
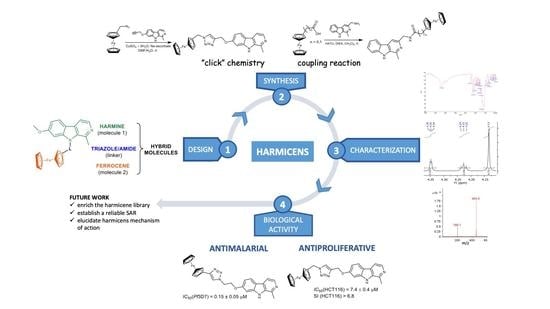
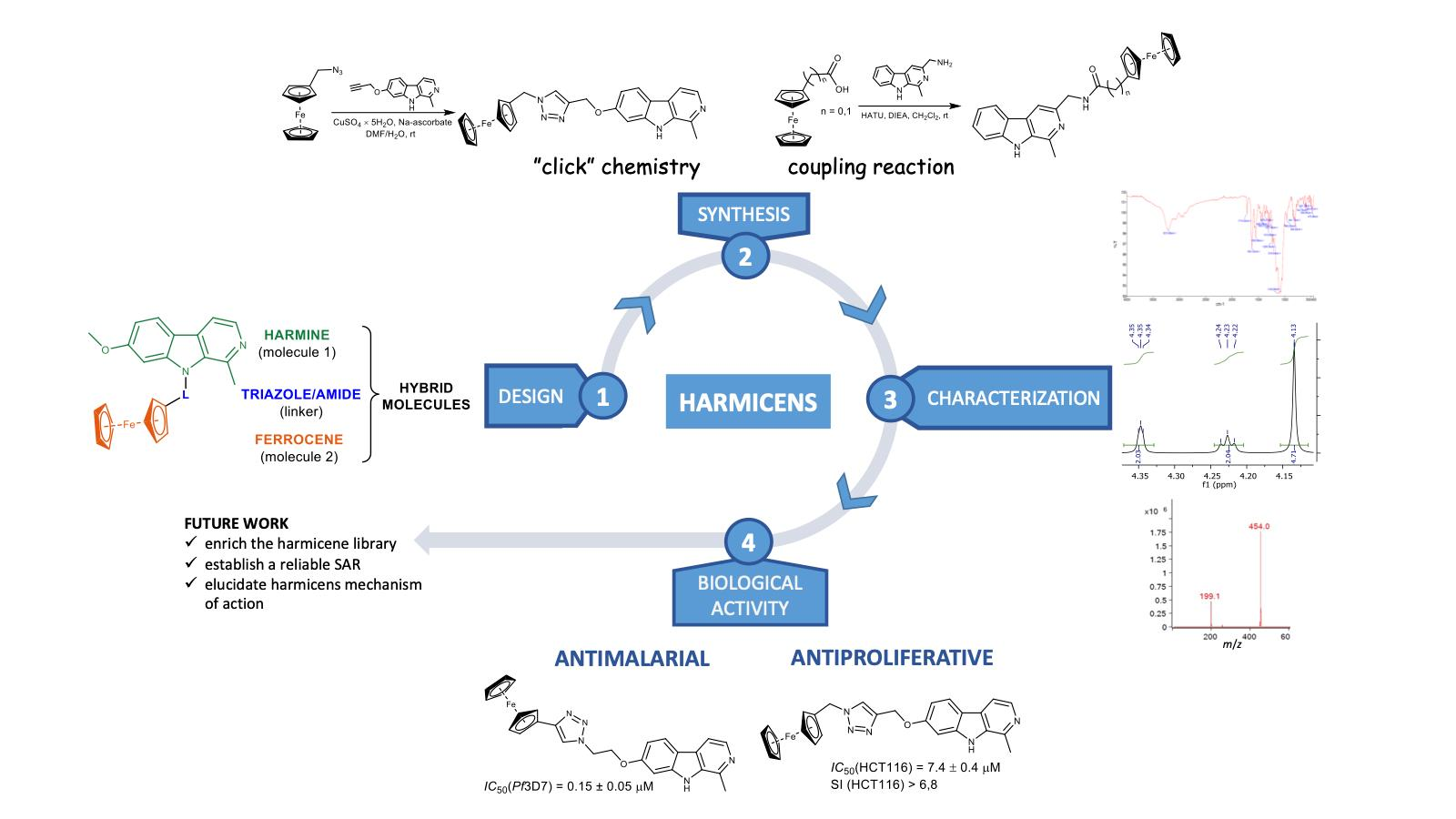
Harmicens, Novel Harmine and Ferrocene Hybrids: Design, Synthesis and Biological Activity

 , , ,
, , ,
Abstract
:
1. Introduction
2. Results and Discussion
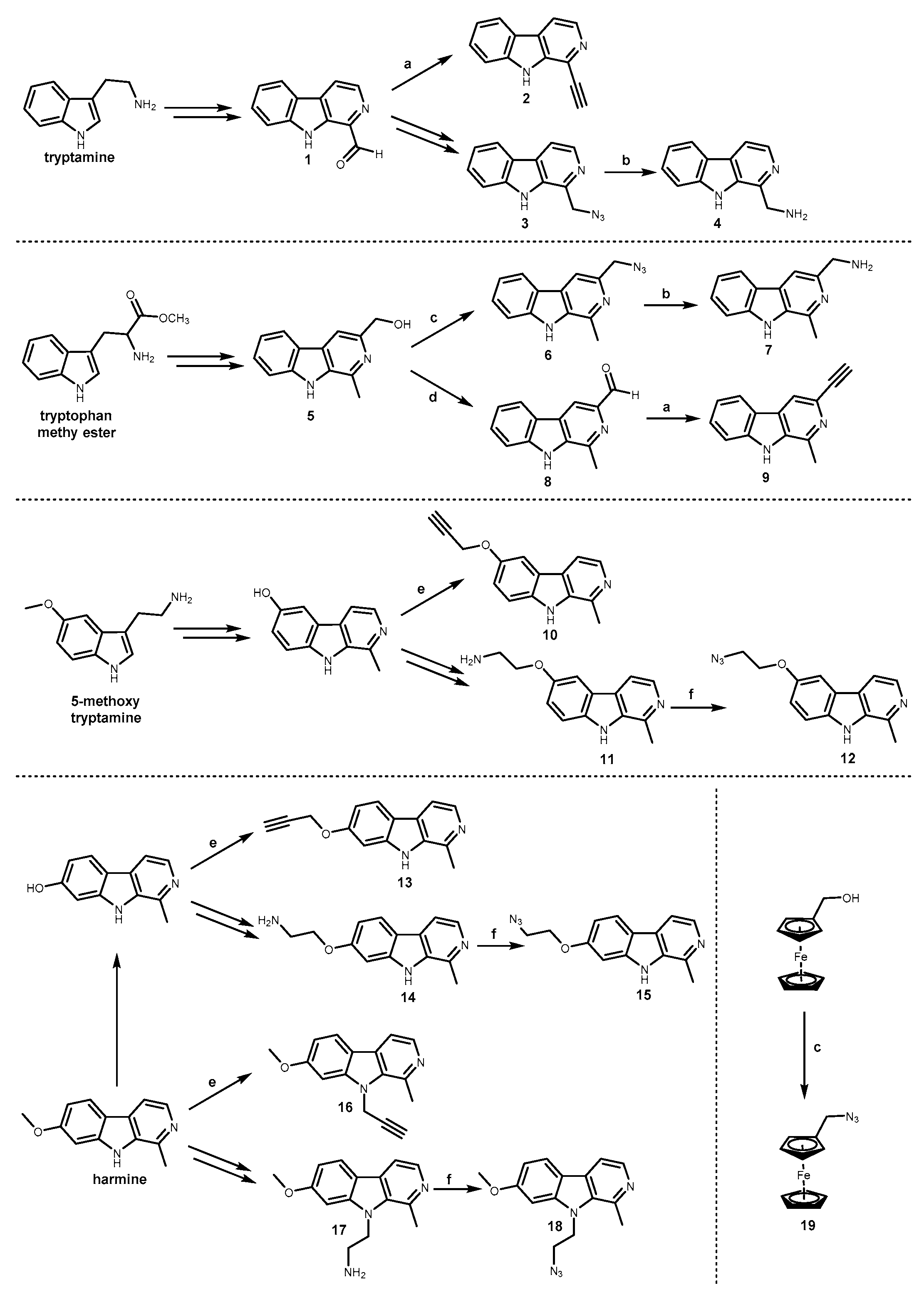
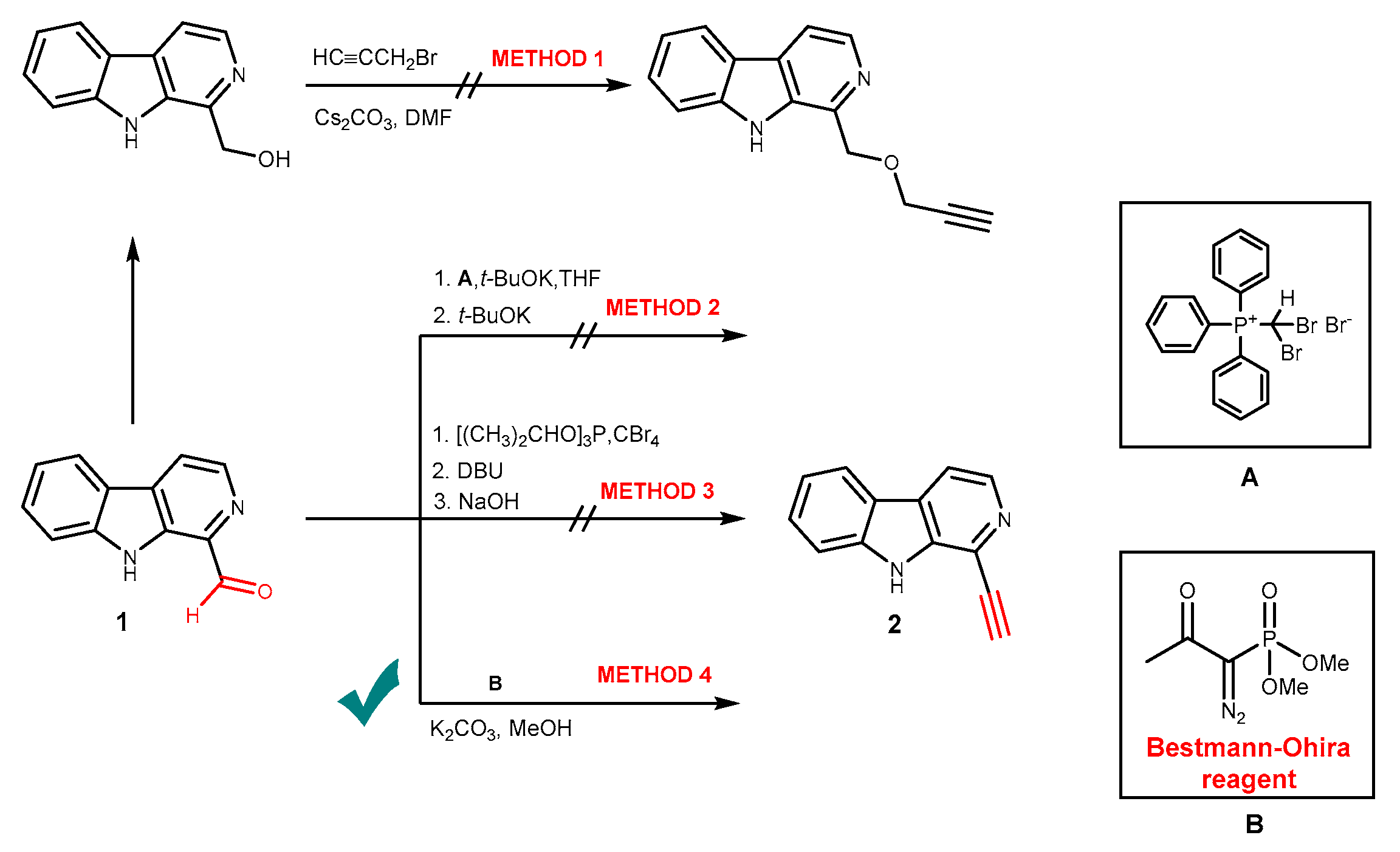
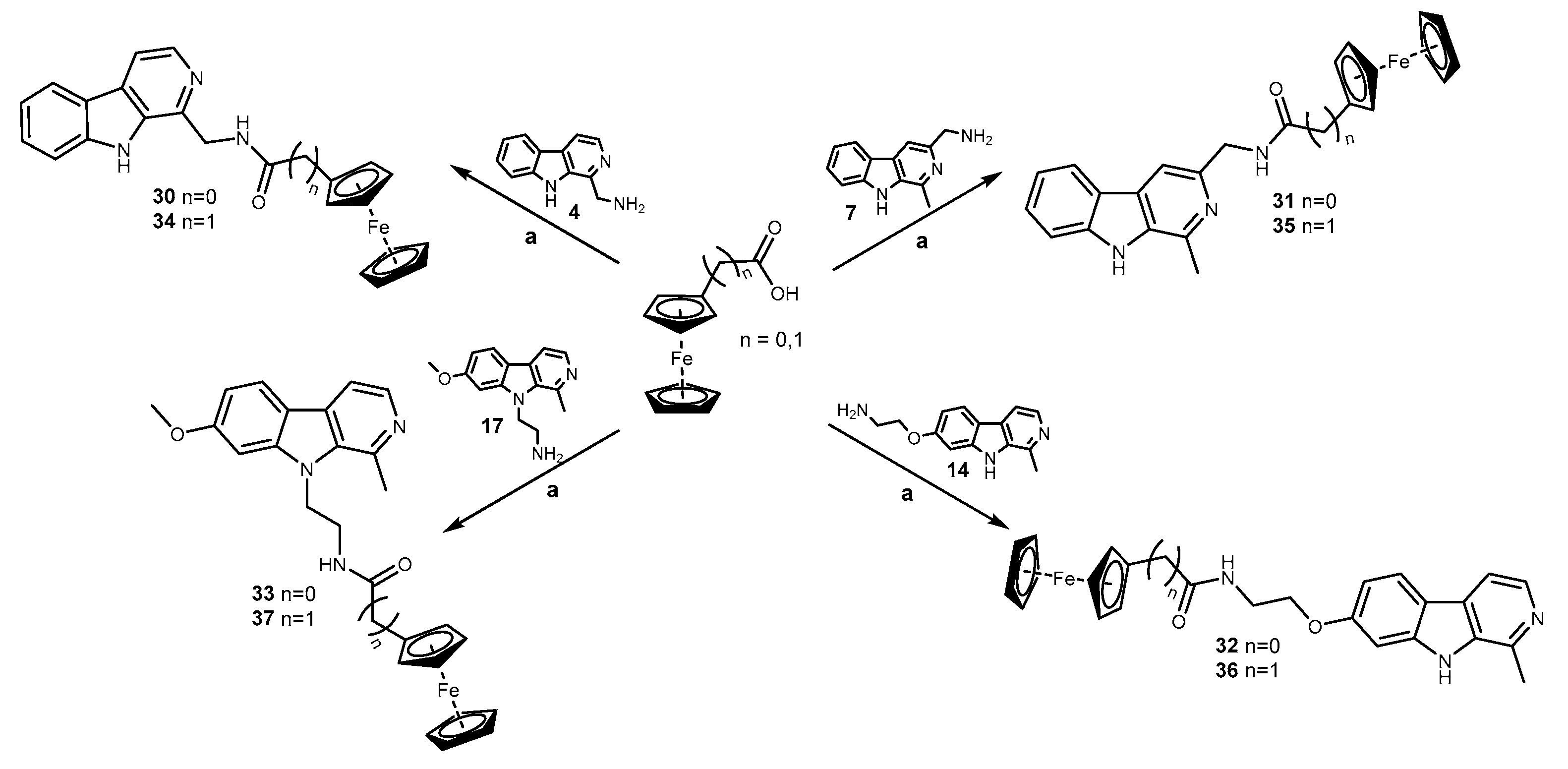
2.1. Chemistry
2.2. Biological Activity
2.2.1. In Vitro Antiplasmodial Activity
2.2.2. In Vitro Antiproliferative Activity
- (1)
- C-1: Harmicens were either selective against HCT116 (TT harmicene 25) or MCF-7 (AT harmicene 30), or exerted very low activity/were inactive.
- (2)
- C-3: Harmicens were mostly selective, especially TT harmicens 21 and 26. Although AT harmicens had lower activity, a similar trend was observed.
- (3)
- O-6: Harmicens exerted very strong activity against the tumor cell lines. Harmicene 27 was also selective against MCF-7 and SW620. Unfortunately, only TT harmicens were prepared, so comparison with AT harmicens was not possible.
- (4)
- O-7: Harmicens showed pronounced and nonselective activity against all cell lines tested, with the exception of TT harmicene 28.
- (5)
- N-9: The activity of harmicens was moderate to low, and some degree of selectivity was observed against the HCT116 and MCF-7 cell lines.
2.2.3. Cell Localization
2.2.4. Cell Cycle Analysis
3. Materials and Methods
3.1. Chemistry
3.1.1. General Information
3.1.2. General Procedure for the Synthesis of Alkynes 2 and 9
1-Ethynyl-9H-pyrido[3,4-b]indole (2)
3-Ethynyl-1-methyl-9H-pyrido[3,4-b]indole (9)
3.1.3. General Procedure for the Synthesis of Azides 12, 15, 18
6-(2-Azidoethoxy)-1-methyl-9H-pyrido[3,4-b]indole (12)
7-(2-Azidoethoxy)-1-methyl-9H-pyrido[3,4-b]indole (15)
9-(2-Azidoethyl)-7-methoxy-1-methyl-9H-pyrido[3,4-b]indole (18)
3.1.4. (Azidomethyl)ferrocene (19)
3.1.5. General Procedure for the Synthesis of TT Harmicens 20–24
1-((4-Ferrocenyl-1H-1,2,3-triazol-1-yl)methyl)-9H-pyrido[3,4-b]indole (20)
1-Methyl-3-((4-ferrocenyl-1H-1,2,3-triazol-1-yl)methyl)-9H-pyrido[3,4-b]indole (21)
1-Methyl-6-(2-(4-ferrocenyl-1H-1,2,3-triazol-1-yl)ethoxy)-9H-pyrido[3,4-b]indole (22)
1-Methyl-7-(2-(4-ferrocenyl-1H-1,2,3-triazol-1-yl)ethoxy)-9H-pyrido[3,4-b]indole (23)
7-Methoxy-1-methyl-9-(2-(4-ferrocenyl-1H-1,2,3-triazol-1-yl)ethyl)-9H-pyrido[3,4-b]indole (24)
3.1.6. General Procedure for the Synthesis of TT Harmicens 25–29
1-(1-(Ferrocenylmethyl)-1H-1,2,3-triazol-4-yl)-9H-pyrido[3,4-b]indole (25)
3-(1-(Ferrocenylmethyl)-1H-1,2,3-triazol-4-yl)-1-methyl-9H-pyrido[3,4-b]indole (26)
6-((1-(Ferrocenylmethyl)-1H-1,2,3-triazol-4-yl)methoxy)-1-methyl-9H-pyrido[3,4-b]indole (27)
7-((1-(Ferrocenylmethyl)-1H-1,2,3-triazol-4-yl)methoxy)-1-methyl-9H-pyrido[3,4-b]indole (28)
9-((1-(Ferocenylmethyl)-1H-1,2,3-triazol-4-yl)methyl)-7-methoxy-1-methyl-9H-pyrido[3,4-b]indole (29)
3.1.7. General Procedure for the Synthesis of AT Harmicens 30–33
N-((9H-Pyrido[3,4-b]indol-1-yl)methyl)ferrocenecarboxamide (30)
N-((1-Methyl-9H-pyrido[3,4-b]indol-3-yl)methyl)ferrocenecarboxamide (31)
N-(2-((1-Methyl-9H-pyrido[3,4-b]indol-7-yl)oxy)ethyl)ferrocenecarboxamide (32)
N-(2-(7-Methoxy-1-methyl-9H-pyrido[3,4-b]indol-9-yl)ethyl)ferrocenecarboxamide (33)
3.1.8. General Procedure for the Synthesis of AT Harmicens 34–37
N-((9H-Pyrido[3,4-b]indol-1-yl)methyl)-2-ferrocenylacetamide (34)
N-((1-Methyl-9H-pyrido[3,4-b]indol-3-yl)methyl)-2-ferrocenylacetamide (35)
N-(2-((1-Methyl-9H-pyrido[3,4-b]indol-7-yl)oxy)ethyl)-2-ferrocenylacetamide (36)
N-(2-(7-Methoxy-1-methyl-9H-pyrido[3,4-b]indol-9-yl)ethyl)-2-ferrocenylacetamide (37)
3.2. Biological Evaluation
3.2.1. In Vitro Drug Sensitivity Assay against Erythrocytic Stages of P. falciparum
3.2.2. Cytotoxicity Assay in Human Cell Lines
3.2.3. Cell Localization
3.2.4. Cell Cycle Analysis
3.2.5. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- World Health Organization. Available online: https://www.who.int/news-room/fact-sheets/detail/cancer (accessed on 20 July 2022).
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Freddie, B. Cancer statistics for the year 2020: An overview. Int. J. Cancer. 2021, 149, 778–789. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. International Agency for Research on Cancer. Available online: https://gco.iarc.fr/today (accessed on 18 February 2022).
- Wang, R.; Chen, H.; Yan, W.; Zheng, M.; Zhang, T.; Zhang, Y. Ferrocene-containing hybrids as potential anticancer agents: Current developments, mechanisms of action and structure-activity relationships. Eur. J. Med. Chem. 2020, 190, 112019. [Google Scholar] [CrossRef] [PubMed]
- Sato, S. Plasmodium—A brief introduction to the parasites causing human malaria and their basic biology. J. Physiol. Anthropol. 2021, 40, 1. [Google Scholar] [CrossRef] [PubMed]
- Yeung, S. Malaria-Update on Antimalarial Resistance and Treatment Approaches. Pediatr. Infect. Dis. J. 2018, 37, 367–369. [Google Scholar] [CrossRef]
- World Health Organization. World Malaria Report 2021. Available online: https://www.who.int/publications/i/item/9789240040496 (accessed on 15 July 2022).
- Nepali, K.; Sharma, S.; Sharma, M.; Bedi, P.M.; Dhar, K.L. Rational approaches, design strategies, structure activity relationship and mechanistic insights for anticancer hybrids. Eur. J. Med. Chem. 2014, 77, 422–487. [Google Scholar] [CrossRef]
- Soni, J.P.; Yeole, Y.; Shankaraiah, N. β-Carboline-based molecular hybrids as anticancer agents: A brief sketch. RSC Med. Chem. 2021, 12, 730–750. [Google Scholar] [CrossRef]
- Walsh, J.J.; Bell, A. Hybrid drugs for malaria. Curr. Pharm. Des. 2009, 15, 2970–2985. [Google Scholar] [CrossRef]
- Cao, R.; Peng, W.; Wang, Z.; Xu, A. beta-Carboline alkaloids: Biochemical and pharmacological functions. Curr. Med. Chem. 2007, 14, 479–500. [Google Scholar] [CrossRef]
- Ishida, J.; Wang, H.K.; Bastow, K.F.; Hu, C.Q.; Lee, K.H. Antitumor agents 201. Cytotoxicity of harmine and β-carboline analogs. Bioorg. Med. Chem. Lett. 1999, 9, 3319–3324. [Google Scholar] [CrossRef]
- Guan, H.; Chen, H.; Peng, W.; Ma, Y.; Cao, R.; Liu, X.; Xu, A. Design of beta-carboline derivatives as DNA-targeting antitumor agents. Eur. J. Med. Chem. 2006, 41, 1167–1179. [Google Scholar] [CrossRef]
- Xu, Q.B.; Chen, X.F.; Feng, J.; Miao, J.F.; Liu, J.; Liu, F.T.; Niu, B.X.; Cai, J.Y.; Huang, C.; Zhang, Y.; et al. Design, synthesis and biological evaluation of hybrids of β-carboline and salicylic acid as potential anticancer and apoptosis inducing agents. Sci. Rep. 2016, 6, 36238. [Google Scholar] [CrossRef]
- Aaghaz, S.; Sharma, K.; Jain, R.; Kamal, A. β-Carbolines as potential anticancer agents. Eur. J. Med. Chem. 2021, 216, 113321. [Google Scholar] [CrossRef]
- Meinguet, C.; Bruyère, C.; Frédérick, R.; Mathieu, V.; Vancraeynest, C.; Pochet, L.; Laloy, J.; Mortier, J.; Wolber, G.; Kiss, R.; et al. 3D-QSAR, design, synthesis and characterization of trisubstituted harmine derivatives with in vitro antiproliferative properties. Eur. J. Med. Chem. 2015, 94, 45–55. [Google Scholar] [CrossRef]
- Kumar, S.; Singh, A.; Kumar, K.; Kumar, V. Recent insights into synthetic β-carbolines with anti-cancer activities. Eur. J. Med. Chem. 2017, 142, 48–73. [Google Scholar] [CrossRef]
- Shahinas, D.; Macmullin, G.; Benedict, C.; Crandall, I.; Pillai, D.R. Harmine is a potent antimalarial targeting Hsp90 and synergizes with chloroquine and artemisinin. Antimicrob. Agents Chemother. 2012, 56, 4207–4213. [Google Scholar] [CrossRef]
- Gorki, V.; Walter, N.S.; Singh, R.; Chauhan, M.; Dhingra, N.; Salunke, D.B.; Kaur, S. β-Carboline Derivatives Tackling Malaria: Biological Evaluation and Docking Analysis. ACS Omega 2020, 5, 17993–18006. [Google Scholar] [CrossRef]
- Kamboj, A.; Sihag, B.; Brar, D.S.; Kaur, A.; Salunke, D.B. Structure activity relationship in β-carboline derived anti-malarial agents. Eur. J. Med. Chem. 2021, 221, 113536. [Google Scholar] [CrossRef]
- Heinze, K.; Lang, H. Ferrocene—Beauty and Function. Organometallics 2013, 32, 5623–5625. [Google Scholar] [CrossRef]
- Sharma, B.; Kumar, V. Has Ferrocene Really Delivered Its Role in Accentuating the Bioactivity of Organic Scaffolds? J. Med. Chem. 2021, 64, 16865–16921. [Google Scholar] [CrossRef]
- Roux, C.; Biot, C. Ferrocene-based antimalarials. Future Med. Chem. 2012, 4, 783–797. [Google Scholar] [CrossRef]
- Peter, S.; Aderibigbe, B.A. Ferrocene-Based Compounds with Antimalaria/Anticancer Activity. Molecules 2019, 24, 3604. [Google Scholar] [CrossRef]
- Ren, S.Z.; Wang, Z.C.; Zhu, D.; Zhu, X.H.; Shen, F.Q.; Wu, S.Y.; Chen, J.J.; Xu, C.; Zhu, H.L. Design, synthesis and biological evaluation of novel ferrocene-pyrazole derivatives containing nitric oxide donors as COX-2 inhibitors for cancer therapy. Eur. J. Med. Chem. 2018, 157, 909–924. [Google Scholar] [CrossRef]
- Chellan, P.; Sadler, P.J. Enhancing the Activity of Drugs by Conjugation to Organometallic Fragments. Chem. Eur. J. 2020, 26, 8676–8688. [Google Scholar] [CrossRef]
- Nguyen, A.; Top, S.; Pigeon, P.; Vessières, A.; Hillard, E.A.; Plamont, M.A.; Huché, M.; Rigamonti, C.; Jaouen, G. Synthesis and structure-activity relationships of ferrocenyl tamoxifen derivatives with modified side chains. Chem. Eur. J. 2009, 15, 684–696. [Google Scholar] [CrossRef]
- Dubar, F.; Khalife, J.; Brocard, J.; Dive, D.; Biot, C. Ferroquine, an ingenious antimalarial drug: Thoughts on the mechanism of action. Molecules 2008, 13, 2900–2907. [Google Scholar] [CrossRef]
- Adoke, Y.; Zoleko-Manego, R.; Ouoba, S.; Tiono, A.B.; Kaguthi, G.; Bonzela, J.E.; Duong, T.T.; Nahum, A.; Bouyou-Akotet, M.; Ogutu, B.; et al. A randomized, double-blind, phase 2b study to investigate the efficacy, safety, tolerability and pharmacokinetics of a single-dose regimen of ferroquine with artefenomel in adults and children with uncomplicated Plasmodium falciparum malaria. Malar. J. 2021, 20, 222. [Google Scholar] [CrossRef]
- Kondratskyi, A.; Kondratska, K.; Vanden Abeele, F.; Gordienko, D.; Dubois, C.; Toillon, R.A.; Slomianny, C.; Lemière, S.; Delcourt, P.; Dewailly, E.; et al. Ferroquine, the next generation antimalarial drug, has antitumor activity. Sci. Rep. 2017, 7, 15896. [Google Scholar] [CrossRef]
- Perković, I.; Raić-Malić, S.; Fontinha, D.; Prudêncio, M.; Pessanha de Carvalho, L.; Held, J.; Tandarić, T.; Vianello, R.; Zorc, B.; Rajić, Z. Harmicines-harmine and cinnamic acid hybrids as novel antiplasmodial hits. Eur. J. Med. Chem. 2020, 187, 111927. [Google Scholar] [CrossRef]
- Marinović, M.; Perković, I.; Fontinha, D.; Prudêncio, M.; Held, J.; Pessanha de Carvalho, L.; Tandarić, T.; Vianello, R.; Zorc, B.; Rajić, Z. Novel Harmicines with Improved Potency against Plasmodium. Molecules 2020, 25, 4376. [Google Scholar] [CrossRef]
- Marinović, M.; Poje, G.; Perković, I.; Fontinha, D.; Prudêncio, M.; Held, J.; Pessanha de Carvalho, L.; Tandarić, T.; Vianello, R.; Rajić, Z. Further investigation of harmicines as novel antiplasmodial agents: Synthesis, structure-activity relationship and insight into the mechanism of action. Eur. J. Med. Chem. 2021, 15, 113687. [Google Scholar] [CrossRef] [PubMed]
- Pavić, K.; Beus, M.; Poje, G.; Uzelac, L.; Kralj, M.; Rajić, Z. Synthesis and Biological Evaluation of Harmirins, Novel Harmine-Coumarin Hybrids as Potential Anticancer Agents. Molecules 2021, 26, 6490. [Google Scholar] [CrossRef] [PubMed]
- Poje, G.; Pessanha de Carvalho, L.; Held, J.; Moita, D.; Prudêncio, M.; Perković, I.; Tandarić, T.; Vianello, R.; Rajić, Z. Design and synthesis of harmiquins, harmine and chloroquine hybrids as potent antiplasmodial agents. Eur. J. Med. Chem. 2022, 238, 114408. [Google Scholar] [CrossRef] [PubMed]
- Allardyce, C.S.; Dorcier, A.; Scolaro, C.; Dyson, P.J. Development of organometallic (organo-transitionmetal) pharmaceuticals. Appl. Organometal. Chem. 2005, 19, 1–10. [Google Scholar] [CrossRef]
- Hartinger, C.G.; Dyson, P.J. Bioorganometallic chemistry--from teaching paradigms to medicinal applications. Chem. Soc. Rev. 2009, 38, 391–401. [Google Scholar] [CrossRef]
- Matos, J.; da Cruz, F.P.; Cabrita, É.; Gut, J.; Nogueira, F.; do Rosário, V.E.; Moreira, R.; Rosenthal, P.J.; Prudêncio, M.; Gomes, P. Novel potent metallocenes against liver stage malaria. Antimicrob. Agents Chemother. 2012, 56, 1564–1570. [Google Scholar] [CrossRef]
- Patra, M.; Gasser, G. The medicinal chemistry of ferrocene and its derivatives. Nat. Rev. Chem. 2017, 1, 66. [Google Scholar] [CrossRef]
- Wani, W.A.; Jameel, E.; Baig, U.; Mumtazuddin, S.; Hun, L.T. Ferroquine and its derivatives: New generation of antimalarial agents. Eur. J. Med. Chem. 2015, 101, 534–551. [Google Scholar] [CrossRef]
- Ludwig, B.S.; Correia, J.D.G.; Kühn, F.E. Ferrocene derivatives as anti-infective agents. Coord. Chem. Rev. 2019, 396, 22–48. [Google Scholar] [CrossRef]
- Habrant, D.; Rauhala, V.; Koskinen, A.M. Conversion of carbonyl compounds to alkynes: General overview and recent developments. Chem. Soc. Rev. 2010, 39, 2007–2017. [Google Scholar] [CrossRef]
- Goddard-Borger, E.D.; Stick, R.V. An efficient, inexpensive, and shelf-stable diazotransfer reagent: Imidazole-1-sulfonyl azide hydrochloride. Org. Lett. 2007, 9, 3797–3800. [Google Scholar] [CrossRef]
- Held, J.; Gebru, T.; Kalesse, M.; Jansen, R.; Gerth, K.; Müller, R.; Mordmüller, B. Antimalarial activity of the myxobacterial macrolide chlorotonil a. Antimicrob. Agents Chemother. 2014, 58, 6378–6384. [Google Scholar] [CrossRef]
- Noedl, H.; Bronnert, J.; Yingyuen, K.; Attlmayr, B.; Kollaritsch, H.; Fukuda, M. Simple histidine-rich protein 2 double-site sandwich enzyme-linked immunosorbent assay for use in malaria drug sensitivity testing. Antimicrob. Agents Chemother. 2005, 49, 3575–3577. [Google Scholar] [CrossRef]
- The R Project for Statistical Computing. Available online: https://www.R-project.org/ (accessed on 20 December 2021).
- Kim, G.D. Harmine Hydrochloride Triggers G2/M Cell Cycle Arrest and Apoptosis in HCT116 Cells through ERK and PI3K/AKT/mTOR Signaling Pathways. Prev. Nutr. Food Sci. 2021, 26, 445–452. [Google Scholar] [CrossRef]
- Ock, C.W.; Kim, G.D. Harmine Hydrochloride Mediates the Induction of G2/M Cell Cycle Arrest in Breast Cancer Cells by Regulating the MAPKs and AKT/FOXO3a Signaling Pathways. Molecules 2021, 26, 6714. [Google Scholar] [CrossRef]
- Mota, N.S.R.S.; Kviecinski, M.R.; Felipe, K.B.; Grinevicius, V.M.A.S.; Siminski, T.; Almeida, G.M.; Zeferino, R.C.; Pich, C.T.; Filho, D.W.; Pedrosa, R.C.; et al. β-carboline alkaloid harmine induces DNA damage and triggers apoptosis by a mitochondrial pathway: Study in silico, in vitro and in vivo. Int. J. Funct. Nutr. 2020, 1, 1. [Google Scholar]
- Zheng, J.; Zeng, L.; Tang, M.; Lin, H.; Pi, C.; Xu, R.; Cui, X. Novel Ferrocene Derivatives Induce G0/G1 Cell Cycle Arrest and Apoptosis through the Mitochondrial Pathway in Human Hepatocellular Carcinoma. Int. J. Mol. Sci. 2021, 22, 3097. [Google Scholar] [CrossRef]
- Zeng, L.; Tang, M.; Pi, C.; Zheng, J.; Gao, S.; Chabanne, T.; Chauvin, R.; Cheng, W.; Lin, H.; Xu, R.; et al. Novel Ferrocene Derivatives Induce Apoptosis through Mitochondria-Dependent and Cell Cycle Arrest via PI3K/Akt/mTOR Signaling Pathway in T Cell Acute Lymphoblastic Leukemia. Cancers 2021, 13, 4677. [Google Scholar] [CrossRef]
- Singh, D.; Hazra, C.K.; Malakar, C.C.; Pandey, S.K.; Kaith, B.S.; Singh, V. Indium-Mediated Domino Allylation-Lactonisation Approach: Diastereoselective Synthesis of β-Carboline C-3 Tethered α-Methylene γ-Butyrolactones. ChemistrySelect 2018, 3, 4859–4864. [Google Scholar] [CrossRef]
- Szabó, T.; Hazai, V.; Volk, B.; Simig, G.; Milen, M. First total synthesis of the β-carboline alkaloids trigonostemine A, trigonostemine B and a new synthesis of pityriacitrin and hyrtiosulawesine. Tetrahedron Lett. 2019, 60, 1471–1475. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compd. | IC50 a (µM) | ||||
|---|---|---|---|---|---|
| Pf3D7 | PfDd2 | Pf3D7 | PfDd2 | ||
| 20 | 9.23 ± 1.96 | 11.20 ± 2.23 | 30 | >56 b | 14.01 ± 1.95 c |
| 21 | 7.14 ± 0.38 | 6.06 ± 1.36 | 31 | 13.24 ± 8.54 | 8.03 ± 0.19 |
| 22 | 0.35 ± 0.19 | 0.89 ± 0.01 | 32 | 0.30 ± 0.09 | 0.66 ± 0.01 |
| 23 | 0.15 ± 0.05 | 1.02 ± 0.52 | 33 | 0.40 ± 0.18 | 2.04 ± 0.29 |
| 24 | 1.35 ± 0.28 | 2.31 ± 0.29 | 34 | 1.34 ± 0.09 | 1.11 ± 0.01 |
| 25 | 3.62 ± 1.08 | 4.37 ± 1.79 | 35 | 7.85 ± 2.18 | 12.99 ± 5.90 |
| 26 | 2.63 ± 0.27 | 4.02 ± 0.61 | 36 | 0.42 ± 0.02 | 1.53 ± 0.06 |
| 27 | 0.31 ± 0.01 | 1.09 ± 0.01 | 37 | 0.83 ± 0.23 | 1.80 ± 0.35 |
| 28 | >56 | >56 | HAR d | 8.25 ± 2.83 | >27.7 |
| 29 | 2.12 ± 0.81 | 2.91 ± 0.05 | CQ e | 0.004 ± 0.002 | 0.29 ± 0.10 |
| Compd. | IC50 a (µM) | SI b (HCT116) | SI (MCF-7) | ||||
|---|---|---|---|---|---|---|---|
| HepG2 | SW620 | HCT116 | MCF-7 | Hek293T | |||
| 20 | 43.9 ± 1.1 | >50 | 29.4 ± 4.6 | 39.4 ± 7.9 | >50 | >1.7 | >1.3 |
| 21 | >50 | 19.7 ± 1.9 | 8.3 ± 1.4 | 17.7 ± 0.6 | >50 | >6 | >2.8 |
| 22 | 8.4 ± 0.9 | 6.5 ± 0.7 | 9 ± 0.7 | 6.9 ± 0.5 | 7.3 ± 2.8 | 0.8 | 1.1 |
| 23 | 9.5 ± 0.5 | 7.6 ± 0.1 | 38.6 ± 2.8 | 8.0 ± 0.2 | 17.1 ± 1.8 | 0.4 | 2.1 |
| 24 | >50 | >50 | >50 | >50 | >50 | >1 | >1 |
| 25 | >50 | 23.1 ± 6.4 | 7.6 ± 0.5 | >50 | >50 | >6.6 | >1 |
| 26 | 12.3 ± 5 | 3.8 ± 0.4 | 35.2 ± 4.7 | 3.7 ± 0.2 | 46.5 ± 1 | 1.3 | 12.6 |
| 27 | 17 ± 0.3 | 8.1 ± 0.2 | >50 | 6.8 ± 0.2 | 43 ± 8.6 | < 0.9 | 6.3 |
| 28 | >50 | >50 | 7.4 ± 0.4 | >50 | >50 | >6.8 | >1 |
| 29 | >50 | >50 | 14.6 ± 3.7 | >50 | >50 | >3.4 | >1 |
| 30 | >50 | 45.1 ± 3.7 | 16.8 ± 0.9 | 8.5 ± 1.5 | >50 | >3 | >5.9 |
| 31 | >50 | >50 | >50 | 19.9 ± 1.1 | >50 | >1 | >2.51 |
| 32 | 15.6 ± 0.3 | 9.9 ± 4 | 6.6 ± 0.8 | 9.1 ± 1.4 | 18.6 ± 0.2 | 2.8 | 2 |
| 33 | >50 | >50 | 16.3 ± 3.2 | 30.5 ± 13.2 | >50 | >1 | >1.7 |
| 34 | >50 | >50 | >50 | >50 | >50 | >1 | >1 |
| 35 | >50 | 25 ± 8.1 | 20.9 ± 1.5 | 36.9 ± 6.8 | >50 | >2.4 | >1.4 |
| 36 | 7.4 ± 0.5 | 4.2 ± 0.3 | 5.9 ± 1.3 | 4.5 ± 0.3 | 8.4 ± 0.3 | 1.4 | 1.9 |
| 37 | 28 ± 2.5 | 9.9 ± 1.1 | 7.5 ± 0.4 | 13.2 ± 0.5 | 30.8 ± 2.4 | 4.1 | 2.3 |
| HAR c | 18.7 ± 0.8 | 4.7 ± 0.6 | 4.0 ± 0.8 | 13.5 ± 1.1 | 12.6 ± 0.8 | 3.2 | 0.9 |
| 5-FU d | 5.5 ± 0.6 | 9.4 ± 0.3 | 5.2 ± 2.8 | 23.9 ± 5.7 | 8.1 ± 0.8 | 1.6 | 0.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poje, G.; Marinović, M.; Pavić, K.; Mioč, M.; Kralj, M.; de Carvalho, L.P.; Held, J.; Perković, I.; Rajić, Z. Harmicens, Novel Harmine and Ferrocene Hybrids: Design, Synthesis and Biological Activity. Int. J. Mol. Sci. 2022, 23, 9315. https://doi.org/10.3390/ijms23169315
Poje G, Marinović M, Pavić K, Mioč M, Kralj M, de Carvalho LP, Held J, Perković I, Rajić Z. Harmicens, Novel Harmine and Ferrocene Hybrids: Design, Synthesis and Biological Activity. International Journal of Molecular Sciences. 2022; 23(16):9315. https://doi.org/10.3390/ijms23169315
Chicago/Turabian StylePoje, Goran, Marina Marinović, Kristina Pavić, Marija Mioč, Marijeta Kralj, Lais Pessanha de Carvalho, Jana Held, Ivana Perković, and Zrinka Rajić. 2022. "Harmicens, Novel Harmine and Ferrocene Hybrids: Design, Synthesis and Biological Activity" International Journal of Molecular Sciences 23, no. 16: 9315. https://doi.org/10.3390/ijms23169315