | ? | 10.4 ± 5.36 | Protocol-Specified Response Criteria at 12w | not met | ESSPRI = | ESSPRI = | ESSDAI = |
| BMS-986142 BTK inhibitor | NCT02843659 Phase II [351] | 2016 ACR/EULAR criteria SSa and/or SSb+ ESSDAI ≥ 6 UWS >0 mL/min | 5+6 | 7 | 51.2 ± 11.41 | ? | ? | ΔESSDAI at 12w | Not published | Secondary endpoint | Secondary endpoint | Secondary endpoint |
| Branebrutinib BTK inhibitor | NCT04186871 Phase II [352] | 2016 ACR/EULAR criteria Moderate to severe pSS | ? | ? | ? | ? | ? | Protocol-Specified Response Criteria at 24w | Estimated Study Completion on June 2022 | na | na | Primary endpoint |
| Baminercept LTβ-R fusion protein | NCT01552681 Phase II [353] | 2016 ACR/EULAR criteria UWS >0.1 mL/min ≥ 1 non-life-threatening systemic manifestation(s) | 33 | 19 | 52.0 ± 11.0 | ? | 3.1 ± 3.4 | ΔSWS at 24w | not met | D-VAS = Schirmer ⇧ UWS = | F-VAS = | ESSDAI = |
| Parsaclisib (INCB050465) PI3Kδ inhibitor | NCT03627065 Phase II [354] | AECG criteria SGUS score > 2 SSa and/or SSb+ ESSDAI ≥ 6 Oral dryness score ≥ 5. | 10 | ? | ? | ? | ΔSGUS score at 12w | Not published | na | na | na | |
| Seletalisib (UCB5857) PI3Kδ inhibitor | NCT02610543 Phase II [355] | AECG criteria FAN ≥ 1:160 SSa and/or SSb+ ESSDAI ≥ 6 | 13 | 14 | ? | ? | ? | ΔESSDAI at 12w | not met | ESSPRI = SWSF = Schirmer = | na | ESSDAI = |
| Leniolisib (CDZ173) PI3Kδ inhibitor | NCT02775916 Phase II [356] | pSS diagnosis SSa and/or SSb+ ESSDAI ≥ 6, ESSPRI ≥ 5 SWS > 0.1 mL/min | 20 | 10 | 47.3 ± 13.07 | ? | ? | ΔESSDAI at 12w SAEs at 12w | not met | ESSPRI = | SF-36 = MFI = | na |
Table 9.
T-cell targeted drugs in pSS: co-stimulation receptors or ligands inhibition.
| DRUG | TRIAL | Inclusion Criteria | Number of Subjects | Age (Years) | Disease Duration (Years) | Mean ESSDAI | Primary Outcome | Results | Effects (Statistically Significant) | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Drg | Ctrl | Sicca Syndrome | Fibro-Like | Systemic | ||||||||
| Abatacept CTLA-4 Ig fusion protein | NCT02915159 Phase III [357] | 2016 ACR/EULAR criteria SSa+ ESSDAI ≥ 5 | 92 | 95 | 52 ± 12.9 | ? | 9.4 ± 4.3 | ΔESSDAI at 169d | Not met | ESSPRI = SWS = | ESSPRI = | ESSDAI = DAS28 ⇩ |
| Phase I/II ASAP [358] | AECG criteria and ESSDAI ≥ 6 Disease duration ≤ 5 years SWS ≥ 0.10 mL/min SSa and/or SSb+ or FR+ Proven by parotid gland biopsy. | 15 | - | 43 (32–51) | 11 (7–36) | 11 (8–14) | ΔESSDAI at 24-48w | met | ESSPRI ⇩ SWS/UWS = Schirmer = BUT ⇧ | ESSPRI ⇩ | ESSDAI ⇩ | |
| NCT02067910 Phase III ASAPIII [359] | AECG criteria and ESSDAI ≥ 5 Time from diagnosis ≤ 7 years | 40 | 39 | 49 ± 16 | 8 (4–14) | ? | ΔESSDAI at 24w | Not met | ESSPRI ⇩ FSFI ⇧ DVAS = UWS = Schirmer = | Fatigue = | ESSDAI = Articular ⇩ | |
| Iscalimab (CFZ533) Anti-CD40 | NCT02291029 Phase IIa [360] | AECG criteria and ESSDAI ≥ 6 SSA+ OR FR+ and FAN ≥ 1:320 SWS ≥ 0 mL/min | 8+21 | 4+11 | 51.3 ± 13.5 | ? | 10.7 ± 4.6 | SAEs at 12w | safe | ESSPRI ⇩ UWS = Schirmer = | MFI = SF-36 = | ESSDAI ⇩ Articular ⇩ |
| NCT03905525 Phase II TWINSS [361] | 2016 ACR/EULAR criteria SSa+ SWS > 0.01 mL/min, P1: ESSDAI ≥ 5 or P2 ESSPRI ≥ 5. | 260 | ? | ? | ? | ΔESSDAI at 24w in P1 ΔESSPRI at 24w in P2 | Estimated Study Completion on June 2022 | Included endpoint | Included endpoint | Included endpoint | ||
| VIB4920 MEDI4920 Anti-CD40L | NCT04129164 Phase II [362] | P1: ESSDAI ≥ 5 P2: ESSDAI < 5 et ESSPRI ≥ 5 | 174 | ? | ? | ? | ΔESSDAI at 169d in P1 ΔESSPRI at 169d in P2 | Estimated Study Completion on April 2022 | Included endpoint | Included endpoint | Included endpoint | |
| Prezalumab (AMG557) (MEDI5872) Anti-B7RP1 | NCT02334306 Phase IIa [363] | AECG criteria and ESSDAI ≥ 5 SSa and/or SSb+ FR+, cryoglobulinemia or hypergammaglobulinemia | 16 | 16 | 50.7 ± 13 | ? | ? | ΔESSDAI at 99d | Not met | ESSPRI = | ESSPRI = | ESSDAI = |
| Lulizumab (BMS-931699) Anti-CD28 | NCT02843659 Phase II [351] | 2016 ACR/EULAR criteria SSa and/or SSb+ ESSDAI ≥ 5 USW > 0.01 mL/min | 5+6 | 7 | 51.2 ± 11.41 | ? | ? | ΔESSDAI at 12w | Not published | Secondary endpoint | Secondary endpoint | Primary endpoint |
Table 10.
T-cell targeted drugs in pSS: therapies preventing autoantigen presentation.
| DRUG | TRIAL (Reference) | Inclusion Criteria | Number of Subjects | Age (Years) | Disease Duration (Years) | Mean ESSDAI | Primary Outcome | Results | Effects (Statistically Significant) | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Drg | Ctrl | Sicca Syndrome | Fibro-Like | Systemic | ||||||||
| Petesicatib RO5459072 Cathepsin S Inhibitor | NCT02701985 Phase IIa [364] | AECG criteria SSa and/or SSb+ ESSDAI ≥ 5 ESSPRI ≥ 5 USW > 0.0 mL/min Oral D-VAS ≥ 5/10 | 38 | 37 | 52.2 ± 12.5 | ? | ? | ΔESSDAI ≥ 3 at 12w | Not met | ESSPRI = | ESSPRI = SF36 = | ESSDAI = |
| Efalizumab Anti-LFA-1 | NCT00344448 Phase II [365] | AECG criteria SSa and/or SSb+ | 6 | 3 | 53 ± 11.2 | ? | ? | Protocol-specified composite score at 12w | Early termination due to serious side effect in other trial | |||
Table 11.
Anti-cytokine targeted drugs in pSS.
| DRUG | TRIAL | Inclusion Criteria | Number of Subjects | Age (Years) | Disease Duration (Years) | Mean ESSDAI | Primary Outcome | Results | Effects (Statistically Significant) | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Drg | Ctrl | Sicca Syndrome | Fibro-Like | Systemic | ||||||||
| Anakinra IL1R antagonist protein | NCT00683345 Phase II [333] | AECG criteria 18–80 years Western European descent No depression or comorbidity | 13 | 13 | 55 (36–80) | 5 (1–17) | ? | Group-wise comparison of the fatigue scores at 4w | not met | na | F-VAS ⇩ | na |
| Tocilizumab Anti-IL-6R | NCT01782235 Phase I/II ETAP [366] | AECG criteria ESSDAI ≥ 5 | 55 | 55 | 50.9 (26–76) | ? | 11.5 (5–25) | ΔESSDAI ≥ 3 at 12W without new item without ⇧ ≥1/10 PGA | not met | ESSPRI = Schirmer = | ESSPRI = | ESSDAI = Articular ⇩ |
| Infliximab Anti-TNF | Phase III TRIPSS [367] | AECG criteria 2/3 D-VAS, F-VAS, P-VAS ≥ 5/10 | 54 | 49 | 54.4 ± 10.4 | 4.0 ± 5.5 | na | ⇧ 30% in 2/3 D-VAS, F-VAS, P-VAS at 10–22w | not met | SWS = Schirmer = | SF-36 = | SJC = TJC = |
| Etanercept TNFR-Ig fusion protein | NCT00001954 Phase II [368] | 1986 and AECG criteria Elevated ESR or IgG levels | 14 | 14 | 55.5 (46, 59) | ? | na | ⇧ 20% in 2/3 pSS domains (protocol-specified) | not met | D-VAS = Schirmer = VB = SWS = | na | na |
| Ustekinumab Anti- IL-12/IL-23 (p40 subunit) | NCT04093531 Phase I [341] | 2016 ACR/EULAR criteria | 15 | - | ? | ? | ? | ΔESSDAI at 24W | Estimated Study Completion on December 2021 | na | Secondary endpoint | Primary endpoint |
| GSK2618960 anti–IL-7Rα | NCT03239600 Phase II [369] | AECG criteria SWS >0.1 mL/min ⇧ Ig or FR+ or ANA ≥ 1:320 D-VAS ≥ 5/10 or Schirmer < 10 mm | 0 | - | - | - | SAEs at 27w | Withdraw The study is stopped for Portfolio prioritization | ||||
Table 12.
Miscellaneous targeted drugs in pSS.
| DRUG | TRIAL (Reference) | Inclusion Criteria | Number of Subjects | Age (years) | Disease Duration (Years) | Mean ESSDAI | Primary Outcome | Results | Effects (Statistically Significant) | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Drg | Ctrl | Sicca Syndrome | Fibro-Like | Systemic | ||||||||
| Daxdilimab VIB7734 Anti-ILT7 | NCT03817424 Phase I [370] | Unspecified | ? | ? | ? | ? | SAEs at 169d AESIs at 169d | June 2020 | na | na | na | na |
| Filgotinib Jak1 inhibitor | NCT03100942 Phase II [342] | AECG criteria SSa and/or SSb+ ESSDAI ≥ 4 | 38 | 37 | 52.2 ± 10.54 | ? | 10.2 ± 6.23 | Protocol-Specified Response Criteria at 12w | not met | ESSPRI = | ESSPRI = | ESSDAI = |
| Lanraplenib (GS-9876) SIK inhibitor | NCT03100942 Phase II [342] | AECG criteria SSa and/or SSb+ ESSDAI ≥ 4 | 38 | 37 | 56.2 ± 9.72 | ? | 10.5 ± 4.89 | Protocol-Specified Response Criteria at 12w | not met | ESSPRI = | ESSPRI = | ESSDAI = |
| RSLV-132 RNase1-Fc fusion protein | NCT03247686 Phase II [371] | AECG criteria SSA+ Interferon signature | 22 | 8 | ? | ? | ? | Interferon gene expression at day99 | Not published | ESSPRI = | mPRO-F ⇧ DSST ⇩ FACIT-F = ESSPRI = | ESSDAI = |
| Low-dose IL-2 T-reg induction | NCT01988506 Phase II Transreg [372] | pSS diagnosis | 84-132 | ? | ? | ? | T-reg percentage | Estimated Study Completion on February 2022 | na | na | na | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Parisis, D.; Chivasso, C.; Perret, J.; Soyfoo, M.S.; Delporte, C. Current State of Knowledge on Primary Sjögren’s Syndrome, an Autoimmune Exocrinopathy. J. Clin. Med. 2020, 9, 2299. https://doi.org/10.3390/jcm9072299
AMA Style
Parisis D, Chivasso C, Perret J, Soyfoo MS, Delporte C. Current State of Knowledge on Primary Sjögren’s Syndrome, an Autoimmune Exocrinopathy. Journal of Clinical Medicine. 2020; 9(7):2299. https://doi.org/10.3390/jcm9072299
Chicago/Turabian StyleParisis, Dorian, Clara Chivasso, Jason Perret, Muhammad Shahnawaz Soyfoo, and Christine Delporte. 2020. "Current State of Knowledge on Primary Sjögren’s Syndrome, an Autoimmune Exocrinopathy" Journal of Clinical Medicine 9, no. 7: 2299. https://doi.org/10.3390/jcm9072299
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.