Reactivity of Coordinated 2-Pyridyl Oximes: Synthesis, Structure, Spectroscopic Characterization and Theoretical Studies of Dichlorodi{(2-Pyridyl)Furoxan}Zinc(II) Obtained from the Reaction between Zinc(II) Nitrate and Pyridine-2-Chloroxime

 , , , ,
, , , ,
Abstract
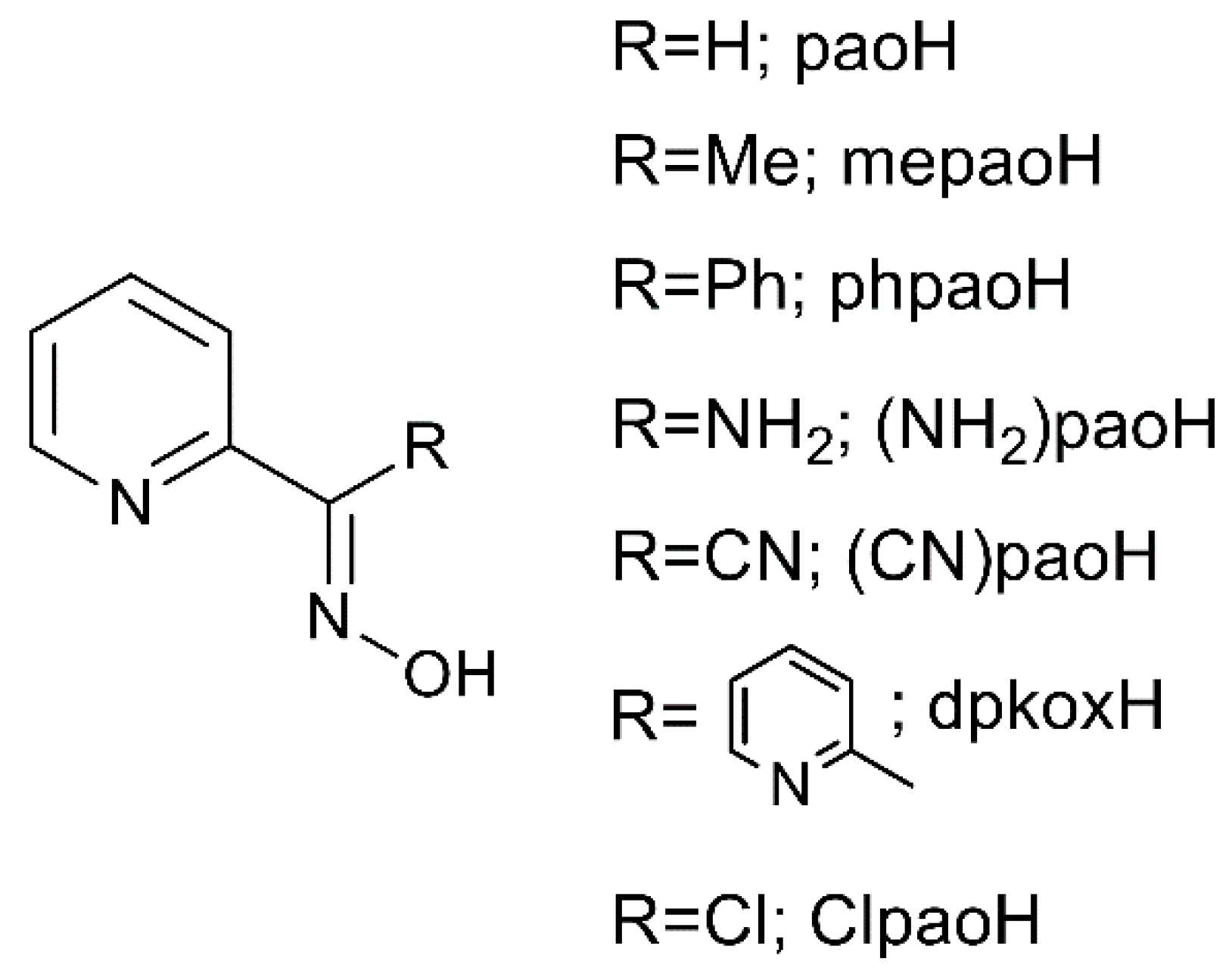
:1. Introduction
2. Results and Discussion
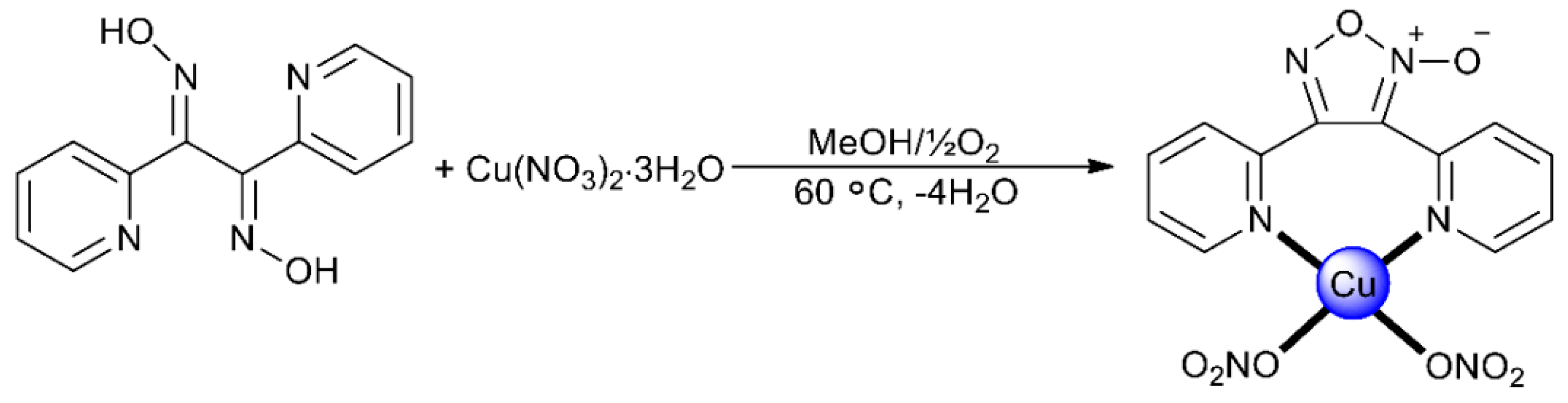
2.1. Synthetic Comments
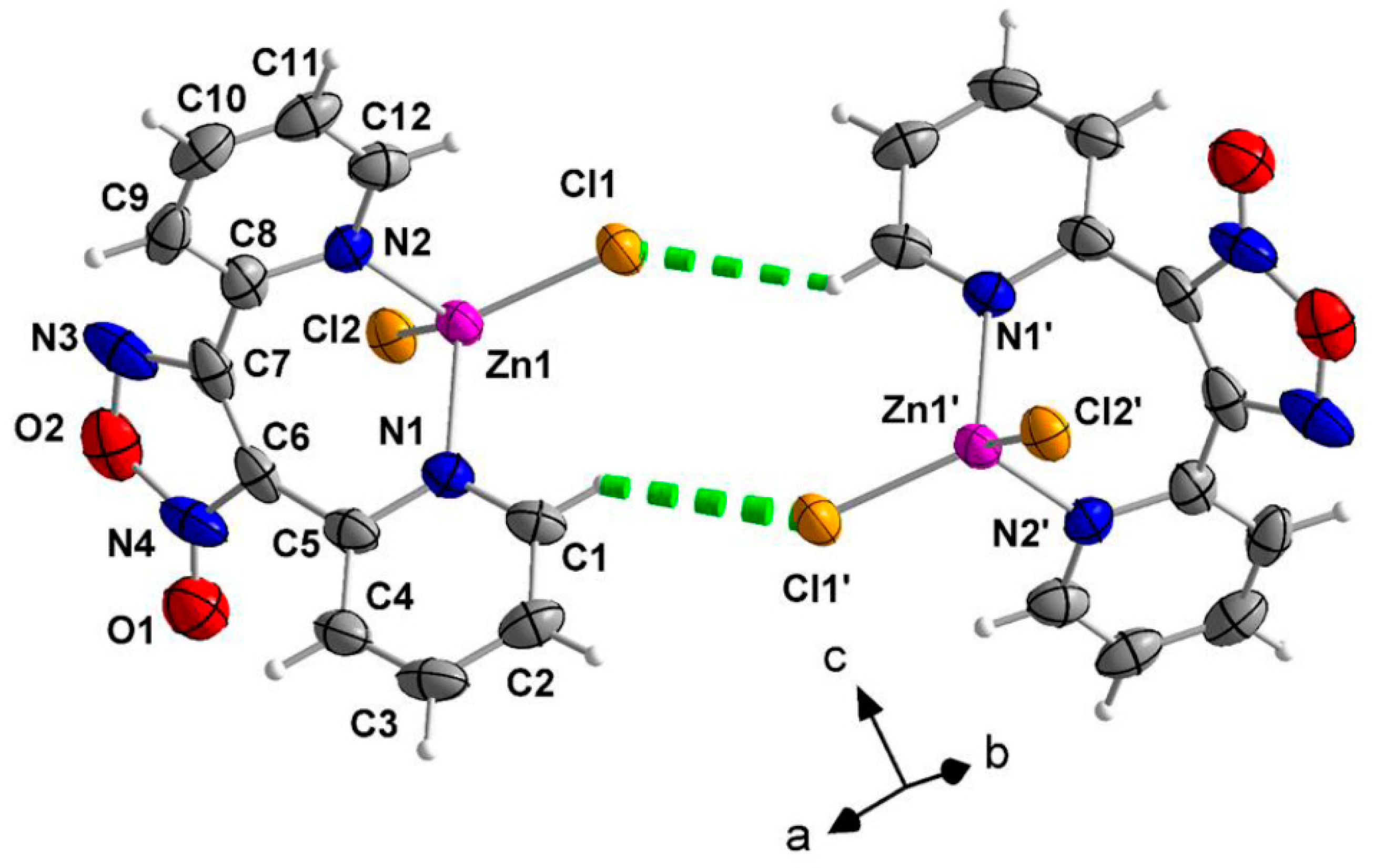
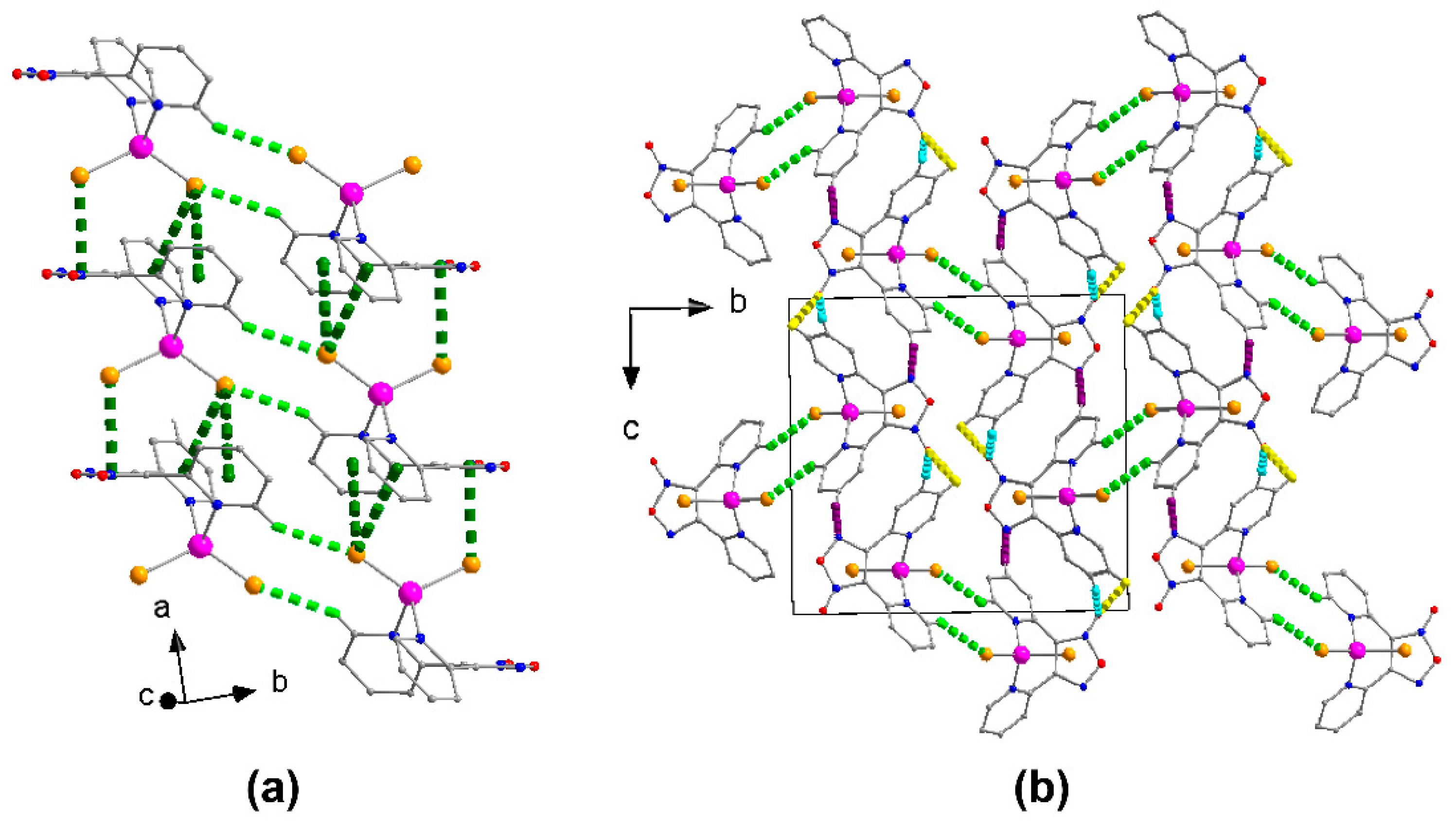
2.2. Description of Structure
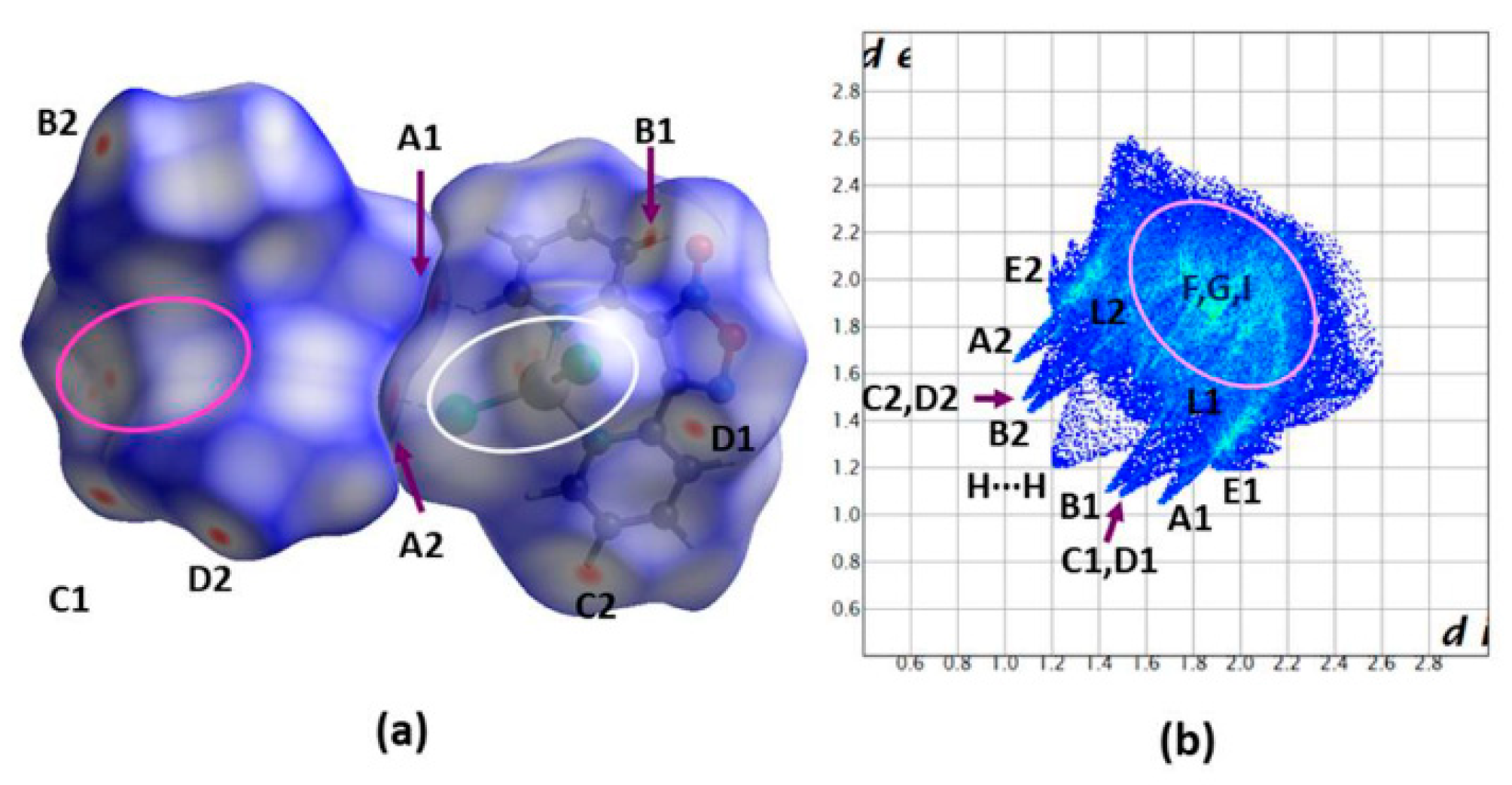
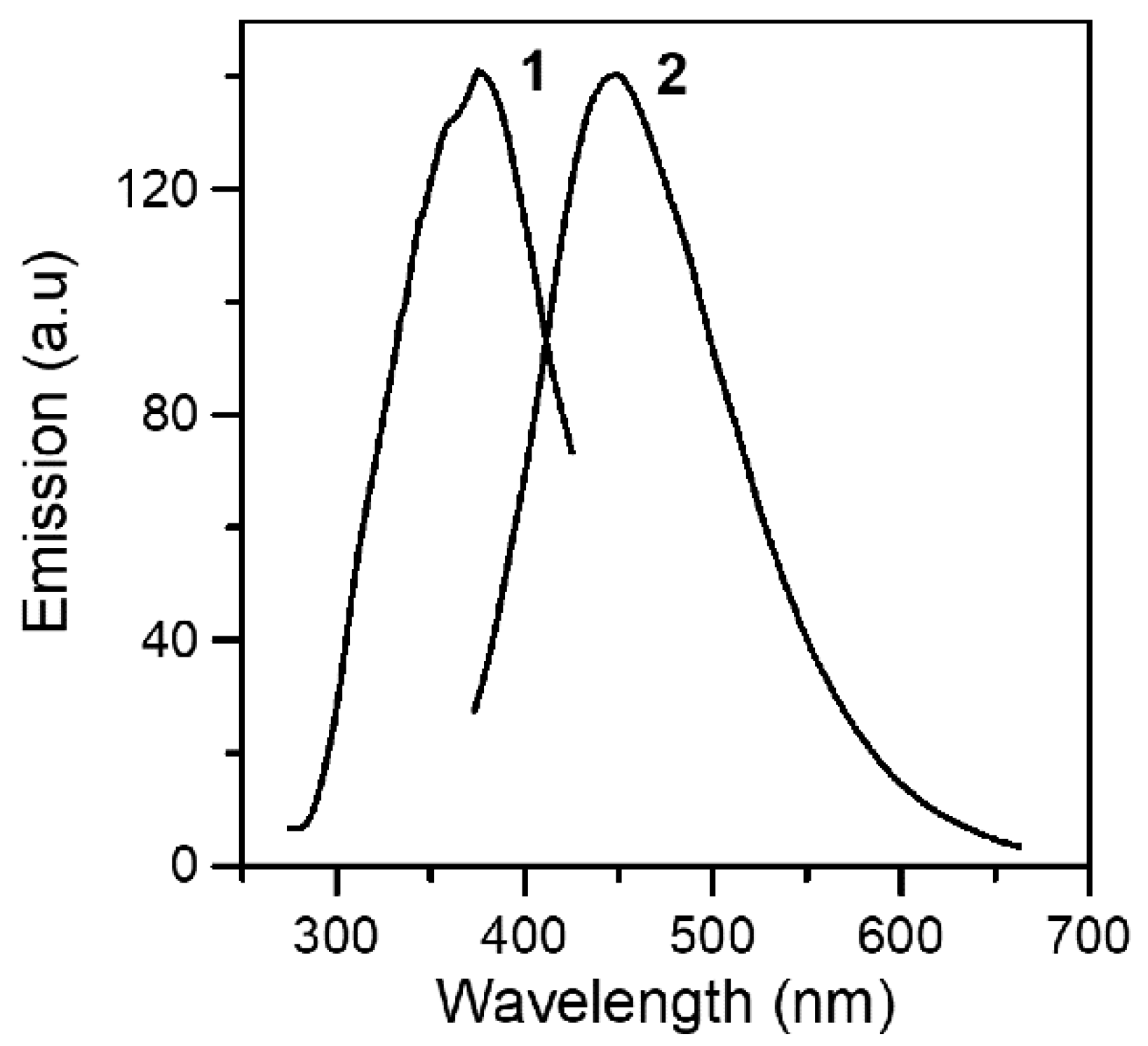
2.3. Spectroscopic Characterization in Brief
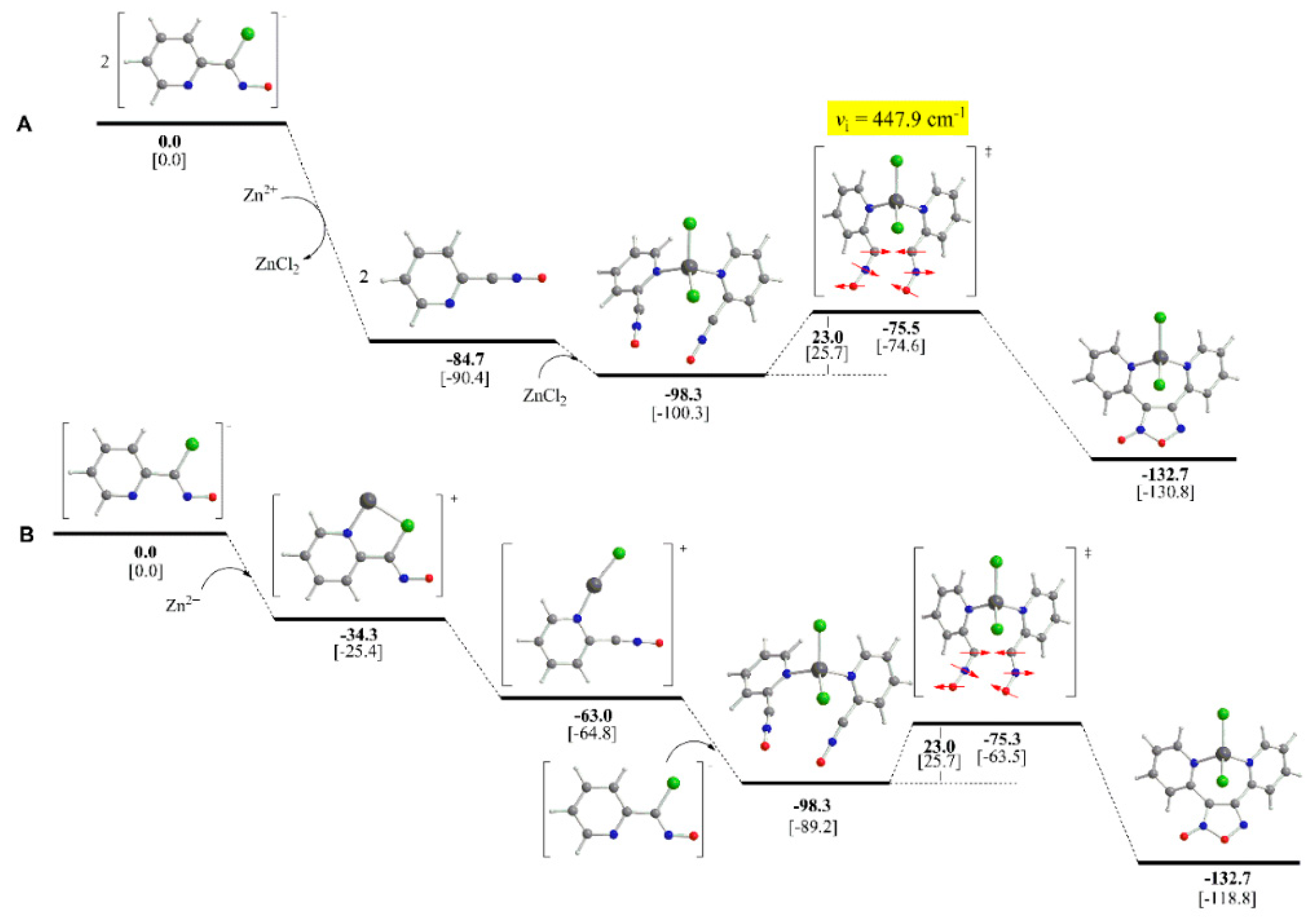
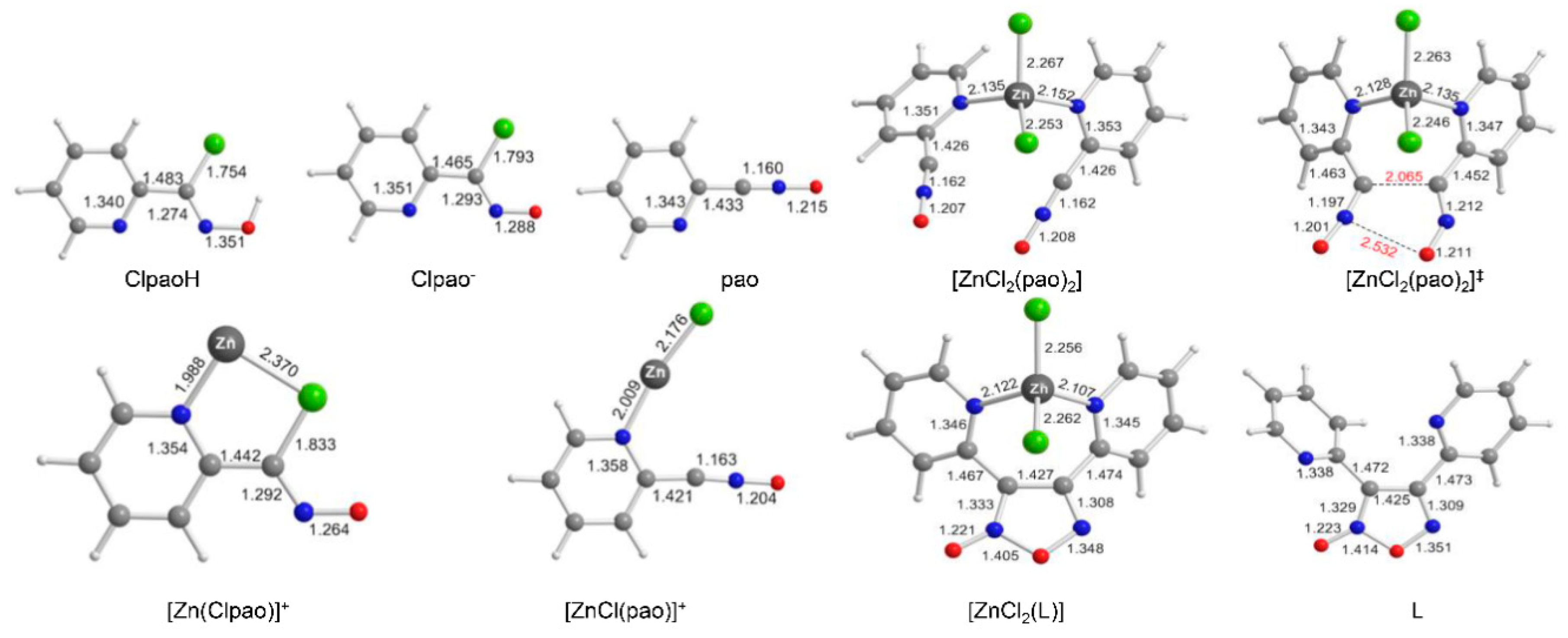
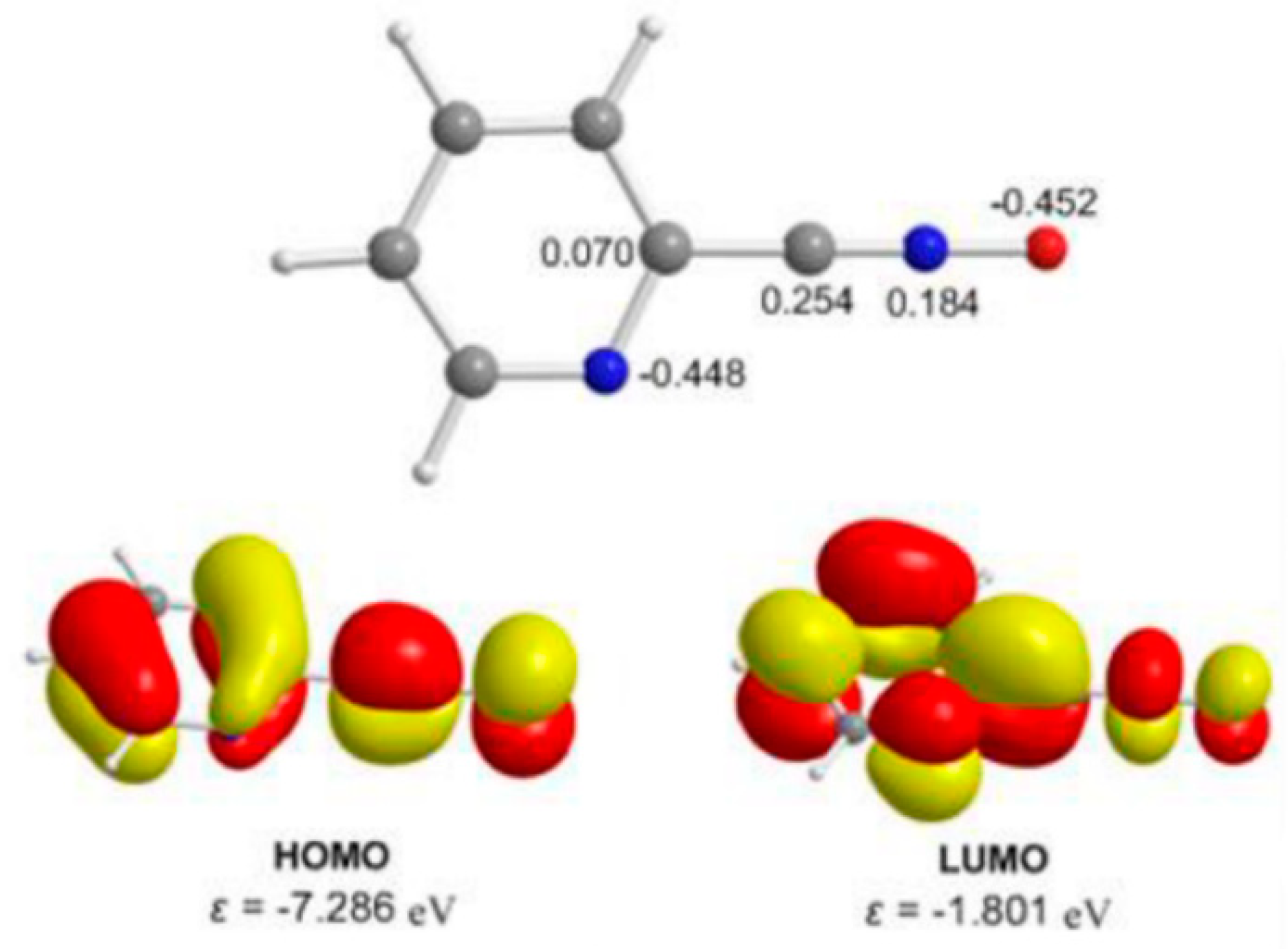
2.4. Theoretical Studies and Mechanistic Aspects
3. Materials and Methods
3.1. Materials
3.2. Physical and Spectroscopic Measurements
3.3. Syntheses of the Complex
3.4. Single-Crystal X-Ray Crystallography
3.5. Computational Details
4. Concluding Comments and Perspectives
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lippard, S.J.; Berg, J.M. Principles of Bioinorganic Chemistry; University Science Books: Mill Valley, CA, USA, 1994; pp. 35–36. [Google Scholar]
- Constable, E.C. Metals and Ligand Reactivity; VCH: Weinheim, Germany, 1996. [Google Scholar]
- Garnovskii, D.A.; Kukushkin, V.Y. Metal-mediated reactions of oximes. Russ. Chem. Rev. 2006, 75, 111–124. [Google Scholar] [CrossRef]
- Kukushkin, V.Y.; Pombeiro, A.J.L. Oxime and oximate metal complexes: Unconventional synthesis and reactivity. Coord. Chem. Rev. 1999, 181, 147–175. [Google Scholar] [CrossRef]
- Kukushkin, V.Y.; Tudela, D.; Pombeiro, A.J.L. Metal-ion assisted reactions of oximes and reactivity of oxime-containing metal complexes. Coord. Chem. Rev. 1996, 156, 333–362. [Google Scholar] [CrossRef]
- Pombeiro, A.J.L.; Kukushkin, V.Y. Reactivity of coordinated oximes. In Comprehensive Coordination Chemistry II; McCleverty, J.A., Meyer, T.J., Eds.; Elsevier: Amsterdam, The Netherlands, 2004; Volume 1, pp. 631–637. [Google Scholar]
- Bolotin, D.S.; Bokach, N.A.; Demakova, M.Y.; Kukushkin, V.Y. Metal-involving synthesis and reactions of oximes. Chem. Rev. 2017, 117, 13039–13122. [Google Scholar] [CrossRef] [PubMed]
- Gerasimchuk, N. Chemistry and applications of cyanoximes and their metal complexes. Dalton Trans. 2019, 48, 7985–8013. [Google Scholar] [CrossRef]
- Lada, Z.G.; Soto Beobide, A.; Savvidou, A.; Raptopoulou, C.P.; Psycharis, V.; Voyiatzis, G.A.; Turnbull, M.M.; Perlepes, S.P. A unique copper(II)-assisted transformation of acetylacetone dioxime in acetone that leads to one-dimensional, quinoxaline-bridged coordination polymers. Dalton Trans. 2017, 46, 260–274. [Google Scholar] [CrossRef]
- Kumar, R.; Chowdhury, B. Comprehensive study for vapor phase Beckmann rearrangement reaction over zeolite systems. Ind. Eng. Chem. Res. 2014, 53, 16587–16599. [Google Scholar] [CrossRef]
- Worek, F.; Thiermann, H.; Wille, T. Oximes in organophosphate poisoning: 60 years of hope and despair. Chem. Biol. Interact. 2016, 259, 93–98. [Google Scholar] [CrossRef]
- Mercey, G.; Verdelet, T.; Renou, J.; Kliachyna, M.; Baati, R.; Nachon, F.; Jean, L.; Renard, P.Y. Reactivators of acetylcholinesterase inhibited by organophosphorus nerve agents. Acc. Chem. Res. 2012, 45, 756–766. [Google Scholar] [CrossRef]
- Letendre, L.; Harriman, J.; Drag, M.; Mullins, A.; Malinski, J.; Rehbein, S. The intravenous and oral pharmacokinetics of Afoxolaner and Milbemycin used as a combination chewable parasiticide for dogs. J. Vet. Pharmacol. Ther. 2017, 40, 35–43. [Google Scholar] [CrossRef]
- Cai, C.; Jiang, H.; Li, L.; Liu, T.; Song, X.; Liu, B. Characterization of the sweet taste receptor Tas1r2 from an old world monkey species Rhesus monkey and species-dependent activation of the monomeric receptor by an intense sweetener Perillartine. PLoS ONE 2016, 11, e0160079. [Google Scholar] [CrossRef] [PubMed]
- Collins, J.; Xiao, Z.; Müllner, M.; Connal, L.A. The emergence of oxime click chemistry and its utility in polymer science. Polym. Chem. 2016, 7, 3812–3826. [Google Scholar] [CrossRef]
- Zhao, Z.; Yang, Y.; Xiao, Y.; Fan, Y. Recovery of gallium from Bayer liquor: A review. Hydrometallurgy 2012, 125–126, 115–124. [Google Scholar] [CrossRef]
- Ilinski, M.; Knorre, G.V. Ueber eine neue methode zur trennung von eisen und aluminium. Ber. Dtsch. Chem. Ges. 1885, 18, S.2728–S.2734. [Google Scholar] [CrossRef] [Green Version]
- Tschugaeff, L. Ueber ein neues, empfindliches reagens auf nickel. Ber. Dtsch. Chem. Ges. 1905, 38, 2520–2522. [Google Scholar] [CrossRef] [Green Version]
- Smith, A.G.; Tasker, P.A.; White, D.J. The structures of phenolic oximes and their complexes. Coord. Chem. Rev. 2003, 241, 61–85. [Google Scholar] [CrossRef]
- Tasker, P.A.; Tong, C.C.; Westra, A.N. Co-extraction of cations and anions in base metal recovery. Coord. Chem. Rev. 2007, 251, 1868–1877. [Google Scholar] [CrossRef]
- Thorpe, J.M.; Beddoes, R.L.; Collison, D.; Garner, C.D.; Helliwell, M.; Holmes, J.M.; Tasker, P.A. Surface coordination chemistry: Corrosion inhibition by tetranuclear cluster formation of iron with salicylaldoxime. Angew. Chem. Int. Ed. 1999, 38, 1119–1121. [Google Scholar] [CrossRef]
- Gerasimchuk, N.; Maher, T.; Durham, P.; Domasevitch, K.V.; Wilking, J.; Mokhir, A. Tin(IV) cyanoximates: Synthesis, characterization, and cytotoxicity. Inorg. Chem. 2007, 46, 7268–7284. [Google Scholar] [CrossRef]
- Karamtzioti, P.; Papastergiou, A.; Stefanakis, J.G.; Koumbis, A.E.; Anastasiou, I.; Koffa, M.; Fylaktakidou, K.C. O-benzoyl pyridine aldoxime and amidoxime derivatives: Novel efficient DNA photo-cleavage agents. Med. Chem. Commun. 2015, 6, 719–726. [Google Scholar] [CrossRef]
- Sahyoun, T.; Arrault, A.; Schneider, R. Amidoximes and oximes: Synthesis, structure, and their key role as NO donors. Molecules 2019, 24, 2470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettenuzzo, A.; Pigot, R.; Ronconi, L. Vitamin B12–metal conjugates for targeted chemotherapy and diagnosis: Current status and future prospects. Eur. J. Inorg. Chem. 2017, 1625–1638. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.-J.; Zhang, Z.-Z.; Lin, S.-B. A review of manganese-based molecular magnets and supramolecular architectures from phenolic oximes. Coord. Chem. Rev. 2015, 289–290, 289–314. [Google Scholar] [CrossRef]
- Chaudhuri, P. Homo- and hetero-polymetallic exchange-coupled metal-oximates. Coord. Chem. Rev. 2003, 243, 143–190. [Google Scholar] [CrossRef]
- Milios, C.J.; Inglis, R.; Vinslava, A.; Bagai, R.; Wernsdorfer, W.; Parsons, S.; Perlepes, S.P.; Christou, G.; Brechin, E.K. Toward a magnetostructural correlation for a family of Mn6 SMMs. J. Am. Chem. Soc. 2007, 129, 12505–12511. [Google Scholar] [CrossRef] [PubMed]
- Kopylovich, M.N.; Kukushkin, V.Y.; Haukka, M.; Fraústo da Silva, J.J.R.; Pombeiro, A.J.L. Zinc(II)/ketoxime system as a simple and efficient catalyst for hydrolysis of organonitriles. Inorg. Chem. 2002, 41, 4798–4804. [Google Scholar] [CrossRef]
- Alonso, D.A.; Nájera, C. Oxime-derived palladacycles as source of palladium nanoparticles. Chem. Soc. Rev. 2010, 39, 2891–2902. [Google Scholar] [CrossRef]
- Anastasiadis, N.C.; Polyzou, C.D.; Kostakis, G.E.; Bekiari, V.; Lan, Y.; Perlepes, S.P.; Konidaris, K.F.; Powell, A.K. Dinuclear lanthanide(III)/zinc(II) complexes with methyl 2-pyridyl ketone oxime. Dalton Trans. 2015, 44, 19791–19795. [Google Scholar] [CrossRef] [Green Version]
- Polyzou, C.D.; Koumousi, E.S.; Lada, Z.G.; Raptopoulou, C.P.; Psycharis, V.; Rouzières, M.; Tsipis, A.C.; Mathonière, C.; Clérac, R.; Perlepes, S.P. “Switching on” the single-molecule magnet properties within a series of dinuclear cobalt(III)–dysprosium(III) 2-pyridyloximate complexes. Dalton Trans. 2017, 46, 14812–14825. [Google Scholar] [CrossRef]
- Konidaris, K.F.; Bekiari, V.; Katsoulakou, E.; Raptopoulou, C.P.; Psycharis, V.; Manessi-Zoupa, E.; Kostakis, G.E.; Perlepes, S.P. Investigation of the zinc(II)-benzoate-2-pyridinealdoxime reaction system. Dalton Trans. 2012, 41, 3797–3806. [Google Scholar] [CrossRef]
- Duros, V.; Sartzi, H.; Teat, S.J.; Sanakis, Y.; Roubeau, O.; Perlepes, S.P. Tris{2,4-bis(2-pyridyl)-1,3,5-triazapentanedienato}manganese(III), a complex derived from a unique metal ion-assisted transformation of pyridine-2-amidoxime. Inorg. Chem. Commun. 2014, 50, 117–121. [Google Scholar] [CrossRef] [Green Version]
- Konidaris, K.F.; Polyzou, C.D.; Kostakis, G.E.; Tasiopoulos, A.J.; Roubeau, O.; Teat, S.J.; Manessi-Zoupa, E.; Powell, A.K.; Perlepes, S.P. Metal ion-assisted transformations of 2-pyridinealdoxime and hexafluorophosphate. Dalton Trans. 2012, 41, 2862–2865. [Google Scholar] [CrossRef]
- Stoumpos, C.C.; Inglis, R.; Roubeau, O.; Sartzi, H.; Kitos, A.A.; Milios, C.J.; Aromí, G.; Tasiopoulos, A.J.; Nastopoulos, V.; Brechin, E.K.; et al. Rare oxidation-state combinations and unusual structural motifs in hexanuclear Mn complexes using 2-pyridyloximate ligands. Inorg. Chem. 2010, 49, 4388–4390. [Google Scholar] [CrossRef]
- Milios, C.J.; Kyritsis, P.; Raptopoulou, C.P.; Terzis, A.; Vicente, R.; Escuer, A.; Perlepes, S.P. Di-2-pyridyl ketone oxime [(py)2CNOH] in manganese carboxylate chemistry: Mononuclear, dinuclear and tetranuclear complexes, and partial transformation of (py)2CNOH to the gem-diolate(2−) derivative of di-2-pyridyl ketone leading to the formation of NO3−. Dalton Trans. 2005, 3, 501–511. [Google Scholar] [CrossRef]
- Milios, C.J.; Kefalloniti, E.; Raptopoulou, C.P.; Terzis, A.; Escuer, A.; Vicente, R.; Perlepes, S.P. 2-pyridinealdoxime [(py)CHNOH] in manganese(II) carboxylate chemistry: Mononuclear, dinuclear, tetranuclear and polymeric complexes, and partial transformation of (py)CHNOH to picolinate(−1). Polyhedron 2004, 23, 83–95. [Google Scholar] [CrossRef]
- Milios, C.J.; Raptopoulou, C.P.; Terzis, A.; Vicente, R.; Escuer, A.; Perlepes, S.P. Di-2-pyridyl ketone oxime in 3d-metal carboxylate cluster chemistry: A new family of mixed-valence MnII2MnIII2 complexes. Inorg. Chem. Commun. 2003, 6, 1056–1060. [Google Scholar] [CrossRef]
- Dash, A.K.; Jaladanki, C.K.; Maiti, D.K.; Singh, D.; Tripathi, A.K.; Gupta, V.K.; Bharatam, P.V.; Mukherjee, D. Tandem gem–dichlorination and nitrile oxide generation from chlorochromene aldoximes: Synthesis of a new class of room temperature fluxional 4-chromanone derivatives. ChemistrySelect 2016, 1, 567–571. [Google Scholar] [CrossRef]
- Voloshin, Y.Z.; Belaya (Makarenko), I.G.; Belov, A.S.; Platonov, V.E.; Maksimov, A.M.; Vologzhanina, A.V.; Starikova, Z.A.; Dolganov, A.V.; Novikov, V.V.; Bubnov, Y.N. Formation of the second superhydrophobic shell around an encapsulated metal ion: Synthesis, X-ray structure and electrochemical study of the clathrochelate and bis-clathrochelate iron(II) and cobalt(II,III) dioximates with ribbed perfluoroarylsulfide substituents. Dalton Trans. 2012, 41, 737–746. [Google Scholar]
- Burdukov, A.B.; Vershinin, M.A.; Pervukhina, N.V.; Kozlova, S.G.; El’tsov, I.V.; Voloshin, Y.Z. Free-radical reactions of the tris-dioximate clathrochelates: Synthesis and X-ray structure of the first cyclohexyl-substituted monoribbed-functionalized macrobicyclic iron(II) complex. Russ. Chem. Bull. 2011, 60, 2504–2509. [Google Scholar] [CrossRef]
- Wiley, R.H.; Wakefield, B.J. Infrared Spectra of the nitrile N-oxides: Some new furoxans. J. Org. Chem. 1960, 25, 546–551. [Google Scholar] [CrossRef]
- Wingard, L.A.; Gazmán, P.E.; Sabatini, J.J. A chlorine gas-free synthesis of dichloroglyoxime. Org. Process Res. Dev. 2016, 20, 1686–1688. [Google Scholar] [CrossRef]
- Stevens, J.; Schweizer, M.; Rauhut, G. Toward an understanding of the furoxan-dinitrosoethylene equilibrium. J. Am. Chem. Soc. 2001, 123, 7326–7333. [Google Scholar] [CrossRef]
- Xu, H.; Ling, Y.; Zou, Z.H.; Huang, W.L.; Yao, C. 5-(6,7-Dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl) -4-phenyl-1,2,5-oxadiazole N-oxide. Acta Crystallogr. Sect. E Struct. Rep. Online 2006, 62, o3130–o3132. [Google Scholar] [CrossRef]
- Das, O.; Paria, S.; Zangrando, E.; Paine, T.K. Copper(II)-mediated oxidative transformation of vic -dioxime to furoxan: Evidence for a copper(II)-dinitrosoalkene intermediate. Inorg. Chem. 2011, 50, 11375–11383. [Google Scholar] [CrossRef]
- Zhai, L.; Qu, X.; Wang, B.; Bi, F.; Chen, S.; Fan, X.; Xie, G.; Wei, Q.; Gao, S. High energy density materials incorporating 4,5-bis(dinitromethyl)-furoxanate and 4,5-bis(dinitromethyl)-3-oxy-furoxanate. ChemPlusChem 2016, 81, 1156–1159. [Google Scholar] [CrossRef]
- Paton, R.M. 1,2,5-oxadiazoles. In Comprehensive Heterocyclic Chemistry II; Katritzky, A.R., Rees, C.W., Scriven, T.V., Eds.; Elsevier: Amsterdam, The Netherlands, 1996; Volume 4, pp. 229–265. [Google Scholar]
- Wang, P.G.; Xian, M.; Tang, X.; Wu, X.; Wen, Z.; Cai, T.; Janczuk, A.J. Nitric oxide donors: Chemical activities and biological applications. Chem. Rev. 2002, 102, 1091–1134. [Google Scholar] [CrossRef]
- Nikonov, G.N.; Bobrov, S. 1,2,5-oxadiazoles. In Comprehensive Heterocyclic Chemistry III; Katritzky, A.R., Ramsden, C.A., Taylor, R.J.K., Eds.; Elsevier: Amsterdam, The Netherlands, 2008; Volume 5, pp. 315–394. [Google Scholar]
- Richardson, C.; Steel, P.J. Di(2-pyridyl)furoxan: Metal complexes and an unusual ruthenium-induced molecular rearrangement. Aust. J. Chem. 2000, 53, 93–97. [Google Scholar] [CrossRef]
- Bikas, R.; Valadbeigi, Y.; Otręba, M.; Lis, T. Mechanistic studies on the in-situ generation of furoxan ring during the formation of Cu(II) coordination compound from dioxime ligand: Theoretical and experimental study. Inorg. Chim. Acta 2020, 510, 119756. [Google Scholar] [CrossRef]
- Xu, Z.D.; Liu, H.; Xiao, S.L.; Yang, M.; Bu, X.H. Synthesis, crystal structure, antitumor activity and DNA-binding study on the Mn(II) complex of 2H-5-hydroxy-1,2,5-oxadiazo[3,4-f]1,10-phenanthroline. J. Inorg. Biochem. 2002, 90, 79–84. [Google Scholar] [CrossRef]
- Lakshman, T.R.; Deb, J.; Paine, T.K. Anti-inflammatory activity and enhanced COX-2 selectivity of nitric oxide-donating zinc(II)-NSAID complexes. Dalton Trans. 2016, 45, 14053–14057. [Google Scholar] [CrossRef]
- Perlepes, S.P.; Kasselouri, S.; Garoufis, A.; Lutz, F.; Bau, R.; Hadjiliadis, N. Reaction of the biheteroaromatic ligand 2-(2′-pyridyl)quinoxaline (L) with zinc(II) and cadmium(II) halides: Preparation and characterization of the 1:1 complexes. Polyhedron 1995, 14, 1461–1470. [Google Scholar] [CrossRef]
- Wolf, S.K.; Grimwood, D.J.; Mckinnon, J.J.; Turne, M.J.; Jayatilaka, D.; Spackman, M.A. Crystal Explorer 3; The University of Western Australia: Crowley, Australia, 2012. [Google Scholar]
- Thallapally, P.K.; Nangia, A. A Cambridge Structural Database analysis of the C–H⋯Cl interaction: C–H⋯Cl− and C–H⋯Cl–M often behave as hydrogen bonds but C–H⋯Cl–C is generally a van der Waals interaction. CrystEngComm 2001, 3, 114–119. [Google Scholar] [CrossRef]
- Aakeröy, C.B.; Evans, T.A.; Seddon, K.R.; Pálinkó, I. The C–H···Cl hydrogen bond: Does it exist? New J. Chem. 1999, 23, 145–152. [Google Scholar] [CrossRef] [Green Version]
- Brammer, L.; Bruton, E.A.; Sherwood, P. Understanding the behavior of halogens as hydrogen bond acceptors. Cryst. Growth Des. 2001, 1, 277–290. [Google Scholar] [CrossRef]
- Balamurugan, V.; Jacob, W.; Mukherjee, J.; Mukherjee, R. Designing neutral coordination networks using inorganic supramolecular synthons: Combination of coordination chemistry and C–H⋯Cl hydrogen bonding. CrystEngComm 2004, 6, 396–400. [Google Scholar] [CrossRef]
- Zukerman-Schpector, J.; Prado, K.E.; Name, L.L.; Cella, R.; Jotani, M.M.; Tiekink, E.R.T. 2-[(4-chlorophenyl)selanyl]-3,4-dihydro-2H-benzo[h]chromene-5,6-dione: Crystal structure and Hirshfeld analysis. Acta Crystallogr. Sect. E 2017, 73, 918–924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riley, S.; Staples, R.J.; Biros, S.M.; Ngassa, F.N. Crystal structure of phenyl 2,4,5-trichloro-benzenesulfonate. Acta Crystallogr. Sect. E 2016, 72, 789–792. [Google Scholar] [CrossRef] [PubMed]
- Imai, Y.N.; Inoue, Y.; Nakanishi, I.; Kitaura, K. Cl-π interactions in protein-ligand complexes. Protein Sci. 2008, 17, 1129–1137. [Google Scholar] [CrossRef] [Green Version]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld surface analysis. CrystEngComm 2009, 11, 19–32. [Google Scholar] [CrossRef]
- Newberry, R.W.; Raines, R.T. The n→π∗ interaction. Acc. Chem. Res. 2017, 50, 1838–1846. [Google Scholar] [CrossRef] [Green Version]
- Chiang, Y.-H. Chlorination of oximes. I. Reaction and mechanism of the chlorination of oximes in commercial chloroform and methylene chloride. J. Org. Chem. 1971, 36, 2146–2155. [Google Scholar] [CrossRef]
- Dollish, F.; Fateley, W.; Bentley, F. Characteristic Raman Frequencies of Organic Compounds; Wiley: New York, NY, USA, 1974; pp. 263–272. [Google Scholar]
- Bellamy, L.J. The Infrared Spectra of Complex Molecules, 2nd ed.; Methuen: London, UK, 1966; pp. 277–281. [Google Scholar]
- Geary, W.J. The use of conductivity measurements in organic solvents for the characterisation of coordination compounds. Coord. Chem. Rev. 1971, 7, 81–122. [Google Scholar] [CrossRef]
- Thomas, S.; Brühl, I.; Heilmann, D.; Kleinpeter, E. 13C NMR chemical shift calculations for some substituted pyridines: A comparative consideration. J. Chem. Inf. Comput. Sci. 1997, 37, 726–730. [Google Scholar] [CrossRef]
- Oszczapowicz, J. Substituent Effects in the 13C-NMR spectra of six-membered nitrogen heteroaromatic compounds. Int. J. Mol. Sci. 2005, 6, 11–17. [Google Scholar] [CrossRef] [Green Version]
- Städeli, W.; von Philipsborn, W. 15N NMR substituent effects in pyridines and pyrimidines. Org. Magn. Reson. 1981, 15, 106–109. [Google Scholar] [CrossRef]
- Lee, C.W.; Hwang, G.T.; Kim, B.H. Novel silicon-bridged macrocycles: Efficient synthesis by quadruple cycloadditive macrocyclization and intramolecular nitrile oxide dimerization. Tetrahedron Lett. 2000, 41, 4177–4180. [Google Scholar] [CrossRef]
- Konidaris, K.F.; Bekiari, V.; Katsoulakou, E.; Raptopoulou, C.P.; Psycharis, V.; Perlepes, S.P.; Stamatatos, T.C.; Manessi-Zoupa, E. Initial employment of pyridine-2-amidoxime in zinc(II) chemistry: Synthetic, structural and spectroscopic studies of mononuclear and dinuclear complexes. Inorg. Chim. Acta 2011, 376, 470–478. [Google Scholar] [CrossRef]
- Katsoulakou, E.; Dermitzaki, D.; Konidaris, K.F.; Moushi, E.E.; Raptopoulou, C.P.; Psycharis, V.; Tasiopoulos, A.J.; Bekiari, V.; Manessi-Zoupa, E.; Perlepes, S.P.; et al. Hexanuclear zinc(II) carboxylate complexes from the use of pyridine-2,6-dimethanol: Synthetic, structural and photoluminescence studies. Polyhedron 2013, 52, 467–475. [Google Scholar] [CrossRef]
- King, A.O.; Okukado, N.; Negishi, E. Highly general stereo-, regio-, and chemo-selective synthesis of terminal and internal conjugated enynes by the Pd-catalysed reaction of alkynylzinc reagents with alkenyl halides. J. Chem. Soc. Chem. Commun. 1977, 683–684. [Google Scholar] [CrossRef]
- Heck, K.F.; Nolley, J.P. Palladium-catalyzed vinylic hydrogen substitution reactions with aryl, benzyl, and styryl halides. J. Org. Chem. 1972, 37, 2320–2322. [Google Scholar] [CrossRef]
- Stille, J.K. The palladium-catalyzed cross-coupling reactions of organotin reagents with organic electrophiles. Angew. Chem. Int. Ed. 1986, 25, 508–524. [Google Scholar] [CrossRef]
- Miyaura, N.; Suzuki, A. Palladium-catalyzed cross-coupling reactions of organoboron compounds. Chem. Rev. 1995, 95, 2457–2483. [Google Scholar] [CrossRef] [Green Version]
- Sonogashira, K. Development of Pd–Cu catalyzed cross-coupling of terminal acetylenes with sp2-carbon halides. J. Organomet. Chem. 2002, 653, 46–49. [Google Scholar] [CrossRef]
- Paul, F.; Patt, J.; Hartwig, J.F. Palladium-catalyzed formation of carbon-nitrogen bonds. Reaction intermediates and catalyst improvements in the hetero cross-coupling of aryl halides and tin amides. J. Am. Chem. Soc. 1994, 116, 5969–5970. [Google Scholar] [CrossRef]
- Guram, A.S.; Buchwald, S.L. Palladium-catalyzed aromatic animations with in situ generated aminostannanes. J. Am. Chem. Soc. 1994, 116, 7901–7902. [Google Scholar] [CrossRef]
- Bernasek, E. Pyridineamidoximes. J. Org. Chem. 1957, 22, 1263. [Google Scholar] [CrossRef]
- CrystalClear; Rigaku: The Woodlands, TX, USA; MSC Inc.: Melville, NY, USA, 2005.
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Diamond, Crystal and MOLECULAR structure Visualization; Version 3.1; Crystal Impact: Bonn, Germany, 2018.
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Ernzerhof, M.; Scuseria, G.E. Assessment of the Perdew-Burke-Ernzerhof exchange-correlation functional. J. Chem. Phys. 1999, 110, 5029–5036. [Google Scholar] [CrossRef] [Green Version]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Vetere, V.; Adamo, C.; Maldivi, P. Performance of the ‘parameter free’ PBE0 functional for the modeling of molecular properties of heavy metals. Chem. Phys. Lett. 2000, 325, 99–105. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef] [PubMed]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Weinhold, F. Natural Bond Orbital Methods. In The Encyclopedia of Computational Chemistry; Schleyer, P.V.R., Allinger, N.L., Clark, T., Gasteiger, J., Kollman, P.A., Schaefer, H.F., Schreiner, P.R., Eds.; Wiley: Chichester, UK, 1998; pp. 1792–1811. [Google Scholar]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | [ZnCl2(L)] (1) |
|---|---|
| Empirical formula | C12H8ZnCl2N4O2 |
| Formula weight | 376.49 |
| Crystal system | monoclinic |
| Space group | P21/n |
| Color | pale yellow |
| Crystal size, mm | 0.34 × 0.10 × 0.09 |
| Crystal habit | parallelepiped |
| a, Å | 6.4893(1) |
| b, Å | 14.9997(3) |
| c, Å | 14.2753(3) |
| α, ° | 90.00(1) |
| β, ° | 90.637(1) |
| γ, ° | 90.00(1) |
| Volume, Å3 | 1389.44(5) |
| Z | 4 |
| Temperature, K | 160 |
| Radiation, Å | Cu Kα, 1.54178 |
| Calculated density, g·cm−3 | 1.800 |
| Absorption coefficient, mm−1 | 6.09 |
| No. of measured, independent and observed [I > 2σ(Ι)] reflections | 15,854, 2338, 1890 |
| Rint | 0.071 |
| Number of parameters | 191 |
| Final R indices [Ι > 2σ(Ι)] a | R1 = 0.0705, wR2 = 0.1514 |
| Goodness-of-fit in F2 | 1.12 |
| Largest differences peak and hole (e·Å−3) | 0.73/−0.60 |
| Bond Lengths (Å) | Bond Angles (°) | ||
|---|---|---|---|
| Zn1–Cl1 | 2.195(2) | Cl1–Zn1–Cl2 | 120.4(1) |
| Zn1–Cl2 | 2.221(2) | Cl1–Zn1–N1 | 111.7(2) |
| Zn1–N1 | 2.054(6) | Cl1–Zn1–N2 | 108.5(2) |
| Zn1–N2 | 2.068(6) | Cl2–Zn1–N1 | 107.3(2) |
| C6–C7 | 1.409(11) | Cl2–Zn1–N2 | 110.5(2) |
| C6–N4 | 1.341(11) | N1–Zn1–N2 | 95.8(2) |
| N4–O2 | 1.427(10) | N3–O2–N4 | 112.7(7) |
| N3–O2 | 1.334(9) | O2–N4–C6 | 107.1(6) |
| N3–C7 | 1.397(11) | O1–N4–O2 | 120.4(8) |
| N4–O1 | 1.170(8) | O1–N4–C6 | 132.5(9) |
| D–H⋯A | d(D⋯A) | d(H⋯A) | <DHA | Symmetry Code of A |
|---|---|---|---|---|
| C1–H1⋯Cl1′ | 3.529(5) | 2.757(2) | 138.5(4) | −x, −y + 2, −z + 1 |
| C10–H10⋯O1‴ | 3.50(1) | 2.621(7) | 154.0(5) | x + 0.5, −y + 1.5, z + 0.5 |
| C3–H3⋯N3i | 3.57(1) | 2.658(7) | 162.0(5) | x + 0.5, −y + 1.5, z − 0.5 |
| C11–H11⋯O1ii a | 3.11(1) | 2.902(7) | 93.9(5) | x−0.5, −y + 1.5, z + 0.5 |
| Reaction | ΔH (Kcal/mol) |
|---|---|
| ClpaoH → paoH+ + Cl− | 43.8 |
| ClpaoH + Et3N → Clpao− + Et3NH+ | 5.5 |
| Clpao− → pao + Cl− | 8.2 |
| 2 Clpao− + Zn2+ → 2pao + ZnCl2 | −84.7 |
| Clpao− + Zn2+ → [Zn(Clpao)]+ | −34.3 |
| Clpao− + Zn2+ → [ZnCl(pao)]+ | −63.0 |
| 2 pao + ZnCl2 → complex 1 | −48.2 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsantis, S.T.; Bekiari, V.; Tzimopoulos, D.I.; Raptopoulou, C.P.; Psycharis, V.; Tsipis, A.; Perlepes, S.P. Reactivity of Coordinated 2-Pyridyl Oximes: Synthesis, Structure, Spectroscopic Characterization and Theoretical Studies of Dichlorodi{(2-Pyridyl)Furoxan}Zinc(II) Obtained from the Reaction between Zinc(II) Nitrate and Pyridine-2-Chloroxime. Inorganics 2020, 8, 47. https://doi.org/10.3390/inorganics8090047
Tsantis ST, Bekiari V, Tzimopoulos DI, Raptopoulou CP, Psycharis V, Tsipis A, Perlepes SP. Reactivity of Coordinated 2-Pyridyl Oximes: Synthesis, Structure, Spectroscopic Characterization and Theoretical Studies of Dichlorodi{(2-Pyridyl)Furoxan}Zinc(II) Obtained from the Reaction between Zinc(II) Nitrate and Pyridine-2-Chloroxime. Inorganics. 2020; 8(9):47. https://doi.org/10.3390/inorganics8090047
Chicago/Turabian StyleTsantis, Sokratis T., Vlasoula Bekiari, Demetrios I. Tzimopoulos, Catherine P. Raptopoulou, Vassilis Psycharis, Athanasios Tsipis, and Spyros P. Perlepes. 2020. "Reactivity of Coordinated 2-Pyridyl Oximes: Synthesis, Structure, Spectroscopic Characterization and Theoretical Studies of Dichlorodi{(2-Pyridyl)Furoxan}Zinc(II) Obtained from the Reaction between Zinc(II) Nitrate and Pyridine-2-Chloroxime" Inorganics 8, no. 9: 47. https://doi.org/10.3390/inorganics8090047