
Oxidative Stress in Traumatic Brain Injury
Yale School of Medicine, Department of Neurology, Yale University, New Haven, CT 06510, USA
Int. J. Mol. Sci. 2022, 23(21), 13000; https://doi.org/10.3390/ijms232113000
Submission received: 7 July 2022
/
Revised: 21 October 2022
/
Accepted: 25 October 2022
/
Published: 27 October 2022
(This article belongs to the Special Issue Oxidative Stress and Brain Injury 3.0)
Abstract
:Traumatic Brain Injury (TBI) remains a major cause of disability worldwide. It involves a complex neurometabolic cascade, including oxidative stress. The products of this manuscript is examining the underlying pathophysiological mechanism, including reactive oxygen species (ROS) and reactive nitrogen species (RNS). This process in turn leads to secondary injury cascade, which includes lipid peroxidation products. These reactions ultimately play a key role in chronic inflammation and synaptic dysfunction in a synergistic fashion. Although there are no FDA approved antioxidant therapy for TBI, there is a number of antioxidant therapies that have been tested and include free radical scavengers, activators of antioxidant systems, inhibitors of free radical generating enzymes, and antioxidant enzymes. Antioxidant therapies have led to cognitive and functional recovery post TBI, and they offer a promising treatment option for patients recovering from TBI. Current major challenges in treatment of TBI symptoms include heterogenous nature of injury, as well as access to timely treatment post injury. The inherent benefits of antioxidant therapies include minimally reported side effects, and relative ease of use in the clinical setting. The current review also provides a highlight of the more studied anti-oxidant regimen with applicability for TBI treatment with potential use in the real clinical setting.
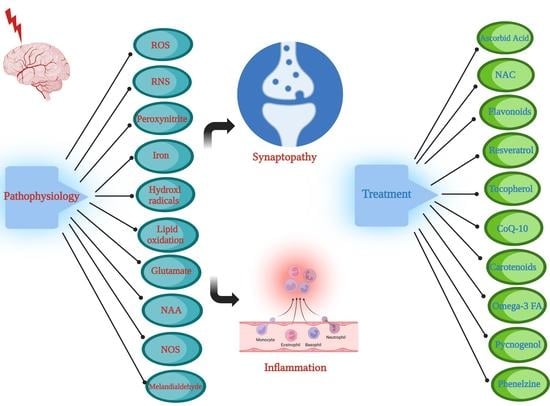
1. Introduction
Traumatic brain injury (TBI) has become a significant cause of morbidity and mortality within the United States and worldwide [1,2]. Approximately 1.7 million individuals experience a TBI, resulting in 52,000 deaths annually. A substantial portion of those injured, estimated to be 124,000, develop chronic disability [1,2,3]. The annual cost of disability has been estimated to be $56 billion a year, creating a significant socioeconomic challenge [4]. Depending on a variety of factors including the severity of the injury, age, and premorbid general health, TBI could lead to wide-ranging neurocognitive, neuroendocrine, and neuropsychiatric sequelae [5,6].
TBI could be conceptualized in two different stages, involving a primary acute process and a subacute/chronic secondary injury neurometabolic cascade. The primary injury is typically due to a mechanical anatomical lesion, involving laceration, contusion, intracranial hemorrhage, and diffuse axonal injury [7]. The secondary injury is a delayed and prolonged complex response, leading to long-term neuropathological changes including metabolic alterations, oxidative stress, neuroinflammation, axonal injury, neurovascular changes, and ultimate neurodegenerative changes, depending on the severity of the injury [8,9]. The brain uses approximately 20% of the body’s total amount of oxygen and demands the highest oxygen supply in the body. An elevated oxygen consumption rate post-injury heightens the probability of producing reactive oxygen species (ROS).
The elevated metabolic rate heightens the probability of the production of reactive oxygen species (ROS). The brain parenchyma also has significant amounts of unsaturated fatty acids and polyunsaturated fatty acids (PUFAs) that are especially vulnerable to free radical reactions and lipid peroxidation. The brain also contains high quantities of iron that can be directly involved with free radical injury [4,10]. The brain also has limited defense mechanisms available against oxidative stress based on its catalase supply compared to other organs such as the liver [11].
This review aims to examine the underlying pathophysiological mechanisms leading to TBI-induced oxidative stress and resultant chronic neuroinflammatory sequelae. Oxidative stress is a product of the orchestration of multiple cellular processes including the activation of oxidative enzymes, nitric oxides, and lipid peroxidation, as well as enhanced excitotoxic activation of glutamatergic neurons [1,12,13]. The resultant oxidative process would ultimately lead to the chronic sequelae of neuroinflammation and synaptopathy. In addition, the discussion will include a number of robust antioxidant regimens reported in preclinical and clinical studies [13].
2. Pathophysiology of Oxidative Stress in TBI
The pathophysiological etiology of TBI is heterogenous and occurs on multiple scales. TBI-induced pathological changes include axonal shearing otherwise known as diffuse axonal injury (DAI), neurovascular compartment damage, and neuronal network disruption, as well as apoptotic and necrotic cellular damage [14,15]. The neuroinflammatory response post-injury involves pro-inflammatory cytokines and free radical formation [16], further elaborated in Section 4 below. Oxidative stress plays a key role in this process, and it involves the production of reactive oxygen species (ROS) and reactive nitrogen species (RNS) as outlined in Figure 1. Oxidative stress is a likely result of disequilibrium between the biochemical processes leading to the production of ROS, and the processes responsible for the removal of ROS, involving the enzymatic and non-enzymatic anti-oxidant cellular defense system [17] listed in Table 1. Oxidative stress has been proposed to lead to the formation of brain edema due to mitochondrial dysfunction, blood–brain barrier (BBB) breakdown, sensory-motor dysfunction, and secondary neuronal injury [18,19,20,21]. This review article examines a range of both preclinical and clinical TBI studies with a focus on the central role of oxidative stress in TBI pathology, as well as potential treatment modalities with real translational clinical applications. There are a number of preclinical TBI models proposed, which include the fluid percussion model, the controlled cortical impact model, Feeney’s weight drop model, Marmarou’s weight drop model, the penetrating ballistic-like brain injury (PBBI) model, and the blast wave model [22,23].
2.1. Oxidative Enzymes
Oxidative stress is a product of the disruption of the equilibrium between the production of oxygen-derived free radicals and the scavenging antioxidant system. The oxidative agents include hydrogen peroxide (H), its high oxidative metabolic demand, limited antioxidant capacity, and repair mechanisms [39].
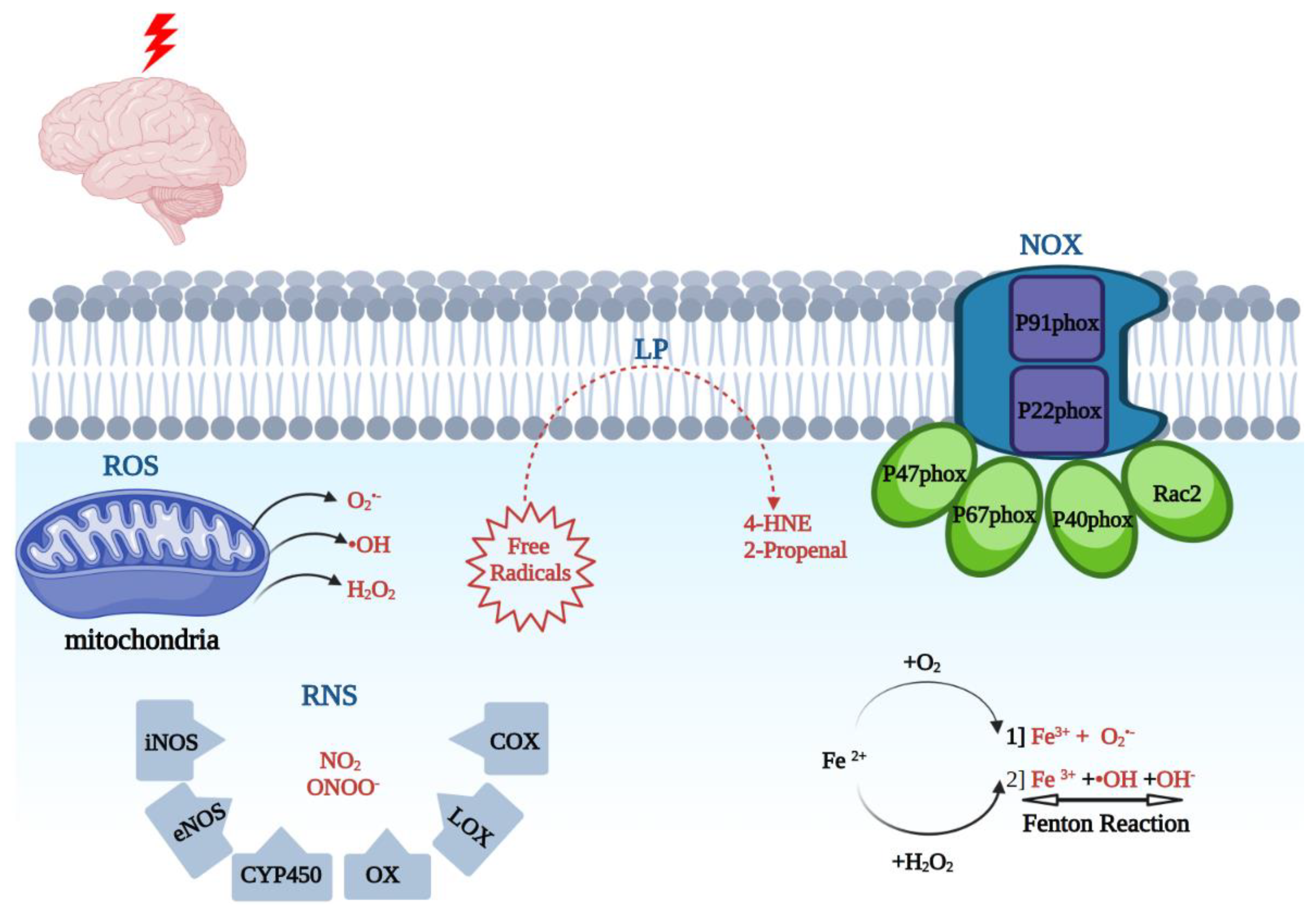
ROS are products of arachidonic acid (AA) cascade activity, mitochondrial leakage, catecholamine oxidation, neutrophils, as well as oxidative stress-inducing enzymes [40]. The oxidative stress-inducing enzymes include NADPH oxidase (NOX). NOX is a multi-component enzyme, which consists of cytoplasmic subunits, which include p47phox, p67phox, p40phox, and Rac2. Once phosphorylated, the NOX enzyme can form a more complex structure, translocate to the plasma membrane, and dock with plasma membrane subunits p91phox and p22phox. NOX catalysis occurs at the p91phox subunit (NOX2). Prior studies have reported that overactivation of NOX2 leads to oxidative damage in ischemic, traumatic, and neurodegenerative conditions [7,18,41].
2.2. ROS and Oxidative Stress
ROS- superoxide (O2●−), hydrogen peroxide (H2O2), peroxyl (ROO−) radical, and the hydroxyl radical (●OH) are the products of 1-electron reduction of oxygen [1]. These are produced by mitochondria and other cellular enzymes as products of cellular oxidative metabolism. These radicals can, in turn, react with lipids, nucleic acids, and proteins, and lead to downstream protein inactivation including enzymes, receptors, and ion channels [24]. With iron present, superoxide and H2O2 can be converted into a more potent oxidative agent, hydroxyl radical. Furthermore, superoxide will react with nitric oxide to generate peroxynitrite (ONOO−). Myeloperoxidase (MPO) produces hypochlorous acid (HOCl−) and inducible nitric oxide synthase (iNOS) produces nitric oxide (NO●). Cyclooxygenases (COX-1 and COX-2) produce prostaglandin intermediates from arachidonic acid through peroxidase activity first to PGG2 and then to PGH2. NADPH oxidase (NOX) enzymes catalyze the production of O2●− and H2O2 from oxygen. These are the major agents of cellular generation of ROS in the cells that can contribute to neuroinflammation [1].
2.3. RNS and Oxidative Stress
Reactive nitrogen species (RNS) include various nitric oxide compounds, such as peroxynitrite (ONOO−) and nitrogen dioxide (NO2). The RNS species generally have longer half-lives than O2●− and ●OH [12,25]. It is important to add that multiple enzymes are involved in the free radical formation, which include nitric oxide synthase (iNOS), endothelial nitric oxide synthase (eNOS), cytochrome P450 (CYP450), cyclooxygenase (COX), lipoxygenase (LOX), and xanthine oxidase (XO) [26,27,42,43]. One of the main species leading to oxidative tissue injury is peroxynitrites (PN; ONOO-). PN is produced by the combination of NO synthases (NOS)-generated NO radical and O2●−. PN can also react with carbon dioxide (CO2) and form nitoperoxocarbonate (ONOOCO2). Each of the PN-derived radicals can cause lipid peroxidation (LP) cellular damage via the transfer of an electron from a hydrogen atom bound to allylic carbon in PUFAs or lead to protein carbonylation due to reacting with particular amino acids [8]. PN is implicated in TBI pathophysiology in multiple ways. The three NOS isoforms (endothelial, neuronal, and inducible) are upregulated during the first 24 h post-TBI, as reported in preclinical studies [21,44]. In addition, prior reports have shown that acute treatment with NOS inhibitors, such as L-NAME, can have a neuroprotective effect and lead to an improved neurological outcome [8,45]. Furthermore, the biochemical marker of PN-mediated damage, based on increased 3-NT levels and ADP ribosylation, has been documented in rodent TBI studies [8,28].
2.4. Iron and Hydroxyl Radicals
Unbound iron (Fe2+) is one of the most abundant ions within the CNS. Iron dyshomeostasis is a major factor involved in various neurodegenerative conditions including Alzheimer’s Disease (AD), Parkinson’s Disease (PD), amyotrophic lateral sclerosis (ALS), Huntington’s disease, and Friedreich’s ataxia [29]. The iron distribution varies in concordance with the sensitivity of various anatomical regions to oxidative stress [8,46]. Under basal physiological conditions, low molecular forms of redox-active iron are maintained at very low levels. The iron transport protein transferrin tightly binds iron in the Fe3+ oxidation state. This ion is stored intracellularly by the iron storage protein ferritin. Post TBI, the pH within injured areas is lowered, in turn leading to the release of iron from its storage sites [8,46,47,48]. The second source of catalytically active iron is hemoglobin. It has been reported that iron released from hemoglobin is responsible for hemoglobin-mediated oxidative damage [48]. Free iron, Fe2+, can in turn undergo the following two reactions: First, autooxidation to form Fe3+ leading to the formation of O2●− (Equation (1)), or the Fenton reaction to form ●OH (Equation (2)):
Fe 2+ + O2 ---> Fe 3+ + O2●−
Fe 2+ + H2O2 ---> Fe 3+ + ●OH + OH− (Fenton reaction)
It is hypothesized that inflammatory cells that are activated post-injury in TBI are mainly responsible for iron deposition in TBI [29,49]. The dysregulation of ceruloplasmin, the main copper-binding protein, can lead to an increase in free copper ions and a decrease in ferroxidase activity, and, in turn, reduce iron clearance from the CNS. Ceruloplasmin-deficient mice subjected to TBI have been reported to have elevated brain iron levels and enhanced infarct volume [29]. Iron chelation therapies, including deferoxamine and deferipone, have been extensively studied as a treatment strategy in intracerebral hemorrhage (ICH) and stroke [30,31], and are a promising potential regimen in TBI [29].
2.5. Lipid Peroxidation
The phenomenon of lipid peroxidation (LP) has been reported in stroke studies [8]. LP has been associated with the loss of cell membrane integrity, increased membrane permeability, and diminished activity in membrane ATPase, leading to further injury. LP involves a process of electron transfer from membrane lipids to free radicals, resulting in cell damage, and most often affects the PUFAs. These compounds contain multiple double bonds and highly reactive hydrogens. The reaction is comprised of three major steps, namely initiation, propagation, and scission. The products of these reactions include lipid peroxyl radical, lipid hydroperoxide, and a peroxyl-fatty acid radical. The final step of the reaction, scission, leads to the formation of neurotoxic aldehydes such as 4-hydroxynonenal (4-HNE) and 2-propenal (acrolein). 4-HNE produces neurotoxicity via binding to basic amino acids, including lysine or histidine, as well as sulfhydryl-containing cysteine residues [8].
2.6. Glutathione Peroxidase
Glutathione peroxidase (GPx) and glutathione reductase (GR) are intracellular antioxidant enzymes. GPx-catalyzed oxidation of glutathione (GSH) produces its oxidized form (GSSG), while GR catalyzes the recycling of GSSG to GSH. Enzymes that remove superoxide and H2O2 protect the cell against oxidative stress. Catalase and GPx, in conjunction with SOD, are the major defense enzymes against superoxide radicals [50]. Due to oxidative stress, the function and transport of mitochondria to synaptic regions are impaired, leading to synaptic dysfunction and neurodegeneration [8,32,51].
2.7. Malondialdehyde
ROS-induced damage is defined by the onset of LP and is reflected by malondialdehyde (MDA) levels. MDA reacts with several functional groups on proteins, lipoproteins, and DNA. MDA levels have been measured to be elevated 1 min post-trauma, with a progressive increase to the maximum in the second hour, and persisting up to 48 h post-trauma [8].
2.8. Glutamate
TBI involves an excitotoxic physiological cascade, marked by excessive glutamate release. Glutamate plays a central role in various neurocognitive functions including cognition, memory, and learning [8,33]. Glutamate also plays a key role in brain development, neuronal signalling leading to cellular survival and differentiation, and the formation and elimination of synapses. TBI results in the excessive release of extracellular glutamate, in turn leading to the overstimulation of metabotropic and ionotropic receptors, which can ultimately lead to neuronal cell death. This process leads to the release of calcium (Ca2+) from internal stores, and in turn, the activation of N-methyl-D-aspartate (NMDA) receptor, 2-amino-3-(3-hydroxy-5-methylisoxazaol-4-yl)proprionic acid receptor (AMPAR), and kainite receptor. AMPAR receptor activation leads to the entry of Na+ into the cell, depolarization, and subsequent cellular edema. NMDA receptor activation also leads to the entry of Na+ and Ca2+ into the cell and subsequent membrane depolarization, and the induction of Ca2+-dependent enzymes, including calpains, protein kinases, endonucleases, and NOS [8,52].
2.9. N-acetylaspartate
N-acetylaspartate (NAA) is the acetylated form of aspartate and is ubiquitous in the central nervous system (CNS). NAA is a robust imaging biomarker in brain H-MRS studies. Using whole-brain NAA measurements by High-Performance Liquid Chromatography (HPLC), the levels of NAA have been correlated with the degree of injury [53]. Utilizing MR spectroscopy, NAA can be used to quantify neuronal damage and has been correlated with post-TBI neurocognitive sequelae [8,35,36].
2.10. Nitric Oxide Synthase
Nitric oxide synthases are a group of enzymes that facilitate the production of NO from L-arginine. TBI leads to the activation of NOS and, in turn, the formation of NO in the brain [54]. NO has been measured in the cerebrospinal fluid (CSF) of TBI patients [55], and the final products of NO and nitrate/nitrite levels have been correlated with TBI severity [56]. NO can be produced by three NOS isoforms: NOS1 (neuronal) and NOS3 (endothelial), which are Ca2+ dependent enzymes, and NOS2 (inducible), which leads to high levels of NO production in inflammatory situations. The role of NOS in TBI has been previously reported [45,57]. Non-selective NOS inhibitors given post-TBI, such as L-NAME and 7-nitroindazole, have been shown to reduce neurological deficits [37,38].
3. Oxidative Stress and Synapse
Oxidative stress has been reported in both clinical and preclinical TBI studies, and has been found to persist up to six months post-injury [4,58,59,60,61]. Synaptic alterations play a key role in the convergence of glutamate toxicity and oxidative stress. NMDA receptor activation and the influx of Ca2+ lead to ROS and RNS generation [62]. Postsynaptic density protein 95 (PSD95) plays a fundamental role in coupling PKCα to NMDA receptors. It also interacts with nitric oxide synthase (nNOS), leading to nitric oxide generation [61,63], which contributes to both inflammation and oxidative stress [8]. The physiological coupling of nNOS and PSD95 is foundational in oxidative stress. Disrupting PSD95 has been shown to be neuroprotective in a preclinical model of TBI [64]. Multiple prior preclinical TBI studies using a systems biology approach have suggested the potential role of nNOS and PSD95 as TBI biomarkers [65]. Prior preclinical TBI studies by Ansari et al. have examined the relationship between oxidative stress and synaptic density alterations in hippocampal and cortical areas [19,66].
A longitudinal analysis of post-TBI tissue demonstrated evidence of increased carbonylation, 3-nitrotyrosine, and acrolein, markers of oxidative stress. The increase in oxidative stress markers proceeded to a decrease in PSD95 level, at 24 h and 48 h time points. The temporal association suggests the potential role of oxidative stress in synaptopathy. There is a pronounced release of glutamate post-TBI, as reported in both clinical and preclinical studies [34,67,68,69]. The rise of glutamate is thought to be due to multiple potential etiologies, which include the extravasation of blood into the parenchymal space [70], the release of structural amino acids via newly developed micropores post-injury [67], dysregulation of astrocytic glutamate transporters GLT-1 and GLAS leading to glutamate uptake dysfunction, and increased synaptic release and resultant overflow from the synaptic terminal [71]. The increased glutamate post0injury has a profound effect on excitotoxicity and resultant synaptic alterations.
Complexins are 15-16 kDa cytosolic proteins that compete with the a-SNAP protein for binding with SNAP receptors (SNARE) in a rapid anti-parallel fashion in the presynaptic terminal [72]. The localization of complexin I in the inhibitory neuron and complexin II in the excitatory neurons in the cerebellum and hippocampus has provided a means of assessing excitatory and inhibitory pathways [73]. Yi et al. were able to demonstrate, using a lateral fluid percussion (FP) injury model in mice, that TBI leads to dynamic changes in complexin I and complexin II levels, and the aforementioned changes are associated with oxidative stress [71].
In another preclinical study by Ansari et al. using a unilateral moderated controlled contusion model of TBI in rats, it was shown that PSD-95, a post-synaptic scaffolding protein, followed a time-dependent decrease, observed at 24 h and perpetuating to 96 h post-injury. Similarly, synapsin-I, a major presynaptic protein, also followed a time-dependent change, with a significant change at 24 h and a continued decline after 96 h post-injury. Synapse-associated protein 97 (SAP-97), a membrane-associated phosphor-protein that interacts with a variety of different AMPA-type glutamate receptors, was shown to decline post-injury, with significant changes at 96 h post-injury [66].
The hippocampus, in charge of episodic memory, is another vulnerable locus of TBI injury due to its intrinsic connectivity and prevalence of high density of excitatory amino acid (glutamate) receptors [69,74]. In an accompanying study by Ansari et al., it was shown that the levels of presynaptic marker protein, synapsin I, SAP-97, and PSD-95 are significantly diminished post-TBI and remain decreased 96 h post-TBI [19].
4. Oxidative Stress and Inflammation
There are a growing number of studies highlighting the roles of cytokine, chemokines, and growth factors in the pathophysiology of TBI. In the acute post-injury period, there is a large production of cytokines, including IL-1β and tumor necrosis factor-α (TNF-α), as well as transforming growth factor-beta (TGF-β), which in turn enhances the brain trauma condition with increased oxidative stress and matrix metalloproteineases (MMPs) [12,75,76,77]. The posttraumatic inflammatory cascade leads to BBB dysfunction and an influx of inflammatory cells from the blood to the brain [78].
IL-1β triggers inflammatory reactions, leading to BBB disruption, edema, and neuronal loss [79,80,81,82]. Heightened IL-1β levels have been reported within the CSF and brain tissue in both humans and experimental animals [12,83,84]. Intracerebroventricular infusion of IL-1β-neutralizing antibody has been shown to reduce cerebral edema, neuronal loss, and lead to cognitive improvement following TBI in mice [85]. TNF-α plays key roles in neuronal development, cell survival, synaptic plasticity, and CNS ionic homeostasis. TNF-α has been measured in the CSF and serum of TBI patients. The TNF-α increase has also been associated with the loss of sensorimotor and cognitive functions in TBI models [86,87,88,89]. Multiple in vivo studies have reported an increased level of TGF-β within the CSF in multiple CNS disorders including TBI, which can be detected within days post-injury [90,91]. High levels of TGF-β within CSF could be due to damaged BBB, and its inhibition has been shown to prevent epilepsy post-injury [92].
Nuclear factor-erythroid-2-related factor (Nrf)-2 is a pleiotropic transcription factor and a master regulator of anti-oxidant defense response. It plays a major part in protecting cells from cytotoxic/oxidative damage [93,94]. The role of Nrf-2 as a protective agent in TBI-induced oxidation and the neuroinflammation cascade is increasingly being investigated [95]. Under basal conditions, Kelch-like ECH-associated protein 1 (KEAP1) binds Nrf2 and sequesters it in the cytoplasm. Post-injury, the increased ROS modifies cysteine residues in KEAP1 and decreases its affinity for Nrf2, and Nrf2 subsequently translocates into the nucleus. It subsequently combines with antioxidant response element (ARE) sequences, leading to activation of the Nrf2/ARE pathway and the gene expression of phase II detoxifying enzymes and antioxidases, such as heme oxygenase 1 (HO-1), SOD-1, and glutathione peroxidase 1 (GPx1). These enzymes protect cells from oxidative stress and a broad range of other toxins [14,96]. Using a preclinical TBI model, Cordaro et al. examined a new compound, Hidrox®, and demonstrated the modulation of oxidative stress and neuroinflammation, partly via reducing the activation of the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB) pathway and associated cytokine activation [14]. TBI leads to a cascade of inflammatory processes, involving NF- kB, which contributes to further neuronal damage and delays in functional recovery [94]. TBI animals treated with Hidrox® had significantly increased IkB-α levels in the cytosol and reduced NFkB expression in the nucleus [14].
5. Treatment Options of Oxidative Stress
There are a variety of preclinical studies focused on the utility of antioxidants in TBI. However, there are multiple challenges that make the clinical interpretation of such studies exceedingly more challenging, and these include the large variability in preclinical TBI models, the antioxidant tested in the study, the time interval of the antioxidant utilized, dosage, and the route of drug administration, as well as the range of neurocognitive parameters used [97].
Antioxidants function to robustly reduce the power of various oxidants, including ROS and RNS [97]. They can be generally divided into hydrophilic (water-soluble) and hydrophobic (fat-soluble) antioxidants. With the exception of reduced glutathione (GSH), uric acid, and coenzyme Q10, the primary sources of antioxidants are exogenous dietary sources [98]. Therefore, the supplementation of nutraceuticals is of key importance to provide an adequate supply of antioxidant protection [99].
Although the importance of the antioxidant regimen in TBI treatment is increasingly being recognized, there is no robust evidence for its efficacy in a clinical setting [97,100]. The reader is referred to an excellent review article written by Di Pietro et al. on this topic [97]. In Table 2, we summarize selected low-molecular-weight antioxidants that have been published in both preclinical and clinical TBI studies.
Oxidative stress is a result of disequilibrium between free radical formation and the antioxidant system. CSF MDA levels can rise within a 2–3 h window post-TBI and remain elevated 7 days post-injury. Moreover, SOD activity has been shown to diminish 24 h post-TBI, and remain so 7 days post severe TBI [8,101]. Antioxidant treatment focused on diminishing oxidative stress levels has been shown to reduce post-TBI neurological deficits, cerebral edema, and brain lesion volume [8]. Various antioxidant regimens have been studied, including polyethylene glycol (PEG)-conjugated SOD (PEG-SOD) [102], melatonin mimicking GPx [103], OPC-14117 (7-hydroxy-1-[4-(3-methoxyphenyl)-1-piperazinyl] [104], acetylamino-2, 2,4, 6-tetramethylindan (a superoxide anion scavenger), and phenyl-tert-butylnitrone (a free radical scavenger) [105], as well as mesylate tirilazad (an LP inhibitor) [28]. L-NAME treatment has been shown to decrease nitrotyrosine levels post-TBI, supporting the role of NO in PN formation. Furthermore, PN scavengers such as penicillamine, penicillamine methylester, and tempol have shown beneficial treatment effects [28].
5.1. Ascorbic Acid (Vitamin C)
Ascorbic Acid (AA) is a highly abundant water-soluble antioxidant and a key cofactor in multiple important enzymatic reactions [106]. As a robust reducing agent, it reacts with a range of ROS and RNS. AA deficiency is linked to debilitating connective tissue disease [107]. AA is not endogenously produced in the body, and exogenous dietary sources are required for the supply needed [108]. AA absorption occurs via SVCT1 Na+-AA transporter activity [109]. It has been reported that the levels of AA decrease dramatically after experimental TBI [110,111,112], and the changes in AA levels are correlated with the TBI severity [113]. In a preclinical study, using closed-head TBI, different doses of AA administered for 2 weeks, either alone or in addition to tocopherol, were shown to reduce the mortality rate, decrease melanoaldehyde (MDA) levels, and increase tissue superoxide dismutase levels [114]. A double-blind controlled clinical trial study by Razmkon et al. reporting on 100 TBI patients showed that a high dose of AA treatment led to the diminished progression of cortical edema [114].
5.2. N-Acetyl-Cysteine (NAC)
Reduced glutathione (GSH) has a fundamental role in various detoxifying reactions, which also includes scavenging ROS and RND, as well as the elimination of xenobiotics [115]. GSH is a low-molecular-weight antioxidant that mammals can endogenously synthesize. GSH is one of the main reducing agents for protein thiol groups and can also modulate inflammatory responses [116]. GSH is the cofactor for the enzymes GSH peroxidase (GPx) and glutathione-S-transferases. The reduction of oxidized GSH (GSSG) is performed by the NADPH-dependent enzyme GSH reductase (GR) [97]. GSH also binds to glutamate NMDA and AMPA receptors and can function as a neuromodulator [117]. A portion of the total cellular GSH content is in the form of S-nitroglutathione (GSNO), produced by a reaction between NO and GSH. GSNO is also a source of NO, another key antioxidant in lipid peroxidation reactions [19]. Prior preclinical studies have reported a decrease in GSH levels following TBI. There is a significant decrease in brain GSH levels following TBI, and more specifically, a decrease in GSH/GSSG levels has been reported in the subacute period post-TBI [20]. There is also a reported negative correlation between the degree of injury and rat brain GSH levels [110].
It has been shown in a preclinical study that i.p. injection of γ-glutamylcysteine ethyl ester (GCEE) in the post-injury period leads to a significant reduction of oxidative/nitrosative damage, highlighting the potential protective role of increasing GSH levels post-injury [118]. The therapeutic potential of N-acetylcysteine (NAC) is increasingly being recognized in both preclinical and clinical studies because of its direct role as a ROS and RNS scavenging agent or cysteine availability and GSH synthesis [119]. In a preclinical model of controlled cortical impact, NAC administration 15 min post-injury led to diminished MDA formation, increased SOD and GPx, and was found to be neuroprotective. Furthermore, NAC in combination with minocycline (MINO), given 12 h post-injury, prevented the TBI-induced reduction in the expression of oligodendrocyte markers CC1, 2′,3′-cyclic-nucleotide 3′-phosphodiesterase, and phospholipid protein in the subacute period post close head injury (CHI) [120]. An amidated form of NAC, namely N-acetylcysteine amide (NACA), can potentially provide more blood–brain barrier permeability, and potentially more robust therapeutic efficacy [121]. Complexin I and complexin II proteins, a family of cytosolic proteins and measures of inhibitory and excitatory synaptic activities, respectively, were shown to be completely blocked by N-acetylcysteine (NAC) administration 5 min post-injury in a preclinical study by Yi et al. [71]. Neuronal loss was also shown to be reduced in the injured hemisphere, post-TBI NAC treatment.
5.3. Flavonoids
Flavonoids are structurally identified by one phenyl ring fused with an oxygen-containing heterocyclic ring and a second phenyl ring. These compounds are ubiquitous in plants and fungi [97,122]. There are multiple subtypes of flavonoids, which include anthocyanidins, anthoxanthins, flavanones, flavanonols, and flavans. Due to their negative oxido-reductive potential, flavonoids can function as robust antioxidants towards various ROS and RNS species [123]. Due to the vast number of potential flavonoid sources, systematic clinical studies have been more difficult to conduct. A small pilot study, comprised of 60 mild TBI patients, using oral enzogenol (1 g daily) for 6 weeks, resulted in significantly fewer self-reported cognitive deficits [123].
5.4. Resveratrol
Resveratrol, a mostly hydrophobic compound, is abundantly existent in the skin of red grapes. Red wines contain an ample amount of resveratrol and have been thought to be the protective agent in the so-called “French paradox”, pertaining to low cardiovascular diseases in the French population, in spite of their high fat consumption [124]. It is proposed to have anti-inflammatory benefits, as well as modulate mitochondrial functions [125]. In a preclinical study, involving a weight drop model of TBI, rats treated with resveratrol (100 mg/kg) given via i.p. injection were found to have lower levels of MDA, xanthine oxidase (XO), and NO and increased levels of GSH as well as diminished parenchymal damage [126]. In another pre-clinical study involving a CCI-TBI preclinical model, resveratrol was shown to increase cell survival by inhibiting GSK-3β-mediated autophagy and apoptosis [127]. As per another preclinical study, resveratrol was found to have anti-inflammatory properties, noted by decreased microglial activity in various brain regions as diminished cytokine levels such as IL-6 and IL-12 levels [128].
5.5. Alpha-Tocopherol (Vitamin E)
Tocopherols are fat-soluble compounds, ubiquitous in vegetable oils, with potent antioxidant properties [97]. They are comprised of four tocopherols and four tocotrienols [129]. They contain a central chromane ring, which has oxidoreductive capacity. The brain, although rich in easily oxidizable fat-soluble compounds, has the lowest content of tocopherols [97]. In a preclinical study, using a mild or severe model of TBI, i.p. administration of alpha-tocopherol (100 mg/kg) was shown to diminish brain levels of MDA produced by oxidative/nitrosative stress-induced lipid peroxidation [129]. In another study using focal moderate TBI, Sprague–Dawley rats received i.p. injection of alpha -tocopherols (600 mg/kg) prior to TBI induction. These rats performed better on neurocognitive tests and showed less evidence of edema, inflammation, and a higher degree of cerebral tissue regeneration based on Nogo-A and NgR levels [130]. In another preclinical study, rats either received a regular diet with 500 IU/kg of -tocopherols for 4 weeks, or without it, prior to receiving focal mild TBI, using a fluid percussion-based injury. The group fed the tocopherol-rich diet had enhanced levels of SOD, Sir2, and BDNF caused by TBI, as well as diminished motor function impairments [131]. There is a relative paucity of clinical studies using tocopherols. According to Razmkon et al. using four groups of TBI patients, IM administration of tocopherol (400 IU for 7 days) was associated with diminished mortality rates and enhanced clinical outcomes [114].
5.6. Coenzyme Q10
Coenzyme Q is a component of the mitochondrial chain of aerobic organisms. It contains a quinone ring, which in turn allows it to exist in fully oxidized, fully reduced, or semi-reduced forms [97]. Coenzyme Q10 (CoQ10) is found in the mitochondrial inner membrane, and actively participates in electron transfer [132]. There are a number of preclinical studies that have shown the efficacy of CoQ10 in the treatment of head trauma. In a preclinical Marmarou TBI model, CoQ10 (10 mg/Kg) was administered immediately after trauma, and after 24 h via gavage. The animals treated with CoQ10 had diminished MDA levels, vascular congestion, neuronal loss, nuclear pyknosis, nuclear hyperchromasia, cytoplasmic eosinophilia, and axonal edema, and increased cerebral SOD [133]. CoQ10 was also found to modify the cerebral expression of genes involved in bioenergetics and oxidative/nitrosative stress [134]. A similar preclinical study involving the administration of CoQ10 infusion before or 30 min after TBI led to diminished brain mitochondrial damage and apoptosis, as well as lower serum levels of serum glial fibrillary acidic protein (GFAP) and ubiquitin C-terminal hydrolase-L1 (UCH-L1) [135].
5.7. Carotenoids
Carotenoids are a group of pigments synthesized by plants, algae, and species of bacteria [97,136]. They are further subdivided into oxygen-containing carotenoids (xantophylls) and a class of oxygen-free carotenoids (carotenes). The use of different carotenoids as a potential therapeutic modality has been studied in depth in various neurodegenerative diseases [136]. In a preclinical model of TBI using astrocytes, astaxanthin diminished apoptosis via the inhibition of NKCC1 expression and reduced the expression of NF-κB-mediated pro-inflammatory factors [137]. In another study using the CCI preclinical model of TBI, astaxanthin of various doses (10, 25, 50, or 100 mg/kg body weight) administered 30 min post-injury decreased TBI-related brain tissue injury by inhibiting AQP4/NKCC1-mediated cerebral edema in a dose-dependent manner and was found to improve neurologic deficits and the BBB permeability [138]. In a separate study, involving a weight drop model of TBI, astaxanthin was found to reduce lesion size in the cortex and increase brain-derived neurotrophic factor (BDNF), growth-associated protein-43 (GAP-43), synapsin, and synaptophysin (SYP), supporting an induction of neuronal survival and plasticity, also evident by enhanced cognitive recovery [139]. Other carotenoids, including fucoxanthin, bexarotene, crocin, and lutein, have shown similar beneficial results in prior preclinical studies [137,140,141,142,143].
5.8. Omega-3 Fatty Acids
Omega-3 fatty acids belong to a group of polyunsaturated fatty acids (PUFA) that are subdivided into essential (linoleic acid) or semi-essential (eicosapentaenoic acid, docosahexaenoic acid) fatty acids for humans. Omega 3 fatty acids are not only foundational components of membrane phospholipids, but also precursors of key biologically active molecules (prostaglandins) [97]. Both docosahexaenoic acid (DHA) and eicosapentaenoic acid (EPA) are abundant in brain phospholipids, with DHA comprising 50% of the weight of neural tissue within the brain [144]. A number of preclinical studies have reported the benefits of omega-3-fatty acids and DHA in preclinical TBI models [97]. However, clinical studies, including a study performed by Matsouoka et al. using 53 TBI patients, with 52% of the participants with mild TBI, and involving the administration of 12 weeks of 1470 mg DHA and 147 mg EPA, did not confer any neurological (including measures of BDNF), cognitive, or affective benefits based on a multitude of neuropsychological batteries [145].
There is a synergistic reaction between dietary DHA and exercise. In a preclinical model using the mild fluid percussion injury (mFPI) model, it was shown that rats that underwent mFPI and were on a diet high in DHA (1.2% DHA), combined with 12 days of exercise, had elevated BDNF and p-TrkB levels, which are involved in synaptic plasticity and cognitive function. These levels were significantly different from the exercise-only group and regular diet-only group, respectively [146].
5.9. Pycnogenol
The neuroprotective effects of pycnogenol (PYC) have been explored in rodent models of TBI [147]. PYC is a combination of bioflavonoids, which are extracted from the bark of the French maritime pine tree (Pinus maritima) and have a robust capacity to scavenge free radicals. Ansari et al., using a well-established preclinical model of TBI, demonstrated the neuroprotective effects of PYC in TBI. PYC treatment was shown to reduce oxidative stress, and rescue both pre- and post-synaptic proteins in the cortex and the hippocampus. The level of neuroprotection was shown to be dose-dependent, with a therapeutic window extending up to 4 h post-trauma. However, there was a therapeutic time effect, with animals treated with PYC at 15 min demonstrating significantly lower levels of oxidative stress compared to a 2 or 4 h delay following injury within the cortex. However, the hippocampus did not show a significant time effect [147].
5.10. Phenelzine
Phenelzine (PZ) is an FDA-approved drug that functions as an MAO inhibitor. PZ has been shown to have aldehyde-scavenging properties and provide partial protection from LP-mediated oxidative injury [148,149,150]. In a preclinical study using a controlled cortical impact injury (CCI) model, PZ was administered subcutaneously (3–30 mg/kg) at different times post-injury. It was shown that PZ treatment preserved both synaptic and non-synaptic mitochondrial bioenergetics at 24 h, and this protection was partially extended to 72 h post-injury. PZ administration was also shown to significantly improve mitochondrial respiration, with a more robust response of synaptic mitochondria compared to the non-synaptic mitochondria [148].

{kind=link}
{kind=link}
Table 2.
Outlines a number of reported therapeutic agents used in treatment of TBI-induced oxidative stress.
Table 2.
Outlines a number of reported therapeutic agents used in treatment of TBI-induced oxidative stress.
| Therapeutic Agent | Mechanism | References |
|---|---|---|
| Ascorbic Acid (Vitamin C) | As a robust reducing agent, it reacts with a range of ROS and RNS. | Castiglione et al. 2018 [108] Parker et al. 2015 [112] |
| N-acetylcysteine (NAC) | direct role as ROS and RNS scavenging agent, or increasing cysteine availability and GSH synthesis, extensively studied in preclinical and clinical settings | Limongi et al. 2019 [115] Koza et al. 2019 [119] |
| Flavonoids | Given their negative oxido-reductive potential, flavonoids can function as robust anti-oxidants towards various ROS and RNS species | Izzi et al. 2012 [122] Theadom et al. 2013 [123] |
| Resveratrol | Has both anti-inflammatory benefits, as well as modulation of mitochondrial functions | Irias-Mata et al. 2017 [128] Gatson et al. 2013 [127] |
| a-Tocopherol (Vitamin E) | Fat soluble compounds, ubiquitous in vegetable oils, with potent anti-oxidant properties | Di Pietro et al. 2020 [97] Razmkon et al. 2011 [114] |
| Coenzyme Q10 | component of the mitochondrial chain aerobic organisms with efficacy in TBI treatment in a few preclinical studies | Pierce et al. 2017 [134] Pierce et al. 2018 [135] |
| Carotenoids | group of pigments synthesized by plants, algae, and species of bacteria, and in preclinical studies have bene shown to diminish apoptosis, and reduced expression of NF-κB-based inflammation | Zhang et al. 2017 [137] Ji et al. 2013 [139] |
| Omega-3 Fatty Acids | group of polyunsaturated fatty acids (PUFA), that are subdivided into essential or semi-essential for humans, and benefits of omega-3-fatty acids and DHA in preclinical TBI models | Matsuoka et al. 2015 [145] Wu et al. 2013 [146] |
| Pycnogenol (PYC) | combination of bioflavonoids with robust capacity to scavenge free radicals | Ansari et al. 2013 [147] |
| Phenelzine | FDA-approved drug, which functions as an MAO inhibitor and has been shown to have aldehyde scavenging properties, and provide partial protection from LP-mediated oxidative injury | Hill et al. 2020 [148] Cebak et al. 2017 [149] |
6. Conclusions
TBI remains a leading cause of morbidity and disability globally as our collective understanding of the disorder continues to evolve. Oxidative stress is a key component of the secondary injury cascade that can often be perpetuating and chronic. As there are currently no FDA-approved regimens for TBI treatment, alternative therapies such as the use of antioxidants described here become a viable treatment option for consideration in the clinical setting. TBI is inherently a heterogeneous disease due to variance in both the mechanism and severity of injuries. Hence, a range of treatment options will be needed to meet the challenge. The preclinical TBI models, although salient from a mechanistic point of view, can be challenging to translate into a real clinical setting. In addition, there is a great deal of temporal variation between the time of injury and clinical assessment. The use of antioxidants may provide a relatively safe option to offer in an acute treatment setting, which in turn would alter the trajectory of the disease process. Future studies, both on preclinical and clinical levels, can be focused on TBI progression in terms of oxidative stress and the potential efficacy of antioxidant treatment on multiple different time scales.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Eastman, C.L.; D’Ambrosio, R.; Ganesh, T. Modulating neuroinflammation and oxidative stress to prevent epilepsy and improve outcomes after traumatic brain injury. Neuropharmacology 2020, 172, 107907. [Google Scholar] [CrossRef] [PubMed]
- Leo, P.; Mccrea, M. Epidemiology. In Translational Research in Traumatic Brain Injury; Laskowitz, D., Grant, G., Eds.; CRC Press/Taylor and Francis Group: Boca Raton, FL, USA, 2016; Chapter 1. [Google Scholar]
- Selassie, A.W.; Zaloshnja, E.; Langlois, J.A.; Miller, T.; Jones, P.; Steiner, C. Incidence of long-term disability following traumatic brain injury hospitalization, United States, 2003. J. Head Trauma Rehabil. 2008, 23, 123–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.-C.; Lin, Y.-J.; Shih, F.-Y.; Chang, H.-W.; Su, Y.-J.; Cheng, B.-C.; Su, C.-M.; Tsai, N.-W.; Chang, Y.-T.; Kwan, A.-L. The role of serial oxidative stress levels in acute traumatic brain injury and as predictors of outcome. World Neurosurg. 2016, 87, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Capizzi, A.; Woo, J.; Verduzco-Gutierrez, M. Traumatic brain injury: An overview of epidemiology, pathophysiology, and medical management. Med. Clin. 2020, 104, 213–238. [Google Scholar]
- Pavlovic, D.; Pekic, S.; Stojanovic, M.; Popovic, V. Traumatic brain injury: Neuropathological, neurocognitive and neurobehavioral sequelae. Pituitary 2019, 22, 270–282. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, A.P.O.; Rodrigues, F.S.; Della-Pace, I.D.; Mota, B.C.; Oliveira, S.M.; de Campos Velho Gewehr, C.; Bobinski, F.; de Oliveira, C.V.; Brum, J.S.; Oliveira, M.S.; et al. The effect of NADPH-oxidase inhibitor apocynin on cognitive impairment induced by moderate lateral fluid percussion injury: Role of inflammatory and oxidative brain damage. Neurochem. Int. 2013, 63, 583–593. [Google Scholar] [CrossRef]
- Cornelius, C.; Crupi, R.; Calabrese, V.; Graziano, A.; Milone, P.; Pennisi, G.; Radak, Z.; Calabrese, E.J.; Cuzzocrea, S. Traumatic brain injury: Oxidative stress and neuroprotection. Antioxid. Redox Signal. 2013, 19, 836–853. [Google Scholar] [CrossRef]
- Greve, M.W.; Zink, B.J. Pathophysiology of traumatic brain injury. Mt. Sinai J. Med. J. Transl. Pers. Med. J. Transl. Pers. Med. 2009, 76, 97–104. [Google Scholar] [CrossRef]
- Herbert, V.; Shaw, S.; Jayatilleke, E.; Stopler-Kasdan, T. Most free-radical injury is iron-related: It is promoted by iron, hemin, holoferritin and vitamin c, and inhibited by desferoxamine and apoferritin. Stem Cells 1994, 12, 289–303. [Google Scholar] [CrossRef]
- Floyd, R.A.; Carney, J.M. Free radical damage to protein and DNA: Mechanisms involved and relevant observations on brain undergoing oxidative stress. Ann. Neurol. 1992, 32, S22–S27. [Google Scholar] [CrossRef]
- Abdul-Muneer, P.; Chandra, N.; Haorah, J. Interactions of oxidative stress and neurovascular inflammation in the pathogenesis of traumatic brain injury. Mol. Neurobiol. 2015, 51, 966–979. [Google Scholar] [CrossRef] [PubMed]
- Crupi, R.; Cordaro, M.; Cuzzocrea, S.; Impellizzeri, D. Management of traumatic brain injury: From present to future. Antioxidants 2020, 9, 297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cordaro, M.; Trovato Salinaro, A.; Siracusa, R.; D’Amico, R.; Impellizzeri, D.; Scuto, M.; Ontario, M.L.; Crea, R.; Cuzzocrea, S.; Di Paola, R.; et al. Hidrox® Roles in Neuroprotection: Biochemical Links between Traumatic Brain Injury and Alzheimer’s Disease. Antioxidants 2021, 10, 818. [Google Scholar] [CrossRef] [PubMed]
- Acosta, S.A.; Tajiri, N.; Sanberg, P.R.; Kaneko, Y.; Borlongan, C.V. Increased Amyloid Precursor Protein and Tau Expression Manifests as Key Secondary Cell Death in Chronic Traumatic Brain Injury. J. Cell. Physiol. 2017, 232, 665–677. [Google Scholar] [CrossRef] [PubMed]
- Shi, K.; Zhang, J.; Dong, J.-f.; Shi, F.-D. Dissemination of brain inflammation in traumatic brain injury. Cell. Mol. Immunol. 2019, 16, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Frati, A.; Cerretani, D.; Fiaschi, A.I.; Frati, P.; Gatto, V.; La Russa, R.; Pesce, A.; Pinchi, E.; Santurro, A.; Fraschetti, F. Diffuse axonal injury and oxidative stress: A comprehensive review. Int. J. Mol. Sci. 2017, 18, 2600. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.-Y.; Wang, H.-D.; Xu, J.-G.; Ding, K.; Li, T. NADPH oxidase inhibition improves neurological outcome in experimental traumatic brain injury. Neurochem. Int. 2014, 69, 14–19. [Google Scholar] [CrossRef]
- Ansari, M.A.; Roberts, K.N.; Scheff, S.W. Oxidative stress and modification of synaptic proteins in hippocampus after traumatic brain injury. Free Radic. Biol. Med. 2008, 45, 443–452. [Google Scholar] [CrossRef] [Green Version]
- Bayir, H.; Kagan, V.E.; Tyurina, Y.Y.; Tyurin, V.; Ruppel, R.A.; Adelson, P.D.; Graham, S.H.; Janesko, K.; Clark, R.S.; Kochanek, P.M. Assessment of antioxidant reserves and oxidative stress in cerebrospinal fluid after severe traumatic brain injury in infants and children. Pediatr. Res. 2002, 51, 571–578. [Google Scholar] [CrossRef] [Green Version]
- Hall, E.D.; Detloff, M.R.; Johnson, K.; Kupina, N.C. Peroxynitrite-mediated protein nitration and lipid peroxidation in a mouse model of traumatic brain injury. J. Neurotrauma 2004, 21, 9–20. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Aravind, A.; Pfister, B.J.; Chandra, N.; Haorah, J. Animal Models of Traumatic Brain Injury and Assessment of Injury Severity. Mol. Neurobiol. 2019, 56, 5332–5345. [Google Scholar] [CrossRef]
- Xiong, Y.; Mahmood, A.; Chopp, M. Animal models of traumatic brain injury. Nat. Rev. Neurosci. 2013, 14, 128–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- CERNAK, I.; SAVIC, V.J.; KOTUR, J.; PROKIC, V.; VELJOVIC, M.; GRBOVIC, D. Characterization of plasma magnesium concentration and oxidative stress following graded traumatic brain injury in humans. J. Neurotrauma 2000, 17, 53–68. [Google Scholar] [CrossRef] [PubMed]
- Hall, E.; Yonkers, P.; Andrus, P.; Cox, J.; Anderson, D. Biochemistry and pharmacology of lipid antioxidants in acute brain and spinal cord injury. J. Neurotrauma 1992, 9, S425–S442. [Google Scholar] [PubMed]
- Ano, Y.; Sakudo, A.; Kimata, T.; Uraki, R.; Sugiura, K.; Onodera, T. Oxidative damage to neurons caused by the induction of microglial NADPH oxidase in encephalomyocarditis virus infection. Neurosci. Lett. 2010, 469, 39–43. [Google Scholar] [CrossRef]
- Pisoschi, A.M.; Pop, A. The role of antioxidants in the chemistry of oxidative stress: A review. Eur. J. Med. Chem. 2015, 97, 55–74. [Google Scholar] [CrossRef]
- Balazy, M.; Nigam, S. Aging, lipid modifications and phospholipases—New concepts. Ageing Res. Rev. 2003, 2, 191–209. [Google Scholar] [CrossRef]
- Brandes, R.P.; Weissmann, N.; Schröder, K. NADPH oxidases in cardiovascular disease. Free Radic. Biol. Med. 2010, 49, 687–706. [Google Scholar] [CrossRef]
- Uttara, B.; Singh, A.V.; Zamboni, P.; Mahajan, R. Oxidative stress and neurodegenerative diseases: A review of upstream and downstream antioxidant therapeutic options. Curr. Neuropharmacol. 2009, 7, 65–74. [Google Scholar] [CrossRef] [Green Version]
- Bedard, K.; Krause, K.-H. The NOX Family of ROS-Generating NADPH Oxidases: Physiology and Pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Andrew, P.J.; Mayer, B. Enzymatic function of nitric oxide synthases. Cardiovasc. Res. 1999, 43, 521–531. [Google Scholar] [CrossRef]
- Hall, E.D.; Wang, J.A.; Miller, D.M. Relationship of nitric oxide synthase induction to peroxynitrite-mediated oxidative damage during the first week after experimental traumatic brain injury. Exp. Neurol. 2012, 238, 176–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mésenge, C.; Verrecchia, C.; Allix, M.; Boulu, R.R.; Plotkine, M. Reduction of the neurological deficit in mice with traumatic brain injury by nitric oxide synthase inhibitors. J. Neurotrauma 1996, 13, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Hall, E.D.; Vaishnav, R.A.; Mustafa, A.G. Antioxidant therapies for traumatic brain injury. Neurotherapeutics 2010, 7, 51–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daglas, M.; Adlard, P.A. The involvement of iron in traumatic brain injury and neurodegenerative disease. Front. Neurosci. 2018, 12, 981. [Google Scholar] [CrossRef] [Green Version]
- Zaleska, M.M.; Floyd, R.A. Regional lipid peroxidation in rat brain in vitro: Possible role of endogenous iron. Neurochem. Res 1985, 10, 397–410. [Google Scholar] [CrossRef]
- Halliwell, B. The wanderings of a free radical. Free Radic. Biol. Med. 2009, 46, 531–542. [Google Scholar] [CrossRef]
- Sadrzadeh, S.M.; Eaton, J.W. Hemoglobin-mediated oxidant damage to the central nervous system requires endogenous ascorbate. J. Clin. Investig. 1988, 82, 1510–1515. [Google Scholar] [CrossRef] [Green Version]
- Andersen, H.H.; Johnsen, K.B.; Moos, T. Iron deposits in the chronically inflamed central nervous system and contributes to neurodegeneration. Cell. Mol. Life Sci. 2014, 71, 1607–1622. [Google Scholar] [CrossRef] [Green Version]
- Zeng, L.; Tan, L.; Li, H.; Zhang, Q.; Li, Y.; Guo, J. Deferoxamine therapy for intracerebral hemorrhage: A systematic review. PLoS ONE 2018, 13, e0193615. [Google Scholar] [CrossRef]
- Hatakeyama, T.; Okauchi, M.; Hua, Y.; Keep, R.F.; Xi, G. Deferoxamine reduces neuronal death and hematoma lysis after intracerebral hemorrhage in aged rats. Transl. Stroke Res. 2013, 4, 546–553. [Google Scholar] [CrossRef] [PubMed]
- Okatani, Y.; Wakatsuki, A.; Kaneda, C. Melatonin increases activities of glutathione peroxidase and superoxide dismutase in fetal rat brain. J. Pineal Res. 2000, 28, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Povlishock, J.; Kontos, H. The role of oxygen radicals in the pathobiology of traumatic brain injury. Hum. Cell 1992, 5, 345–353. [Google Scholar]
- Kushner, D. Mild traumatic brain injury: Toward understanding manifestations and treatment. Arch. Intern. Med. 1998, 158, 1617–1624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riedel, G.; Platt, B.; Micheau, J. Glutamate receptor function in learning and memory. Behav. Brain Res. 2003, 140, 1–47. [Google Scholar] [CrossRef]
- Yi, J.H.; Hazell, A.S. Excitotoxic mechanisms and the role of astrocytic glutamate transporters in traumatic brain injury. Neurochem. Int. 2006, 48, 394–403. [Google Scholar] [CrossRef]
- Friedman, S.D.; Brooks, W.M.; Jung, R.E.; Chiulli, S.J.; Sloan, J.H.; Montoya, B.T.; Hart, B.L.; Yeo, R.A. Quantitative proton MRS predicts outcome after traumatic brain injury. Neurology 1999, 52, 1384–1391. [Google Scholar] [CrossRef]
- Signoretti, S.; Marmarou, A.; Tavazzi, B.; Lazzarino, G.; Beaumont, A.; Vagnozzi, R. N-Acetylaspartate reduction as a measure of injury severity and mitochondrial dysfunction following diffuse traumatic brain injury. J. Neurotrauma 2001, 18, 977–991. [Google Scholar] [CrossRef]
- Tavazzi, B.; Vagnozzi, R.; Di Pierro, D.; Amorini, A.M.; Fazzina, G.; Signoretti, S.; Marmarou, A.; Caruso, I.; Lazzarino, G. Ion-pairing high-performance liquid chromatographic method for the detection of N-acetylaspartate and N-acetylglutamate in cerebral tissue extracts. Anal. Biochem. 2000, 277, 104–108. [Google Scholar] [CrossRef]
- Wada, K.; Chatzipanteli, K.; Busto, R.; Dietrich, W.D. Role of nitric oxide in traumatic brain injury in the rat. J. Neurosurg. 1998, 89, 807–818. [Google Scholar] [CrossRef] [Green Version]
- Cherian, L.; Hlatky, R.; Robertson, C.S. Nitric oxide in traumatic brain injury. Brain Pathol. 2004, 14, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Ahn, M.-J.; Sherwood, E.R.; Prough, D.S.; Yie Lin, C.; DeWitt, D.S. The effects of traumatic brain injury on cerebral blood flow and brain tissue nitric oxide levels and cytokine expression. J. Neurotrauma 2004, 21, 1431–1442. [Google Scholar] [CrossRef]
- Kozlov, A.V.; Bahrami, S.; Redl, H.; Szabo, C. Alterations in nitric oxide homeostasis during traumatic brain injury. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2017, 1863, 2627–2632. [Google Scholar] [CrossRef] [PubMed]
- Besson, V.C. Drug targets for traumatic brain injury from poly (ADP-ribose) polymerase pathway modulation. Br. J. Pharmacol. 2009, 157, 695–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishida, A.; Trescher, W.H.; Lange, M.S.; Johnston, M.V. Prolonged suppression of brain nitric oxide synthase activity by 7-nitroindazole protects against cerebral hypoxic–ischemic injury in neonatal rat. Brain Dev. 2001, 23, 349–354. [Google Scholar] [CrossRef]
- Hall, E.D.; Andrus, P.K.; Yonkers, P.A. Brain hydroxyl radical generation in acute experimental head injury. J. Neurochem. 1993, 60, 588–594. [Google Scholar] [CrossRef] [PubMed]
- Tyurin, V.A.; Tyurina, Y.Y.; Borisenko, G.G.; Sokolova, T.V.; Ritov, V.B.; Quinn, P.J.; Rose, M.; Kochanek, P.; Graham, S.H.; Kagan, V.E. Oxidative stress following traumatic brain injury in rats: Quantitation of biomarkers and detection of free radical intermediates. J. Neurochem. 2000, 75, 2178–2189. [Google Scholar] [CrossRef] [Green Version]
- Licastro, F.; Hrelia, S.; Porcellini, E.; Malaguti, M.; Di Stefano, C.; Angeloni, C.; Carbone, I.; Simoncini, L.; Piperno, R. Peripheral inflammatory markers and antioxidant response during the post-acute and chronic phase after severe traumatic brain injury. Front. Neurol. 2016, 7, 189. [Google Scholar] [CrossRef] [Green Version]
- Jamjoom, A.A.; Rhodes, J.; Andrews, P.J.; Grant, S.G. The synapse in traumatic brain injury. Brain 2021, 144, 18–31. [Google Scholar] [CrossRef]
- Szydlowska, K.; Tymianski, M. Calcium, ischemia and excitotoxicity. Cell Calcium 2010, 47, 122–129. [Google Scholar] [CrossRef]
- Brenman, J.E.; Chao, D.S.; Gee, S.H.; McGee, A.W.; Craven, S.E.; Santillano, D.R.; Wu, Z.; Huang, F.; Xia, H.; Peters, M.F.; et al. Interaction of Nitric Oxide Synthase with the Postsynaptic Density Protein PSD-95 and α1-Syntrophin Mediated by PDZ Domains. Cell 1996, 84, 757–767. [Google Scholar] [CrossRef] [Green Version]
- Bach, A.; Clausen, B.H.; Kristensen, L.K.; Andersen, M.G.; Ellman, D.G.; Hansen, P.B.L.; Hasseldam, H.; Heitz, M.; Özcelik, D.; Tuck, E.J.; et al. Selectivity, efficacy and toxicity studies of UCCB01-144, a dimeric neuroprotective PSD-95 inhibitor. Neuropharmacology 2019, 150, 100–111. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Boutté, A.; Yu, X.; Dutta, B.; Feala, J.D.; Schmid, K.; Dave, J.; Tawa, G.J.; Wallqvist, A.; Reifman, J. A systems biology strategy to identify molecular mechanisms of action and protein indicators of traumatic brain injury. J. Neurosci. Res. 2015, 93, 199–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ansari, M.A.; Roberts, K.N.; Scheff, S.W. A time course of contusion-induced oxidative stress and synaptic proteins in cortex in a rat model of TBI. J. Neurotrauma 2008, 25, 513–526. [Google Scholar] [CrossRef] [PubMed]
- Bullock, R.; Zauner, A.; Woodward, J.J.; Myseros, J.; Choi, S.C.; Ward, J.D.; Marmarou, A.; Young, H.F. Factors affecting excitatory amino acid release following severe human head injury. J. Neurosurg. 1998, 89, 507–518. [Google Scholar] [CrossRef]
- Palmer, A.M.; Marion, D.W.; Botscheller, M.L.; Swedlow, P.E.; Styren, S.D.; DeKosky, S.T. Traumatic brain injury-induced excitotoxicity assessed in a controlled cortical impact model. J. Neurochem. 1993, 61, 2015–2024. [Google Scholar] [CrossRef]
- Faden, A.I.; Demediuk, P.; Panter, S.S.; Vink, R. The role of excitatory amino acids and NMDA receptors in traumatic brain injury. Science 1989, 244, 798–800. [Google Scholar] [CrossRef]
- Yi, J.-H.; Hoover, R.; McIntosh, T.K.; Hazell, A.S. Early, transient increase in complexin I and complexin II in the cerebral cortex following traumatic brain injury is attenuated by N-acetylcysteine. J. Neurotrauma 2006, 23, 86–96. [Google Scholar] [CrossRef]
- Koizumi, H.; Fujisawa, H.; Ito, H.; Maekawa, T.; Di, X.; Bullock, R. Effects of mild hypothermia on cerebral blood flow-independent changes in cortical extracellular levels of amino acids following contusion trauma in the rat. Brain Res. 1997, 747, 304–312. [Google Scholar] [CrossRef]
- Yi, J.-H.; Hazell, A.S. N-acetylcysteine attenuates early induction of heme oxygenase-1 following traumatic brain injury. Brain Res. 2005, 1033, 13–19. [Google Scholar] [CrossRef]
- Pabst, S.; Margittai, M.; Vainius, D.; Langen, R.; Jahn, R.; Fasshauer, D. Rapid and selective binding to the synaptic SNARE complex suggests a modulatory role of complexins in neuroexocytosis. J. Biol. Chem. 2002, 277, 7838–7848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eastwood, S.; Cotter, D.; Harrison, P. Cerebellar synaptic protein expression in schizophrenia. Neuroscience 2001, 105, 219–229. [Google Scholar] [CrossRef]
- Redell, J.B.; Moore, A.N.; Dash, P.K. Expression of the prodynorphin gene after experimental brain injury and its role in behavioral dysfunction. Exp. Biol. Med. 2003, 228, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Lucas, S.M.; Rothwell, N.J.; Gibson, R.M. The role of inflammation in CNS injury and disease. Br. J. Pharmacol. 2006, 147, S232–S240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Werner, C.; Engelhard, K. Pathophysiology of traumatic brain injury. BJA Br. J. Anaesth. 2007, 99, 4–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hans, V.H.; Kossmann, T.; Joller, H.; Otto, V.; Morganti-Kossmann, M.-C. Interleukin-6 and its soluble receptor in serum and cerebrospinal fluid after cerebral trauma. Neuroreport 1999, 10, 409–412. [Google Scholar] [CrossRef]
- Soares, H.D.; Hicks, R.R.; Smith, D.; McIntosh, T.K. Inflammatory leukocytic recruitment and diffuse neuronal degeneration are separate pathological processes resulting from traumatic brain injury. J. Neurosci. 1995, 15, 8223–8233. [Google Scholar] [CrossRef] [Green Version]
- Vecil, G.G.; Larsen, P.H.; Corley, S.M.; Herx, L.M.; Besson, A.; Goodyer, C.G.; Yong, V.W. Interleukin-1 is a key regulator of matrix metalloproteinase-9 expression in human neurons in culture and following mouse brain trauma in vivo. J. Neurosci. Res. 2000, 61, 212–224. [Google Scholar] [CrossRef]
- Lawrence, C.B.; Allan, S.M.; Rothwell, N.J. Interleukin-1β and the interleukin-1 receptor antagonist act in the striatum to modify excitotoxic brain damage in the rat. Eur. J. Neurosci. 1998, 10, 1188–1195. [Google Scholar] [CrossRef]
- Hopkins, S.J.; Rothwell, N.J. Cytokines and the nervous system I: Expression and recognition. Trends Neurosci. 1995, 18, 83–88. [Google Scholar] [CrossRef]
- Rothwell, N.J.; Hopkins, S.J. Cytokines and the nervous system II: Actions and mechanisms of action. Trends Neurosci. 1995, 18, 130–136. [Google Scholar] [CrossRef]
- Lu, K.-T.; Wang, Y.-W.; Yang, J.-T.; Yang, Y.-L.; Chen, H.-I. Effect of interleukin-1 on traumatic brain injury–induced damage to hippocampal neurons. J. Neurotrauma 2005, 22, 885–895. [Google Scholar] [CrossRef] [PubMed]
- Knoblach, S.M.; Faden, A.I. Cortical interleukin-1β elevation after traumatic brain injury in the rat: No effect of two selective antagonists on motor recovery. Neurosci. Lett. 2000, 289, 5–8. [Google Scholar] [CrossRef]
- Clausen, F.; Hånell, A.; Israelsson, C.; Hedin, J.; Ebendal, T.; Mir, A.K.; Gram, H.; Marklund, N. Neutralization of interleukin-1β reduces cerebral edema and tissue loss and improves late cognitive outcome following traumatic brain injury in mice. Eur. J. Neurosci. 2011, 34, 110–123. [Google Scholar] [CrossRef] [PubMed]
- Csuka, E.; Morganti-Kossmann, M.C.; Lenzlinger, P.M.; Joller, H.; Trentz, O.; Kossmann, T. IL-10 levels in cerebrospinal fluid and serum of patients with severe traumatic brain injury: Relationship to IL-6, TNF-α, TGF-β1 and blood–brain barrier function. J. Neuroimmunol. 1999, 101, 211–221. [Google Scholar] [CrossRef]
- Ross, S.A.; Halliday, M.I.; Campbell, G.C.; Byrnes, D.P.; Rowlands, B.J. The presence of tumour necrosis factor in CSF and plasma after severe head injury. Br. J. Neurosurg. 1994, 8, 419–425. [Google Scholar] [CrossRef]
- Khuman, J.; Meehan III, W.P.; Zhu, X.; Qiu, J.; Hoffmann, U.; Zhang, J.; Giovannone, E.; Lo, E.H.; Whalen, M.J. Tumor necrosis factor alpha and Fas receptor contribute to cognitive deficits independent of cell death after concussive traumatic brain injury in mice. J. Cereb. Blood Flow Metab. 2011, 31, 778–789. [Google Scholar] [CrossRef] [Green Version]
- Berrrrpohl, D.; You, Z.; Lo, E.H.; Kim, H.-H.; Whalen, M.J. TNF alpha and Fas mediate tissue damage and functional outcome after traumatic brain injury in mice. J. Cereb. Blood Flow Metab. 2007, 27, 1806–1818. [Google Scholar] [CrossRef] [Green Version]
- Rimaniol, A.-C.; Lekieffre, D.; Serrano, A.; Masson, A.; Benavides, J.; Zavala, F. Biphasic transforming growth factor-beta production flanking the pro-inflammatory cytokine response in cerebral trauma. Neuroreport 1995, 7, 133–136. [Google Scholar] [CrossRef]
- Morganti-Kossmann, M.C.; Rancan, M.; Stahel, P.F.; Kossmann, T. Inflammatory response in acute traumatic brain injury: A double-edged sword. Curr. Opin. Crit. Care 2002, 8, 101–105. [Google Scholar] [CrossRef]
- Cacheaux, L.P.; Ivens, S.; David, Y.; Lakhter, A.J.; Bar-Klein, G.; Shapira, M.; Heinemann, U.; Friedman, A.; Kaufer, D. Transcriptome profiling reveals TGF-β signaling involvement in epileptogenesis. J. Neurosci. 2009, 29, 8927–8935. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Sun, G.; Zhang, J.; Strong, R.; Dash, P.K.; Kan, Y.W.; Grotta, J.C.; Aronowski, J. Transcription factor Nrf2 protects the brain from damage produced by intracerebral hemorrhage. Stroke 2007, 38, 3280–3286. [Google Scholar] [CrossRef] [PubMed]
- Casili, G.; Campolo, M.; Paterniti, I.; Lanza, M.; Filippone, A.; Cuzzocrea, S.; Esposito, E. Dimethyl fumarate attenuates neuroinflammation and neurobehavioral deficits induced by experimental traumatic brain injury. J. Neurotrauma 2018, 35, 1437–1451. [Google Scholar] [CrossRef]
- Jin, W.; Wang, H.-D.; Hu, Z.-g.; Yan, W.; Chen, G.; Yin, H.-X. Transcription factor Nrf2 plays a pivotal role in protection against traumatic brain injury-induced acute intestinal mucosal injury in mice. J. Surg. Res. 2009, 157, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.-G.; Zhang, G.-D.; Shi, P.-Q.; Du, B.-S. Expression and antioxidation of Nrf2/ARE pathway in traumatic brain injury. Asian Pac. J. Trop. Med. 2013, 6, 305–310. [Google Scholar] [CrossRef]
- Di Pietro, V.; Yakoub, K.M.; Caruso, G.; Lazzarino, G.; Signoretti, S.; Barbey, A.K.; Tavazzi, B.; Lazzarino, G.; Belli, A.; Amorini, A.M. Antioxidant therapies in traumatic brain injury. Antioxidants 2020, 9, 260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazzarino, G.; Listorti, I.; Bilotta, G.; Capozzolo, T.; Amorini, A.M.; Longo, S.; Caruso, G.; Lazzarino, G.; Tavazzi, B.; Bilotta, P. Water- and Fat-Soluble Antioxidants in Human Seminal Plasma and Serum of Fertile Males. Antioxidants 2019, 8, 96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Gregorio, R.; Simal-Gandara, J. A critical review of bioactive food components, and of their functional mechanisms, biological effects and health outcomes. Curr. Pharm. Des. 2017, 23, 2731–2741. [Google Scholar] [CrossRef]
- Rigg, J.L.; Elovic, E.P.; Greenwald, B.D. A Review of the Effectiveness of Antioxidant Therapy to Reduce Neuronal Damage in Acute Traumatic Brain Injury. J. Head Trauma Rehabil. 2005, 20, 389–391. [Google Scholar] [CrossRef]
- DeKosky, S.T.; Taffe, K.M.; Abrahamson, E.E.; Dixon, C.E.; Kochanek, P.M.; Ikonomovic, M.D. Time course analysis of hippocampal nerve growth factor and antioxidant enzyme activity following lateral controlled cortical impact brain injury in the rat. J. Neurotrauma 2004, 21, 491–500. [Google Scholar] [CrossRef]
- Veronese, F.M.; Caliceti, P.; Schiavon, O.; Sergi, M. Polyethylene glycol–superoxide dismutase, a conjugate in search of exploitation. Adv. Drug Deliv. Rev. 2002, 54, 587–606. [Google Scholar] [CrossRef]
- Ding, K.; Wang, H.; Xu, J.; Li, T.; Zhang, L.; Ding, Y.; Zhu, L.; He, J.; Zhou, M. Melatonin stimulates antioxidant enzymes and reduces oxidative stress in experimental traumatic brain injury: The Nrf2–ARE signaling pathway as a potential mechanism. Free Radic. Biol. Med. 2014, 73, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, N.; Katayama, Y.; Kawamata, T.; Maeda, T.; Mori, T.; Yamamoto, T.; Kikuchi, T.; Uwahodo, Y. Effects of antioxidant, OPC-14117, on secondary cellular damage and behavioral deficits following cortical contusion in the rat. Brain Res. 2002, 934, 117–124. [Google Scholar] [CrossRef]
- Almli, L.M.; Hamrick, S.E.; Koshy, A.A.; Täuber, M.G.; Ferriero, D.M. Multiple pathways of neuroprotection against oxidative stress and excitotoxic injury in immature primary hippocampal neurons. Dev. Brain Res. 2001, 132, 121–129. [Google Scholar] [CrossRef]
- Granger, M.; Eck, P. Dietary vitamin C in human health. Adv. Food Nutr. Res. 2018, 83, 281–310. [Google Scholar]
- Hirschmann, J.; Raugi, G.J. Adult scurvy. J. Am. Acad. Dermatol. 1999, 41, 895–910. [Google Scholar] [CrossRef]
- Castiglione, D.; Platania, A.; Conti, A.; Falla, M.; D’Urso, M.; Marranzano, M. Dietary Micronutrient and Mineral Intake in the Mediterranean Healthy Eating, Ageing, and Lifestyle (MEAL) Study. Antioxidants 2018, 7, 79. [Google Scholar] [CrossRef] [Green Version]
- Bürzle, M.; Suzuki, Y.; Ackermann, D.; Miyazaki, H.; Maeda, N.; Clémençon, B.; Burrier, R.; Hediger, M.A. The sodium-dependent ascorbic acid transporter family SLC23. Mol. Asp. Med. 2013, 34, 436–454. [Google Scholar] [CrossRef]
- Di Pietro, V.; Lazzarino, G.; Amorini, A.M.; Tavazzi, B.; D’Urso, S.; Longo, S.; Vagnozzi, R.; Signoretti, S.; Clementi, E.; Giardina, B.; et al. Neuroglobin expression and oxidant/antioxidant balance after graded traumatic brain injury in the rat. Free Radic. Biol. Med. 2014, 69, 258–264. [Google Scholar] [CrossRef]
- Grünewald, R.A. Ascorbic acid in the brain. Brain Res. Rev. 1993, 18, 123–133. [Google Scholar] [CrossRef]
- Parker, W.H.; Qu, Z.-c.; May, J.M. Ascorbic acid transport in brain microvascular pericytes. Biochem. Biophys. Res. Commun. 2015, 458, 262–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tavazzi, B.; Signoretti, S.; Lazzarino, G.; Amorini, A.M.; Delfini, R.; Cimatti, M.; Marmarou, A.; Vagnozzi, R. Cerebral oxidative stress and depression of energy metabolism correlate with severity of diffuse brain injury in rats. Neurosurgery 2005, 56, 582–589. [Google Scholar] [CrossRef] [PubMed]
- Razmkon, A.; Sadidi, A.; Sherafat-Kazemzadeh, E.; Mehrafshan, A.; Jamali, M.; Malekpour, B.; Saghafinia, M. Administration of vitamin C and vitamin E in severe head injury: A randomized double-blind controlled trial. Neurosurgery 2011, 58, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Limongi, D.; Baldelli, S.; Checconi, P.; Marcocci, M.E.; De Chiara, G.; Fraternale, A.; Magnani, M.; Ciriolo, M.R.; Palamara, A.T. GSH-C4 Acts as Anti-inflammatory Drug in Different Models of Canonical and Cell Autonomous Inflammation through NFκB Inhibition. Front. Immunol. 2019, 10, 155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steullet, P.; Neijt, H.C.; Cuénod, M.; Do, K.Q. Synaptic plasticity impairment and hypofunction of NMDA receptors induced by glutathione deficit: Relevance to schizophrenia. Neuroscience 2006, 137, 807–819. [Google Scholar] [CrossRef]
- Varga, V.; Jenei, Z.; Janáky, R.; Saransaari, P.; Oja, S.S. Glutathione is an endogenous ligand of rat brain N-methyl-D-aspartate (NMDA) and 2-amino-3-hydroxy-5-methyl-4-isoxazolepropionate (AMPA) receptors. Neurochem. Res. 1997, 22, 1165–1171. [Google Scholar] [CrossRef] [PubMed]
- Reed, T.T.; Owen, J.; Pierce, W.M.; Sebastian, A.; Sullivan, P.G.; Butterfield, D.A. Proteomic identification of nitrated brain proteins in traumatic brain-injured rats treated postinjury with gamma-glutamylcysteine ethyl ester: Insights into the role of elevation of glutathione as a potential therapeutic strategy for traumatic brain injury. J. Neurosci. Res. 2009, 87, 408–417. [Google Scholar] [CrossRef]
- Koza, L.; Linseman, D.A. Glutathione precursors shield the brain from trauma. Neural Regen. Res. 2019, 14, 1701–1702. [Google Scholar] [CrossRef]
- Sangobowale, M.; Nikulina, E.; Bergold, P.J. Minocycline plus N-acetylcysteine protect oligodendrocytes when first dosed 12 hours after closed head injury in mice. Neurosci. Lett. 2018, 682, 16–20. [Google Scholar] [CrossRef]
- Bhatti, J.; Nascimento, B.; Akhtar, U.; Rhind, S.G.; Tien, H.; Nathens, A.; da Luz, L.T. Systematic Review of Human and Animal Studies Examining the Efficacy and Safety of N-Acetylcysteine (NAC) and N-Acetylcysteine Amide (NACA) in Traumatic Brain Injury: Impact on Neurofunctional Outcome and Biomarkers of Oxidative Stress and Inflammation. Front. Neurol. 2018, 8, 744. [Google Scholar] [CrossRef] [Green Version]
- Izzi, V.; Masuelli, L.; Tresoldi, I.; Sacchetti, P.; Modesti, A.; Galvano, F.; Bei, R. The effects of dietary flavonoids on the regulation of redox inflammatory networks. Front. Biosci.-Landmark 2012, 17, 2396–2418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theadom, A.; Mahon, S.; Barker-Collo, S.; McPherson, K.; Rush, E.; Vandal, A.C.; Feigin, V.L. Enzogenol for cognitive functioning in traumatic brain injury: A pilot placebo-controlled RCT. Eur. J. Neurol. 2013, 20, 1135–1144. [Google Scholar] [CrossRef]
- Tsai, H.-Y.; Ho, C.-T.; Chen, Y.-K. Biological actions and molecular effects of resveratrol, pterostilbene, and 3′-hydroxypterostilbene. J. Food Drug Anal. 2017, 25, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Ates, O.; Cayli, S.; Altinoz, E.; Gurses, I.; Yucel, N.; Sener, M.; Kocak, A.; Yologlu, S. Neuroprotection by resveratrol against traumatic brain injury in rats. Mol. Cell. Biochem. 2007, 294, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.J.; Chen, T.H.; Yang, L.Y.; Shih, C.M. Resveratrol protects astrocytes against traumatic brain injury through inhibiting apoptotic and autophagic cell death. Cell Death Dis. 2014, 5, e1147. [Google Scholar] [CrossRef] [Green Version]
- Gatson, J.W.; Liu, M.-M.; Abdelfattah, K.; Wigginton, J.G.; Smith, S.; Wolf, S.; Minei, J.P. Resveratrol decreases inflammation in the brain of mice with mild traumatic brain injury. J. Trauma Acute Care Surg. 2013, 74, 470–475. [Google Scholar] [CrossRef]
- Irías-Mata, A.; Stuetz, W.; Sus, N.; Hammann, S.; Gralla, K.; Cordero-Solano, A.; Vetter, W.; Frank, J. Tocopherols, Tocomonoenols, and Tocotrienols in Oils of Costa Rican Palm Fruits: A Comparison between Six Varieties and Chemical versus Mechanical Extraction. J. Agric. Food Chem. 2017, 65, 7476–7482. [Google Scholar] [CrossRef]
- Inci, S.; Özcan, O.E.; Kilinç, K. Time-Level Relationship for Lipid Peroxidation and the Protective Effect of α-Tocopherol in Experimental Mild and Severe Brain Injury. Neurosurgery 1998, 43, 330–335. [Google Scholar] [CrossRef]
- Yang, J.; Han, Y.; Ye, W.; Liu, F.; Zhuang, K.; Wu, G. Alpha tocopherol treatment reduces the expression of Nogo-A and NgR in rat brain after traumatic brain injury. J. Surg. Res. 2013, 182, e69–e77. [Google Scholar] [CrossRef]
- Wu, A.; Ying, Z.; Gomez-Pinilla, F. Vitamin E Protects Against Oxidative Damage and Learning Disability After Mild Traumatic Brain Injury in Rats. Neurorehabil. Neural Repair 2010, 24, 290–298. [Google Scholar] [CrossRef] [Green Version]
- Jorat, M.V.; Tabrizi, R.; Kolahdooz, F.; Akbari, M.; Salami, M.; Heydari, S.T.; Asemi, Z. The effects of coenzyme Q10 supplementation on biomarkers of inflammation and oxidative stress in among coronary artery disease: A systematic review and meta-analysis of randomized controlled trials. Inflammopharmacology 2019, 27, 233–248. [Google Scholar] [CrossRef] [PubMed]
- Kalayci, M.; Unal, M.M.; Gul, S.; Acikgoz, S.; Kandemir, N.; Hanci, V.; Edebali, N.; Acikgoz, B. Effect of Coenzyme Q10 on ischemia and neuronal damage in an experimental traumatic brain-injury model in rats. BMC Neurosci. 2011, 12, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pierce, J.D.; Shen, Q.; Peltzer, J.; Thimmesch, A.; Hiebert, J.B. A pilot study exploring the effects of ubiquinol on brain genomics after traumatic brain injury. Nurs. Outlook 2017, 65, S44–S52. [Google Scholar] [CrossRef] [PubMed]
- Pierce, J.D.; Gupte, R.; Thimmesch, A.; Shen, Q.; Hiebert, J.B.; Brooks, W.M.; Clancy, R.L.; Diaz, F.J.; Harris, J.L. Ubiquinol treatment for TBI in male rats: Effects on mitochondrial integrity, injury severity, and neurometabolism. J. Neurosci. Res. 2018, 96, 1080–1092. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.S.; Shin, M.; Kim, S.; Lee, S.B. Recent Advances in Studies on the Therapeutic Potential of Dietary Carotenoids in Neurodegenerative Diseases. Oxidative Med. Cell. Longev. 2018, 2018, 4120458. [Google Scholar] [CrossRef]
- Zhang, M.; Cui, Z.; Cui, H.; Wang, Y.; Zhong, C. Astaxanthin protects astrocytes against trauma-induced apoptosis through inhibition of NKCC1 expression via the NF-κB signaling pathway. BMC Neurosci. 2017, 18, 42. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Cui, Z.; Cui, H.; Cao, Y.; Wang, Y.; Zhong, C. Astaxanthin alleviates cerebral edema by modulating NKCC1 and AQP4 expression after traumatic brain injury in mice. BMC Neurosci. 2016, 17, 60. [Google Scholar] [CrossRef] [Green Version]
- Ji, X.; Peng, D.; Zhang, Y.; Zhang, J.; Wang, Y.; Gao, Y.; Lu, N.; Tang, P. Astaxanthin improves cognitive performance in mice following mild traumatic brain injury. Brain Res. 2017, 1659, 88–95. [Google Scholar] [CrossRef]
- Zhong, J.; Jiang, L.; Huang, Z.; Zhang, H.; Cheng, C.; Liu, H.; He, J.; Wu, J.; Darwazeh, R.; Wu, Y.; et al. The long non-coding RNA Neat1 is an important mediator of the therapeutic effect of bexarotene on traumatic brain injury in mice. Brain Behav. Immun. 2017, 65, 183–194. [Google Scholar] [CrossRef]
- Zhong, J.; Cheng, C.; Liu, H.; Huang, Z.; Wu, Y.; Teng, Z.; He, J.; Zhang, H.; Wu, J.; Cao, F.; et al. Bexarotene protects against traumatic brain injury in mice partially through apolipoprotein E. Neuroscience 2017, 343, 434–448. [Google Scholar] [CrossRef]
- Wang, K.; Zhang, L.; Rao, W.; Su, N.; Hui, H.; Wang, L.; Peng, C.; Tu, Y.; Zhang, S.; Fei, Z. Neuroprotective effects of crocin against traumatic brain injury in mice: Involvement of notch signaling pathway. Neurosci. Lett. 2015, 591, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.; Yu, X.; Chen, M.; Chen, J.; Xu, J. Lutein protects against severe traumatic brain injury through anti-inflammation and antioxidative effects via ICAM-1/Nrf-2. Mol. Med. Rep. 2017, 16, 4235–4240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauritzen, L.; Hansen, H.S.; Jørgensen, M.H.; Michaelsen, K.F. The essentiality of long chain n-3 fatty acids in relation to development and function of the brain and retina. Prog. Lipid Res. 2001, 40, 1–94. [Google Scholar] [CrossRef]
- Matsuoka, Y.; Nishi, D.; Tanima, Y.; Itakura, M.; Kojima, M.; Hamazaki, K.; Noguchi, H.; Hamazaki, T. Serum pro-BDNF/BDNF as a treatment biomarker for response to docosahexaenoic acid in traumatized people vulnerable to developing psychological distress: A randomized controlled trial. Transl. Psychiatry 2015, 5, e596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, A.; Ying, Z.; Gomez-Pinilla, F. Exercise facilitates the action of dietary DHA on functional recovery after brain trauma. Neuroscience 2013, 248, 655–663. [Google Scholar] [CrossRef] [Green Version]
- Ansari, M.A.; Roberts, K.N.; Scheff, S.W. Dose-and time-dependent neuroprotective effects of Pycnogenol® following traumatic brain injury. J. Neurotrauma 2013, 30, 1542–1549. [Google Scholar] [CrossRef] [Green Version]
- Hill, R.L.; Singh, I.N.; Wang, J.A.; Kulbe, J.R.; Hall, E.D. Protective effects of phenelzine administration on synaptic and non-synaptic cortical mitochondrial function and lipid peroxidation-mediated oxidative damage following TBI in young adult male rats. Exp. Neurol. 2020, 330, 113322. [Google Scholar] [CrossRef]
- Cebak, J.E.; Singh, I.N.; Hill, R.L.; Wang, J.A.; Hall, E.D. Phenelzine protects brain mitochondrial function in vitro and in vivo following traumatic brain injury by scavenging the reactive carbonyls 4-hydroxynonenal and acrolein leading to cortical histological neuroprotection. J. Neurotrauma 2017, 34, 1302–1317. [Google Scholar] [CrossRef] [Green Version]
- Singh, I.N.; Gilmer, L.K.; Miller, D.M.; Cebak, J.E.; Wang, J.A.; Hall, E.D. Phenelzine Mitochondrial Functional Preservation and Neuroprotection after Traumatic Brain Injury Related to Scavenging of the Lipid Peroxidation-Derived Aldehyde 4-Hydroxy-2-Nonenal. J. Cereb. Blood Flow Metab. 2013, 33, 593–599. [Google Scholar] [CrossRef]
Figure 1.
Schematic illustration of the main cellular components involved in the post-TBI oxidative stress process. The NAPDH oxidase (NOX) with its two membrane subunits, P91phox and P22phox, and cytoplasmic components, P47phox, P67phox, P40phox, and Rac2, is a key enzymatic unit involved in this process. The reactive oxygen species (ROS), comprised of superoxide (O2●−), hydrogen peroxide (H2O2), and the hydroxyl radical (●OH), are products of 1-electron reduction of oxygen. The reactive nitrogen species (RNS), including various nitric oxide compounds, such as peroxynitrite (ONOO-) and nitrogen dioxide (NO2), are the products of multiple enzymes including nitric oxide synthase (iNOS), endothelial nitric oxide synthase (eNOS), cytochrome P450 (CYP450), cyclooxygenase (COX), lipoxygenase (LOX), and xanthine oxidase (XO). Free iron (Fe2+), an abundant ion within the CNS, participates in two main reactions, including the Fenton reaction, leading to the production of Fe3+, O2●−, ●OH +, and OH−. The phenomena of lipid peroxidation (LP) involves electron transfer from membrane lipids to free radicals, involving a three-step process, and ultimate production of 4-HNE and 2-propenal (acrolein). The free radicals are depicted as red vs. the enzymes and proteins are in black. Images created with BioRender.com.
Figure 1.
Schematic illustration of the main cellular components involved in the post-TBI oxidative stress process. The NAPDH oxidase (NOX) with its two membrane subunits, P91phox and P22phox, and cytoplasmic components, P47phox, P67phox, P40phox, and Rac2, is a key enzymatic unit involved in this process. The reactive oxygen species (ROS), comprised of superoxide (O2●−), hydrogen peroxide (H2O2), and the hydroxyl radical (●OH), are products of 1-electron reduction of oxygen. The reactive nitrogen species (RNS), including various nitric oxide compounds, such as peroxynitrite (ONOO-) and nitrogen dioxide (NO2), are the products of multiple enzymes including nitric oxide synthase (iNOS), endothelial nitric oxide synthase (eNOS), cytochrome P450 (CYP450), cyclooxygenase (COX), lipoxygenase (LOX), and xanthine oxidase (XO). Free iron (Fe2+), an abundant ion within the CNS, participates in two main reactions, including the Fenton reaction, leading to the production of Fe3+, O2●−, ●OH +, and OH−. The phenomena of lipid peroxidation (LP) involves electron transfer from membrane lipids to free radicals, involving a three-step process, and ultimate production of 4-HNE and 2-propenal (acrolein). The free radicals are depicted as red vs. the enzymes and proteins are in black. Images created with BioRender.com.

Table 1.
Outlines the main pathological agents involved in TBI oxidative stress, and their corresponding proposed mechanisms.
Table 1.
Outlines the main pathological agents involved in TBI oxidative stress, and their corresponding proposed mechanisms.
| Pathological Agent | Mechanism | References |
|---|---|---|
| Reactive Oxygen Species (ROS) | ROS includes superoxide (O2●−), hydrogen peroxide (H2O2), and the hydroxyl radical (●OH), produced by mitochondria and product of cellular oxidative metabolism | Eastman et al. 2020 [1] Pisochi et al. 2015 [24] |
| Reactive Nitrogen Species (RNS) | RNS Includes nitric oxide compounds, such as peroxynitrie (ONOO−), and nitrogen dioxide (NO2), and have longer half-lives than ROS | Balazy et al. 2003 [25] Brandes et al. 2010 [26] Uttara et al. 2009 [27] |
| Peroxynitrite (PN) | PN can also react with carbon dioxide (CO2), and form nitoperoxocarbonate (ONOOCO2) & subsequent cellular damage via transfer of electron from hydrogen atom bound to allylic carbon in polyunsaturated fatty acids (PUFAs) | Hall et al. 2012 [28] Hall et al. 2004 [21] |
| Iron and Hydroxyl Radicals | Post TBI, the PH within injured areas are lowered, in turn leading to release of iron from its storage sites with an additional source of catalytically active iron being hemoglobin, in turn responsible for hemoglobin mediated oxidative damage | Daglas et al. 2018 [29] Hatakeyama et al. 2013 [30] Zeng et al. 2018 [31] |
| Lipid Peroxidation (LP) | LP has been associated with loss of cell membrane integrity, increased membrane permeability, diminished activity in membrane ATPase, in turn leading to further injury | Cornelius et al. 2013 [8] |
| Glutathione Peroxidase (GPx) | GPx reactions lead to oxidation of glutathione (GSH) to oxidized form (GSSG) | Kushner et al. 1998 [32] |
| Malondialdehyde (MDA) | MDA reacts with several functional groups on proteins, lipoproteins and DNA, leading to further oxidative damage | Cornelius et al. 2013 [8] |
| Glutamate | TBI involves excessive release of extracellular glutamate, overstimulation of metabotropic and ionotropic receptors, and neuronal cell death | Riedel at al. 2003 [33] Yi et al. 2006 [34] |
| N-acetylaspartate (NAA) | N-acetylaspartate (NAA) is the acetylated form of aspartate, ubiquitous in CNS and a robust injury marker | Signoretti et al. 2001 [35] Tavazzi et al. 2000 [36] |
| Nitric oxide synthase (NOS) | TBI leads to activation of NOS, in turn the formation of NO in the brain | Besson et al. 2009 [37] Ishida et al. 2001 [38] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Fesharaki-Zadeh, A. Oxidative Stress in Traumatic Brain Injury. Int. J. Mol. Sci. 2022, 23, 13000. https://doi.org/10.3390/ijms232113000
AMA Style
Fesharaki-Zadeh A. Oxidative Stress in Traumatic Brain Injury. International Journal of Molecular Sciences. 2022; 23(21):13000. https://doi.org/10.3390/ijms232113000
Chicago/Turabian StyleFesharaki-Zadeh, Arman. 2022. "Oxidative Stress in Traumatic Brain Injury" International Journal of Molecular Sciences 23, no. 21: 13000. https://doi.org/10.3390/ijms232113000
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.