
Defining the Emergence of New Immunotherapy Approaches in Breast Cancer: Role of Myeloid-Derived Suppressor Cells
 , , , and
, , , and
Abstract
:1. Introduction
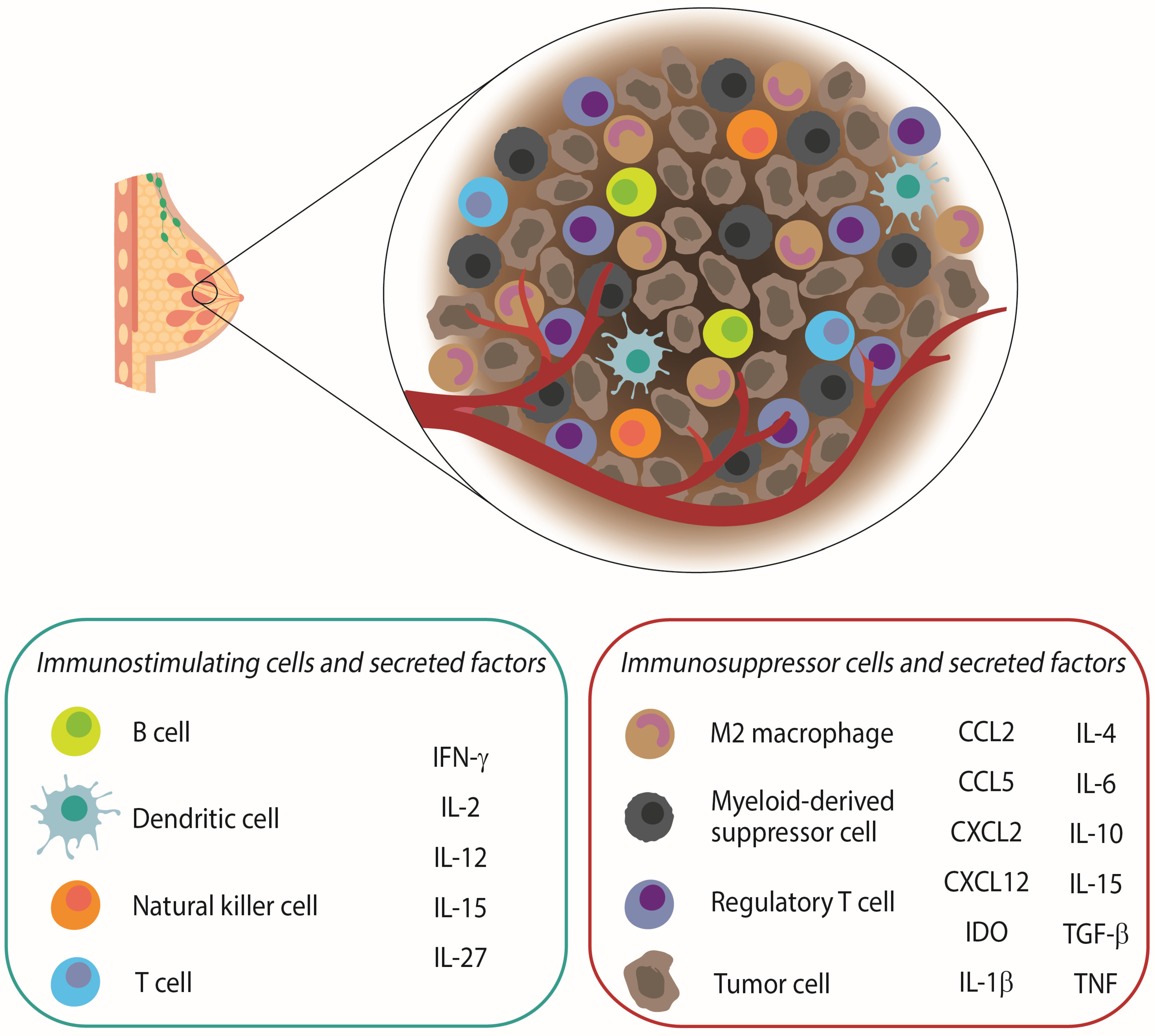
2. The Role of the Immune System in Breast Cancer
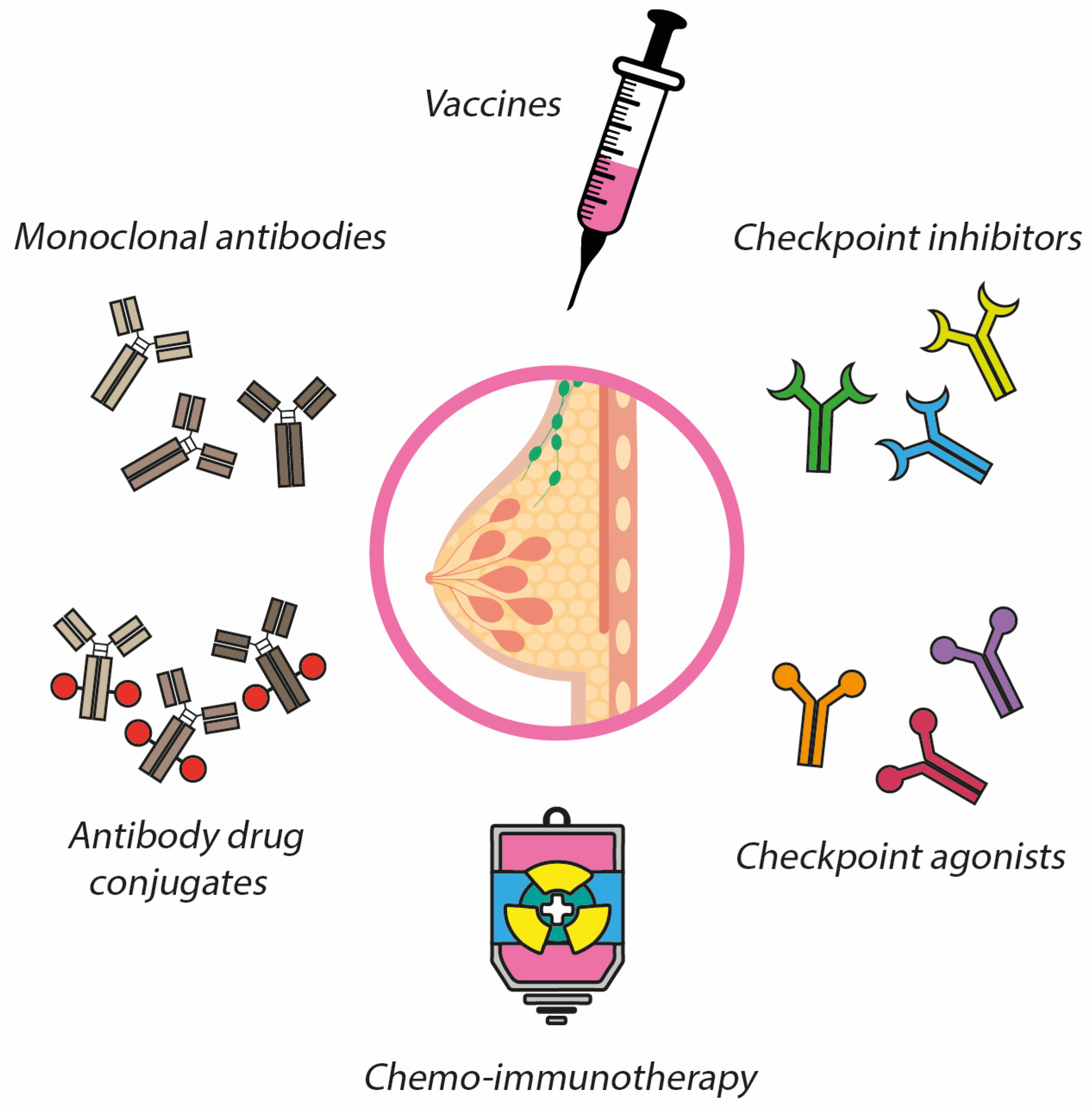
3. Immunotherapy in Breast Cancer
3.1. Vaccines
3.1.1. Peptide Vaccines
3.1.2. Protein-Based Vaccines
3.1.3. Tumor Cell Vaccines
3.1.4. Dendritic Cell-Based Vaccines
3.1.5. DNA-Based Vaccines
3.1.6. Carbohydrate Antigen Vaccines
3.2. Monoclonal Antibodies (mAbs)
3.3. Antibody Drug Conjugates (ADC)
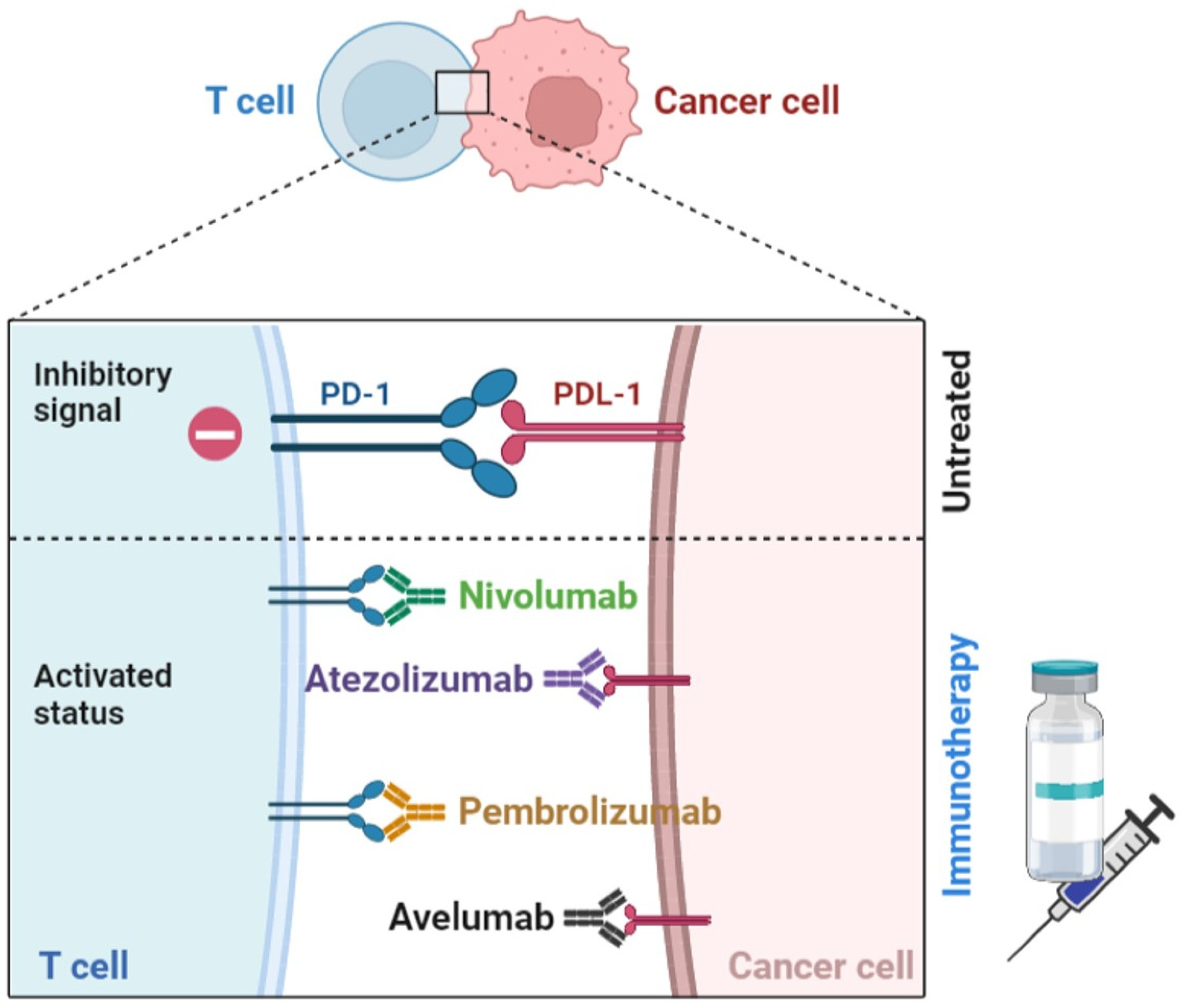
3.4. Immune Checkpoints Blockers (ICB)
3.5. Stimulatory Molecule Agonist Antibodies
4. Myeloid-Derived Suppressor Cells as a Therapeutic Target in Breast Cancer
MDSC-Targeted Immunotherapies in Breast Cancer
5. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattiuzzi, C.; Lippi, G. Current Cancer Epidemiology. J. Epidemiol. Glob. Health 2019, 9, 217–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukasiewicz, S.; Czeczelewski, M.; Forma, A.; Baj, J.; Sitarz, R.; Stanislawek, A. Breast Cancer-Epidemiology, Risk Factors, Classification, Prognostic Markers, and Current Treatment Strategies-An Updated Review. Cancers 2021, 13, 4287. [Google Scholar] [CrossRef] [PubMed]
- Zubair, M.; Wang, S.; Ali, N. Advanced Approaches to Breast Cancer Classification and Diagnosis. Front. Pharmacol. 2020, 11, 632079. [Google Scholar] [CrossRef] [PubMed]
- Erber, R.; Hartmann, A. Histology of Luminal Breast Cancer. Breast Care 2020, 15, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Cejalvo, J.M.; Martinez de Duenas, E.; Galvan, P.; Garcia-Recio, S.; Burgues Gasion, O.; Pare, L.; Antolin, S.; Martinello, R.; Blancas, I.; Adamo, B.; et al. Intrinsic Subtypes and Gene Expression Profiles in Primary and Metastatic Breast Cancer. Cancer Res. 2017, 77, 2213–2221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russnes, H.G.; Lingjaerde, O.C.; Borresen-Dale, A.L.; Caldas, C. Breast Cancer Molecular Stratification: From Intrinsic Subtypes to Integrative Clusters. Am. J. Pathol. 2017, 187, 2152–2162. [Google Scholar] [CrossRef]
- Prat, A.; Pineda, E.; Adamo, B.; Galvan, P.; Fernandez, A.; Gaba, L.; Diez, M.; Viladot, M.; Arance, A.; Munoz, M. Clinical implications of the intrinsic molecular subtypes of breast cancer. Breast 2015, 24 (Suppl. 2), S26–S35. [Google Scholar] [CrossRef] [Green Version]
- Parsa, Y.; Mirmalek, S.A.; Kani, F.E.; Aidun, A.; Salimi-Tabatabaee, S.A.; Yadollah-Damavandi, S.; Jangholi, E.; Parsa, T.; Shahverdi, E. A Review of the Clinical Implications of Breast Cancer Biology. Electron. Physician 2016, 8, 2416–2424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fragomeni, S.M.; Sciallis, A.; Jeruss, J.S. Molecular Subtypes and Local-Regional Control of Breast Cancer. Surg. Oncol. Clin. North Am. 2018, 27, 95–120. [Google Scholar] [CrossRef]
- Zhao, S.; Zuo, W.J.; Shao, Z.M.; Jiang, Y.Z. Molecular subtypes and precision treatment of triple-negative breast cancer. Ann. Transl. Med. 2020, 8, 499. [Google Scholar] [CrossRef] [PubMed]
- Bou Zerdan, M.; Ghorayeb, T.; Saliba, F.; Allam, S.; Bou Zerdan, M.; Yaghi, M.; Bilani, N.; Jaafar, R.; Nahleh, Z. Triple Negative Breast Cancer: Updates on Classification and Treatment in 2021. Cancers 2022, 14, 1253. [Google Scholar] [CrossRef]
- Akram, M.; Iqbal, M.; Daniyal, M.; Khan, A.U. Awareness and current knowledge of breast cancer. Biol. Res. 2017, 50, 33. [Google Scholar] [CrossRef] [Green Version]
- Hortobagyi, G.N.; Edge, S.B.; Giuliano, A. New and Important Changes in the TNM Staging System for Breast Cancer. Am. Soc. Clin. Oncol. Educ. Book 2018, 38, 457–467. [Google Scholar] [CrossRef] [PubMed]
- Fisusi, F.A.; Akala, E.O. Drug Combinations in Breast Cancer Therapy. Pharm. Nanotechnol. 2019, 7, 3–23. [Google Scholar] [CrossRef] [PubMed]
- Fassl, A.; Geng, Y.; Sicinski, P. CDK4 and CDK6 kinases: From basic science to cancer therapy. Science 2022, 375, eabc1495. [Google Scholar] [CrossRef]
- Magnoni, F.; Alessandrini, S.; Alberti, L.; Polizzi, A.; Rotili, A.; Veronesi, P.; Corso, G. Breast Cancer Surgery: New Issues. Curr. Oncol. 2021, 28, 4053–4066. [Google Scholar] [CrossRef] [PubMed]
- Moo, T.A.; Sanford, R.; Dang, C.; Morrow, M. Overview of Breast Cancer Therapy. PET Clin. 2018, 13, 339–354. [Google Scholar] [CrossRef]
- DeNardo, D.G.; Andreu, P.; Coussens, L.M. Interactions between lymphocytes and myeloid cells regulate pro- versus anti-tumor immunity. Cancer Metastasis Rev. 2010, 29, 309–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, B.Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colotta, F.; Allavena, P.; Sica, A.; Garlanda, C.; Mantovani, A. Cancer-related inflammation, the seventh hallmark of cancer: Links to genetic instability. Carcinogenesis 2009, 30, 1073–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The immunobiology of cancer immunosurveillance and immunoediting. Immunity 2004, 21, 137–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gil Del Alcazar, C.R.; Huh, S.J.; Ekram, M.B.; Trinh, A.; Liu, L.L.; Beca, F.; Zi, X.; Kwak, M.; Bergholtz, H.; Su, Y.; et al. Immune Escape in Breast Cancer During In Situ to Invasive Carcinoma Transition. Cancer Discov. 2017, 7, 1098–1115. [Google Scholar] [CrossRef] [Green Version]
- Tower, H.; Ruppert, M.; Britt, K. The Immune Microenvironment of Breast Cancer Progression. Cancers 2019, 11, 1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittal, D.; Gubin, M.M.; Schreiber, R.D.; Smyth, M.J. New insights into cancer immunoediting and its three component phases--elimination, equilibrium and escape. Curr. Opin. Immunol. 2014, 27, 16–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Sullivan, T.; Saddawi-Konefka, R.; Vermi, W.; Koebel, C.M.; Arthur, C.; White, J.M.; Uppaluri, R.; Andrews, D.M.; Ngiow, S.F.; Teng, M.W.L.; et al. Cancer immunoediting by the innate immune system in the absence of adaptive immunity. J. Exp. Med. 2012, 209, 1869–1882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quezada, S.A.; Peggs, K.S.; Simpson, T.R.; Allison, J.P. Shifting the equilibrium in cancer immunoediting: From tumor tolerance to eradication. Immunol. Rev. 2011, 241, 104–118. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, M.; Mount, N. Genetically modified T cells in cancer therapy: Opportunities and challenges. Dis. Model Mech. 2015, 8, 337–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef] [Green Version]
- Ostrand-Rosenberg, S.; Sinha, P. Myeloid-derived suppressor cells: Linking inflammation and cancer. J. Immunol. 2009, 182, 4499–4506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Safarzadeh, E.; Hashemzadeh, S.; Duijf, P.H.G.; Mansoori, B.; Khaze, V.; Mohammadi, A.; Kazemi, T.; Yousefi, M.; Asadi, M.; Mohammadi, H.; et al. Circulating myeloid-derived suppressor cells: An independent prognostic factor in patients with breast cancer. J. Cell. Physiol. 2019, 234, 3515–3525. [Google Scholar] [CrossRef] [PubMed]
- Soysal, S.D.; Tzankov, A.; Muenst, S.E. Role of the Tumor Microenvironment in Breast Cancer. Pathobiology 2015, 82, 142–152. [Google Scholar] [CrossRef]
- De la Cruz-Merino, L.; Chiesa, M.; Caballero, R.; Rojo, F.; Palazon, N.; Carrasco, F.H.; Sánchez-Margalet, V. Breast Cancer Immunology and Immunotherapy: Current Status and Future Perspectives. Int. Rev. Cell Mol. Biol. 2017, 331, 1–53. [Google Scholar] [PubMed]
- De la Cruz-Merino, L.; Barco-Sanchez, A.; Henao Carrasco, F.; Nogales Fernandez, E.; Vallejo Benitez, A.; Brugal Molina, J.; Martínez Peinado, A.; Grueso López, A.; Ruiz Borrego, M.; de Villena, M.C.M.; et al. New insights into the role of the immune microenvironment in breast carcinoma. Clin. Dev. Immunol. 2013, 2013, 785317. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Chan, T.A.; Kroemer, G.; Wolchok, J.D.; Lopez-Soto, A. The hallmarks of successful anticancer immunotherapy. Sci. Transl. Med. 2018, 10, eaat7807. [Google Scholar] [CrossRef]
- Ye, Y.; Xu, C.; Chen, F.; Liu, Q.; Cheng, N. Targeting Innate Immunity in Breast Cancer Therapy: A Narrative Review. Front. Immunol. 2021, 12, 771201. [Google Scholar] [CrossRef] [PubMed]
- Davodabadi, F.; Sarhadi, M.; Arabpour, J.; Sargazi, S.; Rahdar, A.; Diez-Pascual, A.M. Breast cancer vaccines: New insights into immunomodulatory and nano-therapeutic approaches. J. Control. Release 2022, 349, 844–875. [Google Scholar] [CrossRef]
- Zhu, S.Y.; Yu, K.D. Breast Cancer Vaccines: Disappointing or Promising? Front. Immunol. 2022, 13, 828386. [Google Scholar] [CrossRef] [PubMed]
- Coulie, P.G.; Van den Eynde, B.J.; van der Bruggen, P.; Boon, T. Tumour antigens recognized by T lymphocytes: At the core of cancer immunotherapy. Nat. Rev. Cancer 2014, 14, 135–146. [Google Scholar] [CrossRef]
- Van der Burg, S.H. Correlates of immune and clinical activity of novel cancer vaccines. Semin. Immunol. 2018, 39, 119–136. [Google Scholar] [CrossRef] [PubMed]
- Hammerl, D.; Smid, M.; Timmermans, A.M.; Sleijfer, S.; Martens, J.W.M.; Debets, R. Breast cancer genomics and immuno-oncological markers to guide immune therapies. Semin. Cancer Biol. 2018, 52, 178–188. [Google Scholar] [CrossRef] [PubMed]
- Solinas, C.; Aiello, M.; Migliori, E.; Willard-Gallo, K.; Emens, L.A. Breast cancer vaccines: Heeding the lessons of the past to guide a path forward. Cancer Treat. Rev. 2020, 84, 101947. [Google Scholar] [CrossRef] [PubMed]
- Melero, I.; Gaudernack, G.; Gerritsen, W.; Huber, C.; Parmiani, G.; Scholl, S.; Thatcher, N.; Wagstaff, J.; Zielinski, C.; Faulkner, I.; et al. Therapeutic vaccines for cancer: An overview of clinical trials. Nat. Rev. Clin. Oncol. 2014, 11, 509–524. [Google Scholar] [CrossRef]
- Clements, C.J.; Griffiths, E. The global impact of vaccines containing aluminium adjuvants. Vaccine 2002, 20 (Suppl. 3), S24–S33. [Google Scholar] [CrossRef]
- Jager, E.; Ringhoffer, M.; Dienes, H.P.; Arand, M.; Karbach, J.; Jager, D.; Ilsemann, C.; Hagedorn, M.; Oesch, F.; Knuth, A. Granulocyte-macrophage-colony-stimulating factor enhances immune responses to melanoma-associated peptides in vivo. Int. J. Cancer 1996, 67, 54–62. [Google Scholar] [CrossRef]
- Hailemichael, Y.; Dai, Z.; Jaffarzad, N.; Ye, Y.; Medina, M.A.; Huang, X.F.; Dorta-Estremera, S.M.; Greeley, N.R.; Nitti, G.; Peng, W.; et al. Persistent antigen at vaccination sites induces tumor-specific CD8(+) T cell sequestration, dysfunction and deletion. Nat. Med. 2013, 19, 465–472. [Google Scholar] [CrossRef] [Green Version]
- Anderson, K.S. Tumor vaccines for breast cancer. Cancer Investig. 2009, 27, 361–368. [Google Scholar] [CrossRef]
- Chew, V.; Toh, H.C.; Abastado, J.P. Immune microenvironment in tumor progression: Characteristics and challenges for therapy. J. Oncol. 2012, 2012, 608406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schutz, F.; Marme, F.; Domschke, C.; Sohn, C.; von Au, A. Immunooncology in Breast Cancer: Active and Passive Vaccination Strategies. Breast Care 2018, 13, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Luiten, R.M.; Kueter, E.W.; Mooi, W.; Gallee, M.P.; Rankin, E.M.; Gerritsen, W.R.; Clift, S.M.; Nooijen, W.J.; Weder, P.; van de Kasteele, W.F.; et al. Immunogenicity, including vitiligo, and feasibility of vaccination with autologous GM-CSF-transduced tumor cells in metastatic melanoma patients. J. Clin. Oncol. 2005, 23, 8978–8991. [Google Scholar] [CrossRef] [Green Version]
- Peoples, G.E.; Gurney, J.M.; Hueman, M.T.; Woll, M.M.; Ryan, G.B.; Storrer, C.E.; Fisher, C.; Shriver, C.D.; Ioannides, C.G.; Ponniah, S. Clinical trial results of a HER2/neu (E75) vaccine to prevent recurrence in high-risk breast cancer patients. J. Clin. Oncol. 2005, 23, 7536–7545. [Google Scholar] [CrossRef] [PubMed]
- Schneble, E.J.; Berry, J.S.; Trappey, F.A.; Clifton, G.T.; Ponniah, S.; Mittendorf, E.; Peoples, G.E. The HER2 peptide nelipepimut-S (E75) vaccine (NeuVax) in breast cancer patients at risk for recurrence: Correlation of immunologic data with clinical response. Immunotherapy 2014, 6, 519–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brossart, P.; Stuhler, G.; Flad, T.; Stevanovic, S.; Rammensee, H.G.; Kanz, L.; Brugger, W. Her-2/neu-derived peptides are tumor-associated antigens expressed by human renal cell and colon carcinoma lines and are recognized by in vitro induced specific cytotoxic T lymphocytes. Cancer Res. 1998, 58, 732–736. [Google Scholar] [PubMed]
- Fisk, B.; Blevins, T.L.; Wharton, J.T.; Ioannides, C.G. Identification of an immunodominant peptide of HER-2/neu protooncogene recognized by ovarian tumor-specific cytotoxic T lymphocyte lines. J. Exp. Med. 1995, 181, 2109–2117. [Google Scholar] [CrossRef] [Green Version]
- Fisk, B.; Anderson, B.W.; Gravitt, K.R.; A O’Brian, C.; Kudelka, A.P.; Murray, J.L.; Wharton, J.T.; Ioannides, C.G. Identification of naturally processed human ovarian peptides recognized by tumor-associated CD8+ cytotoxic T lymphocytes. Cancer Res. 1997, 57, 87–93. [Google Scholar]
- Lustgarten, J.; Theobald, M.; Labadie, C.; LaFace, D.; Peterson, P.; Disis, M.L.; Cheever, M.A.; Sheran, L.A. Identification of Her-2/Neu CTL epitopes using double transgenic mice expressing HLA-A2.1 and human CD.8. Hum. Immunol. 1997, 52, 109–118. [Google Scholar] [CrossRef]
- Murray, J.L.; E Gillogly, M.; Przepiorka, N.; Brewer, H.; Ibrahim, N.K.; Booser, D.J.; Hortobagyi, G.N.; Kudelka, A.P.; Grabstein, K.H.; A Cheever, M.; et al. Toxicity, immunogenicity, and induction of E75-specific tumor-lytic CTLs by HER-2 peptide E75 (369-377) combined with granulocyte macrophage colony-stimulating factor in HLA-A2+ patients with metastatic breast and ovarian cancer. Clin. Cancer Res. 2002, 8, 3407–3418. [Google Scholar] [PubMed]
- Pallerla, S.; Abdul, A.; Comeau, J.; Jois, S. Cancer Vaccines, Treatment of the Future: With Emphasis on HER2-Positive Breast Cancer. Int. J. Mol. Sci. 2021, 22, 779. [Google Scholar] [CrossRef]
- You, Z.; Zhou, W.; Weng, J.; Feng, H.; Liang, P.; Li, Y.; Shi, F. Application of HER2 peptide vaccines in patients with breast cancer: A systematic review and meta-analysis. Cancer Cell Int. 2021, 21, 489. [Google Scholar] [CrossRef] [PubMed]
- Mittendorf, E.A.; Storrer, C.E.; Foley, R.J.; Harris, K.; Jama, Y.; Shriver, C.D.; Ponniah, S.; Peoples, G.E. Evaluation of the HER2/neu-derived peptide GP2 for use in a peptide-based breast cancer vaccine trial. Cancer 2006, 106, 2309–2317. [Google Scholar] [CrossRef]
- Humphreys, R.E.; Adams, S.; Koldzic, G.; Nedelescu, B.; von Hofe, E.; Xu, M. Increasing the potency of MHC class II-presented epitopes by linkage to Ii-Key peptide. Vaccine 2000, 18, 2693–2697. [Google Scholar] [CrossRef]
- Mittendorf, E.A.; Ardavanis, A.; Symanowski, J.; Murray, J.L.; Shumway, N.M.; Litton, J.K.; Hale, D.; Perez, S.; Anastasopoulou, E.; Pistamaltzian, N.; et al. Primary analysis of a prospective, randomized, single-blinded phase II trial evaluating the HER2 peptide AE37 vaccine in breast cancer patients to prevent recurrence. Ann. Oncol. 2016, 27, 1241–1248. [Google Scholar] [CrossRef]
- Harrington, L.; Zhou, W.; McPhail, T.; Oulton, R.; Yeung, D.S.; Mar, V.; Bass, M.B.; Robinson, M.O. Human telomerase contains evolutionarily conserved catalytic and structural subunits. Genes Dev. 1997, 11, 3109–3115. [Google Scholar] [CrossRef] [Green Version]
- Kim, N.W.; Piatyszek, M.A.; Prowse, K.R.; Harley, C.B.; West, M.D.; Ho, P.L.; Coviello, G.M.; Wright, W.E.; Weinrich, S.L.; Shay, J.W. Specific association of human telomerase activity with immortal cells and cancer. Science 1994, 266, 2011–2015. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Pankhong, P.; Shin, T.H.; Obeng-Adjei, N.; Morrow, M.P.; Walters, J.N.; Khan, A.S.; Sardesai, N.Y.; Weiner, D.B. Highly optimized DNA vaccine targeting human telomerase reverse transcriptase stimulates potent antitumor immunity. Cancer Immunol. Res. 2013, 1, 179–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Zhou, X.; Sha, H.; Xie, L.; Liu, B. Recent Progress on Therapeutic Vaccines for Breast Cancer. Front. Oncol. 2022, 12, 905832. [Google Scholar] [CrossRef] [PubMed]
- Costa, R.L.B.; Soliman, H.; Czerniecki, B.J. The clinical development of vaccines for HER2(+) breast cancer: Current landscape and future perspectives. Cancer Treat. Rev. 2017, 61, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Disis, M.L.; Schiffman, K.; Guthrie, K.; Salazar, L.G.; Knutson, K.L.; Goodell, V.; dela Rosa, C.; Cheever, M.A. Effect of dose on immune response in patients vaccinated with an her-2/neu intracellular domain protein-based vaccine. J. Clin. Oncol. 2004, 22, 1916–1925. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, E.; Blackwell, K.; Hobeika, A.C.; Clay, T.M.; Broadwater, G.; Ren, X.R.; Chen, W.; Castro, H.; Lehmann, F.; Spector, N.; et al. Phase 1 clinical trial of HER2-specific immunotherapy with concomitant HER2 kinase inhibition [corrected]. J. Transl. Med. 2012, 10, 28. [Google Scholar] [CrossRef] [Green Version]
- Li, J.J.; Tsang, J.Y.; Tse, G.M. Tumor Microenvironment in Breast Cancer-Updates on Therapeutic Implications and Pathologic Assessment. Cancers 2021, 13, 4233. [Google Scholar] [CrossRef]
- Foy, T.M.; Fanger, G.R.; Hand, S.; Gerard, C.; Bruck, C.; Cheever, M.A. Designing HER2 vaccines. Semin. Oncol. 2002, 29 (Suppl. 11), 53–61. [Google Scholar] [CrossRef] [PubMed]
- Curigliano, G.; Romieu, G.; Campone, M.; Dorval, T.; Duck, L.; Canon, J.L.; Roemer-Becuwe, C.; Roselli, M.; Neciosup, S.; Burny, W.; et al. A phase I/II trial of the safety and clinical activity of a HER2-protein based immunotherapeutic for treating women with HER2-positive metastatic breast cancer. Breast Cancer Res. Treat. 2016, 156, 301–310. [Google Scholar] [CrossRef]
- Simons, J.W.; Jaffee, E.M.; Weber, C.E.; Levitsky, H.I.; Nelson, W.G.; Carducci, M.A.; Lazenby, A.J.; Cohen, L.K.; Finn, C.C.; Clift, S.M.; et al. Bioactivity of autologous irradiated renal cell carcinoma vaccines generated by ex vivo granulocyte-macrophage colony-stimulating factor gene transfer. Cancer Res. 1997, 57, 1537–1546. [Google Scholar] [PubMed]
- Ahlert, T.; Sauerbrei, W.; Bastert, G.; Ruhland, S.; Bartik, B.; Simiantonaki, N.; Schumacher, J.; Häcker, B.; Schumacher, M.; Schirrmacher, V. Tumor-cell number and viability as quality and efficacy parameters of autologous virus-modified cancer vaccines in patients with breast or ovarian cancer. J. Clin. Oncol. 1997, 15, 1354–1366. [Google Scholar] [CrossRef]
- Jiang, X.P.; Yang, D.C.; Elliott, R.L.; Head, J.F. Vaccination with a mixed vaccine of autogenous and allogeneic breast cancer cells and tumor associated antigens CA15-3, CEA and CA125—Results in immune and clinical responses in breast cancer patients. Cancer Biother. Radiopharm. 2000, 15, 495–505. [Google Scholar] [CrossRef]
- Steinman, R.M. The dendritic cell system and its role in immunogenicity. Annu. Rev. Immunol. 1991, 9, 271–296. [Google Scholar] [CrossRef]
- Mempel, T.R.; Henrickson, S.E.; Von Andrian, U.H. T-cell priming by dendritic cells in lymph nodes occurs in three distinct phases. Nature 2004, 427, 154–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kugler, A.; Stuhler, G.; Walden, P.; Zoller, G.; Zobywalski, A.; Brossart, P.; Trefzer, U.; Ullrich, S.; Müller, C.A.; Becker, V.; et al. Regression of human metastatic renal cell carcinoma after vaccination with tumor cell-dendritic cell hybrids. Nat. Med. 2000, 6, 332–336. [Google Scholar] [CrossRef]
- Avigan, D.; Vasir, B.; Gong, J.; Borges, V.; Wu, Z.; Uhl, L.; Atkins, M.; Mier, J.; McDermott, D.; Smith, T.; et al. Fusion cell vaccination of patients with metastatic breast and renal cancer induces immunological and clinical responses. Clin. Cancer Res. 2004, 10, 4699–4708. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Yi, S.; Li, X.; Liu, R.; Jiang, H.; Huang, Z.; Liu, Y.; Wu, J.; Huang, Y. Preparation of triple-negative breast cancer vaccine through electrofusion with day-3 dendritic cells. PLoS ONE 2014, 9, e102197. [Google Scholar] [CrossRef]
- Sakai, Y.; Morrison, B.J.; Burke, J.D.; Park, J.M.; Terabe, M.; Janik, J.E.; Forni, G.; Berzofsky, J.A.; Morris, J.C. Vaccination by genetically modified dendritic cells expressing a truncated neu oncogene prevents development of breast cancer in transgenic mice. Cancer Res. 2004, 64, 8022–8028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manthorpe, M.; Cornefert-Jensen, F.; Hartikka, J.; Felgner, J.; Rundell, A.; Margalith, M.; Dwarki, V. Gene therapy by intramuscular injection of plasmid DNA: Studies on firefly luciferase gene expression in mice. Hum. Gene Ther. 1993, 4, 419–431. [Google Scholar] [CrossRef]
- Williams, J.A.; Carnes, A.E.; Hodgson, C.P. Plasmid DNA vaccine vector design: Impact on efficacy, safety and upstream production. Biotechnol. Adv. 2009, 27, 353–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norell, H.; Poschke, I.; Charo, J.; Wei, W.Z.; Erskine, C.; Piechocki, M.P.; Knutson, K.L.; Bergh, J.; Lidbrink, E.; Kiessling, R. Vaccination with a plasmid DNA encoding HER-2/neu together with low doses of GM-CSF and IL-2 in patients with metastatic breast carcinoma: A pilot clinical trial. J. Transl. Med. 2010, 8, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.W.; Goedegebuure, P.; Gillanders, W.E. Mammaglobin-A is a target for breast cancer vaccination. Oncoimmunology 2016, 5, e1069940. [Google Scholar] [CrossRef] [Green Version]
- Miles, D.W.; Towlson, K.E.; Graham, R.; Reddish, M.; Longenecker, B.M.; Taylor-Papadimitriou, J.; Rubens, R.D. A randomised phase II study of sialyl-Tn and DETOX-B adjuvant with or without cyclophosphamide pretreatment for the active specific immunotherapy of breast cancer. Br. J. Cancer 1996, 74, 1292–1296. [Google Scholar] [CrossRef]
- Miles, D.; Roche, H.; Martin, M.; Perren, T.J.; Cameron, D.A.; Glaspy, J.; Dodwell, D.; Parker, J.; Mayordomo, J.; Tres, A.; et al. Phase III multicenter clinical trial of the sialyl-TN (STn)-keyhole limpet hemocyanin (KLH) vaccine for metastatic breast cancer. Oncologist 2011, 16, 1092–1100. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, N.K.; Murray, J.L.; Zhou, D.; Mittendorf, E.A.; Sample, D.; Tautchin, M.; Miles, D. Survival Advantage in Patients with Metastatic Breast Cancer Receiving Endocrine Therapy plus Sialyl Tn-KLH Vaccine: Post Hoc Analysis of a Large Randomized Trial. J. Cancer 2013, 4, 577–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, T.J.; Chan, J.J.; Kamis, S.; Dent, R.A. What is the role of immunotherapy in breast cancer? Chin. Clin. Oncol. 2018, 7, 13. [Google Scholar] [CrossRef] [PubMed]
- Makhoul, I.; Atiq, M.; Alwbari, A.; Kieber-Emmons, T. Breast Cancer Immunotherapy: An Update. Breast Cancer 2018, 12, 1178223418774802. [Google Scholar] [CrossRef]
- Arab, A.; Yazdian-Robati, R.; Behravan, J. HER2-Positive Breast Cancer Immunotherapy: A Focus on Vaccine Development. Arch. Immunol. Ther. Exp. 2020, 68, 2. [Google Scholar] [CrossRef] [PubMed]
- Kay, C.; Martinez-Perez, C.; Meehan, J.; Gray, M.; Webber, V.; Dixon, J.M.; Turnbull, A.K. Current trends in the treatment of HR+/HER2+ breast cancer. Future Oncol. 2021, 17, 1665–1681. [Google Scholar] [CrossRef] [PubMed]
- Vu, T.; Claret, F.X. Trastuzumab: Updated mechanisms of action and resistance in breast cancer. Front. Oncol. 2012, 2, 62. [Google Scholar] [CrossRef] [Green Version]
- Kreutzfeldt, J.; Rozeboom, B.; Dey, N.; De, P. The trastuzumab era: Current and upcoming targeted HER2+ breast cancer therapies. Am. J. Cancer Res. 2020, 10, 1045–1067. [Google Scholar]
- Musolino, A.; Gradishar, W.J.; Rugo, H.S.; Nordstrom, J.L.; Rock, E.P.; Arnaldez, F.; Pegram, M.D. Role of Fcgamma receptors in HER2-targeted breast cancer therapy. J. Immunother. Cancer 2022, 10, e003171. [Google Scholar] [CrossRef] [PubMed]
- Cassatella, M.A.; Anegon, I.; Cuturi, M.C.; Griskey, P.; Trinchieri, G.; Perussia, B. Fc gamma R(CD16) interaction with ligand induces Ca2+ mobilization and phosphoinositide turnover in human natural killer cells. Role of Ca2+ in Fc gamma R(CD16)-induced transcription and expression of lymphokine genes. J. Exp. Med. 1989, 169, 549–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mando, P.; Rivero, S.G.; Rizzo, M.M.; Pinkasz, M.; Levy, E.M. Targeting ADCC: A different approach to HER2 breast cancer in the immunotherapy era. Breast 2021, 60, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Mercogliano, M.F.; Bruni, S.; Elizalde, P.V.; Schillaci, R. Tumor Necrosis Factor alpha Blockade: An Opportunity to Tackle Breast Cancer. Front. Oncol. 2020, 10, 584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salgado, R.; Denkert, C.; Demaria, S.; Sirtaine, N.; Klauschen, F.; Pruneri, G.; Wienert, S.; Van den Eynden, G.; Baehner, F.L.; Penault-Llorca, F.; et al. The evaluation of tumor-infiltrating lymphocytes (TILs) in breast cancer: Recommendations by an International TILs Working Group 2014. Ann. Oncol. 2015, 26, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Ingold Heppner, B.; Untch, M.; Denkert, C.; Pfitzner, B.M.; Lederer, B.; Schmitt, W.; Eidtmann, H.; Fasching, P.A.; Tesch, H.; Solbach, C.; et al. Tumor-Infiltrating Lymphocytes: A Predictive and Prognostic Biomarker in Neoadjuvant-Treated HER2-Positive Breast Cancer. Clin. Cancer Res. 2016, 22, 5747–5754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carbognin, L.; Pilotto, S.; Nortilli, R.; Brunelli, M.; Nottegar, A.; Sperduti, I.; Giannarelli, D.; Bria, E.; Tortora, G. Predictive and Prognostic Role of Tumor-Infiltrating Lymphocytes for Early Breast Cancer According to Disease Subtypes: Sensitivity Analysis of Randomized Trials in Adjuvant and Neoadjuvant Setting. Oncologist 2016, 21, 283–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bense, R.D.; Sotiriou, C.; Piccart-Gebhart, M.J.; Haanen, J.; van Vugt, M.; de Vries, E.G.E.; Schröder, C.P.; Fehrmann, R.S.N. Relevance of Tumor-Infiltrating Immune Cell Composition and Functionality for Disease Outcome in Breast Cancer. J. Natl. Cancer Inst. 2017, 109, djw192. [Google Scholar] [CrossRef] [Green Version]
- Plitas, G.; Konopacki, C.; Wu, K.; Bos, P.D.; Morrow, M.; Putintseva, E.V.; Chudakov, D.M.; Rudensky, A.Y. Regulatory T Cells Exhibit Distinct Features in Human Breast Cancer. Immunity 2016, 45, 1122–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behravan, J.; Razazan, A.; Behravan, G. Towards Breast Cancer Vaccines, Progress and Challenges. Curr. Drug Discov. Technol. 2019, 16, 251–258. [Google Scholar] [CrossRef]
- Yamashita-Kashima, Y.; Shu, S.; Yorozu, K.; Moriya, Y.; Harada, N. Mode of action of pertuzumab in combination with trastuzumab plus docetaxel therapy in a HER2-positive breast cancer xenograft model. Oncol. Lett. 2017, 14, 4197–4205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nami, B.; Maadi, H.; Wang, Z. Mechanisms Underlying the Action and Synergism of Trastuzumab and Pertuzumab in Targeting HER2-Positive Breast Cancer. Cancers 2018, 10, 342. [Google Scholar] [CrossRef] [Green Version]
- Kunte, S.; Abraham, J.; Montero, A.J. Novel HER2-targeted therapies for HER2-positive metastatic breast cancer. Cancer 2020, 126, 4278–4288. [Google Scholar] [CrossRef] [PubMed]
- Nordstrom, J.L.; Gorlatov, S.; Zhang, W.; Yang, Y.; Huang, L.; Burke, S.; Li, H.; Ciccarone, V.; Zhang, T.; Stavenhagen, J.; et al. Anti-tumor activity and toxicokinetics analysis of MGAH22, an anti-HER2 monoclonal antibody with enhanced Fcgamma receptor binding properties. Breast Cancer Res. 2011, 13, R123. [Google Scholar] [CrossRef] [Green Version]
- Bang, Y.J.; Giaccone, G.; Im, S.A.; Oh, D.Y.; Bauer, T.M.; Nordstrom, J.L.; Li, H.; Chichili, G.; Moore, P.; Hong, S.; et al. First-in-human phase 1 study of margetuximab (MGAH22), an Fc-modified chimeric monoclonal antibody, in patients with HER2-positive advanced solid tumors. Ann. Oncol. 2017, 28, 855–861. [Google Scholar] [CrossRef]
- Rugo, H.S.; Im, S.A.; Cardoso, F.; Cortes, J.; Curigliano, G.; Musolino, A.; Pegram, M.D.; Wright, G.S.; Saura, C.; Escrivá-de-Romaní, S.; et al. Efficacy of Margetuximab vs Trastuzumab in Patients With Pretreated ERBB2-Positive Advanced Breast Cancer: A Phase 3 Randomized Clinical Trial. JAMA Oncol. 2021, 7, 573–584. [Google Scholar] [CrossRef]
- Barroso-Sousa, R.; Tolaney, S.M. Clinical Development of New Antibody-Drug Conjugates in Breast Cancer: To Infinity and Beyond. BioDrugs 2021, 35, 159–174. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhang, D.; Liu, B.; Lv, D.; Zhai, J.; Guan, X.; Yi, Z.; Ma, F. Antibody-drug conjugates in HER2-positive breast cancer. Chin. Med. J. 2021, 135, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Vranic, S.; Beslija, S.; Gatalica, Z. Targeting HER2 expression in cancer: New drugs and new indications. Bosn. J. Basic Med. Sci. 2021, 21, 1–4. [Google Scholar] [CrossRef]
- Hunter, F.W.; Barker, H.R.; Lipert, B.; Rothe, F.; Gebhart, G.; Piccart-Gebhart, M.J.; Sotiriou, C.; Jamieson, S.M.F. Mechanisms of resistance to trastuzumab emtansine (T-DM1) in HER2-positive breast cancer. Br. J. Cancer 2020, 122, 603–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartsch, R. SABCS 2020: Update on triple-negative and metastatic HER2-positive breast cancer. Memo 2021, 14, 247–251. [Google Scholar] [CrossRef]
- Indini, A.; Rijavec, E.; Grossi, F. Trastuzumab Deruxtecan: Changing the Destiny of HER2 Expressing Solid Tumors. Int. J. Mol. Sci. 2021, 22, 4774. [Google Scholar] [CrossRef] [PubMed]
- Doi, T.; Shitara, K.; Naito, Y.; Shimomura, A.; Fujiwara, Y.; Yonemori, K.; Shimizu, C.; Shimoi, T.; Kuboki, Y.; Matsubara, N.; et al. Safety, pharmacokinetics, and antitumour activity of trastuzumab deruxtecan (DS-8201), a HER2-targeting antibody-drug conjugate, in patients with advanced breast and gastric or gastro-oesophageal tumours: A phase 1 dose-escalation study. Lancet Oncol. 2017, 18, 1512–1522. [Google Scholar] [CrossRef]
- Lee, J.; Park, Y.H. Trastuzumab deruxtecan for HER2+ advanced breast cancer. Future Oncol. 2022, 18, 7–19. [Google Scholar] [CrossRef]
- Vranic, S.; Gatalica, Z. Trop-2 protein as a therapeutic target: A focused review on Trop-2-based antibody-drug conjugates and their predictive biomarkers. Bosn. J. Basic Med. Sci. 2022, 22, 14–21. [Google Scholar] [CrossRef]
- Spring, L.M.; Nakajima, E.; Hutchinson, J.; Viscosi, E.; Blouin, G.; Weekes, C.; Rugo, H.; Moy, B.; Bardia, A. Sacituzumab Govitecan for Metastatic Triple-Negative Breast Cancer: Clinical Overview and Management of Potential Toxicities. Oncologist 2021, 26, 827–834. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, P.; Scatena, C.; Ghilli, M.; Bargagna, I.; Lorenzini, G.; Nicolini, A. Molecular Mechanisms, Biomarkers and Emerging Therapies for Chemotherapy Resistant TNBC. Int. J. Mol. Sci. 2022, 23, 1665. [Google Scholar] [CrossRef] [PubMed]
- Adams, E.; Wildiers, H.; Neven, P.; Punie, K. Sacituzumab govitecan and trastuzumab deruxtecan: Two new antibody-drug conjugates in the breast cancer treatment landscape. ESMO Open 2021, 6, 100204. [Google Scholar] [CrossRef] [PubMed]
- Sussman, D.; Smith, L.M.; Anderson, M.E.; Duniho, S.; Hunter, J.H.; Kostner, H.; Miyamoto, J.B.; Nesterova, A.; Westendorf, L.; Van Epps, H.A.; et al. SGN-LIV1A: A novel antibody-drug conjugate targeting LIV-1 for the treatment of metastatic breast cancer. Mol. Cancer Ther. 2014, 13, 2991–3000. [Google Scholar] [CrossRef] [Green Version]
- Rose, A.A.; Grosset, A.A.; Dong, Z.; Russo, C.; Macdonald, P.A.; Bertos, N.R.; St-Pierre, Y.; Simantov, R.; Hallett, M.; Park, M.; et al. Glycoprotein nonmetastatic B is an independent prognostic indicator of recurrence and a novel therapeutic target in breast cancer. Clin. Cancer Res. 2010, 16, 2147–2156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rose, A.A.; Pepin, F.; Russo, C.; Abou Khalil, J.E.; Hallett, M.; Siegel, P.M. Osteoactivin promotes breast cancer metastasis to bone. Mol. Cancer Res. 2007, 5, 1001–1014. [Google Scholar] [CrossRef] [Green Version]
- Okita, Y.; Kimura, M.; Xie, R.; Chen, C.; Shen, L.T.; Kojima, Y.; Suzuki, H.; Muratani, M.; Saitoh, M.; Semba, K.; et al. The transcription factor MAFK induces EMT and malignant progression of triple-negative breast cancer cells through its target GPNMB. Sci. Signal. 2017, 10, eaak9397. [Google Scholar] [CrossRef] [PubMed]
- Vahdat, L.T.; Schmid, P.; Forero-Torres, A.; Blackwell, K.; Telli, M.L.; Melisko, M.; Möbus, V.; Cortes, J.; Montero, A.J.; Ma, C.; et al. Glembatumumab vedotin for patients with metastatic, gpNMB overexpressing, triple-negative breast cancer (“METRIC”): A randomized multicenter study. NPJ Breast Cancer 2021, 7, 57. [Google Scholar] [CrossRef]
- Yao, H.P.; Suthe, S.R.; Hudson, R.; Wang, M.H. Antibody-drug conjugates targeting RON receptor tyrosine kinase as a novel strategy for treatment of triple-negative breast cancer. Drug Discov. Today 2020, 25, 1160–1173. [Google Scholar] [CrossRef]
- Suthe, S.R.; Yao, H.P.; Weng, T.H.; Hu, C.Y.; Feng, L.; Wu, Z.G.; Wang, M.-H. RON Receptor Tyrosine Kinase as a Therapeutic Target for Eradication of Triple-Negative Breast Cancer: Efficacy of Anti-RON ADC Zt/g4-MMAE. Mol. Cancer Ther. 2018, 17, 2654–2664. [Google Scholar] [CrossRef] [Green Version]
- Weng, T.H.; Yao, M.Y.; Xu, X.M.; Hu, C.Y.; Yao, S.H.; Liu, Y.Z.; Wu, Z.-G.; Tang, T.-M.; Fu, P.-F.; Wang, M.-H.; et al. RON and MET Co-overexpression Are Significant Pathological Characteristics of Poor Survival and Therapeutic Targets of Tyrosine Kinase Inhibitors in Triple-Negative Breast Cancer. Cancer Res. Treat. 2020, 52, 973–986. [Google Scholar] [CrossRef]
- Purcell, J.W.; Tanlimco, S.G.; Hickson, J.; Fox, M.; Sho, M.; Durkin, L.; Uziel, T.; Powers, R.; Foster, K.; McGonigal, T.; et al. LRRC15 Is a Novel Mesenchymal Protein and Stromal Target for Antibody-Drug Conjugates. Cancer Res. 2018, 78, 4059–4072. [Google Scholar] [CrossRef] [Green Version]
- Ray, U.; Pathoulas, C.L.; Thirusangu, P.; Purcell, J.W.; Kannan, N.; Shridhar, V. Exploiting LRRC15 as a Novel Therapeutic Target in Cancer. Cancer Res. 2022, 82, 1675–1681. [Google Scholar] [CrossRef] [PubMed]
- Demetri, G.D.; Luke, J.J.; Hollebecque, A.; Powderly, J.D., 2nd; Spira, A.I.; Subbiah, V.; Naumovski, L.; Chen, C.; Fang, H.; Lai, D.W.; et al. First-in-Human Phase I Study of ABBV-085, an Antibody-Drug Conjugate Targeting LRRC15, in Sarcomas and Other Advanced Solid Tumors. Clin. Cancer Res. 2021, 27, 3556–3566. [Google Scholar] [CrossRef] [PubMed]
- Miglietta, F.; Cona, M.S.; Dieci, M.V.; Guarneri, V.; La Verde, N. An overview of immune checkpoint inhibitors in breast cancer. Explor Target Antitumor Ther. 2020, 1, 452–472. [Google Scholar] [CrossRef]
- De la Cruz-Merino, L.; Palazon-Carrion, N.; Henao-Carrasco, F.; Nogales-Fernandez, E.; Alamo-de la Gala, M.; Vallejo-Benitez, A.; Chiesa, M.; Sánchez-Margalet, V.; GEICAM (Spanish Breast Cancer Research Group); GÉTICA (Spanish Group for Cancer Immuno-Biotherapy). New horizons in breast cancer: The promise of immunotherapy. Clin. Transl. Oncol. 2019, 21, 117–125. [Google Scholar] [CrossRef] [Green Version]
- Tomioka, N.; Azuma, M.; Ikarashi, M.; Yamamoto, M.; Sato, M.; Watanabe, K.I.; Yamashiro, K.; Takahashi, M. The therapeutic candidate for immune checkpoint inhibitors elucidated by the status of tumor-infiltrating lymphocytes (TILs) and programmed death ligand 1 (PD-L1) expression in triple negative breast cancer (TNBC). Breast Cancer 2018, 25, 34–42. [Google Scholar] [CrossRef]
- Kitano, A.; Ono, M.; Yoshida, M.; Noguchi, E.; Shimomura, A.; Shimoi, T.; Kodaira, M.; Yunokawa, M.; Yonemori, K.; Shimizu, C.; et al. Tumour-infiltrating lymphocytes are correlated with higher expression levels of PD-1 and PD-L1 in early breast cancer. ESMO Open 2017, 2, e000150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Kim, Y.; Kim, H.Y.; Goo, J.M.; Lim, J.; Lee, C.T.; Jang, S.H.; Lee, W.-C.; Lee, C.W.; Choi, K.S.; et al. Feasibility of implementing a national lung cancer screening program: Interim results from the Korean Lung Cancer Screening Project (K-LUCAS). Transl. Lung Cancer Res. 2021, 10, 723–736. [Google Scholar] [CrossRef]
- Sharma, P.; Allison, J.P. Immune checkpoint targeting in cancer therapy: Toward combination strategies with curative potential. Cell 2015, 161, 205–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veluswamy, P.; Wacker, M.; Scherner, M.; Wippermann, J. Delicate Role of PD-L1/PD-1 Axis in Blood Vessel Inflammatory Diseases: Current Insight and Future Significance. Int. J. Mol. Sci. 2020, 21, 8159. [Google Scholar] [CrossRef] [PubMed]
- Okla, K.; Rajtak, A.; Czerwonka, A.; Bobinski, M.; Wawruszak, A.; Tarkowski, R.; Bednarek, W.; Szumiło, J.; Kotarski, J. Correction to: Accumulation of blood-circulating PD-L1-expressing M-MDSCs and monocytes/macrophages in pretreatment ovarian cancer patients is associated with soluble PD-L1. J. Transl. Med. 2020, 18, 258. [Google Scholar] [CrossRef]
- Zhang, J.Y.; Yan, Y.Y.; Li, J.J.; Adhikari, R.; Fu, L.W. PD-1/PD-L1 Based Combinational Cancer Therapy: Icing on the Cake. Front. Pharmacol. 2020, 11, 722. [Google Scholar] [CrossRef] [PubMed]
- Uchimiak, K.; Badowska-Kozakiewicz, A.M.; Sobiborowicz-Sadowska, A.; Deptala, A. Current State of Knowledge on the Immune Checkpoint Inhibitors in Triple-Negative Breast Cancer Treatment: Approaches, Efficacy, and Challenges. Clin. Med. Insights Oncol. 2022, 16, 11795549221099869. [Google Scholar] [CrossRef] [PubMed]
- Marhelava, K.; Pilch, Z.; Bajor, M.; Graczyk-Jarzynka, A.; Zagozdzon, R. Targeting Negative and Positive Immune Checkpoints with Monoclonal Antibodies in Therapy of Cancer. Cancers 2019, 11, 1756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Johnson, K.C.C.; Gatti-Mays, M.E.; Li, Z. Emerging strategies in targeting tumor-resident myeloid cells for cancer immunotherapy. J. Hematol. Oncol. 2022, 15, 118. [Google Scholar] [CrossRef] [PubMed]
- Heeke, A.L.; Tan, A.R. Checkpoint inhibitor therapy for metastatic triple-negative breast cancer. Cancer Metastasis Rev. 2021, 40, 537–547. [Google Scholar] [CrossRef]
- Rizzo, A.; Cusmai, A.; Acquafredda, S.; Giovannelli, F.; Rinaldi, L.; Misino, A.; Palmiotti, G. KEYNOTE-522, IMpassion031 and GeparNUEVO: Changing the paradigm of neoadjuvant immune checkpoint inhibitors in early triple-negative breast cancer. Future Oncol. 2022, 18, 2301–2309. [Google Scholar] [CrossRef]
- Kwapisz, D. Pembrolizumab and atezolizumab in triple-negative breast cancer. Cancer Immunol. Immunother. 2021, 70, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Howard, F.M.; Villamar, D.; He, G.; Pearson, A.T.; Nanda, R. The emerging role of immune checkpoint inhibitors for the treatment of breast cancer. Expert Opin. Investig. Drugs 2022, 31, 531–548. [Google Scholar] [CrossRef]
- Howard, F.M.; Pearson, A.T.; Nanda, R. Clinical trials of immunotherapy in triple-negative breast cancer. Breast Cancer Res. Treat. 2022, 195, 1–15. [Google Scholar] [CrossRef]
- Zhou, Z.; Li, M. Evaluation of BRCA1 and BRCA2 as Indicators of Response to Immune Checkpoint Inhibitors. JAMA Netw. Open 2021, 4, e217728. [Google Scholar] [CrossRef] [PubMed]
- Samstein, R.M.; Lee, C.H.; Shoushtari, A.N.; Hellmann, M.D.; Shen, R.; Janjigian, Y.Y.; Barron, D.A.; Zehir, A.; Jordan, E.J.; Omuro, A.; et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 2019, 51, 202–206. [Google Scholar] [CrossRef]
- Nolan, E.; Savas, P.; Policheni, A.N.; Darcy, P.K.; Vaillant, F.; Mintoff, C.P.; Dushyanthen, S.; Mansour, M.; Pang, J.-M.B.; Fox, S.B.; et al. Combined immune checkpoint blockade as a therapeutic strategy for BRCA1-mutated breast cancer. Sci. Transl. Med. 2017, 9, eaal4922. [Google Scholar] [CrossRef] [PubMed]
- Bianchini, G.; Balko, J.M.; Mayer, I.A.; Sanders, M.E.; Gianni, L. Triple-negative breast cancer: Challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol. 2016, 13, 674–690. [Google Scholar] [CrossRef]
- Domchek, S.M.; Postel-Vinay, S.; Im, S.A.; Park, Y.H.; Delord, J.P.; Italiano, A.; Alexandre, J.; You, B.; Bastian, S.; Krebs, M.G.; et al. Olaparib and durvalumab in patients with germline BRCA-mutated metastatic breast cancer (MEDIOLA): An open-label, multicentre, phase 1/2, basket study. Lancet Oncol. 2020, 21, 1155–1164. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. BRCAness revisited. Nat. Rev. Cancer 2016, 16, 110–120. [Google Scholar] [CrossRef]
- Vinayak, S.; Tolaney, S.M.; Schwartzberg, L.; Mita, M.; McCann, G.; Tan, A.R.; Wahner-Hendrickson, A.E.; Forero, A.; Anders, C.; Wulf, G.M.; et al. Open-label Clinical Trial of Niraparib Combined With Pembrolizumab for Treatment of Advanced or Metastatic Triple-Negative Breast Cancer. JAMA Oncol. 2019, 5, 1132–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chabanon, R.M.; Soria, J.C.; Lord, C.J.; Postel-Vinay, S. Beyond DNA repair: The novel immunological potential of PARP inhibitors. Mol. Cell. Oncol. 2019, 6, 1585170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sisirak, V.; Faget, J.; Gobert, M.; Goutagny, N.; Vey, N.; Treilleux, I.; Renaudineau, S.; Poyet, G.; Labidi-Galy, S.I.; Goddard-Leon, S.; et al. Impaired IFN-alpha production by plasmacytoid dendritic cells favors regulatory T-cell expansion that may contribute to breast cancer progression. Cancer Res. 2012, 72, 5188–5197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, R.; Al-Khadairi, G.; Decock, J. Immune Checkpoint Inhibitors in Triple Negative Breast Cancer Treatment: Promising Future Prospects. Front. Oncol. 2020, 10, 600573. [Google Scholar] [CrossRef]
- De La Cruz, L.M.; Czerniecki, B.J. Immunotherapy for Breast Cancer is Finally at the Doorstep: Immunotherapy in Breast Cancer. Ann. Surg. Oncol. 2018, 25, 2852–2857. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I. Myeloid-Derived Suppressor Cells. Cancer Immunol. Res. 2017, 5, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Millrud, C.R.; Bergenfelz, C.; Leandersson, K. On the origin of myeloid-derived suppressor cells. Oncotarget 2017, 8, 3649–3665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostrand-Rosenberg, S.; Fenselau, C. Myeloid-Derived Suppressor Cells: Immune-Suppressive Cells That Impair Antitumor Immunity and Are Sculpted by Their Environment. J. Immunol. 2018, 200, 422–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bronte, V.; Brandau, S.; Chen, S.H.; Colombo, M.P.; Frey, A.B.; Greten, T.F.; Mandruzzato, S.; Murray, P.J.; Ochoa, A.; Ostrand-Rosenberg, S.; et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat. Commun. 2016, 7, 12150. [Google Scholar] [CrossRef] [Green Version]
- Bergenfelz, C.; Leandersson, K. The Generation and Identity of Human Myeloid-Derived Suppressor Cells. Front. Oncol. 2020, 10, 109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jimenez-Cortegana, C.; Palazon-Carrion, N.; Martin Garcia-Sancho, A.; Nogales-Fernandez, E.; Carnicero-Gonzalez, F.; Rios-Herranz, E.; de la Cruz-Vicente, F.; Rodríguez-García, G.; Fernández-Álvarez, R.; Dominguez, A.R.; et al. Circulating myeloid-derived suppressor cells and regulatory T cells as immunological biomarkers in refractory/relapsed diffuse large B-cell lymphoma: Translational results from the R2-GDP-GOTEL trial. J. Immunother. Cancer 2021, 9, e002323. [Google Scholar] [CrossRef]
- Sica, A.; Bronte, V. Altered macrophage differentiation and immune dysfunction in tumor development. J. Clin. Investig. 2007, 117, 1155–1166. [Google Scholar] [CrossRef] [Green Version]
- Shou, D.; Wen, L.; Song, Z.; Yin, J.; Sun, Q.; Gong, W. Suppressive role of myeloid-derived suppressor cells (MDSCs) in the microenvironment of breast cancer and targeted immunotherapies. Oncotarget 2016, 7, 64505–64511. [Google Scholar] [CrossRef] [Green Version]
- Cha, Y.J.; Koo, J.S. Role of Tumor-Associated Myeloid Cells in Breast Cancer. Cells 2020, 9, 1785. [Google Scholar] [CrossRef]
- De Cicco, P.; Ercolano, G.; Ianaro, A. The New Era of Cancer Immunotherapy: Targeting Myeloid-Derived Suppressor Cells to Overcome Immune Evasion. Front. Immunol. 2020, 11, 1680. [Google Scholar] [CrossRef]
- Markowitz, J.; Wesolowski, R.; Papenfuss, T.; Brooks, T.R.; Carson, W.E., 3rd. Myeloid-derived suppressor cells in breast cancer. Breast Cancer Res. Treat. 2013, 140, 13–21. [Google Scholar] [CrossRef] [Green Version]
- Bergenfelz, C.; Larsson, A.M.; von Stedingk, K.; Gruvberger-Saal, S.; Aaltonen, K.; Jansson, S.; Jernström, H.; Janols, H.; Wullt, M.; Bredberg, A.; et al. Systemic Monocytic-MDSCs Are Generated from Monocytes and Correlate with Disease Progression in Breast Cancer Patients. PLoS ONE 2015, 10, e0127028. [Google Scholar] [CrossRef] [Green Version]
- Palazon-Carrion, N.; Jimenez-Cortegana, C.; Sanchez-Leon, M.L.; Henao-Carrasco, F.; Nogales-Fernandez, E.; Chiesa, M.; Caballero, R.; Rojo, F.; Nieto-García, M.A.; Sánchez-Margalet, V.; et al. Circulating immune biomarkers in peripheral blood correlate with clinical outcomes in advanced breast cancer. Sci. Rep. 2021, 11, 14426. [Google Scholar] [PubMed]
- De la Cruz-Merino, L.; Gion, M.; Cruz, J.; Alonso-Romero, J.L.; Quiroga, V.; Moreno, F.; Andrés, R.; Santisteban, M.; Ramos, M.; Holgado, E.; et al. Pembrolizumab in combination with gemcitabine for patients with HER2-negative advanced breast cancer: GEICAM/2015-04 (PANGEA-Breast) study. BMC Cancer 2022, 22, 1258. [Google Scholar] [CrossRef] [PubMed]
- Tu, S.P.; Jin, H.; Shi, J.D.; Zhu, L.M.; Suo, Y.; Lu, G.; Liu, A.; Wang, T.C.; Yang, C.S. Curcumin induces the differentiation of myeloid-derived suppressor cells and inhibits their interaction with cancer cells and related tumor growth. Cancer Prev. Res. 2012, 5, 205–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banik, U.; Parasuraman, S.; Adhikary, A.K.; Othman, N.H. Curcumin: The spicy modulator of breast carcinogenesis. J. Exp. Clin. Cancer Res. 2017, 36, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farghadani, R.; Naidu, R. Curcumin: Modulator of Key Molecular Signaling Pathways in Hormone-Independent Breast Cancer. Cancers 2021, 13, 3427. [Google Scholar] [CrossRef]
- Cao, Y.; Slaney, C.Y.; Bidwell, B.N.; Parker, B.S.; Johnstone, C.N.; Rautela, J.; Eckhardt, B.L.; Anderson, R.L. BMP4 inhibits breast cancer metastasis by blocking myeloid-derived suppressor cell activity. Cancer Res. 2014, 74, 5091–5102. [Google Scholar] [CrossRef] [Green Version]
- Roland, C.L.; Lynn, K.D.; Toombs, J.E.; Dineen, S.P.; Udugamasooriya, D.G.; Brekken, R.A. Cytokine levels correlate with immune cell infiltration after anti-VEGF therapy in preclinical mouse models of breast cancer. PLoS ONE 2009, 4, e7669. [Google Scholar] [CrossRef] [PubMed]
- Richard, V.; Kindt, N.; Saussez, S. Macrophage migration inhibitory factor involvement in breast cancer (Review). Int. J. Oncol. 2015, 47, 1627–1633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simpson, K.D.; Templeton, D.J.; Cross, J.V. Macrophage migration inhibitory factor promotes tumor growth and metastasis by inducing myeloid-derived suppressor cells in the tumor microenvironment. J. Immunol. 2012, 189, 5533–5540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forghani, P.; Khorramizadeh, M.R.; Waller, E.K. Silibinin inhibits accumulation of myeloid-derived suppressor cells and tumor growth of murine breast cancer. Cancer Med. 2014, 3, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Thakur, A.; Schalk, D.; Sarkar, S.H.; Al-Khadimi, Z.; Sarkar, F.H.; Lum, L.G. A Th1 cytokine-enriched microenvironment enhances tumor killing by activated T cells armed with bispecific antibodies and inhibits the development of myeloid-derived suppressor cells. Cancer Immunol. Immunother. 2012, 61, 497–509. [Google Scholar] [CrossRef]
- Kmieciak, M.; Basu, D.; Payne, K.K.; Toor, A.; Yacoub, A.; Wang, X.Y.; Smith, L.; Bear, H.D.; Manjili, M.H. Activated NKT cells and NK cells render T cells resistant to myeloid-derived suppressor cells and result in an effective adoptive cellular therapy against breast cancer in the FVBN202 transgenic mouse. J. Immunol. 2011, 187, 708–717. [Google Scholar] [CrossRef] [Green Version]
- Chandra, D.; Jahangir, A.; Quispe-Tintaya, W.; Einstein, M.H.; Gravekamp, C. Myeloid-derived suppressor cells have a central role in attenuated Listeria monocytogenes-based immunotherapy against metastatic breast cancer in young and old mice. Br. J. Cancer 2013, 108, 2281–2290. [Google Scholar] [CrossRef] [Green Version]
- Batalha, S.; Ferreira, S.; Brito, C. The Peripheral Immune Landscape of Breast Cancer: Clinical Findings and In Vitro Models for Biomarker Discovery. Cancers 2021, 13, 1305. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar]
- Miller, L.D.; Chou, J.A.; Black, M.A.; Print, C.; Chifman, J.; Alistar, A.; Putti, T.; Zhou, X.; Bedognetti, D.; Hendrickx, W.; et al. Immunogenic Subtypes of Breast Cancer Delineated by Gene Classifiers of Immune Responsiveness. Cancer Immunol. Res. 2016, 4, 600–610. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Li, M.; Jiang, Z.; Wang, X. A Comprehensive Immunologic Portrait of Triple-Negative Breast Cancer. Transl. Oncol. 2018, 11, 311–329. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| PARPi + Immunotherapy | Class | Settings | Clinical Trial Phase | Status | Clinical Trials Reference |
|---|---|---|---|---|---|
| Talazoparib + Avelumab | PARPi + anti-PD-L1 | Metastatic solid tumor (TNBC, NSCLC, UC, CRPC) | II | Completed * | NCT03330405 |
| Niraparib + Pembrolizumab | PARPi + anti-PD-1 | TNBC or recurrent ovarian cancer | I/II | Completed Has Results | NCT02657889 (TOPACIO) |
| Olaparib + Atezolizumab | PARPi + anti-PDL-1 | Non-HER2positive mBC | II | Active, not recruiting | NCT02849496 |
| Olaparib + Durvalumab | PARPi + anti-PDL-1 | mTBNC | II | Completed Has Results | NCT03167619 (DORA) |
| Olaparib + MEDI4736 | PARPi + anti-PDL-1 | gBRCAm HER2 negative mBC | I/II | Active, not recruiting | NCT02734004 |
| Immunotherapy | Treatment | Additional Treatments | Settings | Clinical Trial Phase | Status | Clinical Trial Reference |
|---|---|---|---|---|---|---|
| Vaccines | Talimogene laherparepvec | Paclitaxel | TNBC | I/II | Active, not recruiting | NCT02779855 |
| Mammaglobin-A DNA | Neoadjuvant hormonal therapy | HR+ BC | IB | Recruiting | NCT02204098 | |
| PVX-410 | Alone/Combined with Durvalumab | Stage II/ III TNBC | IB | Active, not recruiting | NCT02826434 | |
| Nelipepimut-S + GM-CSF | Trastuzumab | HER2+ BC high risk | II | Completed * | NCT02297698 | |
| Monoclonal antibodies/antibody drug conjugates | QL1209 | Trastuzumab, docetaxel | Early/locally advanced ER/PR- BC | III | Not yet recruiting | NCT04629846 |
| Pyrotinib | Epirrubicin, cyclophosphamide͢͢͢, taxanes, trastuzumab | Moderate/high risk early BC | II/III | Not yet recruiting | NCT04290793 | |
| Trastuzumab Deruxtecan | Pertuzumab, placebo | Metastatic | III | Recruiting | NCT04784715 | |
| Pyrotinib | Trastuzumab, docetaxel | Metastatic | III | Recruiting | NCT03863223 | |
| Margetuximab | Capecitabine, vinorelbine, Gemcitabine, Eribulin | Metastatic | III | Active, not recruiting | NCT02492711 | |
| Paclitaxel, pertuzumab | Neoadjuvant | II | Recruiting | NCT04425018 | ||
| Anti-PD-1 | Nivolumab | Nanoparticle albumin-bound paclitaxel ± Gemcitabine or Carboplatin | Metastatic | II | Recruiting | NCT02309177 |
| Nivolumab | Paclitaxel, cyclophosphamide, endrocine therapy, anthracycline | Metastatic | III | Active, not recruiting | NCT0419066 | |
| Nivolumab | Low dose doxorubicin, cisplatin | Metastatic | II | Recruiting | NCT04159818 | |
| Pembrolizumab | Carboplatin | Metastatic | II | Recruiting | NCT03213041 | |
| Pembrolizumab | Abemaciclib | Metastatic | Ib | Active, not recruiting | NCT02779751 | |
| Pembrolizumab | Trastuzumab emtansine | Metastatic | Ib | Active, not recruiting | NCT03032107 | |
| Anti-PD-L1 | Atezolizumab | Carboplatin | Metastatic | II | Active, not recruiting | NCT03206203 |
| Durvalumab | Hypofractionated RT, Tremelimumab | Metastatic | I | Active, not recruiting | NCT02639026 | |
| Avelumab | Palbociclib and Fulvestrant | Metastatic | II | Active, not recruiting | NCT03147287 |
| Therapy | Combination Therapy | MDSC Target | Additional Tumors Tested | Clinical Trial Phase | Status | Clinical Trial Reference |
|---|---|---|---|---|---|---|
| Entinostat | Nivolumab | Class I HDCA | - | I | Active, not recruiting | NCT02453620 |
| IPI-549 | Nivolumab | PI3K | NSCLC, SCCHN, AdC, MEL, MES | I/Ib | Active, not recruiting | NCT02637531 |
| IPI-549 | Tecentriq + Abraxane | PI3K | - | II | Active, not recruiting | NCT03961698 |
| Reparixin | Paclitaxel | CXCR2 | - | II | Completed | NCT02370238 |
| AB928 | IPI-549, PLD, NP | AzaR and AzbR | Ovarian | I/Ib | Completed | NCT03719326 |
| PD-0360324 | Avelumab | CSF-1 | A variety of advanced tumors (including BC) | Ib/II | Active, not recruiting | NCT02554812 |
| Ciclophosphamide/Decitabine/carboplatn/paclitaxel/Doxorubicin | Pembrolizumab | PD-1 | - | II | Active, not recruiting | NCT02957968 |
| Leronlimab | Carboplatin | CCR5 | - | Ib/II | Active, not recruiting | NCT03838367 |
| Imiquimod | Paclitaxel | TLR7 | - | II | Completed | NCT00821964 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sánchez-León, M.L.; Jiménez-Cortegana, C.; Silva Romeiro, S.; Garnacho, C.; de la Cruz-Merino, L.; García-Domínguez, D.J.; Hontecillas-Prieto, L.; Sánchez-Margalet, V. Defining the Emergence of New Immunotherapy Approaches in Breast Cancer: Role of Myeloid-Derived Suppressor Cells. Int. J. Mol. Sci. 2023, 24, 5208. https://doi.org/10.3390/ijms24065208
Sánchez-León ML, Jiménez-Cortegana C, Silva Romeiro S, Garnacho C, de la Cruz-Merino L, García-Domínguez DJ, Hontecillas-Prieto L, Sánchez-Margalet V. Defining the Emergence of New Immunotherapy Approaches in Breast Cancer: Role of Myeloid-Derived Suppressor Cells. International Journal of Molecular Sciences. 2023; 24(6):5208. https://doi.org/10.3390/ijms24065208
Chicago/Turabian StyleSánchez-León, María Luisa, Carlos Jiménez-Cortegana, Silvia Silva Romeiro, Carmen Garnacho, Luis de la Cruz-Merino, Daniel J. García-Domínguez, Lourdes Hontecillas-Prieto, and Víctor Sánchez-Margalet. 2023. "Defining the Emergence of New Immunotherapy Approaches in Breast Cancer: Role of Myeloid-Derived Suppressor Cells" International Journal of Molecular Sciences 24, no. 6: 5208. https://doi.org/10.3390/ijms24065208