
Pharmacological Modulation by Low Molecular Weight Heparin of Purinergic Signaling in Cardiac Cells Prevents Arrhythmia and Lethality Induced by Myocardial Infarction
 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
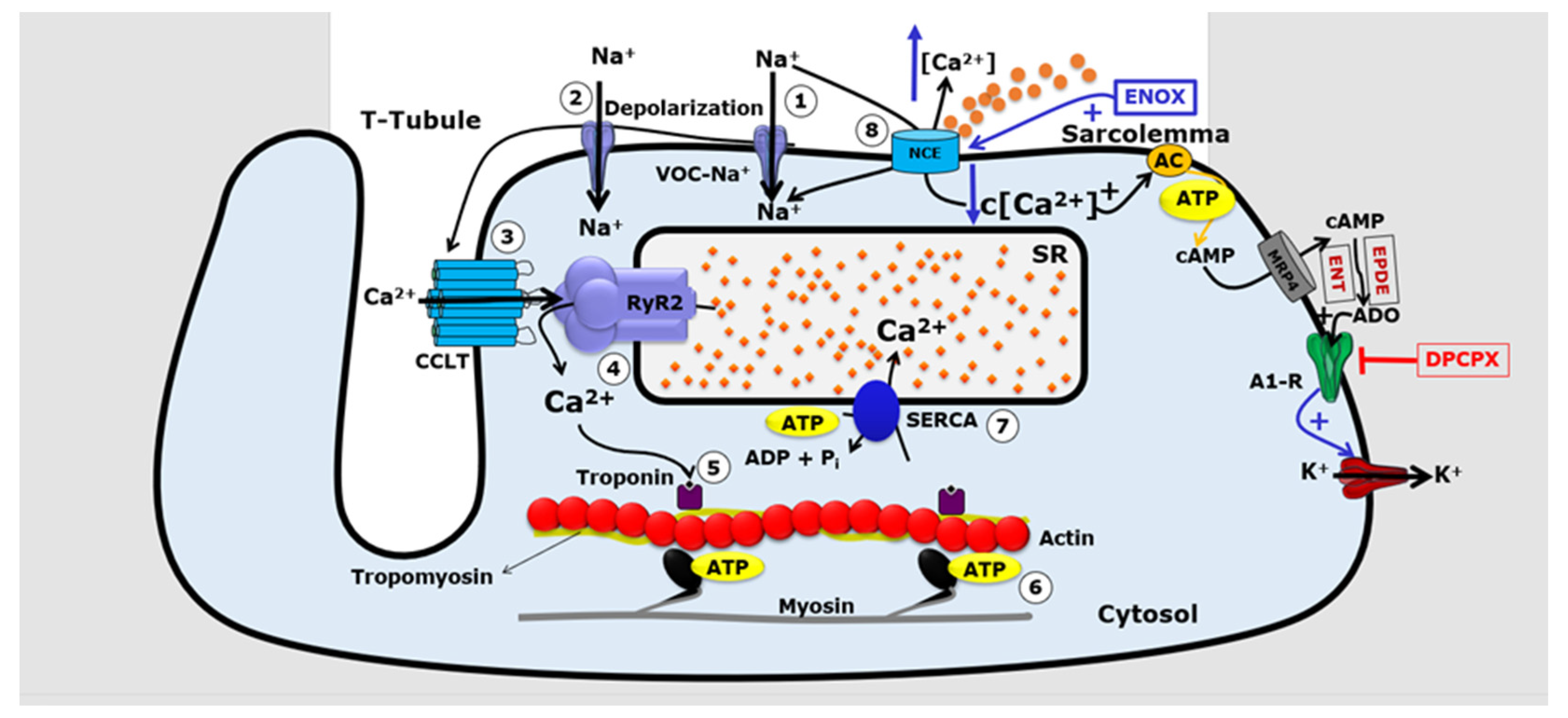{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
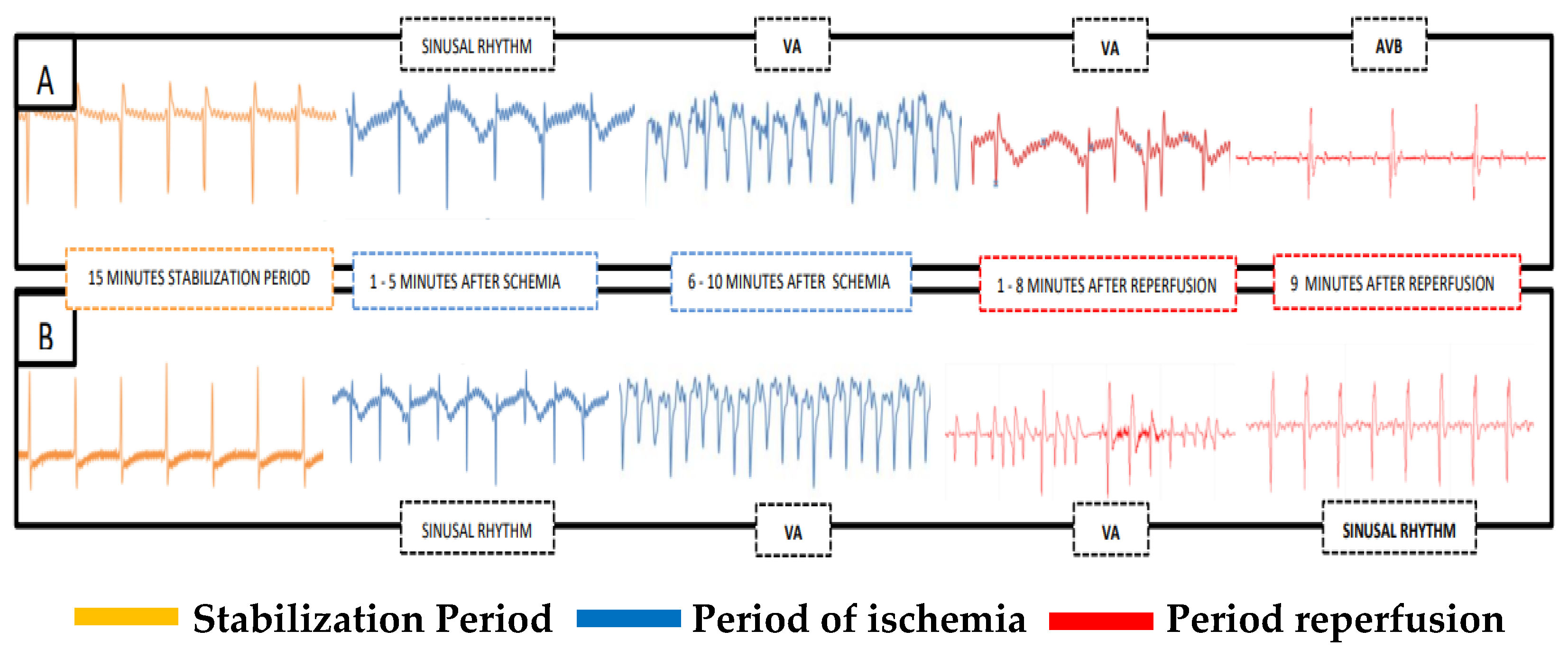
2.2. Induction of Cardiac Ischemia and Reperfusion (CIR)
2.3. Evaluation of Cardiac Activity during CIR
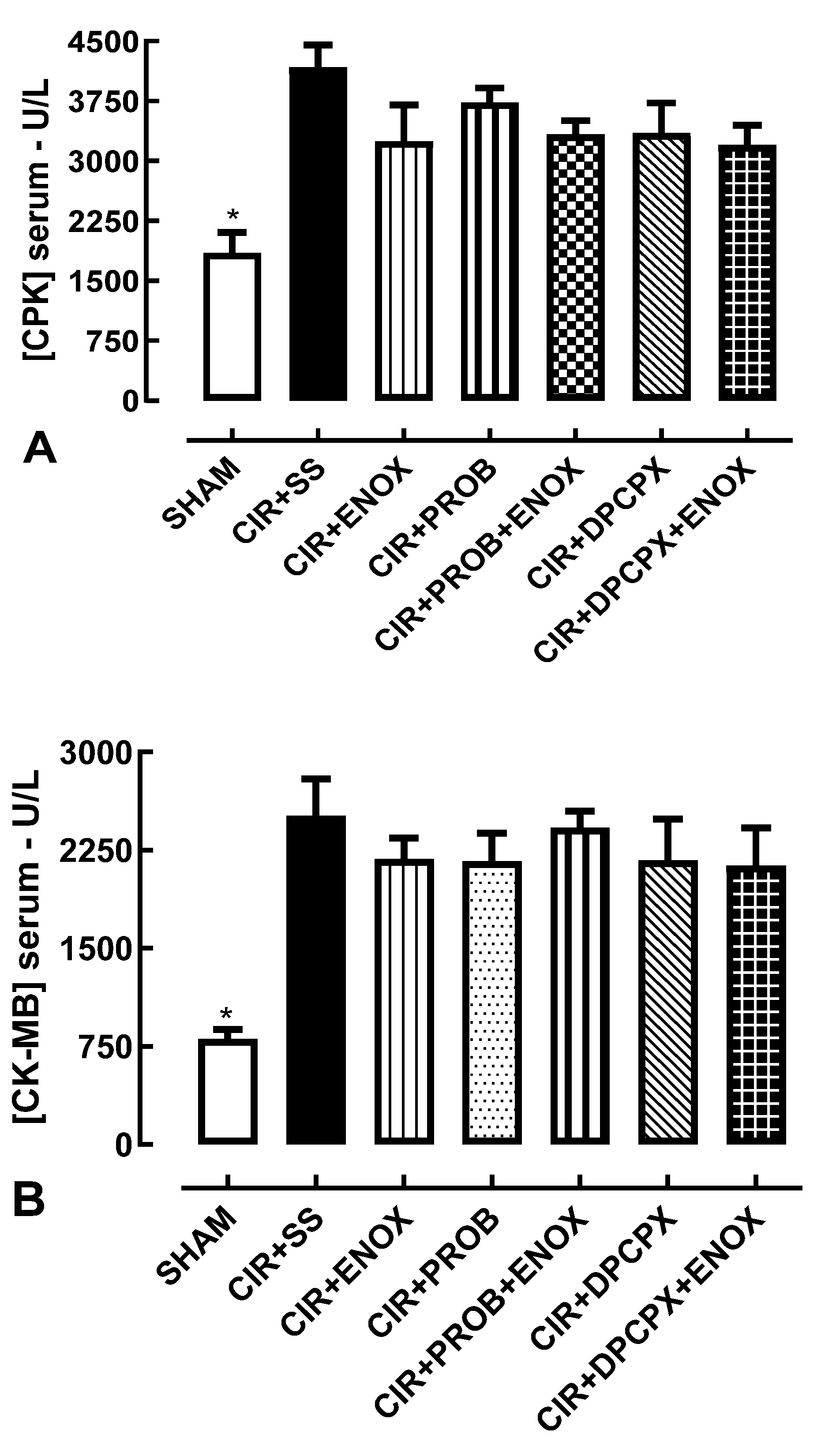
2.4. Biochemical Evaluation of Biomarkers of Cardiac Lesions
2.5. Pharmacological Treatments
- (1)
- SHAM-operated: Rats were submitted to the previously described procedure; however, the nylon thread was only passed under the left coronary artery and the coronary artery was not blockaded; therefore, neither ischemia nor reperfusion was induced (n = 10);
- (2)
- CIR + SS: control rats that received the 0.9% SS vehicle (IV) immediately before cardiac ischemia (n = 24);
- (3)
- CIR + ENOX: rats treated with ENOX (1 mg/kg, IV) immediately before cardiac ischemia (n = 12);
- (4)
- CIR + PROB: rats treated with PROB (100 mg/kg, IV) five minutes before cardiac ischemia (n = 12);
- (5)
- CIR + DPCPX: rats treated with DPCPX (100 µg/kg, IV) five minutes before cardiac ischemia (n = 12);
- (6)
- CIR + ENOX + PROB: rats treated with PROB (100 mg/kg, IV) five minutes before cardiac ischemia, and treated with ENOX (1 mg/kg, IV) immediately before cardiac ischemia (n = 12);
- (7)
- CIR + ENOX + DPCPX: rats treated with DPCPX (100 µg/kg, IV) five minutes before cardiac ischemia, and treated with ENOX (1 mg/kg, IV) immediately before cardiac ischemia (n = 12).
2.6. Statistical Analysis
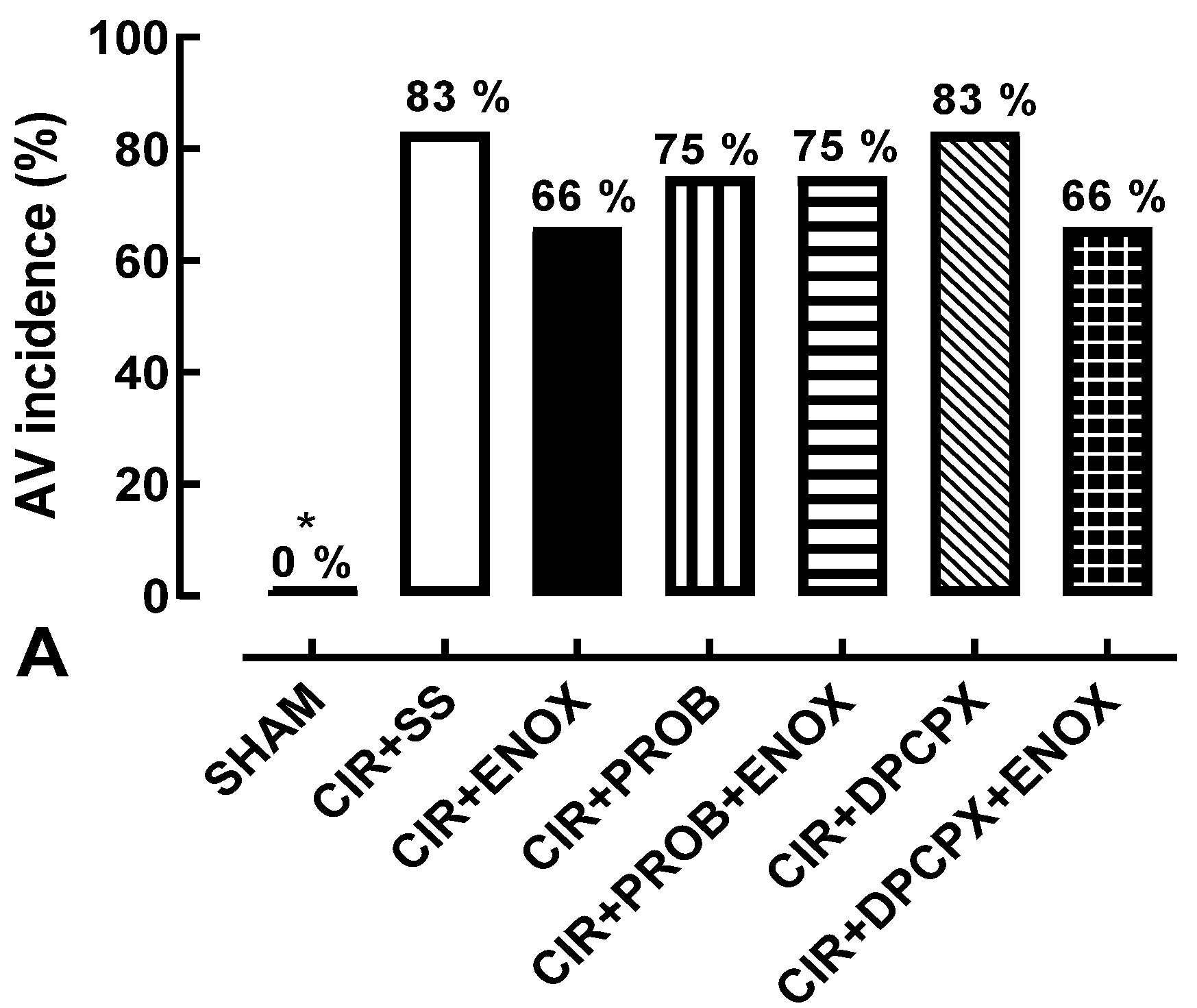
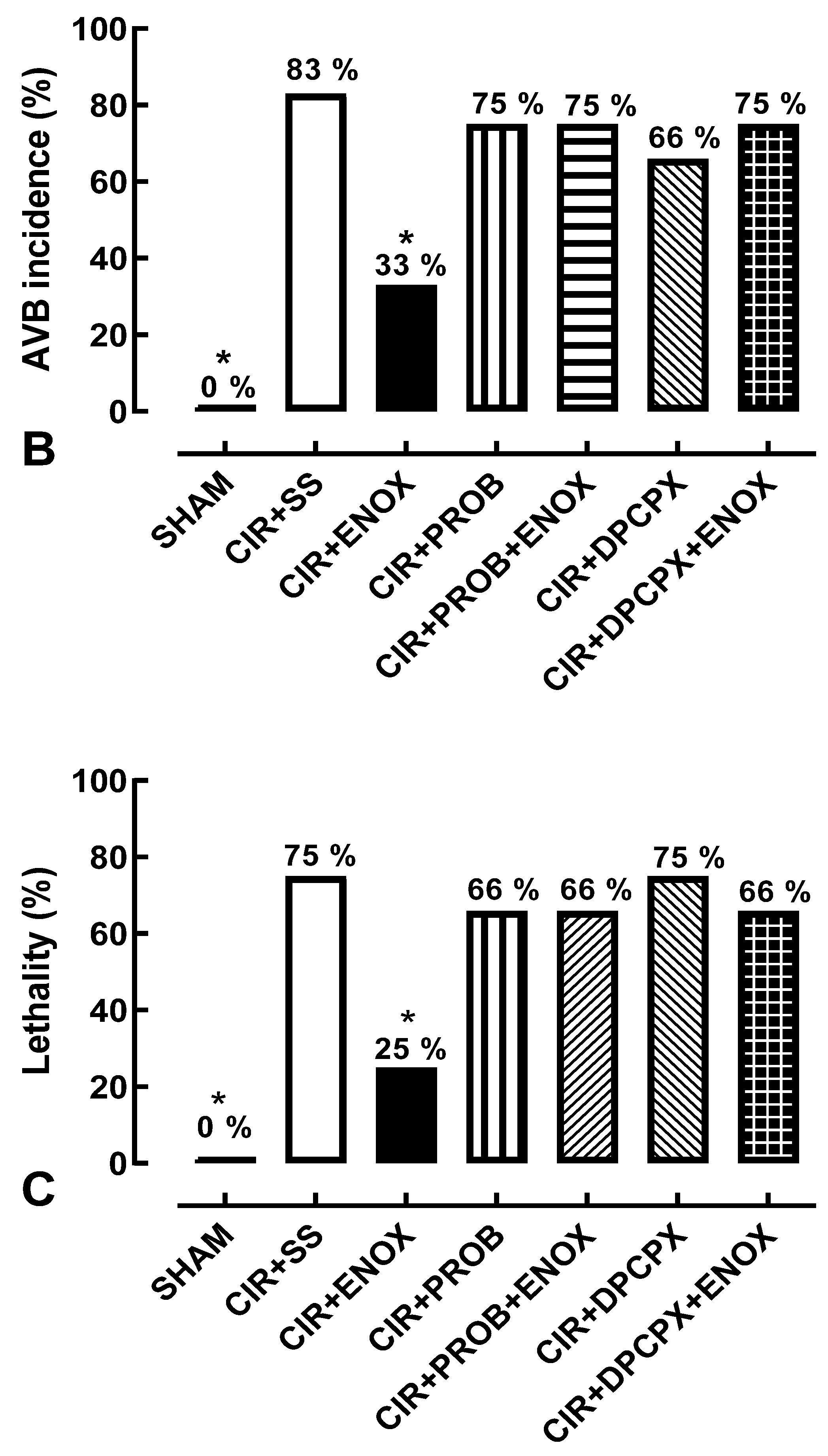
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Joseph, P.; Leong, D.; McKee, M.; Anand, S.S.; Schwalm, J.-D.; Teo, K.; Mente, A.; Yusuf, S. Reducing the Global Burden of Cardiovascular Disease, Part 1: The Epidemiology and Risk Factors. Circ. Res. 2017, 121, 677–694. [Google Scholar] [CrossRef] [PubMed]
- Stary, H.C.; Chandler, A.B.; Dinsmore, R.E.; Fuster, V.; Glagov, S.; Insull, W.; Rosenfeld, M.E.; Schwartz, C.J.; Wagner, W.D.; Wissler, R.W. A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Circulation 1995, 92, 1355–1374. [Google Scholar] [CrossRef] [PubMed]
- Tosaki, A. ArrhythmoGenoPharmacoTherapy. Front. Pharmacol. 2020, 11, 616. [Google Scholar] [CrossRef] [PubMed]
- Menon, V.; Harrington, R.A.; Hochman, J.S.; Cannon, C.P.; Goodman, S.D.; Wilcox, R.G.; Schünemann, H.J.; Ohman, E.M. Thrombolysis and adjunctive therapy in acute myocardial infarction: The Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest 2004, 126, 549S–575S. [Google Scholar] [CrossRef] [Green Version]
- Chin, K.Y.; Qin, C.; May, L.; Ritchie, R.H.; Woodman, O.L. New Pharmacological Approaches to the Prevention of Myocardial Ischemia- Reperfusion Injury. Curr. Drug Targets 2017, 18, 1689–1711. [Google Scholar] [CrossRef]
- Dhalla, N.S.; Shah, A.K.; Adameova, A.; Bartekova, M. Role of Oxidative Stress in Cardiac Dysfunction and Subcellular Defects Due to Ischemia-Reperfusion Injury. Biomedicines 2022, 10, 1473. [Google Scholar] [CrossRef]
- Lekli, I.; Haines, D.D.; Balla, G.; Tosaki, A. Autophagy: An adaptive physiological countermeasure to cellular senescence and ischaemia/reperfusion-associated cardiac arrhythmias. J. Cell. Mol. Med. 2017, 21, 1058–1072. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Zhong, J.; Wang, D.; Xu, J.; Su, H.; An, C.; Zhu, H.; Yan, J. Increasing glutamate promotes ischemia-reperfusion-induced ventricular arrhythmias in rats in vivo. Pharmacology 2014, 93, 4–9. [Google Scholar] [CrossRef]
- Said, M.; Becerra, R.; Valverde, C.A.; Kaetzel, M.A.; Dedman, J.R.; Mundiña-Weilenmann, C.; Wehrens, X.H.; Vittone, L.; Mattiazzi, A. Calcium-calmodulin dependent protein kinase II (CaMKII): A main signal responsible for early reperfusion arrhythmias. J. Mol. Cell. Cardiol. 2011, 51, 936–944. [Google Scholar] [CrossRef] [Green Version]
- Becerra, R.; Román, B.; Di Carlo, M.N.; Mariangelo, J.I.; Salas, M.; Sanchez, G.; Donoso, P.; Schinella, G.R.; Vittone, L.; Wehrens, X.H.; et al. Reversible redox modifications of ryanodine receptor ameliorate ventricular arrhythmias in the ischemic-reperfused heart. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, H713–H724. [Google Scholar] [CrossRef] [Green Version]
- Ashpole, N.M.; Herren, A.W.; Ginsburg, K.S.; Brogan, J.D.; Johnson, D.E.; Cummins, T.R.; Bers, D.M.; Hudmon, A. Ca2+/calmodulin-dependent protein kinase II (CaMKII) regulates cardiac sodium channel NaV1.5 gating by multiple phosphorylation sites. J. Biol. Chem. 2012, 287, 19856–19869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, S.; Li, G.; Huang, C.L.-H.; Lei, M.; Wu, L. Late sodium current associated cardiac electrophysiological and mechanical dysfunction. Pflugers Arch. 2018, 470, 461–469. [Google Scholar] [CrossRef]
- Makielski, J.C. Late sodium current: A mechanism for angina, heart failure, and arrhythmia. J. Cardiovasc. Pharmacol. 2009, 54, 279–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menezes-Rodrigues, F.S.; Errante, P.R.; Araújo, E.A.; Fernandes, M.P.P.; Silva, M.M.D.; Pires-Oliveira, M.; Scorza, C.A.; Scorza, F.A.; Taha, M.O.; Caricati-Neto, A. Cardioprotection stimulated by resveratrol and grape products prevents lethal cardiac arrhythmias in an animal model of ischemia and reperfusion. Acta Cir. Bras. 2021, 36, e360306. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Luo, D.; Liao, X.; He, J.; Liu, C.; Yang, C.; Ma, H. Ang-(1-7) offers cytoprotection against ischemia-reperfusion injury by restoring intracellular calcium homeostasis. J. Cardiovasc. Pharmacol. 2014, 63, 259–264. [Google Scholar] [CrossRef]
- Yang, J.; Yin, H.-S.; Cao, Y.-J.; Jiang, Z.-A.; Li, Y.-J.; Song, M.-C.; Wang, Y.-F.; Wang, Z.-H.; Yang, R.; Jiang, Y.-F.; et al. Arctigenin Attenuates Ischemia/Reperfusion Induced Ventricular Arrhythmias by Decreasing Oxidative Stress in Rats. Cell. Physiol. Biochem. 2018, 49, 728–742. [Google Scholar] [CrossRef]
- Caricati-Neto, A.; Errante, P.R.; Menezes-Rodrigues, F.S. Recent Advances in Pharmacological and Non-Pharmacological Strategies of Cardioprotection. Int. J. Mol. Sci. 2019, 20, 4002. [Google Scholar] [CrossRef] [Green Version]
- De Godoy, C.M.G.; Vasques, Ê.R.; Caricati-Neto, A.; Tavares, J.G.P.P.; Alves, B.J.; Duarte, J.; Miranda-Ferreira, R.; Lima, M.A.; Nader, H.B.; Tersariol, I.L.D.S. Heparin Oligosaccharides Have Antiarrhythmic Effect by Accelerating the Sodium-Calcium Exchanger. Front. Cardiovasc. Med. 2018, 5, 67. [Google Scholar] [CrossRef] [Green Version]
- Guarini, S.; Martini, M.C.; Bertolini, A. Reperfusion-induced arrhythmias and lethality are reduced by a 2KDa heparin fragment. Life Sci. 1995, 57, 967–972. [Google Scholar] [CrossRef]
- Menezes-Rodrigues, F.S.; Padrão Tavares, J.G.; Pires de Oliveira, M.; Guzella de Carvalho, R.; Ruggero Errante, P.; Omar Taha, M.; José Fagundes, D.; Caricati-Neto, A. Anticoagulant and antiarrhythmic effects of heparin in the treatment of COVID-19 patients. J. Thromb. Haemost. 2020, 18, 2073–2075. [Google Scholar] [CrossRef]
- Burnstock, G.; Pelleg, A. Cardiac purinergic signalling in health and disease. Purinergic Signal. 2015, 11, 1–46. [Google Scholar] [CrossRef] [PubMed]
- Menezes-Rodrigues, F.S.; Tavares, J.G.P.; Vasques, E.R.; Errante, P.R.; Araújo, E.A.D.; Pires-Oliveira, M.; Scorza, C.A.; Scorza, F.A.; Taha, M.O.; Caricati-Neto, A. Cardioprotective effects of pharmacological blockade of the mitochondrial calcium uniporter on myocardial ischemia-reperfusion injury. Acta Cir. Bras. 2020, 35, e202000306. [Google Scholar] [CrossRef] [PubMed]
- Menezes-Rodrigues, F.S.; Errante, P.R.; Ferreira, R.M.; Tavares, J.G.P.; de Paula, L.; Araújo, E.A.; Govato, T.C.P.; Tikazawa, E.H.; Reis, M.D.C.M.; Luna-Filho, B.; et al. Cardioprotective effect of lipstatin derivative orlistat on normotensive rats submitted to cardiac ischemia and reperfusion. Acta Cir. Bras. 2018, 33, 524–532. [Google Scholar] [CrossRef] [PubMed]
- Tavares, J.G.P.; Errante, P.R.; Govato, T.C.P.; Vasques, Ê.R.; Ferraz, R.R.N.; Taha, M.O.; Menezes-Rodrigues, F.S.; Caricati-Neto, A. Cardioprotective effect of preconditioning is more efficient than postconditioning in rats submitted to cardiac ischemia and reperfusion1. Acta Cir. Bras. 2018, 33, 588–596. [Google Scholar] [CrossRef] [Green Version]
- Menezes-Rodrigues, F.S.; Errante, P.R.; Tavares, J.G.P.; Ferraz, R.R.N.; Gomes, W.J.; Taha, M.O.; Scorza, C.A.; Scorza, F.A.; Caricati-Neto, A. Pharmacological modulation of b-adrenoceptors as a new cardioprotective strategy for therapy of myocardial dysfunction induced by ischemia and reperfusion. Acta Cir. Bras. 2019, 34, e201900505. [Google Scholar] [CrossRef]
- Tavares, J.G.P.; Vasques, E.R.; Arida, R.M.; Cavalheiro, E.A.; Cabral, F.R.; Torres, L.B.; Menezes-Rodrigues, F.S.; Jurkiewicz, A.; Caricati-Neto, A.; Godoy, C.M.G.; et al. Epilepsy-induced electrocardiographic alterations following cardiac ischemia and reperfusion in rats. Brazilian J. Med. Biol. Res. = Rev. Bras. Pesqui. medicas e Biol. 2015, 48, 140–145. [Google Scholar] [CrossRef] [Green Version]
- Coates, J.; Sheehan, M.J.; Strong, P. 1,3-Dipropyl-8-cyclopentyl xanthine (DPCPX): A useful tool for pharmacologists and physiologists? Gen. Pharmacol. 1994, 25, 387–394. [Google Scholar] [CrossRef]
- Lohse, M.J.; Klotz, K.N.; Lindenborn-Fotinos, J.; Reddington, M.; Schwabe, U.; Olsson, R.A. 8-Cyclopentyl-1,3-dipropylxanthine (DPCPX)--a selective high affinity antagonist radioligand for A1 adenosine receptors. Naunyn. Schmiedebergs. Arch. Pharmacol. 1987, 336, 204–210. [Google Scholar] [CrossRef]
- Yusuf, S.; Mehta, S.R.; Xie, C.; Ahmed, R.J.; Xavier, D.; Pais, P.; Zhu, J.; Liu, L. CREATE Trial Group Investigators Effects of reviparin, a low-molecular-weight heparin, on mortality, reinfarction, and strokes in patients with acute myocardial infarction presenting with ST-segment elevation. JAMA 2005, 293, 427–435. [Google Scholar]
- Harrington, J.; Petrie, M.C.; Anker, S.D.; Bhatt, D.L.; Jones, W.S.; Udell, J.A.; Hernandez, A.F.; Butler, J. Evaluating the Application of Chronic Heart Failure Therapies and Developing Treatments in Individuals with Recent Myocardial Infarction: A Review. JAMA Cardiol. 2022, 7, 1067–1075. [Google Scholar] [CrossRef]
- Beurskens, D.M.H.; Huckriede, J.P.; Schrijver, R.; Hemker, H.C.; Reutelingsperger, C.P.; Nicolaes, G.A.F. The Anticoagulant and Nonanticoagulant Properties of Heparin. Thromb. Haemost. 2020, 120, 1371–1383. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Takahashi, K.; Onishi, M.; Suzuki, T.; Tanaka, Y.; Ota, T.; Yoshida, S.; Nakaike, S.; Matsuda, T.; Baba, A. Effects of SEA0400, a novel inhibitor of the Na+/Ca2+ exchanger, on myocardial stunning in anesthetized dogs. Eur. J. Pharmacol. 2004, 505, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Dhalla, N.S.; Hryshko, L.V. Therapeutic potential of novel Na+-Ca2+ exchange inhibitors in attenuating ischemia-reperfusion injury. Can. J. Cardiol. 2005, 21, 509–516. [Google Scholar] [PubMed]
- Brieger, D.; Collet, J.-P.; Silvain, J.; Landivier, A.; Barthélémy, O.; Beygui, F.; Bellemain-Appaix, A.; Mercadier, A.; Choussat, R.; Vignolles, N.; et al. Heparin or enoxaparin anticoagulation for primary percutaneous coronary intervention. Catheter. Cardiovasc. Interv. 2011, 77, 182–190. [Google Scholar] [CrossRef] [PubMed]
- Iscan, S.; Eygi, B.; Besir, Y.; Yurekli, I.; Cakir, H.; Yilik, L.; Gokalp, O.; Gurbuz, A. Inflammation, Atrial Fibrillation and Cardiac Surgery: Current Medical and Invasive Approaches for the Treatment of Atrial Fibrillation. Curr. Pharm. Des. 2018, 24, 310–322. [Google Scholar] [CrossRef]
- Antoons, G.; Sipido, K.R. Targeting calcium handling in arrhythmias. Europace 2008, 10, 1364–1369. [Google Scholar] [CrossRef]
- Barry, W.H.; Zhang, X.Q.; Halkos, M.E.; Vinten-Johansen, J.; Saegusa, N.; Spitzer, K.W.; Matsuoka, N.; Sheets, M.; Rao, N.V.; Kennedy, T.P. Nonanticoagulant heparin reduces myocyte Na+ and Ca2+ loading during simulated ischemia and decreases reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H102–H111. [Google Scholar] [CrossRef] [Green Version]
- Knaus, H.G.; Moshammer, T.; Friedrich, K.; Kang, H.C.; Haugland, R.P.; Glossman, H. In vivo labeling of L-type Ca2+ channels by fluorescent dihydropyridines: Evidence for a functional, extracellular heparin-binding site. Proc. Natl. Acad. Sci. USA 1992, 89, 3586–3590. [Google Scholar] [CrossRef] [Green Version]
- Bers, D.M. Calcium cycling and signaling in cardiac myocytes. Annu. Rev. Physiol. 2008, 70, 23–49. [Google Scholar] [CrossRef] [Green Version]
- Hoppensteadt, D.A.; Gray, A.; Jeske, W.P.; Walenga, J.M.; Fareed, J. Anticoagulant and Antithrombotic Actions of AVE5026, an Enriched Anti-Xa Hemisynthetic Ultra-Low-Molecular-Weight Heparin. Clin. Appl. Thromb. Hemost. 2014, 20, 621–628. [Google Scholar] [CrossRef]
- Ander, B.P.; Hurtado, C.; Raposo, C.S.; Maddaford, T.G.; Deniset, J.F.; Hryshko, L.V.; Pierce, G.N.; Lukas, A. Differential sensitivities of the NCX1.1 and NCX1.3 isoforms of the Na+-Ca2+ exchanger to alpha-linolenic acid. Cardiovasc. Res. 2007, 73, 395–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurtado, C.; Prociuk, M.; Maddaford, T.G.; Dibrov, E.; Mesaeli, N.; Hryshko, L.V.; Pierce, G.N. Cells expressing unique Na+/Ca2+ exchange (NCX1) splice variants exhibit different susceptibilities to Ca2+ overload. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H2155–H2162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seki, S.; Taniguchi, M.; Takeda, H.; Nagai, M.; Taniguchi, I.; Mochizuki, S. Inhibition by KB-r7943 of the reverse mode of the Na+/Ca2+ exchanger reduces Ca2+ overload in ischemic-reperfused rat hearts. Circ. J. 2002, 66, 390–396. [Google Scholar] [CrossRef] [Green Version]
- Komuro, I.; Ohtsuka, M. Forefront of Na+/Ca2+ exchanger studies: Role of Na+/Ca2+ exchanger--lessons from knockout mice. J. Pharmacol. Sci. 2004, 96, 23–26. [Google Scholar] [CrossRef] [Green Version]
- Guinamard, R.; Bouvagnet, P.; Hof, T.; Liu, H.; Simard, C.; Sallé, L. TRPM4 in cardiac electrical activity. Cardiovasc. Res. 2015, 108, 21–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menezes-Rodrigues, F.S.; Padrao Tavares, J.G.; Errante, P.R.; Vasques, Ê.R.; Maia Reis, M.D.C.; Luna Filho, B.; Scorza, F.A.; Caricati-Neto, A.; Bergantin, L.B. Role of the Extracellular Ca2+/cyclic AMP- Adenosine Signaling Pathways in Cardioprotection. J. Thromb. Circ. 2017, 3, 1000e106. [Google Scholar]
- Sassi, Y.; Ahles, A.; Truong, D.J.J.; Baqi, Y.; Lee, S.Y.; Husse, B.; Hulot, J.S.; Foinquinos, A.; Thum, T.; Müller, C.E.; et al. Cardiac myocyte-secreted cAMP exerts paracrine action via adenosine receptor activation. J. Clin. Invest. 2014, 124, 5385–5397. [Google Scholar] [CrossRef] [Green Version]
- Sellers, Z.M.; Naren, A.P.; Xiang, Y.; Best, P.M. MRP4 and CFTR in the regulation of cAMP and β-adrenergic contraction in cardiac myocytes. Eur. J. Pharmacol. 2012, 681, 80–87. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, J.Q.D.; da Silva, E.D.; de Magalhães Galvão, K.; Miranda-Ferreira, R.; Caricati-Neto, A.; Jurkiewicz, N.H.; Garcia, A.G.; Jurkiewicz, A. Differential regulation of atrial contraction by P1 and P2 purinoceptors in normotensive and spontaneously hypertensive rats. Hypertens. Res. 2014, 37, 210–219. [Google Scholar] [CrossRef]
- Camara, H.; Rodrigues, J.Q.D.; Alves, G.A.; da Silva Junior, E.D.; Caricati-Neto, A.; Garcia, A.G.; Jurkiewicz, A. Would calcium or potassium channels be responsible for cardiac arrest produced by adenosine and ATP in the right atria of Wistar rats? Eur. J. Pharmacol. 2015, 768, 199–206. [Google Scholar] [CrossRef] [Green Version]
- Halestrap, A.P.; Clarke, S.J.; Khaliulin, I. The role of mitochondria in protection of the heart by preconditioning. Biochim. Biophys. Acta 2007, 1767, 1007–1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasques, E.R.; Vasques, E.R.; Tersariol, I.L.; Nader, H.B.; Godoy, C.M. Enoxaparin Reverses Ventricular Tachycardia and Torsades de Pointes in Rat Isolated Heart Model. Circ. Res. 2019, 125, A820. [Google Scholar] [CrossRef]
- Yu, H.J.; Ma, H.; Green, R.D. Calcium entry via L-type calcium channels acts as a negative regulator of adenylyl cyclase activity and cyclic AMP levels in cardiac myocytes. Mol. Pharmacol. 1993, 44, 689–693. [Google Scholar]
- Ukena, D.; Schudt, C.; Sybrecht, G.W. Adenosine receptor-blocking xanthines as inhibitors of phosphodiesterase isozymes. Biochem. Pharmacol. 1993, 45, 847–851. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Filho, C.E.B.; Barbosa, A.H.P.; Nicolau, L.A.D.; Medeiros, J.V.R.; Pires-Oliveira, M.; dos Santos Póvoa, R.M.; Govato, T.C.P.; Júnior, H.J.F.; de Carvalho, R.G.; Luna-Filho, B.; et al. Pharmacological Modulation by Low Molecular Weight Heparin of Purinergic Signaling in Cardiac Cells Prevents Arrhythmia and Lethality Induced by Myocardial Infarction. J. Cardiovasc. Dev. Dis. 2023, 10, 103. https://doi.org/10.3390/jcdd10030103
Filho CEB, Barbosa AHP, Nicolau LAD, Medeiros JVR, Pires-Oliveira M, dos Santos Póvoa RM, Govato TCP, Júnior HJF, de Carvalho RG, Luna-Filho B, et al. Pharmacological Modulation by Low Molecular Weight Heparin of Purinergic Signaling in Cardiac Cells Prevents Arrhythmia and Lethality Induced by Myocardial Infarction. Journal of Cardiovascular Development and Disease. 2023; 10(3):103. https://doi.org/10.3390/jcdd10030103
Chicago/Turabian StyleFilho, Carlos Eduardo Braga, Adriano Henrique Pereira Barbosa, Lucas Antonio Duarte Nicolau, Jand Venes Rolim Medeiros, Marcelo Pires-Oliveira, Rui Manuel dos Santos Póvoa, Tânia Carmen Penãranda Govato, Hézio Jadir Fernandes Júnior, Rafael Guzella de Carvalho, Bráulio Luna-Filho, and et al. 2023. "Pharmacological Modulation by Low Molecular Weight Heparin of Purinergic Signaling in Cardiac Cells Prevents Arrhythmia and Lethality Induced by Myocardial Infarction" Journal of Cardiovascular Development and Disease 10, no. 3: 103. https://doi.org/10.3390/jcdd10030103