
Developing 3D Organoid Raft Cultures from Patient-Derived Xenografts as Rapid Models to Screen Efficacy of Experimental Therapeutics
 , and
, and 
Abstract
:1. Introduction
2. Results
2.1. Organoid Raft Cultures Retain the Morphological Features of Colorectal Cancer
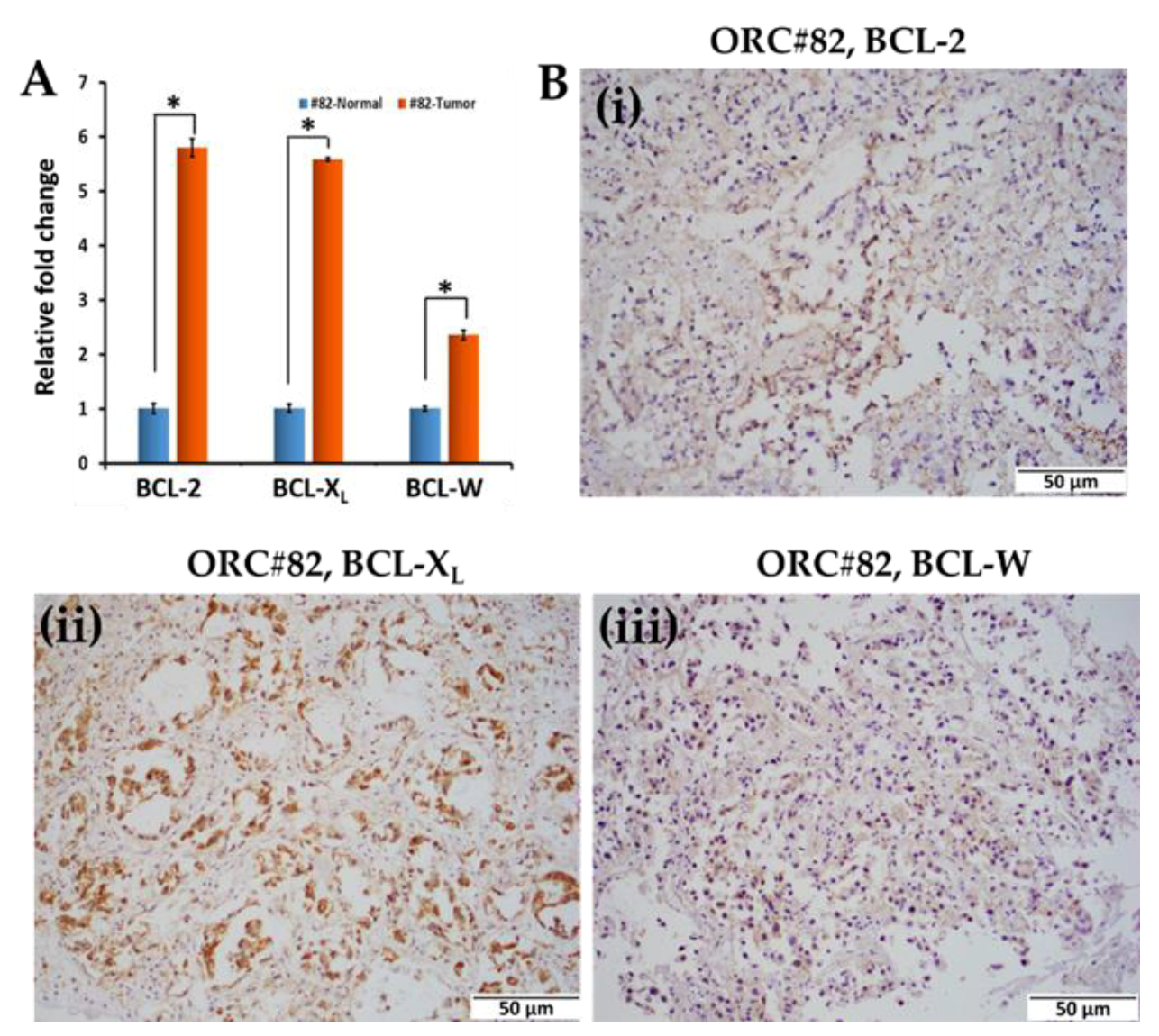
2.2. Colorectal Primary Tissues Express Varying Levels of BCL-2 Family Proteins
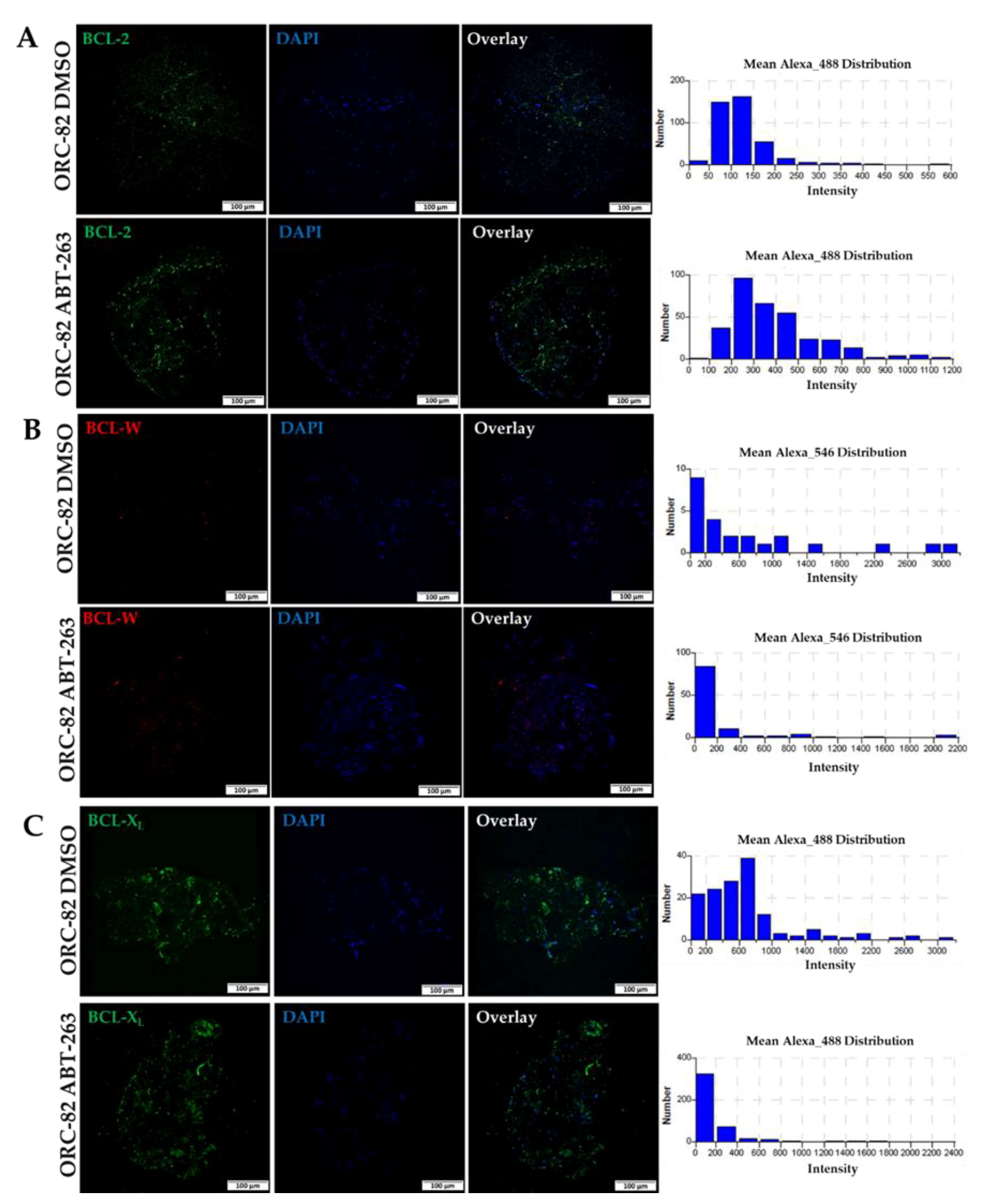
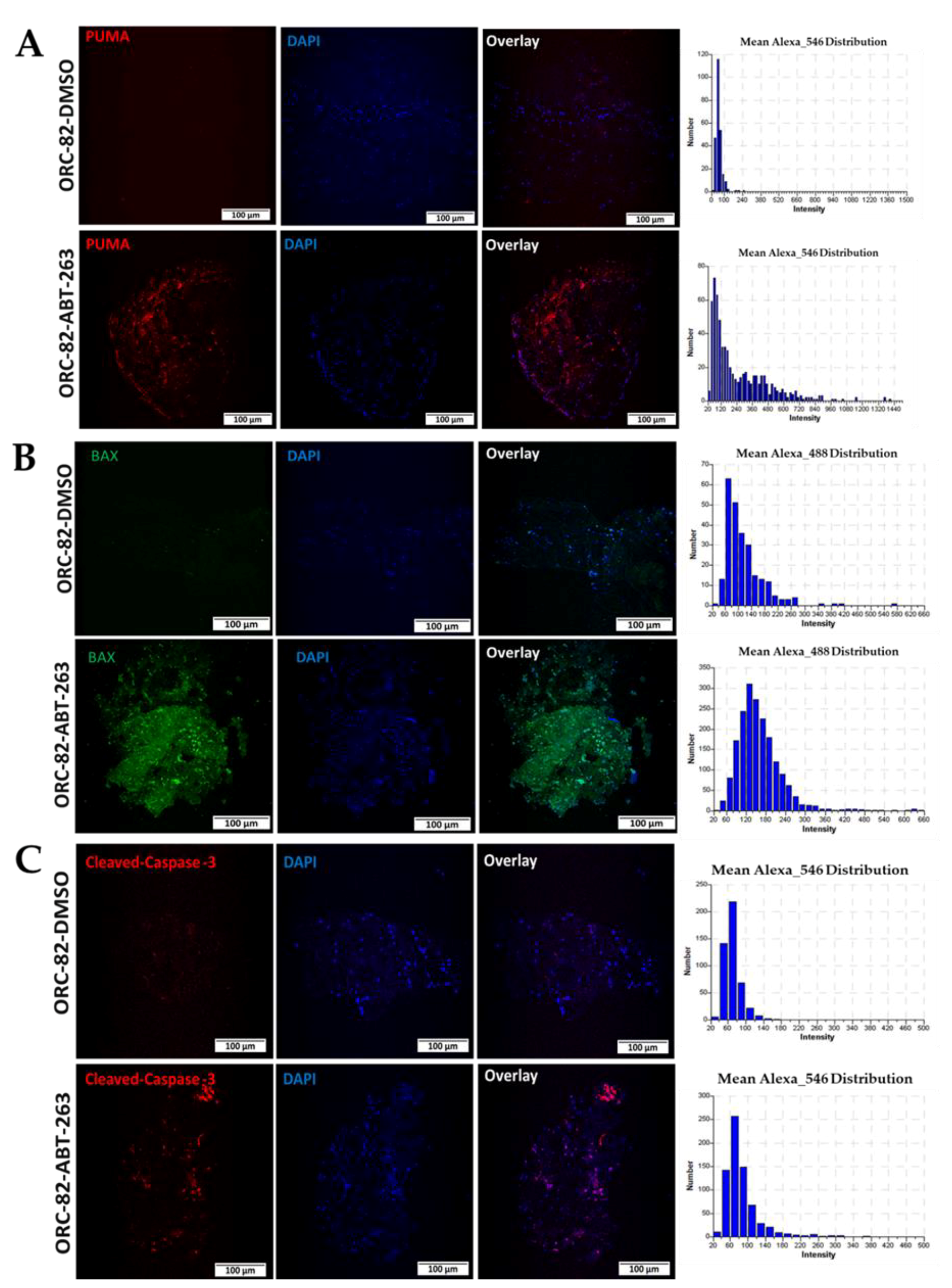
2.3. Effect of ABT-263 Treatment on BCL-2 Family Proteins in Organoid Raft Cultures
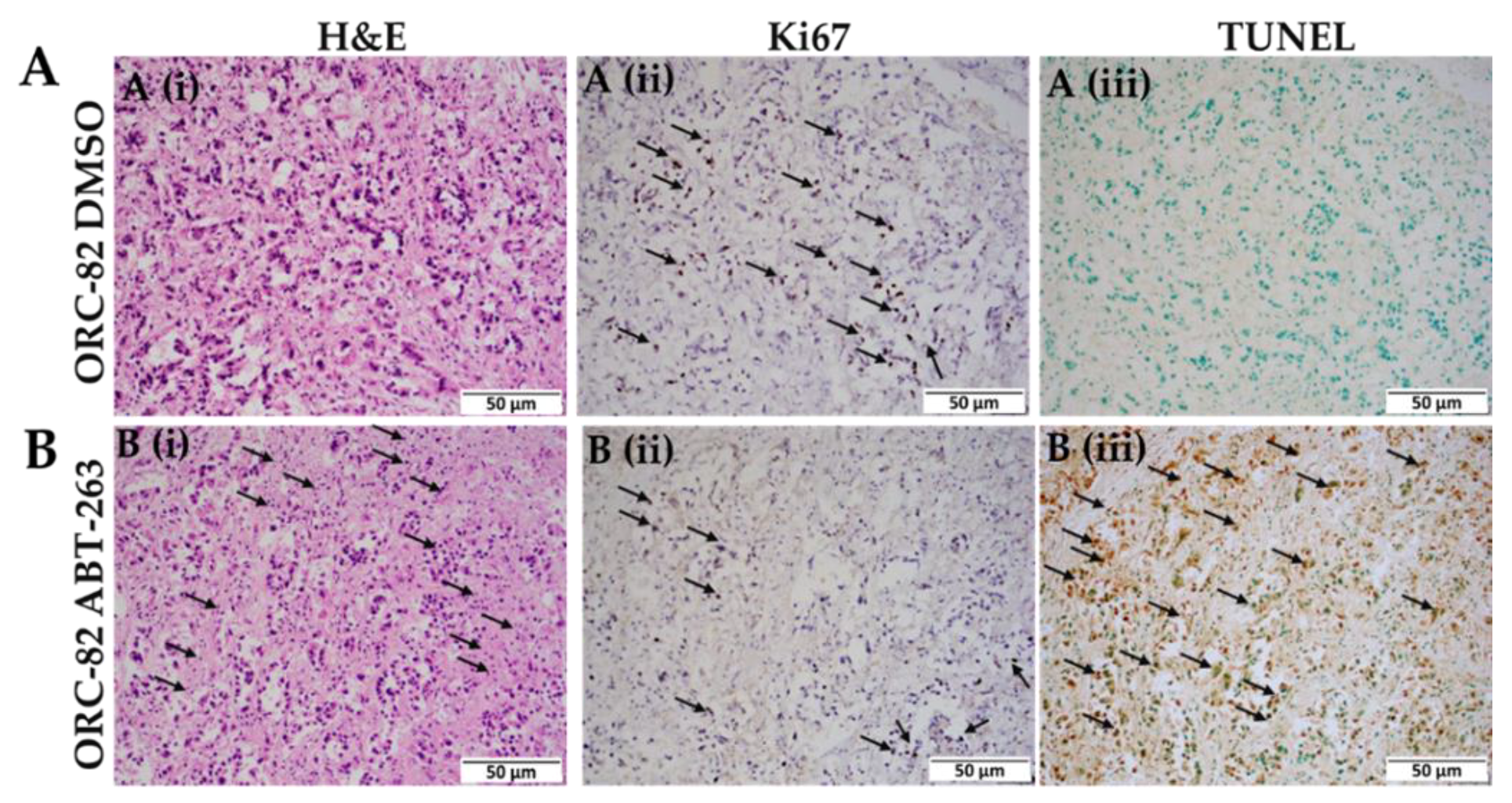
2.4. ABT-263 Treatment of Organoid Raft Cultures Decreases Cell Proliferation and Increases Apoptosis
3. Discussion
4. Materials and Methods
4.1. Materials Required for Organoid Raft Cultures (ORCs)
- Dulbecco’s modified Eagle’s medium (DMEM), supplemented with 10% fetal bovine serum and 1% Penicillin Streptomycin Antibiotic (100×, Corning, Cat # 30-002-CI, Fisher Scientific, Hampton, NH) was used for culturing feeder cells.
- We used Ham’s F12:F12 mix (1X) (1 L, package, GIBCO, Cat # 21700-0075, Thermo Fisher Scientific, Waltham, MA, USA). For reconstitution, directions on the package were followed, and 1.176 g of sodium bicarbonate was added. The pH was adjusted to 7.0 with 1 N HCl before filtration. F12, not more than a month old, should be used, as the pH fluctuates swiftly.
- Insulin stock (1000X) (Sigma–Aldrich, Cat # I-1882, Millipore Sigma, Burlington, MA, USA) was made by dissolving 100 mg in 20 mL sterile ultrapure water inside a cell culture hood. Aliquots were stored at −20 °C.
- Apo-transferrin stock (1000X) (Sigma–Aldrich, Cat # T1147, Millipore Sigma, Burlington, MA, USA) was made by dissolving 100 mg in 20 mL of sterile ultrapure water inside a cell culture hood. Aliquots were stored at −20 °C.
- Hydrocortisone-21-hemisuccinate stock (10,000X) (Sigma–Aldrich, Cat # H-488, Millipore Sigma, Burlington, MA, USA) was made by dissolving 100 mg in 25 mL of 100% ethanol inside a cell culture hood. Aliquots were stored at −20 °C.
- Human epidermal growth factor stock (10,000X) MilliporeSigma Calbiochem, Cat # 32-483-1200UG, Fisher Scientific, Hampton, NH, USA) was made by diluting 100 µL in 20 mL of sterile ultrapure water inside a cell culture hood. Aliquots were stored at −20 °C.
- Cholera toxin stock (10,000X, 1 mM) (Cayman Chemical, Cat # NC1425699, Fisher Scientific, Hampton, NH) was made by dissolving 1 mg in 2 mL of sterile ultrapure water inside a cell culture hood. Of this solution, 174 µL was added to 826 µL of sterile ultrapure water. Aliquots were stored at 4 °C. This solution should not be frozen.
- We used Type I Rat Tail Collagen (Corning, Cat # CB-40236, Fisher Scientific, Hampton, NH, USA).
- F12 stock (10X) (GIBCO, Cat # 21700-0075, Thermo Fisher Scientific, Waltham, MA, USA) was made by dissolving one 1-L package of Ham’s F12 medium in 100 mL of sterile ultrapure water. In this stock, pH was not adjusted, and sodium bicarbonate was not added. Filter-sterilized aliquots were then stored at −20 °C.
- We used phosphate-buffered saline (PBS) (10X) (Fisher BioReagents, Cat # BP39920, Fisher Scientific, Hampton, NH, USA).
- We used Antibiotic and Antimycotic mixture (Anti–Anti) (100X) (GIBCO, Cat # 15240062 Thermo Fisher Scientific, Waltham, MA, USA.
- We used 3T3-J2 mouse fibroblasts cells (Cat#EF303, Kerafast, Boston, MA, USA).
- Stainless steel grids (Edward J. Darby Inc, Philadelphia, PA, USA) measuring 1”× 1” (L × B) consisted of squares of wire mesh with square pores (size, 0.25 mm). The corners of the wired mesh were bent to make rafts/boats (approximate height 2 mm).
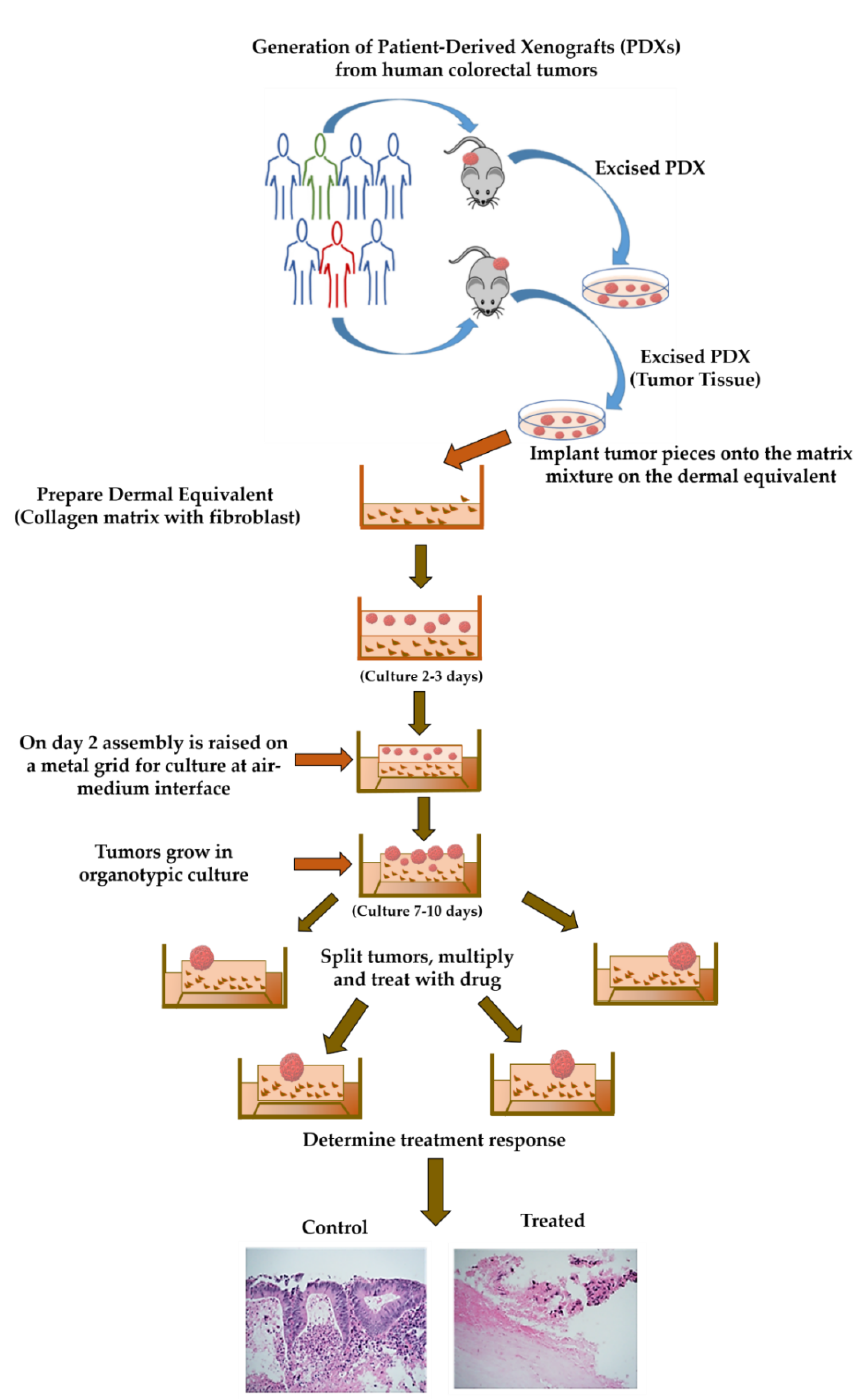
4.2. Generation of Patient-Derived Xenografts (PDXs)
4.3. Collection of the Patient-Derived Xenograft Tissue
4.4. Preparing Collagen Dermal Equivalent
- 400 µL of 10× Reconstitution Buffer (RB Buffer: 2.2 gm sodium bicarbonate, 0.2 gm sodium hydroxide (Sigma, Cat # 1310-73-2, Millipore Sigma, Burlington, MA, USA), and 4.76 gm HEPES (free acid; Sigma, Cat# H 3375) per 100 mL. Aliquots of 10 mL were stored at −20 °C);
- 400 µL 10× F12;
- 4 mL Type-1 Rat tail collagen.
4.5. Seeding on Collagen Bed/Boat
4.6. Generation of Organoid Raft Cultures (ORCs)
4.7. Treatment with Navitoclax (ABT-263)
4.8. Harvesting the Organoid Raft Cultures (ORCs)
4.9. Immunohistochemical (IHC) Analysis
4.10. RNA Extraction and Quantitative-PCR
4.11. TUNEL Staining
4.12. Immunofluorescence Microscopy
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Drost, J.; Clevers, H. Organoids in cancer research. Nat. Rev. Cancer 2018, 18, 407–418. Available online: https://www.ncbi.nlm.nih.gov/pubmed/29692415 (accessed on 4 April 2022). [CrossRef] [PubMed]
- Ooft, S.N.; Weeber, F.; Dijkstra, K.K.; McLean, C.M.; Kaing, S.; van Werkhoven, E.; Schipper, L.; Hoes, L.; Vis, D.J.; van de Haar, J.; et al. Patient-derived organoids can predict response to chemotherapy in metastatic colorectal cancer patients. Sci. Transl. Med. 2019, 11, eaay2574. Available online: https://www.ncbi.nlm.nih.gov/pubmed/31597751 (accessed on 4 April 2022). [CrossRef]
- Sato, T.; Stange, D.E.; Ferrante, M.; Vries, R.G.; Van Es, J.H.; Van den Brink, S.; Van Houdt, W.J.; Pronk, A.; Van Gorp, J.; Siersema, P.D.; et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology 2011, 141, 1762–1772. Available online: https://www.ncbi.nlm.nih.gov/pubmed/21889923 (accessed on 13 May 2022). [CrossRef]
- Weeber, F.; van de Wetering, M.; Hoogstraat, M.; Dijkstra, K.K.; Krijgsman, O.; Kuilman, T.; Gadellaa-van Hooijdonk, C.G.; van der Velden, D.L.; Peeper, D.S.; Cuppen, E.P.; et al. Preserved genetic diversity in organoids cultured from biopsies of human colorectal cancer metastases. Proc. Natl. Acad. Sci. USA 2015, 112, 13308–13311. Available online: https://www.ncbi.nlm.nih.gov/pubmed/26460009 (accessed on 14 April 2022). [CrossRef] [PubMed] [Green Version]
- Meyers, B.M.; Cosby, R.; Quereshy, F.; Jonker, D. Adjuvant systemic chemotherapy for stages II and III colon cancer after complete resection: A clinical practice guideline. Curr. Oncol. 2016, 23, 418–424. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5176375/ (accessed on 4 April 2022). [CrossRef] [PubMed] [Green Version]
- Sobrero, A.; Lonardi, S.; Rosati, G.; Di Bartolomeo, M.; Ronzoni, M.; Pella, N.; Scartozzi, M.; Banzi, M.; Zampino, M.G.; Pasini, F.; et al. FOLFOX or CAPOX in Stage II to III Colon Cancer: Efficacy Results of the Italian Three or Six Colon Adjuvant Trial. J. Clin. Oncol. 2018, 36, 1478–1485. Available online: https://www.ncbi.nlm.nih.gov/pubmed/29620994 (accessed on 4 April 2022). [CrossRef] [PubMed]
- Aparo, S.; Goel, S. Evolvement of the treatment paradigm for metastatic colon cancer. From chemotherapy to targeted therapy. Crit. Rev. Oncol./Hematol. 2012, 83, 47–58. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3849106/ (accessed on 4 April 2022). [CrossRef] [Green Version]
- Gonzalo Recondo, E.D.-C., Jr.; de la Vega, M.; Greco, M.; Gonzalo Recondo, M.E.V., Sr. Advances and new perspectives in the treatment of metastatic colon cancer. World J. Gastrointest. Oncol. 2014, 6, 211. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4092338/ (accessed on 4 April 2022). [CrossRef] [PubMed]
- Ramesh, P.; Medema, J.P. BCL-2 family deregulation in colorectal cancer: Potential for BH3 mimetics in therapy. Apoptosis 2020, 25, 305–320. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7244464/ (accessed on 4 April 2022). [CrossRef] [PubMed] [Green Version]
- Bedi, A.; Pasricha, P.J.; Akhtar, A.J.; Barber, J.P.; Bedi, G.C.; Giardiello, F.M.; Zehnbauer, B.A.; Hamilton, S.R.; Jones, R.J. Inhibition of apoptosis during development of colorectal cancer. Cancer Res. 1995, 55, 1811–1816. Available online: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=7728743 (accessed on 4 April 2022). [PubMed]
- Reed, J. Bcl-2 and the regulation of programmed cell death. J. Cell Biol. 1994, 124, 1–6. Available online: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=8294493 (accessed on 4 April 2022). [CrossRef] [PubMed]
- Manne, U.; Myers, R.B.; Moron, C.; Poczatek, R.B.; Dillard, S.; Weiss, H.; Brown, D.; Srivastava, S.; Grizzle, W.E. Prognostic significance of Bcl-2 expression and p53 nuclear accumulation in colorectal adenocarcinoma. Int. J. Cancer 1997, 74, 346–358. Available online: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=9221816 (accessed on 14 April 2022). [CrossRef]
- Corcoran, R.B.; Cheng, K.A.; Hata, A.N.; Faber, A.C.; Ebi, H.; Coffee, E.M.; Greninger, P.; Brown, R.D.; Godfrey, J.T.; Cohoon, T.J.; et al. Synthetic lethal interaction of combined BCL-XL and MEK inhibition promotes tumor regressions in KRAS mutant cancer models. Cancer Cell 2013, 23, 121–128. Available online: https://www.ncbi.nlm.nih.gov/pubmed/23245996 (accessed on 4 April 2022). [CrossRef] [PubMed] [Green Version]
- Shao, H.; Jing, K.; Mahmoud, E.; Huang, H.; Fang, X.; Yu, C. Apigenin Sensitizes Colon Cancer Cells to Antitumor Activity of ABT-263. Mol. Cancer Ther. 2013, 12, 2640–2650. Available online: http://mct.aacrjournals.org/content/molcanther/12/12/2640.full.pdf (accessed on 4 April 2022). [CrossRef] [PubMed] [Green Version]
- Balakrishnan, K.; Gandhi, V. Bcl-2 antagonists: A proof of concept for CLL therapy. Investig. New Drugs 2013, 31, 1384–1394. Available online: https://www.ncbi.nlm.nih.gov/pubmed/23907405 (accessed on 4 April 2022). [CrossRef] [PubMed] [Green Version]
- Tse, C.; Shoemaker, A.R.; Adickes, J.; Anderson, M.G.; Chen, J.; Jin, S.; Johnson, E.F.; Marsh, K.C.; Mitten, M.J.; Nimmer, P.; et al. ABT-263: A potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008, 68, 3421–3428. Available online: https://www.ncbi.nlm.nih.gov/pubmed/18451170 (accessed on 14 April 2022). [CrossRef] [PubMed] [Green Version]
- Chen, Q.; Song, S.; Wei, S.; Liu, B.; Honjo, S.; Scott, A.; Jin, J.; Ma, L.; Zhu, H.; Skinner, H.D.; et al. ABT-263 induces apoptosis and synergizes with chemotherapy by targeting stemness pathways in esophageal cancer. Oncotarget 2015, 6, 25883–25896. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4694873/ (accessed on 4 April 2022). [CrossRef] [Green Version]
- Rysanek, D.; Vasicova, P.; Kolla, J.N.; Sedlak, D.; Andera, L.; Bartek, J.; Hodny, Z. Synergism of BCL-2 family inhibitors facilitates selective elimination of senescent cells. Aging 2022, 14, 6381–6414. Available online: https://www.ncbi.nlm.nih.gov/pubmed/35951353 (accessed on 14 April 2022). [CrossRef]
- Place, T.L.; Domann, F.E.; Case, A.J. Limitations of oxygen delivery to cells in culture: An underappreciated problem in basic and translational research. Free Radic. Biol. Med. 2017, 113, 311–322. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5699948/ (accessed on 4 April 2022). [CrossRef]
- Kety, S.S. The theory and applications of the exchange of inert gas at the lungs and tissues. Pharmacol. Rev. 1951, 3, 1–41. Available online: https://pharmrev.aspetjournals.org/content/pharmrev/3/1/1.full.pdf (accessed on 4 April 2022).
- Krogh, A. The rate of diffusion of gases through animal tissues, with some remarks on the coefficient of invasion. J. Physiol. 1919, 52, 391–408. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1402717/ (accessed on 4 April 2022). [CrossRef] [PubMed]
- Peniche Silva, C.J.; Liebsch, G.; Meier, R.J.; Gutbrod, M.S.; Balmayor, E.R.; van Griensven, M. A New Non-invasive Technique for Measuring 3D-Oxygen Gradients in Wells During Mammalian Cell Culture. Front. Bioeng. Biotechnol. 2020, 8, 595. Available online: https://www.frontiersin.org/articles/10.3389/fbioe.2020.00595 (accessed on 14 April 2022). [CrossRef] [PubMed]
- Al-Ani, A.; Toms, D.; Kondro, D.; Thundathil, J.; Yu, Y.; Ungrin, M. Oxygenation in cell culture: Critical parameters for reproducibility are routinely not reported. PLoS ONE 2018, 13, e0204269. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6191109/ (accessed on 4 April 2022). [CrossRef] [PubMed] [Green Version]
- Wenger, R.H. Mammalian oxygen sensing, signalling and gene regulation. J. Exp. Biol. 2000, 203 Pt 8, 1253–1263. Available online: https://journals.biologists.com/jeb/article/203/8/1253/8520/Mammalian-oxygen-sensing-signalling-and-gene (accessed on 4 April 2022). [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CRC Patient # | Sex | Age | Body Mass Index | Sample Origin | Tumor (T), Nodes (N), and Metastases (M) | Degree of Differentiation | |
|---|---|---|---|---|---|---|---|
| 1 | CRC-18 | Female | 73 | 21.92 | Colon | T3N0Mx | Moderate |
| 2 | CRC-59 | Male | 40 | 26.42 | Sigmoid Colon | T3N0 | Moderate |
| 3 | CRC-82 | Male | 65 | 34.94 | Ascending Colon | T3N1 | Moderate |
| Genes | Primer Sequence (5′→ 3′) | |
|---|---|---|
| 1 | BCL2-F | GGA TTG TGG CCT TCT TTG AG |
| 2 | BCL2-R | GCC GGT TCA GGT ACT CAG TC |
| 3 | BCL-XL-F | AGT TTG AAC TGC GGT ACC GG |
| 4 | BCL-XL-R | GCA TTG TTC CCA TAG AGT TC |
| 5 | BCL W-F | GCG GAG TTC ACAGCT CTA TAC |
| 6 | BCL W-R | AAA AGG CCC CTA CAG TTA CCA |
| 7 | β-Actin-F | CTG CTT GCT GAT CCA CAT CTG |
| 8 | β-Actin-R | ATC AAG ATC ATT GCT CCT CCT GAG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bajpai, P.; Banerjee, N.S.; Moore, D.W.; Kim, H.-G.; Afaq, F.; Contreras, C.M.; Heslin, M.J.; Reddy, V.B.; Peter, S.; Varambally, S.; et al. Developing 3D Organoid Raft Cultures from Patient-Derived Xenografts as Rapid Models to Screen Efficacy of Experimental Therapeutics. Int. J. Mol. Sci. 2022, 23, 14392. https://doi.org/10.3390/ijms232214392
Bajpai P, Banerjee NS, Moore DW, Kim H-G, Afaq F, Contreras CM, Heslin MJ, Reddy VB, Peter S, Varambally S, et al. Developing 3D Organoid Raft Cultures from Patient-Derived Xenografts as Rapid Models to Screen Efficacy of Experimental Therapeutics. International Journal of Molecular Sciences. 2022; 23(22):14392. https://doi.org/10.3390/ijms232214392
Chicago/Turabian StyleBajpai, Prachi, Nilam Sanjib Banerjee, Dianne W. Moore, Hyung-Gyoon Kim, Farrukh Afaq, Carlo M. Contreras, Martin J. Heslin, Vishnu B. Reddy, Shajan Peter, Sooryanarayana Varambally, and et al. 2022. "Developing 3D Organoid Raft Cultures from Patient-Derived Xenografts as Rapid Models to Screen Efficacy of Experimental Therapeutics" International Journal of Molecular Sciences 23, no. 22: 14392. https://doi.org/10.3390/ijms232214392