

The J Domain of Sacsin Disrupts Intermediate Filament Assembly

 and
and 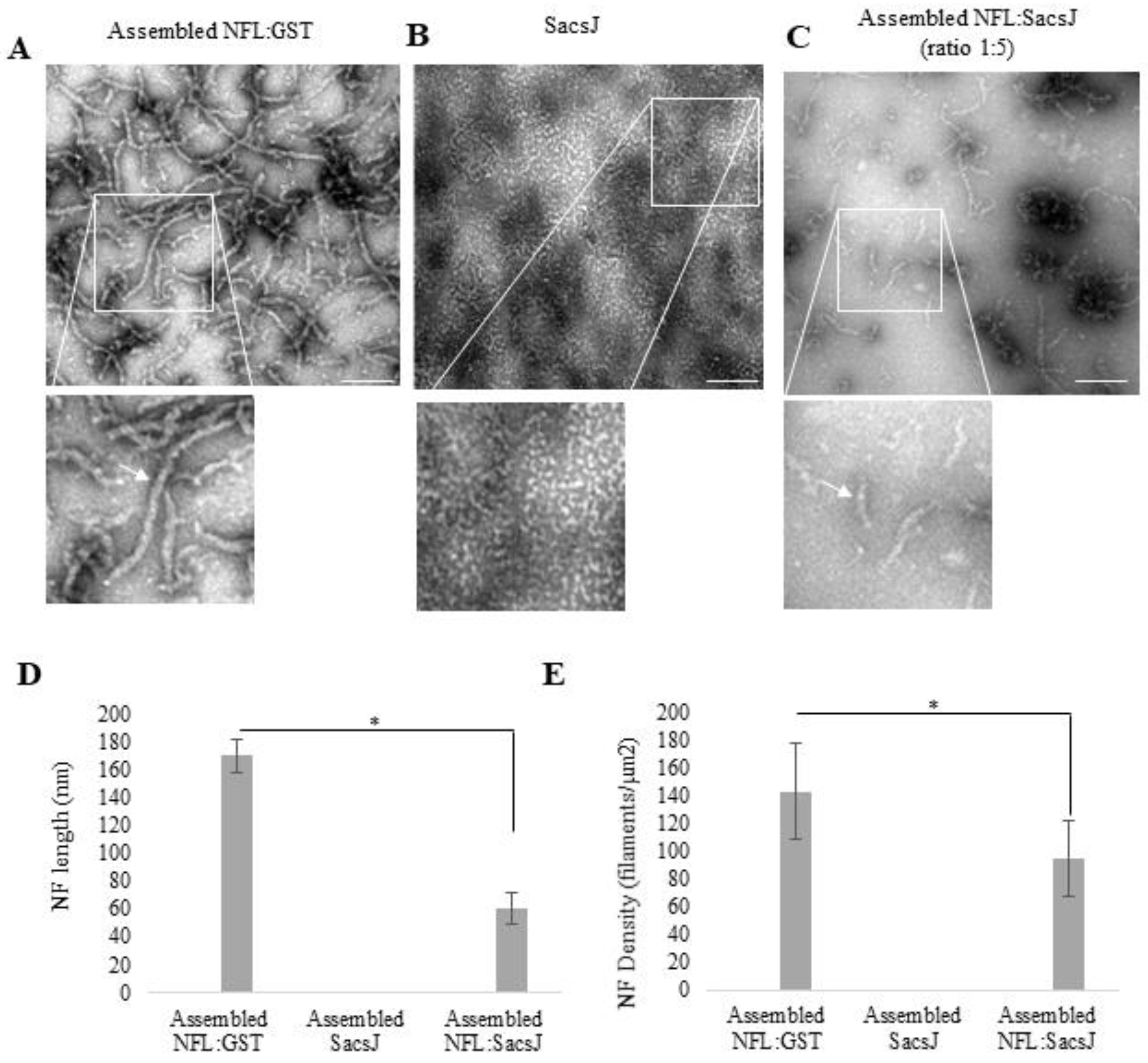{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. SacsJ Disassembled NFL Filaments In Vitro
2.2. SacsJ Prevented In Vitro Assembly of NFL into Filaments
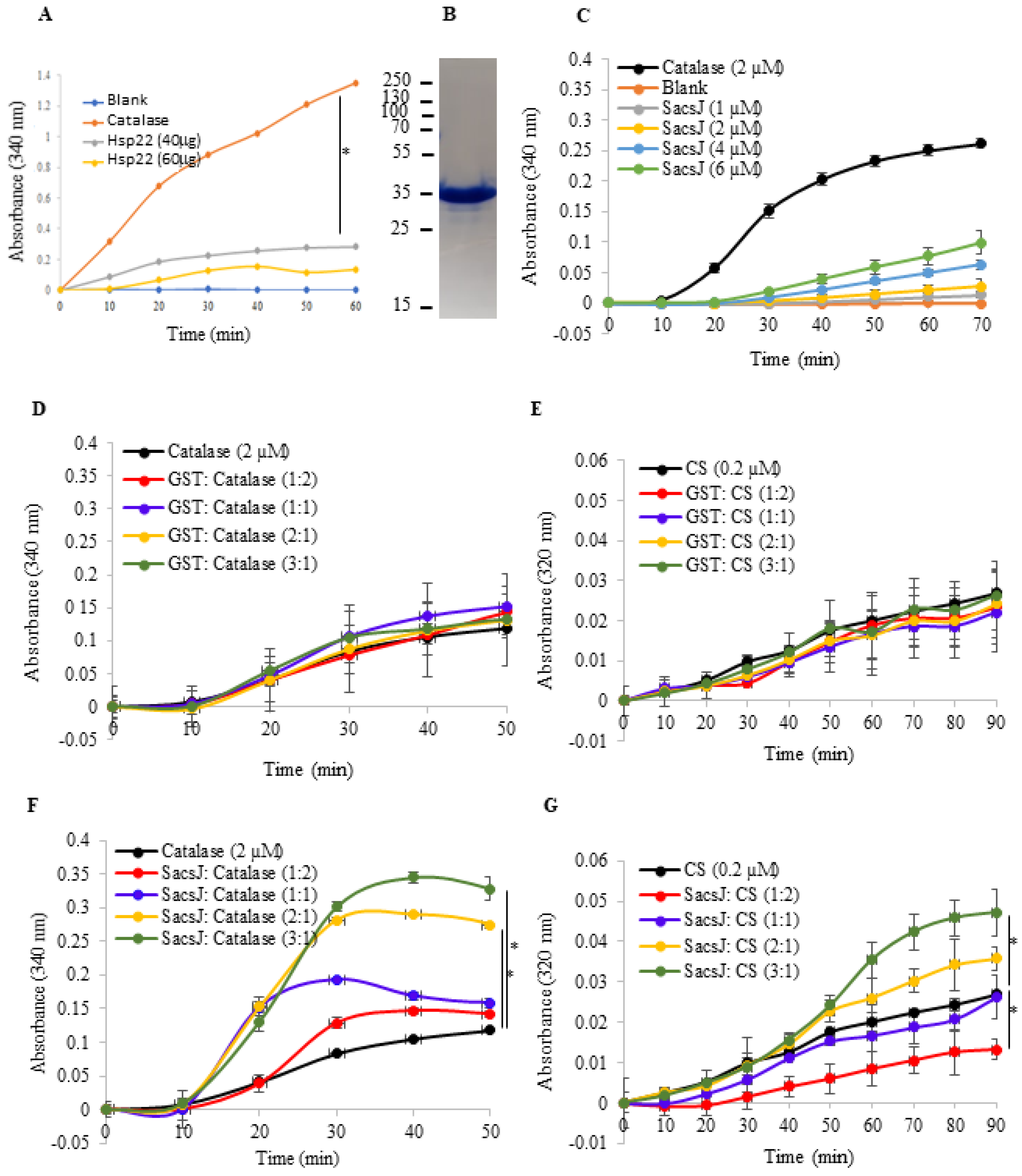
2.3. The SacsJ Domain Lacks General Chaperoning Activity In Vitro
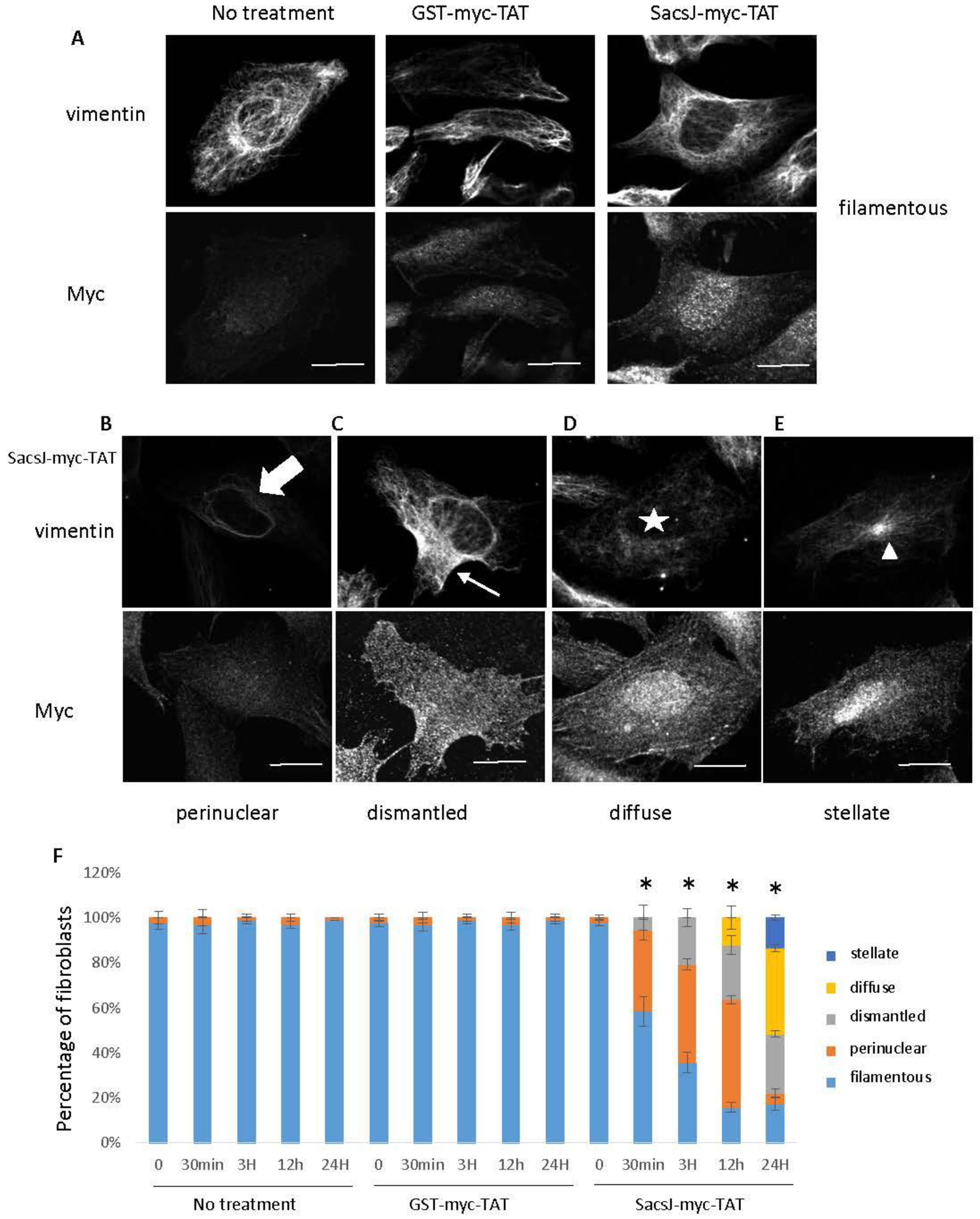
2.4. A Cell-Permeant SacsJ Peptide Disassembld Vimentin Filaments in Fibroblasts
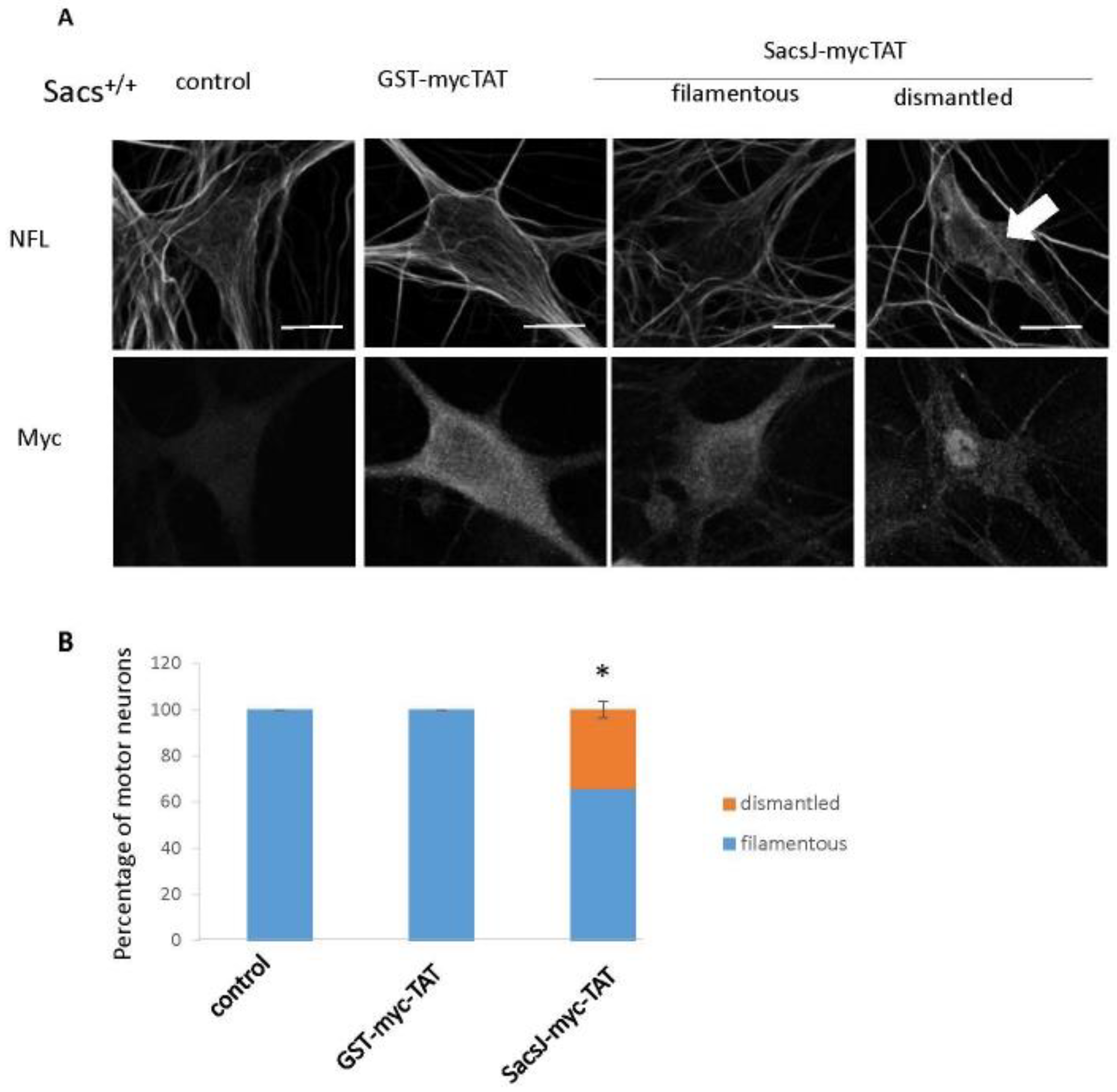
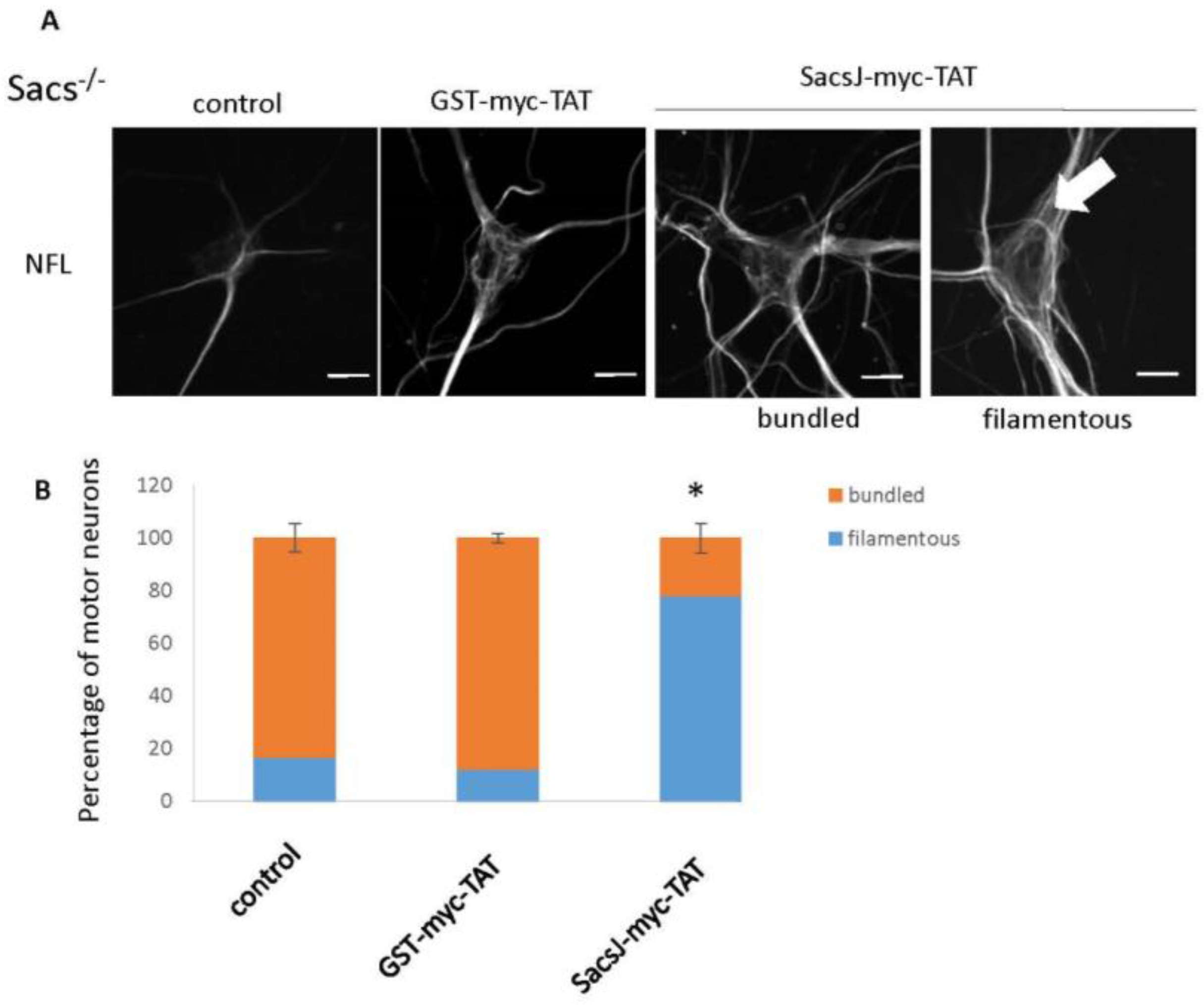
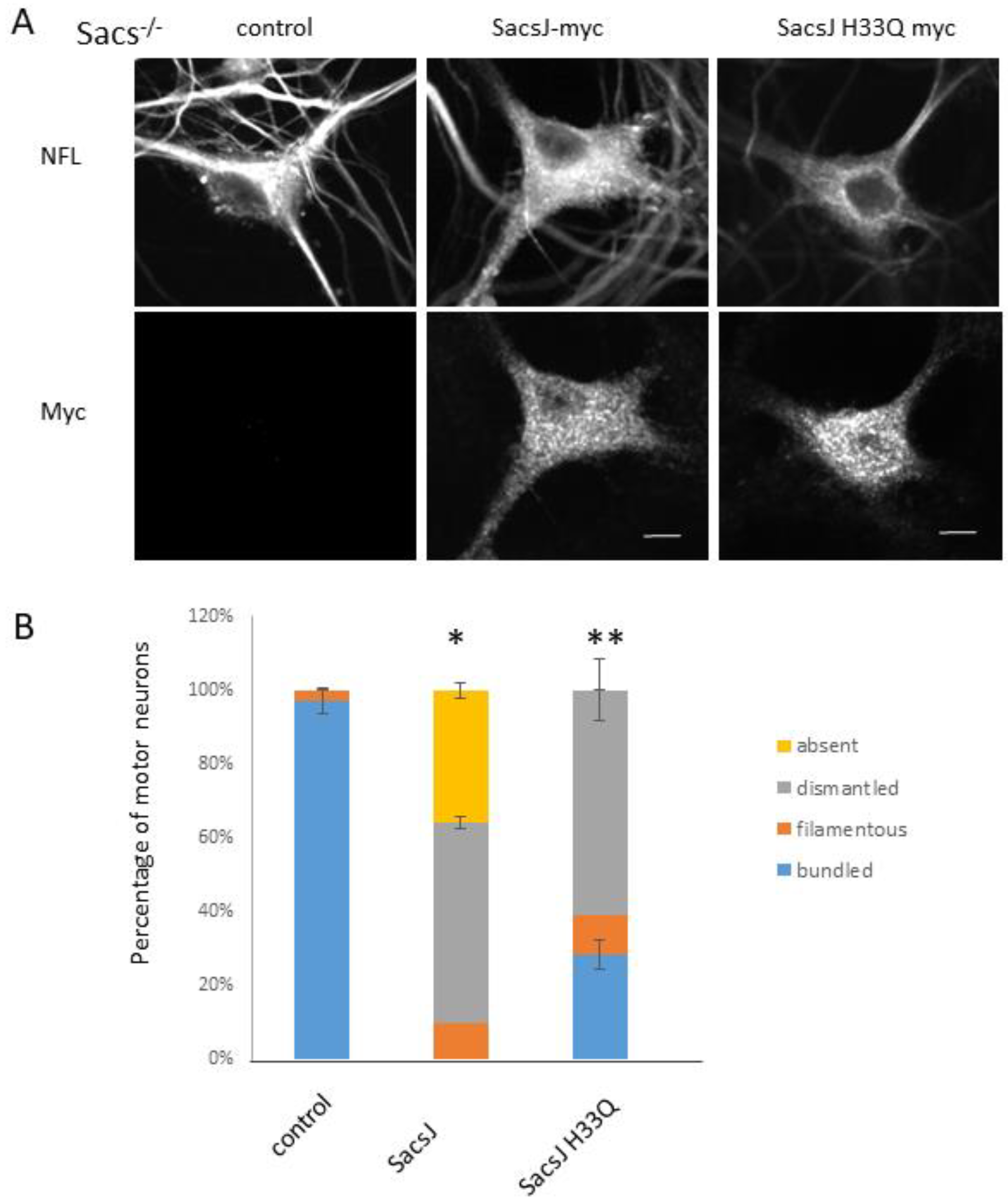
2.5. SacsJ Disassembled NF in Mouse Motor Neurons in Culture
3. Discussion
4. Materials and Methods
4.1. Cloning, Protein Production and Purification
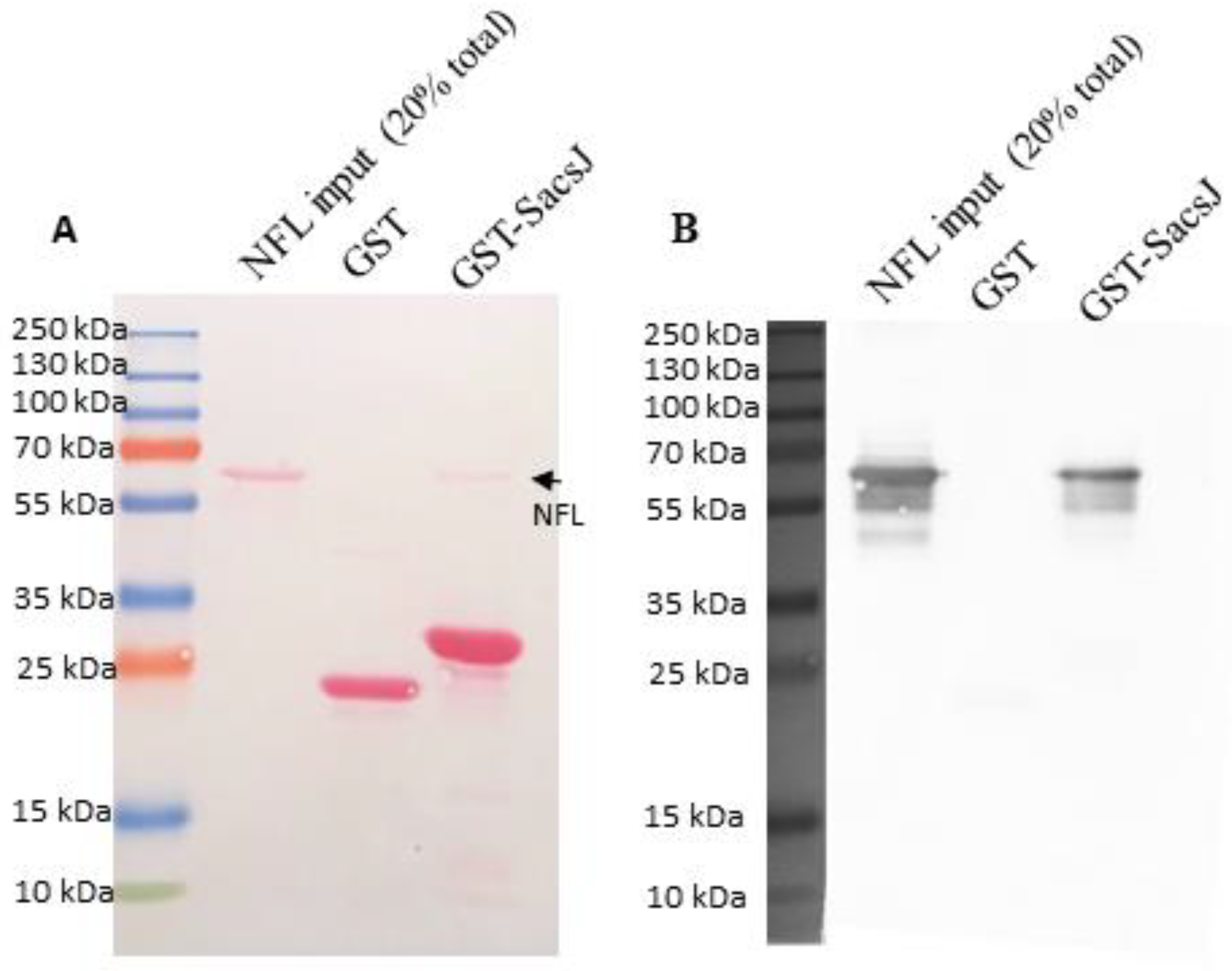
4.2. In Vitro Assembly of NFL and Pulldown Assay
4.3. Negative Staining and Electron Microscopy
4.4. Cell Culture
4.5. In Vitro Chaperone Assay
4.6. Statistics
4.7. Ethics Statement
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vingolo, E.M.; Di Fabio, R.; Salvatore, S.; Grieco, G.; Bertini, E.; Leuzzi, V.; Nesti, C.; Filla, A.; Tessa, A.; Pierelli, F.; et al. Myelinated retinal fibers in autosomal recessive spastic ataxia of Charlevoix-Saguenay. Eur. J. Neurol. 2011, 18, 1187–1190. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Martin, E.; Pablo, L.E.; Gazulla, J.; Polo, V.; Ferreras, A.; Larrosa, J.M. Retinal nerve fibre layer thickness in ARSACS: Myelination or hypertrophy? Br. J. Ophthalmol. 2013, 97, 238–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girard, M.; Larivière, R.; Parfitt, D.A.; Deane, E.C.; Gaudet, R.; Nossova, N.; Blondeau, F.; Prenosil, G.; Vermeulen, E.G.M.; Duchen, M.R.; et al. Mitochondrial dysfunction and Purkinje cell loss in autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS). Proc. Natl. Acad. Sci. USA 2012, 109, 1661–1666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baets, J.; Deconinck, T.; Smets, K.; Goossens, D.; Van den Bergh, P.; Dahan, K.; Schmedding, E.; Santens, P.; Rasic, V.M.; Van Damme, P.; et al. Mutations in SACS cause atypical and late-onset forms of ARSACS. Neurology 2010, 75, 1181–1188. [Google Scholar] [CrossRef]
- Bouchard, J.P.; Barbeau, A.; Bouchard, R.; Bouchard, R.W. Electromyography and nerve conduction studies in Friedreich’s ataxia and autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS). Can. J. Neurol. Sci. 1979, 6, 185–189. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Li, X.B.; Zi, X.H.; Shen, L.; Hu Zh, M.; Huang Sh, X.; Yu, D.L.; Li, H.B.; Xia, K.; Tang, B.S.; et al. A novel hemizygous SACS mutation identified by whole exome sequencing and SNP array analysis in a Chinese ARSACS patient. J. Neurol. Sci. 2016, 362, 111–114. [Google Scholar] [CrossRef]
- Thiffault, I.; Dicaire, M.J.; Tetreault, M.; Huang, K.N.; Demers-Lamarche, J.; Bernard, G.; Duquette, A.; Lariviere, R.; Gehring, K.; Montpetit, A.; et al. Diversity of ARSACS mutations in French-Canadians. Can. J. Neurol. Sci. 2013, 40, 61–66. [Google Scholar] [CrossRef] [Green Version]
- Ouyang, Y.; Segers, K.; Bouquiaux, O.; Wang, F.C.; Janin, N.; Andris, C.; Shimazaki, H.; Sakoe, K.; Nakano, I.; Takiyama, Y. Novel SACS mutation in a Belgian family with sacsin-related ataxia. J. Neurol. Sci. 2008, 264, 73–76. [Google Scholar] [CrossRef]
- Vermeer, S.; Meijer, R.P.P.; Pijl, B.J.; Timmermans, J.; Cruysberg, J.R.M.; Bos, M.M.; Schelhaas, H.J.; van de Warrenburg, B.P.C.; Knoers, N.; Scheffer, H.; et al. ARSACS in the Dutch population: A frequent cause of early-onset cerebellar ataxia. Neurogenetics 2008, 9, 207–214. [Google Scholar] [CrossRef] [Green Version]
- Lariviere, R.; Gaudet, R.; Gentil, B.J.; Girard, M.; Conte, T.C.; Minotti, S.; Leclerc-Desaulniers, K.; Gehring, K.; McKinney, R.A.; Shoubridge, E.A.; et al. Sacs knockout mice present pathophysiological defects underlying autosomal recessive spastic ataxia of Charlevoix-Saguenay. Hum. Mol. Genet. 2015, 24, 727–739. [Google Scholar] [CrossRef]
- Romano, A.; Tessa, A.; Barca, A.; Fattori, F.; de Leva, M.F.; Terracciano, A.; Storelli, C.; Santorelli, F.M.; Verri, T. Comparative analysis and functional mapping of SACS mutations reveal novel insights into sacsin repeated architecture. Hum. Mutat. 2013, 34, 525–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, J.F.; Siller, E.; Barral, J.M. The neurodegenerative-disease-related protein sacsin is a molecular chaperone. J. Mol. Biol. 2011, 411, 870–880. [Google Scholar] [CrossRef] [PubMed]
- Parfitt, D.A.; Michael, G.J.; Vermeulen, E.G.; Prodromou, N.V.; Webb, T.R.; Gallo, J.M.; Cheetham, M.E.; Nicoll, W.S.; Blatch, G.L.; Chapple, J.P. The ataxia protein sacsin is a functional co-chaperone that protects against polyglutamine-expanded ataxin-1. Hum. Mol. Genet. 2009, 18, 1556–1565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, J.F.; Siller, E.; Barral, J.M. The sacsin repeating region (SRR): A novel Hsp90-related supra-domain associated with neurodegeneration. J. Mol. Biol. 2010, 400, 665–674. [Google Scholar] [CrossRef]
- Kozlov, G.; Denisov, A.Y.; Girard, M.; Dicaire, M.J.; Hamlin, J.; McPherson, P.S.; Brais, B.; Gehring, K. Structural basis of defects in the sacsin HEPN domain responsible for autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS). J. Biol. Chem. 2011, 286, 20407–20412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greer, P.L.; Hanayama, R.; Bloodgood, B.L.; Mardinly, A.R.; Lipton, D.M.; Flavell, S.W.; Kim, T.K.; Griffith, E.C.; Waldon, Z.; Maehr, R.; et al. The Angelman Syndrome protein Ube3A regulates synapse development by ubiquitinating arc. Cell 2010, 140, 704–716. [Google Scholar] [CrossRef] [Green Version]
- Morani, F.; Doccini, S.; Sirica, R.; Paterno, M.; Pezzini, F.; Ricca, I.; Simonati, A.; Delledonne, M.; Santorelli, F.M. Functional Transcriptome Analysis in ARSACS KO Cell Model Reveals a Role of Sacsin in Autophagy. Sci. Rep. 2019, 9, 11878. [Google Scholar] [CrossRef] [Green Version]
- Duncan, E.J.; Lariviere, R.; Bradshaw, T.Y.; Longo, F.; Sgarioto, N.; Hayes, M.J.; Romano, L.E.L.; Nethisinghe, S.; Giunti, P.; Bruntraeger, M.B.; et al. Altered organization of the intermediate filament cytoskeleton and relocalization of proteostasis modulators in cells lacking the ataxia protein sacsin. Hum. Mol. Genet. 2017, 26, 3130–3143. [Google Scholar] [CrossRef] [Green Version]
- Kaushik, S.; Cuervo, A.M. Chaperones in autophagy. Pharm. Res. 2012, 66, 484–493. [Google Scholar] [CrossRef] [Green Version]
- Bradshaw, T.Y.; Romano, L.E.; Duncan, E.J.; Nethisinghe, S.; Abeti, R.; Michael, G.J.; Giunti, P.; Vermeer, S.; Chapple, J.P. A reduction in Drp1-mediated fission compromises mitochondrial health in autosomal recessive spastic ataxia of Charlevoix Saguenay. Hum. Mol. Genet. 2016, 25, 3232–3244. [Google Scholar] [CrossRef]
- Gentil, B.J.; Mushynski, W.E.; Durham, H.D. Heterogeneity in the properties of NEFL mutants causing Charcot-Marie-Tooth disease results in differential effects on neurofilament assembly and susceptibility to intervention by the chaperone-inducer, celastrol. Int. J. BioChem. Cell Biol. 2013, 45, 1499–1508. [Google Scholar] [CrossRef] [PubMed]
- Gentil, B.J.; Lai, G.T.; Menade, M.; Lariviere, R.; Minotti, S.; Gehring, K.; Chapple, J.P.; Brais, B.; Durham, H.D. Sacsin, mutated in the ataxia ARSACS, regulates intermediate filament assembly and dynamics. FASEB J. 2019, 33, 2982–2994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuchs, E.; Weber, K. Intermediate filaments: Structure, dynamics, function, and disease. Annu. Rev. Biochem. 1994, 63, 345–382. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, H.; Aebi, U. Intermediate filaments: Molecular structure, assembly mechanism, and integration into functionally distinct intracellular Scaffolds. Annu. Rev. Biochem. 2004, 73, 749–789. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, H.; Kreplak, L.; Aebi, U. Isolation, characterization, and in vitro assembly of intermediate filaments. Methods Cell Biol. 2004, 78, 3–24. [Google Scholar] [CrossRef]
- Herrmann, H.; Häner, M.; Brettel, M.; Ku, N.O.; Aebi, U. Characterization of distinct early assembly units of different intermediate filament proteins. J. Mol. Biol. 1999, 286, 1403–1420. [Google Scholar] [CrossRef]
- Snider, N.T.; Omary, M.B. Post-translational modifications of intermediate filament proteins: Mechanisms and functions. Nat. Rev. Mol. Cell Biol. 2014, 15, 163–177. [Google Scholar] [CrossRef] [Green Version]
- Nefedova, V.V.; Sudnitsyna, M.V.; Gusev, N.B. Interaction of small heat shock proteins with light component of neurofilaments (NFL). Cell Stress Chaperones 2017, 22, 467–479. [Google Scholar] [CrossRef] [Green Version]
- Perng, M.D.; Cairns, L.; van den, I.P.; Prescott, A.; Hutcheson, A.M.; Quinlan, R.A. Intermediate filament interactions can be altered by HSP27 and alphaB-crystallin. J. Cell Sci. 1999, 112 Pt 13, 2099–2112. [Google Scholar] [CrossRef]
- Perng, M.D.; Huang, Y.S.; Quinlan, R.A. Purification of Protein Chaperones and Their Functional Assays with Intermediate Filaments. Methods Enzym. 2016, 569, 155–175. [Google Scholar] [CrossRef]
- Perng, M.D.; Wen, S.F.; van den, I.P.; Prescott, A.R.; Quinlan, R.A. Desmin aggregate formation by R120G alphaB-crystallin is caused by altered filament interactions and is dependent upon network status in cells. Mol. Biol. Cell 2004, 15, 2335–2346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kayser, J.; Haslbeck, M.; Dempfle, L.; Krause, M.; Grashoff, C.; Buchner, J.; Herrmann, H.; Bausch, A.R. The small heat shock protein Hsp27 affects assembly dynamics and structure of keratin intermediate filament networks. Biophys. J. 2013, 105, 1778–1785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izawa, I.; Nishizawa, M.; Ohtakara, K.; Ohtsuka, K.; Inada, H.; Inagaki, M. Identification of Mrj, a DnaJ/Hsp40 family protein, as a keratin 8/18 filament regulatory protein. J. Biol. Chem. 2000, 275, 34521–34527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, X.B.; Shao, Y.M.; Miao, S.; Wang, L. The diversity of the DnaJ/Hsp40 family, the crucial partners for Hsp70 chaperones. Cell Mol. Life Sci. 2006, 63, 2560–2570. [Google Scholar] [CrossRef]
- Leung, C.L.; Liem, R.K. Isolation of intermediate filaments. Curr. Protoc. Cell Biol. 2006, 31, 3–23. [Google Scholar] [CrossRef]
- Lee, G.J.; Pokala, N.; Vierling, E. Structure and in vitro molecular chaperone activity of cytosolic small heat shock proteins from pea. J. Biol. Chem. 1995, 270, 10432–10438. [Google Scholar] [CrossRef] [Green Version]
- Frankel, A.D.; Pabo, C.O. Cellular uptake of the tat protein from human immunodeficiency virus. Cell 1988, 55, 1189–1193. [Google Scholar] [CrossRef]
- Green, M.; Loewenstein, P.M. Autonomous functional domains of chemically synthesized human immunodeficiency virus tat trans-activator protein. Cell 1988, 55, 1179–1188. [Google Scholar] [CrossRef]
- Eriksson, J.E.; He, T.; Trejo-Skalli, A.V.; Harmala-Brasken, A.S.; Hellman, J.; Chou, Y.H.; Goldman, R.D. Specific in vivo phosphorylation sites determine the assembly dynamics of vimentin intermediate filaments. J. Cell Sci. 2004, 117, 919–932. [Google Scholar] [CrossRef] [Green Version]
- Blose, S.H.; Meltzer, D.I. Visualization of the 10-NM filament vimentin rings in vascular endothelial cells in situ: Close resemblance to vimentin cytoskeletons found in monolayers in vitro. Exp. Cell Res. 1981, 135, 299–309. [Google Scholar] [CrossRef]
- Blose, S.H.; Chacko, S. Rings of intermediate (100 A) filament bundles in the perinuclear region of vascular endothelial cells. Their mobilization by colcemid and mitosis. J. Cell Biol. 1976, 70, 459–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dellagi, K.; Brouet, J.C. Redistribution of intermediate filaments during capping of lymphocyte surface molecules. Nature 1982, 298, 284–286. [Google Scholar] [CrossRef] [PubMed]
- Zhai, J.; Lin, H.; Julien, J.P.; Schlaepfer, W.W. Disruption of neurofilament network with aggregation of light neurofilament protein: A common pathway leading to motor neuron degeneration due to Charcot-Marie-Tooth disease-linked mutations in NFL and HSPB1. Hum. Mol. Genet. 2007, 16, 3103–3116. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, S.; Uchiumi, A.; Katagata, Y. Hsp40 regulates the amount of keratin proteins via ubiquitin-proteasome pathway in cultured human cells. Int. J. Mol. Med. 2012, 29, 165–168. [Google Scholar] [CrossRef]
- Terriac, E.; Schutz, S.; Lautenschlager, F. Vimentin Intermediate Filament Rings Deform the Nucleus during the First Steps of Adhesion. Front. Cell Dev. Biol. 2019, 7, 106. [Google Scholar] [CrossRef] [Green Version]
- Herrmann, H.; Haner, M.; Brettel, M.; Muller, S.A.; Goldie, K.N.; Fedtke, B.; Lustig, A.; Franke, W.W.; Aebi, U. Structure and assembly properties of the intermediate filament protein vimentin: The role of its head, rod and tail domains. J. Mol. Biol. 1996, 264, 933–953. [Google Scholar] [CrossRef]
- Balin, B.J.; Clark, E.A.; Trojanowski, J.Q.; Lee, V.M. Neurofilament reassembly in vitro: Biochemical, morphological and immuno-electron microscopic studies employing monoclonal antibodies to defined epitopes. Brain Res. 1991, 556, 181–195. [Google Scholar] [CrossRef]
- Roy, J.; Minotti, S.; Dong, L.; Figlewicz, D.A.; Durham, H.D. Glutamate potentiates the toxicity of mutant Cu/Zn-superoxide dismutase in motor neurons by postsynaptic calcium-dependent mechanisms. J. Neurosci. 1998, 18, 9673–9684. [Google Scholar] [CrossRef] [Green Version]
- Rajaraman, K.; Raman, B.; Ramakrishna, T.; Rao, C.M. Interaction of human recombinant alphaA- and alphaB-crystallins with early and late unfolding intermediates of citrate synthase on its thermal denaturation. FEBS Lett. 2001, 497, 118–123. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dabbaghizadeh, A.; Paré, A.; Cheng-Boivin, Z.; Dagher, R.; Minotti, S.; Dicaire, M.-J.; Brais, B.; Young, J.C.; Durham, H.D.; Gentil, B.J. The J Domain of Sacsin Disrupts Intermediate Filament Assembly. Int. J. Mol. Sci. 2022, 23, 15742. https://doi.org/10.3390/ijms232415742
Dabbaghizadeh A, Paré A, Cheng-Boivin Z, Dagher R, Minotti S, Dicaire M-J, Brais B, Young JC, Durham HD, Gentil BJ. The J Domain of Sacsin Disrupts Intermediate Filament Assembly. International Journal of Molecular Sciences. 2022; 23(24):15742. https://doi.org/10.3390/ijms232415742
Chicago/Turabian StyleDabbaghizadeh, Afrooz, Alexandre Paré, Zacharie Cheng-Boivin, Robin Dagher, Sandra Minotti, Marie-Josée Dicaire, Bernard Brais, Jason C. Young, Heather D. Durham, and Benoit J. Gentil. 2022. "The J Domain of Sacsin Disrupts Intermediate Filament Assembly" International Journal of Molecular Sciences 23, no. 24: 15742. https://doi.org/10.3390/ijms232415742