Insulin Resistance Associated Disorders Pivoting Long-Term Hepatitis B Surface Antigen Decline During Entecavir Therapy

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Experimental Section
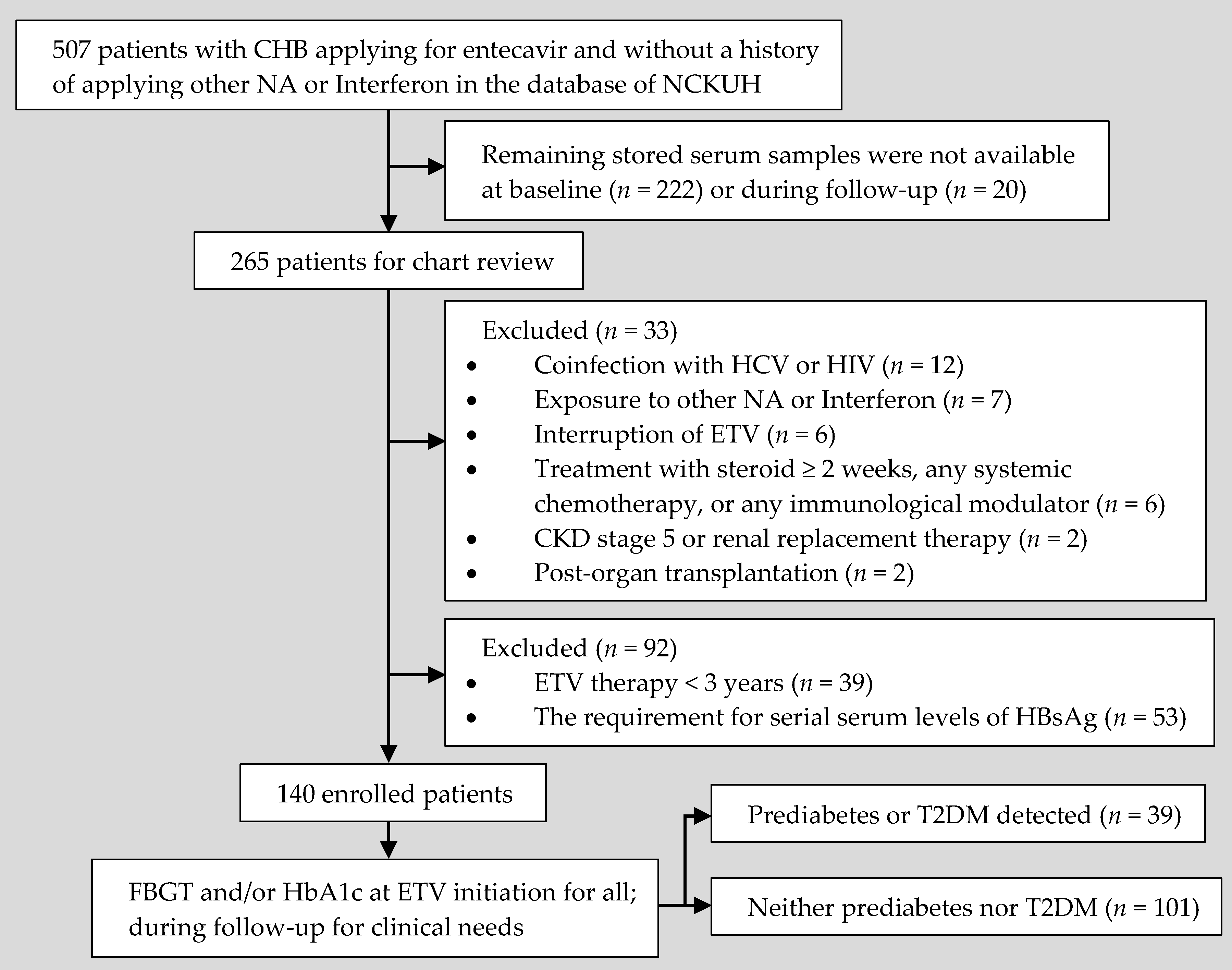
2.1. Study Design and Data Source
2.2. Follow-Up and Data Collection
2.3. Terms and Definitions of Hepatic Events, Virology, Prediabetes, T2DM, and Other Chronic Comorbidities
2.4. Laboratory Measurements
2.5. Statistical Analyses
3. Results
3.1. Characteristics of the Enrolled Patients and Their Clinical Results
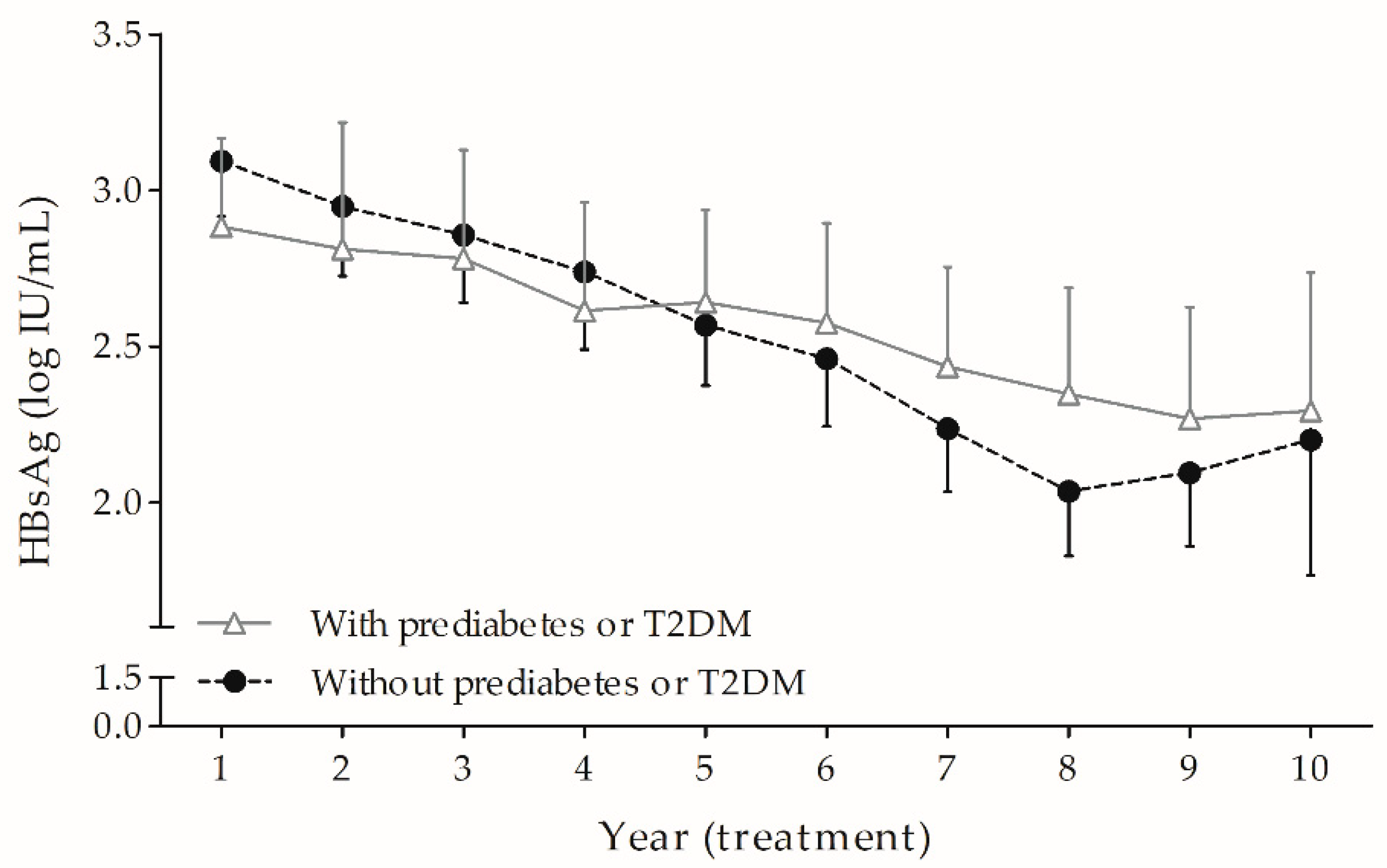
3.2. HBsAg Kinetics During Long-Term ETV: Prediabetes or T2DM Hindered the Fading of HBsAg over Time Independently and Markedly
3.3. Cytokine Profiles: Higher IP-10 Levels in Patients with Prediabetes or T2DM at the Sixth Year of ETV Therapy
3.4. FIB-4 Score: Highest in Subjects with T2DM in the In-Depth Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lampertico, P.; Agarwal, K.; Berg, T.; Buti, M.; Janssen, H.L.A.; Papatheodoridis, G.; Zoulim, F.; Tacke, F. EASL 2017 clinical practice guidelines on the management of hepatitis B virus infection. J. Hepatol. 2017, 67, 370–398. [Google Scholar] [CrossRef] [PubMed]
- Nassal, M. HBV cccDNA: Viral persistence reservoir and key obstacle for a cure of chronic hepatitis B. Gut 2015, 64, 1972–1984. [Google Scholar] [CrossRef] [PubMed]
- Bertoletti, A.; Ferrari, C. Adaptive immunity in HBV infection. J. Hepatol. 2016, 64, S71–S83. [Google Scholar] [CrossRef] [PubMed]
- Zmora, N.; Bashiardes, S.; Levy, M.; Elinav, E. The role of the immune system in metabolic health and disease. Cell Metab. 2017, 25, 506–521. [Google Scholar] [CrossRef]
- Lin, T.C.; Chiu, Y.C.; Chiu, H.C.; Liu, W.C.; Cheng, P.N.; Chen, C.Y.; Chang, T.T.; Wu, I.C. Clinical utility of hepatitis B surface antigen kinetics in treatment-naive chronic hepatitis B patients during long-term entecavir therapy. World J. Gastroenterol. 2018, 24, 725–736. [Google Scholar] [CrossRef]
- Papatheodoridis, G.; Goulis, J.; Manolakopoulos, S.; Margariti, A.; Exarchos, X.; Kokkonis, G.; Hadziyiannis, E.; Papaioannou, C.; Manesis, E.; Pectasides, D.; et al. Changes of HBsAg and interferon-inducible protein 10 serum levels in naive HBeAg-negative chronic hepatitis B patients under 4-year entecavir therapy. J. Hepatol. 2014, 60, 62–68. [Google Scholar] [CrossRef]
- Cornberg, M.; Wong, V.W.; Locarnini, S.; Brunetto, M.; Janssen, H.L.; Chan, H.L. The role of quantitative hepatitis B surface antigen revisited. J. Hepatol. 2016, 66, 398–411. [Google Scholar] [CrossRef]
- Tseng, T.C.; Liu, C.J.; Yang, H.C.; Su, T.H.; Wang, C.C.; Chen, C.L.; Hsu, C.A.; Kuo, S.F.; Liu, C.H.; Chen, P.J.; et al. Serum hepatitis B surface antigen levels help predict disease progression in patients with low hepatitis B virus loads. Hepatology 2013, 57, 441–450. [Google Scholar] [CrossRef]
- Tseng, T.C.; Liu, C.J.; Yang, H.C.; Su, T.H.; Wang, C.C.; Chen, C.L.; Kuo, S.F.; Liu, C.H.; Chen, P.J.; Chen, D.S.; et al. High levels of hepatitis B surface antigen increase risk of hepatocellular carcinoma in patients with low HBV load. Gastroenterology 2012, 142, 1140–1149. [Google Scholar] [CrossRef]
- Marra, F.; Tacke, F. Roles for chemokines in liver disease. Gastroenterology 2014, 147, 577–594. [Google Scholar] [CrossRef]
- Liu, M.; Guo, S.; Hibbert, J.M.; Jain, V.; Singh, N.; Wilson, N.O.; Stiles, J.K. CXCL10/IP-10 in infectious diseases pathogenesis and potential therapeutic implications. Cytokine Growth Factor Rev. 2011, 22, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Maini, M.K.; Gehring, A.J. The role of innate immunity in the immunopathology and treatment of HBV infection. J. Hepatol. 2016, 64, S60–S70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winer, D.A.; Luck, H.; Tsai, S.; Winer, S. The intestinal immune system in obesity and insulin resistance. Cell Metab. 2016, 23, 413–426. [Google Scholar] [CrossRef] [PubMed]
- Hueso, L.; Ortega, R.; Selles, F.; Wu-Xiong, N.Y.; Ortega, J.; Civera, M.; Ascaso, J.F.; Sanz, M.J.; Real, J.T.; Piqueras, L. Upregulation of angiostatic chemokines IP-10/CXCL10 and I-TAC/CXCL11 in human obesity and their implication for adipose tissue angiogenesis. Int. J. Obes. 2018, 42, 1406–1417. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Moschen, A.R.; Roden, M. NAFLD and diabetes mellitus. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 32–42. [Google Scholar] [CrossRef]
- Anstee, Q.M.; Targher, G.; Day, C.P. Progression of NAFLD to diabetes mellitus, cardiovascular disease or cirrhosis. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 330–344. [Google Scholar] [CrossRef] [PubMed]
- Birkenfeld, A.L.; Shulman, G.I. Nonalcoholic fatty liver disease, hepatic insulin resistance, and type 2 diabetes. Hepatology 2014, 59, 713–723. [Google Scholar] [CrossRef]
- Khalili, M.; Lombardero, M.; Chung, R.T.; Terrault, N.A.; Ghany, M.G.; Kim, W.R.; Lau, D.; Lisker-Melman, M.; Sanyal, A.; Lok, A.S.; et al. Diabetes and prediabetes in patients with hepatitis B residing in North America. Hepatology 2015, 62, 1364–1374. [Google Scholar] [CrossRef] [Green Version]
- Sell, H.; Habich, C.; Eckel, J. Adaptive immunity in obesity and insulin resistance. Nat. Rev. Endocrinol. 2012, 8, 709–716. [Google Scholar] [CrossRef]
- Joo, E.J.; Yeom, J.S.; Kwon, M.J.; Chang, Y.; Ryu, S. Insulin resistance increases loss of antibody to hepatitis B surface antigen in nondiabetic healthy adults. J. Viral Hepat. 2016, 23, 889–896. [Google Scholar] [CrossRef]
- Yu, M.W.; Lin, C.L.; Liu, C.J.; Yang, S.H.; Tseng, Y.L.; Wu, C.F. Influence of metabolic risk factors on risk of hepatocellular carcinoma and liver-related death in men with chronic hepatitis B: A large cohort study. Gastroenterology 2017, 153, 1006–1017. [Google Scholar] [CrossRef] [PubMed]
- Yip, T.C.F.; Wong, V.W.S.; Chan, H.L.Y.; Tse, Y.K.; Kong, A.P.S.; Lam, K.L.Y.; Lui, G.C.Y.; Wong, G.L.H. Effects of diabetes and glycemic control on risk of hepatocellular carcinoma after seroclearance of hepatitis B surface antigen. Clin. Gastroenterol. Hepatol. 2018, 16, 765–773. [Google Scholar]
- Tseng, T.C.; Liu, C.J.; Su, T.H.; Yang, W.T.; Chen, C.L.; Yang, H.C.; Wang, C.C.; Kuo, S.F.; Liu, C.H.; Chen, P.J.; et al. Fibrosis-4 index helps identify HBV carriers with the lowest risk of hepatocellular carcinoma. Am. J. Gastroenterol. 2017, 112, 1564–1574. [Google Scholar] [CrossRef] [PubMed]
- Xiao, G.; Yang, J.; Yan, L. Comparison of diagnostic accuracy of aspartate aminotransferase to platelet ratio index and fibrosis-4 index for detecting liver fibrosis in adult patients with chronic hepatitis B virus infection: A systemic review and meta-analysis. Hepatology 2015, 61, 292–302. [Google Scholar] [CrossRef]
- Park, S.H.; Kim, D.J.; Lee, H.Y. Insulin resistance is not associated with histologic severity in nondiabetic, noncirrhotic patients with chronic hepatitis B virus infection. Am. J. Gastroenterol. 2009, 104, 1135–1139. [Google Scholar] [CrossRef]
- Wang, C.C.; Hsu, C.S.; Liu, C.J.; Kao, J.H.; Chen, D.S. Association of chronic hepatitis B virus infection with insulin resistance and hepatic steatosis. J. Gastroenterol. Hepatol. 2008, 23, 779–782. [Google Scholar] [CrossRef]
- Wong, V.W.; Wong, G.L.; Chu, W.C.; Chim, A.M.; Ong, A.; Yeung, D.K.; You, K.K.; Chu, S.H.; Chan, H.Y.; Woo, J.; et al. Hepatitis B virus infection and fatty liver in the general population. J. Hepatol. 2012, 56, 533–540. [Google Scholar] [CrossRef]
- Joo, E.J.; Chang, Y.; Yeom, J.S.; Ryu, S. Hepatitis B virus infection and decreased risk of nonalcoholic fatty liver disease: A cohort study. Hepatology 2017, 65, 828–835. [Google Scholar] [CrossRef]
- Chien, C.H.; Chen, L.W.; Lin, C.L.; Chang, S.W.; Shyu, Y.C.; Chen, K.F.; Chen, S.W.; Hu, C.C.; Yu, C.Y.; Chien, R.N. Unawareness of hepatitis B virus infection confers on higher rate of metabolic syndrome: A community-based study. Sci. Rep. 2017, 7, 9869. [Google Scholar] [CrossRef]
- Lee, J.G.; Lee, S.; Kim, Y.J.; Cho, B.M.; Park, J.S.; Kim, H.H.; Cheong, J.; Jeong, D.W.; Lee, Y.H.; Cho, Y.H.; et al. Association of chronic viral hepatitis B with insulin resistance. World J. Gastroenterol. 2012, 18, 6120–6126. [Google Scholar] [CrossRef]
- Chen, Y.C.; Su, Y.C.; Li, C.Y.; Hung, S.K. 13-year nationwide cohort study of chronic kidney disease risk among treatment-naive patients with chronic hepatitis B in Taiwan. BMC Nephrol. 2015, 16, 110. [Google Scholar] [CrossRef] [PubMed]
- Stauder, R.; Valent, P.; Theurl, I. Anemia at older age: Etiologies, clinical implications, and management. Blood 2018, 131, 505–514. [Google Scholar] [CrossRef] [PubMed]
- Albillos, A.; Lario, M.; Alvarez-Mon, M. Cirrhosis-associated immune dysfunction: Distinctive features and clinical relevance. J. Hepatol. 2014, 61, 1385–1396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finucane, M.M.; Samet, J.H.; Horton, N.J. Translational methods in biostatistics: Linear mixed effect regression models of alcohol consumption and HIV disease progression over time. Epidemiol. Perspect. Innov. 2007, 4, 8. [Google Scholar] [CrossRef] [PubMed]
- McMahon, B.J.; Bruden, D.L.; Petersen, K.M.; Bulkow, L.R.; Parkinson, A.J.; Nainan, O.; Khristova, M.; Zanis, C.; Peters, H.; Margolis, H.S. Antibody levels and protection after hepatitis B vaccination: Results of a 15-year follow-up. Ann. Intern. Med. 2005, 142, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Ruppert, K.; Kuo, S.; DiMartini, A.; Balan, V. In a 12-year study, sustainability of quality of life benefits after liver transplantation varies with pretransplantation diagnosis. Gastroenterology 2010, 139, 1619–1629. [Google Scholar] [CrossRef]
- De Jager, W.; Bourcier, K.; Rijkers, G.T.; Prakken, B.J.; Seyfert-Margolis, V. Prerequisites for cytokine measurements in clinical trials with multiplex immunoassays. BMC Immunol. 2009, 10, 52. [Google Scholar] [CrossRef]
- Manowsky, J.; Camargo, R.G.; Kipp, A.P.; Henkel, J.; Puschel, G.P. Insulin-induced cytokine production in macrophages causes insulin resistance in hepatocytes. Am. J. Physiol. Endocrinol. Metab. 2016, 310, E938–E946. [Google Scholar] [CrossRef]
- Tsai, S.; Clemente-Casares, X.; Zhou, A.C.; Lei, H.; Ahn, J.J.; Chan, Y.T.; Choi, O.; Luck, H.; Woo, M.; Dunn, S.E.; et al. Insulin receptor-mediated stimulation boosts T cell immunity during inflammation and infection. Cell Metab. 2018, 28, 922–934. [Google Scholar] [CrossRef]
- Chevaliez, S.; Hezode, C.; Bahrami, S.; Grare, M.; Pawlotsky, J.M. Long-term hepatitis B surface antigen (HBsAg) kinetics during nucleoside/nucleotide analogue therapy: Finite treatment duration unlikely. J. Hepatol. 2013, 58, 676–683. [Google Scholar] [CrossRef]
- Raimondi, S.; Maisonneuve, P.; Bruno, S.; Mondelli, M.U. Is response to antiviral treatment influenced by hepatitis B virus genotype? J. Hepatol. 2010, 52, 441–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajoriya, N.; Combet, C.; Zoulim, F.; Janssen, H.L.A. How viral genetic variants and genotypes influence disease and treatment outcome of chronic hepatitis B. Time for an individualised approach? J. Hepatol. 2017, 67, 1281–1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuen, M.F.; Wong, D.K.; Sablon, E.; Yuan, H.J.; Sum, S.M.; Hui, C.K.; Chan, A.O.; Wong, B.C.; Lai, C.L. Hepatitis B virus genotypes B and C do not affect the antiviral response to lamivudine. Antivir. Ther. 2003, 8, 531–534. [Google Scholar] [PubMed]
- Ono, A.; Suzuki, F.; Kawamura, Y.; Sezaki, H.; Hosaka, T.; Akuta, N.; Kobayashi, M.; Suzuki, Y.; Saitou, S.; Arase, Y.; et al. Long-term continuous entecavir therapy in nucleos(t)ide-naive chronic hepatitis B patients. J. Hepatol. 2012, 57, 508–514. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.M.; Liaw, Y.F. Hepatitis B surface antigen seroclearance during chronic HBV infection. Antiviral. Ther. 2010, 15, 133–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.C.; Wu, C.L.; Su, W.W.; Shih, K.L.; Tarng, D.C.; Chou, C.T.; Chen, T.Y.; Kor, C.T.; Wu, H.M. Interferon gamma-induced protein 10 is associated with insulin resistance and incident diabetes in patients with nonalcoholic fatty liver disease. Sci. Rep. 2015, 5, 10096. [Google Scholar] [CrossRef]
- Wong, G.L.; Chan, H.L.; Chan, H.Y.; Tse, C.H.; Chim, A.M.; Lo, A.O.; Wong, V.W. Serum interferon-inducible protein 10 levels predict hepatitis B s antigen seroclearance in patients with chronic hepatitis B. Aliment. Pharmacol Ther. 2016, 43, 145–153. [Google Scholar] [CrossRef]
- Lin, C.-F.; Chang, Y.-H.; Chien, S.-C.; Lin, Y.-H.; Yeh, H.-Y. Epidemiology of dyslipidemia in the Asia Pacific region. Int. J. Gerontol. 2018, 12, 2–6. [Google Scholar] [CrossRef]
- Orsi, E.; Grancini, V.; Menini, S.; Aghemo, A.; Pugliese, G. Hepatogenous diabetes: Is it time to separate it from type 2 diabetes? Liver Int. 2017, 37, 950–962. [Google Scholar] [CrossRef]
- Xia, Y.; Protzer, U. Control of hepatitis B virus by cytokines. Viruses 2017, 9, 18. [Google Scholar] [CrossRef]
- Cheng, X.; Xia, Y.; Serti, E.; Block, P.D.; Chung, M.; Chayama, K.; Rehermann, B.; Liang, T.J. Hepatitis B virus evades innate immunity of hepatocytes but activates cytokine production by macrophages. Hepatology 2017, 66, 1779–1793. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Characteristics | Total (n = 140) | Prediabetes or T2DM (n = 39) | Neither of Them (n = 101) | p Value |
|---|---|---|---|---|
| Age (year) | 51.82 ± 11.55 | 57.74 ± 10.55 | 49.53 ± 11.14 | 0.0001 |
| Male, n (%) | 93 (66.4) | 27 (69.2) | 66 (65.3) | 0.66 |
| Treatment time (year) | 6.90 (4.47–9.01) | 7.03 (5.35–9.23) | 6.83 (4.32–8.99) | 0.57 c |
| Cirrhosis, n (%) | 46 (32.9) | 15 (38.5) | 31 (30.7) | 0.38 |
| HCC a, n (%) | 18 (12.9) | 5 (12.8) | 13 (12.9) | 0.99 |
| HBeAg negative, n (%) | 96 (68.6) | 34 (87.2) | 62 (61.4) | 0.003 |
| HBV genotype b: B vs. C, n (%) | 64:57 (52.9:47.1) | 21:11 (65.6:34.4) | 43:46 (48.3:51.7) | 0.09 |
| HBV DNA (log IU/mL) | 5.83 ± 1.74 | 5.57 ± 1.65 | 5.93 ± 1.77 | 0.27 |
| HBsAg (log IU/mL) | 3.18 ± 0.79 | 2.98 ± 0.58 | 3.26 ± 0.85 | 0.032 |
| ALT (× ULN) | 1.68 (1.00–3.57) | 2.04 (1.04–3.77) | 1.56 (0.98–3.37) | 0.36 c |
| Anemia, n (%) | 22 (15.7) | 5 (12.8) | 17 (16.8) | 0.56 |
| CKD stage 2–4: yes vs. no, n (%) | 35:105 (25:75) | 12:27 (30.8:69.2) | 23:78 (22.8:77.2) | 0.33 |
| Dyslipidemia, n (%) | 69 (49.3) | 28 (71.8) | 41 (40.6) | 0.001 |
| Advanced fatty liver, n (%) | 23 (16.4) | 10 (25.6) | 13 (12.9) | 0.07 |
| Model Components | Multivariate Analysis (the General Model) | ||
|---|---|---|---|
| Estimate of Coefficient | Standard Error | p Value | |
| Time (year) | −0.27 | 0.04 | <0.0001 |
| Baseline variables | |||
| Age (year) | −0.00001 | 0.008 | 1.00 |
| Sex (female vs. male a) | −0.02 | 0.15 | 0.87 |
| Cirrhosis (yes vs. no a) | −0.19 | 0.17 | 0.29 |
| HCC (yes vs. no a) | −0.06 | 0.22 | 0.79 |
| HBeAg (positive vs. negative a) | 0.27 | 0.18 | 0.14 |
| HBV genotype (C vs. B a) | 0.37 | 0.13 | 0.007 |
| HBV DNA (log IU/mL) | −0.04 | 0.07 | 0.58 |
| HBsAg (log IU/mL) | 0.55 | 0.12 | <0.0001 |
| ALT (× ULN): ≥2 vs. <2 a | −0.28 | 0.16 | 0.07 |
| Anemia (yes vs. no a) | −0.31 | 0.19 | 0.10 |
| CKD stage 2-4 (yes vs. no a) | −0.15 | 0.15 | 0.30 |
| Prediabetes or T2DM (yes vs. no a) | 0.01 | 0.18 | 0.94 |
| Dyslipidemia (yes vs. no a) | −0.14 | 0.14 | 0.32 |
| Advanced fatty liver (yes vs. no a) | 0.18 | 0.23 | 0.44 |
| Interaction terms | |||
| Time × Age (year) | 0.003 | 0.001 | 0.0001 |
| Time × Prediabetes or T2DM (yes vs. no a) | 0.08 | 0.02 | <0.0001 |
| Time × Advanced fatty liver (yes vs. no a) | −0.07 | 0.02 | 0.001 |
| Intercept | 1.66 | 0.61 | 0.007 |
| Baseline | 1st Year | 3rd Year | 6th Year | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Neither of Them (n = 18) | Prediabetes or T2DM (n = 4) | p Value | Neither of Them (n = 26) | Prediabetes or T2DM (n = 10) | p Value | Neither of Them (n = 40) | Prediabetes or T2DM (n = 17) | p Value | Neither of Them (n = 42) | Prediabetes or T2DM (n = 21) | p Value | |
| IP-10 | 189.85 (134.15–249.17) | 148.38 (123.95–177.69) | 0.17 | 152.97 (124.88–206.46) | 162.11 (119.00–210.40) | 0.83 | 136.54 (111.99–179.11) | 143.72 (130.23–199.41) | 0.30 | 134.61 (94.30–176.84) | 174.26 (156.12–200.77) | 0.013 |
| IFN-γ | 16.32 (8.41–23.57) | 13.41 (11.87–16.08) | 0.67 | 11.05 (4.64–21.45) | 13.11 (5.74–21.64) | 0.92 | 13.31 (5.58–18.81) | 8.02 (2.61–13.25) | 0.11 | 7.86 (3.01–13.55) | 4.82 (3.42–10.27) | 0.36 |
| TGF-β1 | 5295.80 (4245.30–5832.37) | 5713.57 (4874.61–7310.43) | 0.44 | 5836.38 (4534.38–7119.89) | 5461.05 (4605.60–7601.74) | 0.89 | 6172.21 (4524.55–7104.49) | 5648.16 (4180.75–6996.47) | 0.57 | 5704.12 (4553.30–7030.16) | 5297.83 (4079.52–6514.09) | 0.19 |
| IL-1α | 0.00 (0.00–0.25) | 0.00 (0.00–0.40) | 0.57 | 0.00 (0.00–0.44) | 0.00 (0.00–0.95) | 0.56 | 0.00 (0.00–0.00) | 0.00 (0.00–0.00) | 0.61 | 0.00 (0.00–0.00) | 0.00 (0.00–0.05) | 0.26 |
| IL-4 | 0.54 (0.34–0.66) | 0.33 (0.13–0.81) | 0.35 | 0.24 (0.00–0.43) | 0.23 (0.00–0.63) | 0.80 | 0.00 (0.00–0.45) | 0.00 (0.00–0.31) | 0.70 | 0.00 (0.00–0.36) | 0.00 (0.00–0.18) | 0.30 |
| IL-6 | 1.98 (0.74–3.79) | 1.01 (0.16–1.43) | 0.15 | 1.56 (0.77–3.83) | 1.76 (1.35–3.20) | 0.72 | 1.44 (0.88–2.42) | 2.13 (1.21–3.90) | 0.19 | 1.55 (0.70–2.61) | 1.73 (1.29–3.49) | 0.13 |
| IL-10 | 0.78 (0.00–1.81) | 0.45 (0.00–1.48) | 0.54 | 0.00 (0.00–0.65) | 0.58 (0.09–1.14) | 0.08 | 0.57 (0.00–1.33) | 0.63 (0.00–1.59) | 0.87 | 0.77 (0.00–1.24) | 1.03 (0.24–1.62) | 0.12 |
| IL-12p70 | 0.00 (0.00–0.23) | 0.00 (0.00–1.01) | 0.96 | 0.00 (0.00–0.21) | 0.00 (0.00–0.51) | 0.39 | 0.00 (0.00–0.53) | 0.00 (0.00–0.25) | 0.38 | 0.00 (0.00–0.61) | 0.00 (0.00–0.21) | 0.53 |
| IL-17A | 0.83 (0.00–1.66) | 1.44 (0.39–3.09) | 0.30 | 0.38 (0.00–0.76) | 0.37 (0.19–1.13) | 0.42 | 0.00 (0.00–0.71) | 0.00 (0.00–0.95) | 0.75 | 0.00 (0.00–0.67) | 0.00 (0.00–0.58) | 0.72 |
| IL-21 | 0.00 (0.00–0.00) | 0.00 (0.00–6.03) | 0.75 | 0.00 (0.00–0.00) | 0.00 (0.00–2.39) | 0.56 | 0.00 (0.00–0.00) | 0.00 (0.00–0.00) | 0.33 | 0.00 (0.00–0.00) | 0.00 (0.00–19.13) | 0.23 |
| Variable | Neither of Them (n = 95) | Prediabetes (n = 12) | T2DM (n = 27) | p Value | Post hoc Analysis |
|---|---|---|---|---|---|
| FIB-4 | 0.25 (0.16–0.40) | 0.28 (0.20–0.62) | 0.45 (0.26–0.76) | 0.007 b | T2DM > Neither of them b |
| Age (year) | 49.72 ± 11.33 | 54.61 ± 10.19 | 59.14 ± 10.60 | 0.001 ° | T2DM > Neither of them c |
| Male, n (%) | 62 (65.3) | 9 (75.0) | 18 (66.7) | 0.87 d | − |
| Treatment time (year) | 6.74 (4.24–8.88) | 6.18 (3.65–8.37) | 7.47 (5.80–9.35) | 0.18 b | − |
| AST (× ULN) | 1.16 (0.80–1.80) | 1.00 (0.82–2.64) | 1.57 (0.96–2.57) | 0.35 b | − |
| ALT (× ULN) | 1.56 (0.96–3.34) | 1.33 (1.01–3.52) | 2.34 (1.04–3.92) | 0.38 b | − |
| Platelet (109/L) | 195.17 ± 66.89 | 165.00 ± 35.06 | 156.67 ± 58.67 | 0.012 c | Neither of them > T2DM c |
| Cirrhosis, n (%) | 31 (32.6) | 3 (25.0) | 12 (44.4) | 0.42 d | − |
| HCC, n (%) | 13 (13.7) | 2 (16.7) | 3 (11.1) | 0.84 d | − |
| HBeAg negative, n (%) | 58 (61.1) | 9 (75.0) | 25 (92.6) | 0.007 d | T2DM > Neither of them d |
| HBV genotype a: B vs. C, n (%) | 38:46 (45.2:54.8) | 3:7 (30.0:70.0) | 18:4 (81.8:18.2) | 0.003 d | T2DM > Neither of them; T2DM > Prediabetes d (% of genotype B) |
| HBV DNA (log IU/mL) | 5.93 ± 1.76 | 6.24 ± 1.50 | 5.27 ± 1.65 | 0.15 c | − |
| HBsAg (log IU/mL) | 3.28 ± 0.82 | 3.18 ± 0.47 | 2.90 ± 0.61 | 0.07 c | − |
| Anemia, n (%) | 17 (17.9) | 1 (8.3) | 4 (14.8) | 0.86 d | − |
| CKD stage 2-4: yes vs. no, n (%) | 20 (21.1) | 2 (16.7) | 10 (37.0) | 0.21 d | − |
| Dyslipidemia, n (%) | 40 (42.1) | 7 (58.3) | 21 (77.8) | 0.004 d | T2DM > Neither of them d |
| Advanced fatty liver, n (%) | 12 (12.6) | 0 (0.0) | 10 (37.0) | 0.004 d | T2DM > Neither of them d; T2DM > Prediabetes d |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, T.-C.; Liu, W.-C.; Hsu, Y.-H.; Lin, J.-J.; Chiu, Y.-C.; Chiu, H.-C.; Cheng, P.-N.; Chen, C.-Y.; Chang, T.-T.; Wu, I.-C. Insulin Resistance Associated Disorders Pivoting Long-Term Hepatitis B Surface Antigen Decline During Entecavir Therapy. J. Clin. Med. 2019, 8, 1892. https://doi.org/10.3390/jcm8111892
Lin T-C, Liu W-C, Hsu Y-H, Lin J-J, Chiu Y-C, Chiu H-C, Cheng P-N, Chen C-Y, Chang T-T, Wu I-C. Insulin Resistance Associated Disorders Pivoting Long-Term Hepatitis B Surface Antigen Decline During Entecavir Therapy. Journal of Clinical Medicine. 2019; 8(11):1892. https://doi.org/10.3390/jcm8111892
Chicago/Turabian StyleLin, Tien-Ching, Wen-Chun Liu, Yu-Hsiang Hsu, Jia-Jhen Lin, Yen-Cheng Chiu, Hung-Chih Chiu, Pin-Nan Cheng, Chiung-Yu Chen, Ting-Tsung Chang, and I-Chin Wu. 2019. "Insulin Resistance Associated Disorders Pivoting Long-Term Hepatitis B Surface Antigen Decline During Entecavir Therapy" Journal of Clinical Medicine 8, no. 11: 1892. https://doi.org/10.3390/jcm8111892