
Laparoscopic Sleeve Gastrectomy in Patients with Severe Obesity Restores Adaptive Responses Leading to Nonalcoholic Steatohepatitis

 , , , , ,
, , , , ,  , , , and add
Show full author list
, , , and add
Show full author list
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
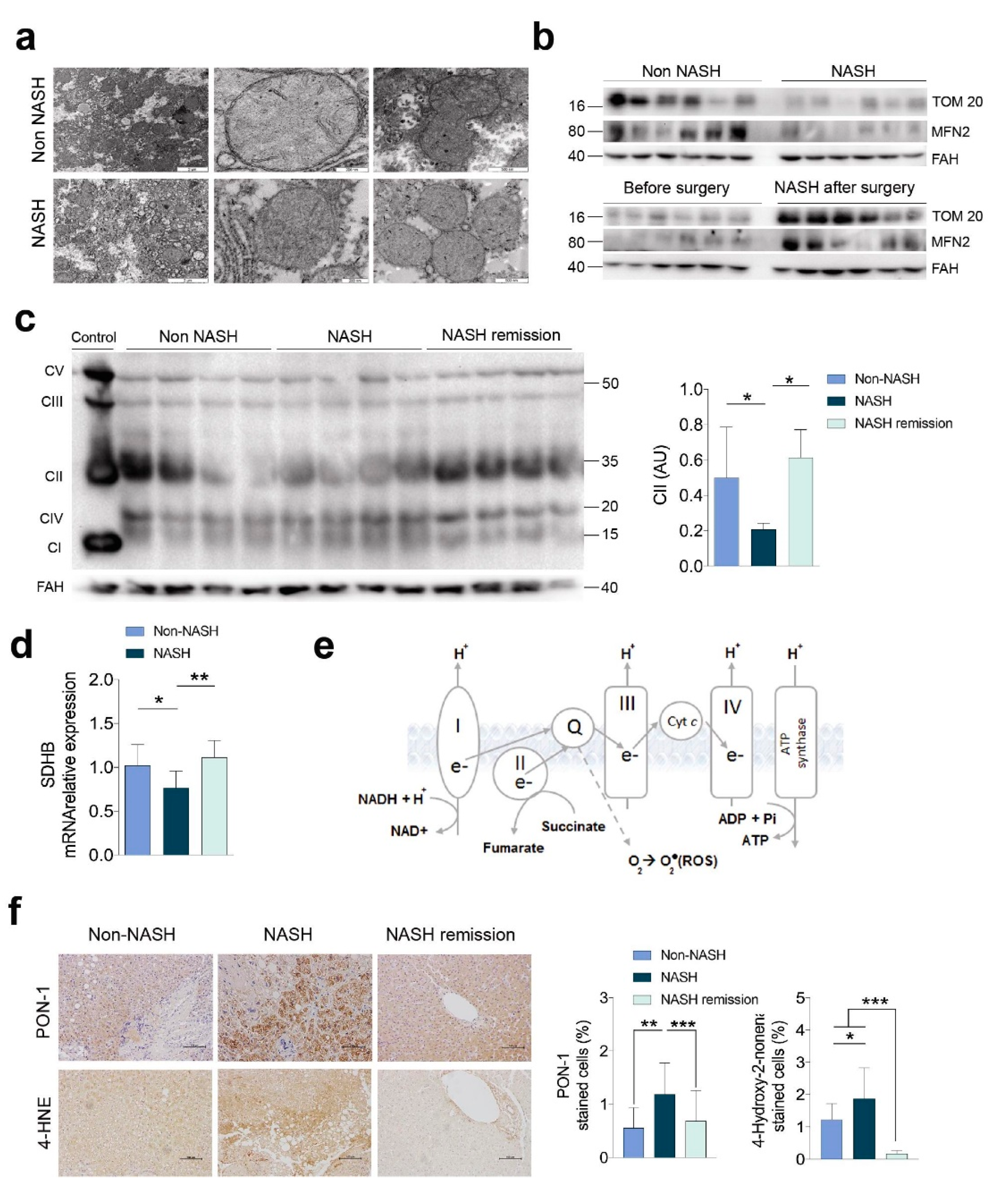
2.1. NASH Remission Included the Reversal of the Altered Liver Mitochondrial Status and Oxidative Stress
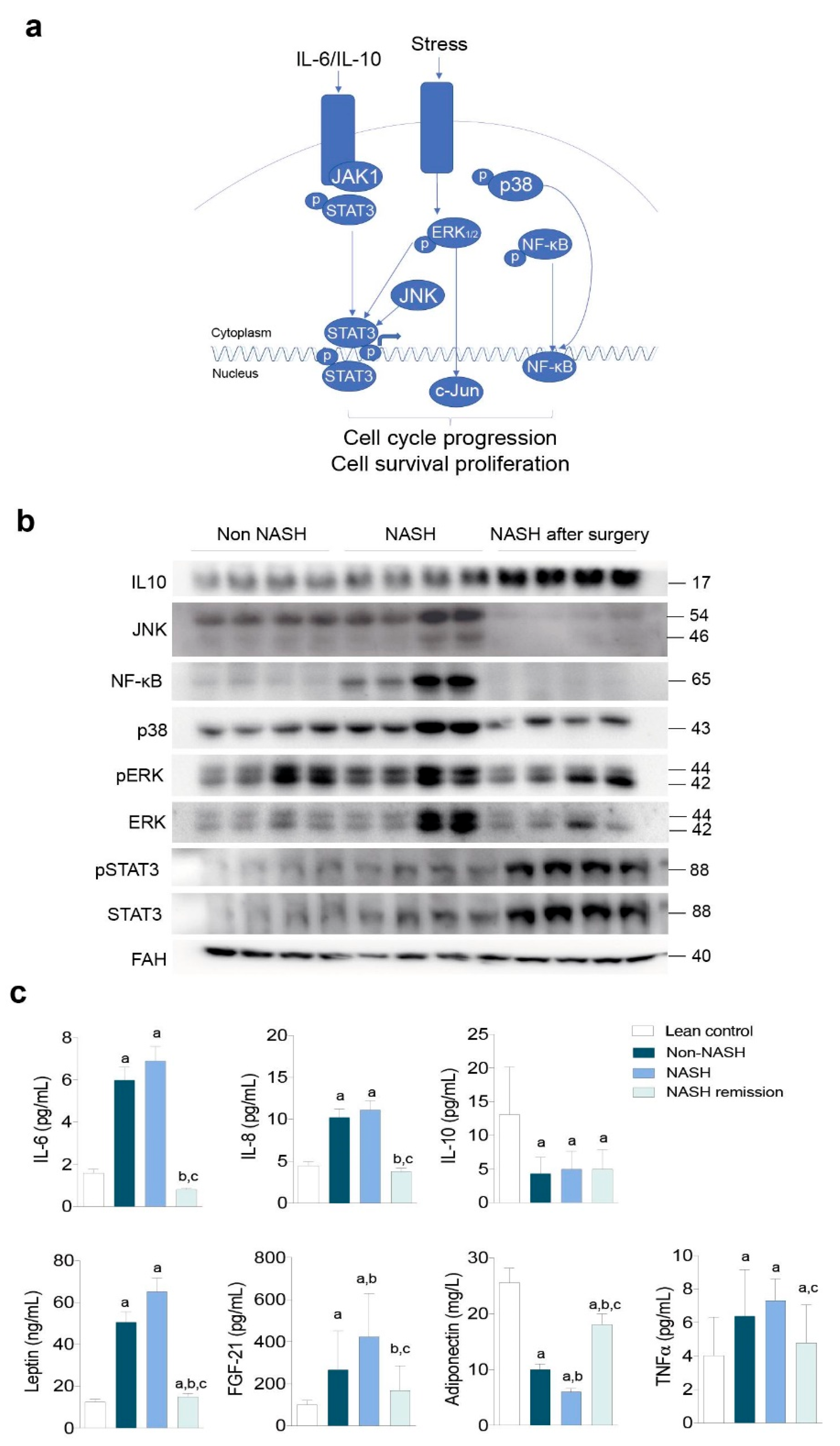
2.2. NASH Reflects an Impact on Multiple Stress-Responsive Pathways
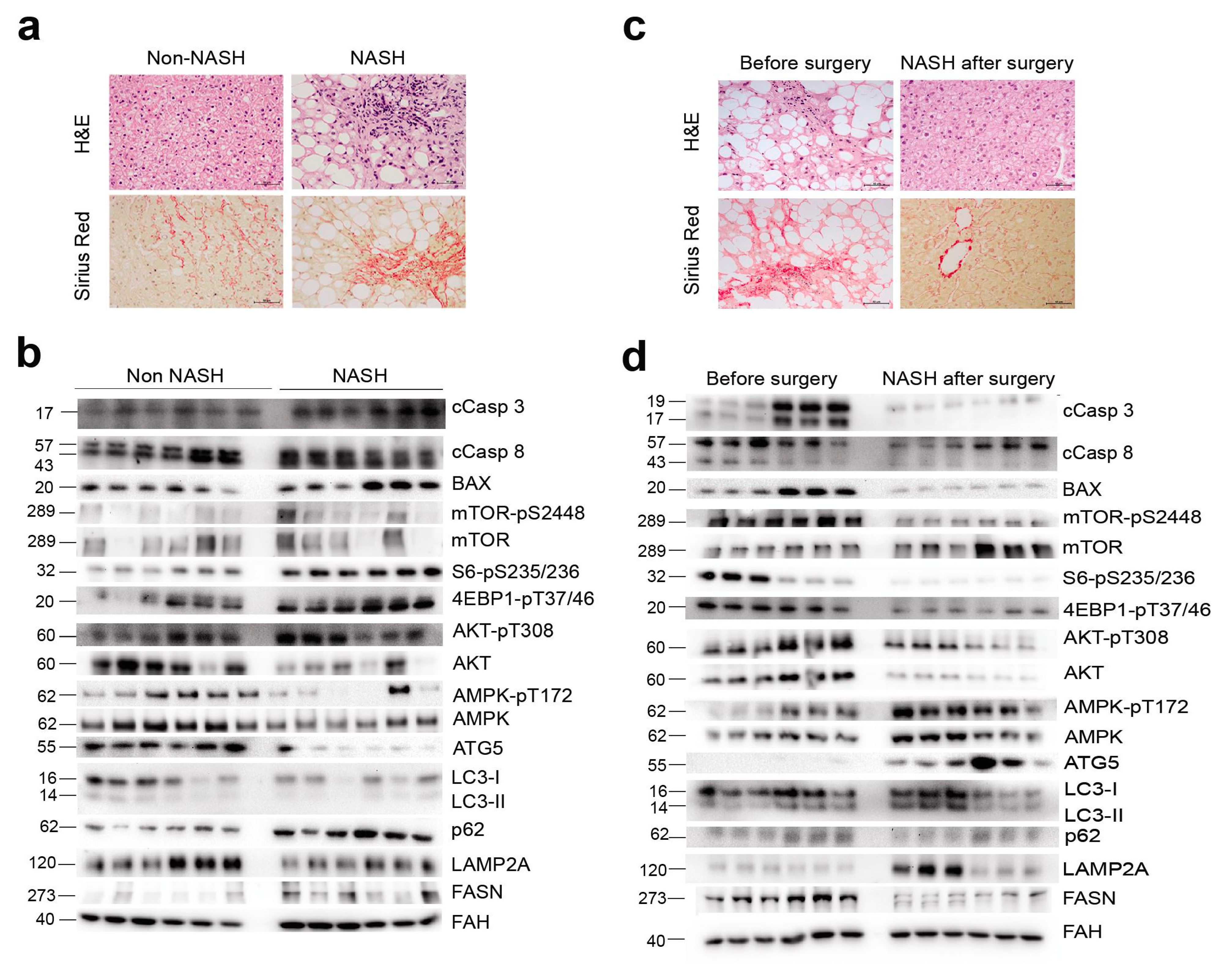
2.3. NASH Remission Reversed Apoptosis and Reactivated Autophagy via mTORC1 Signalling
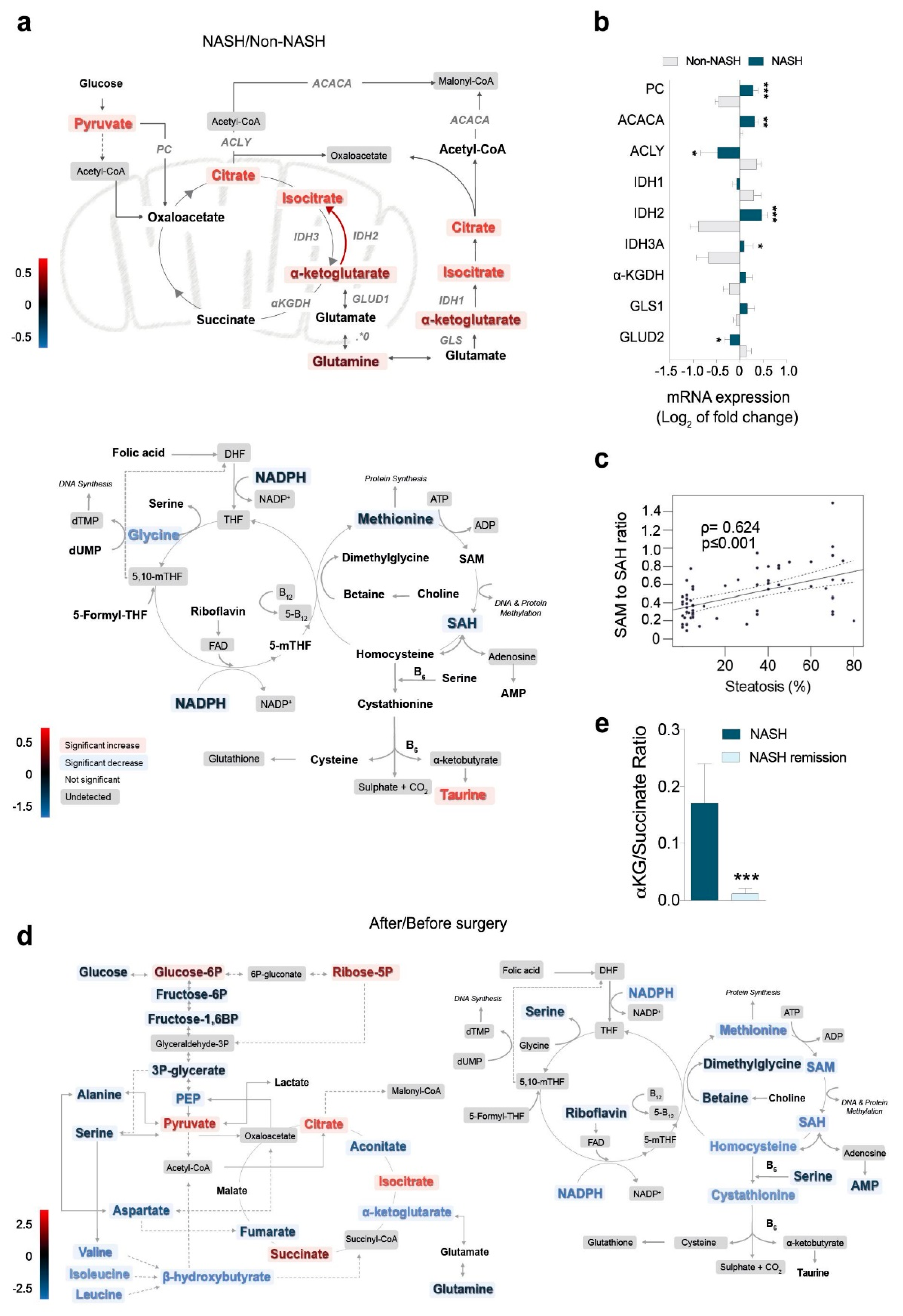
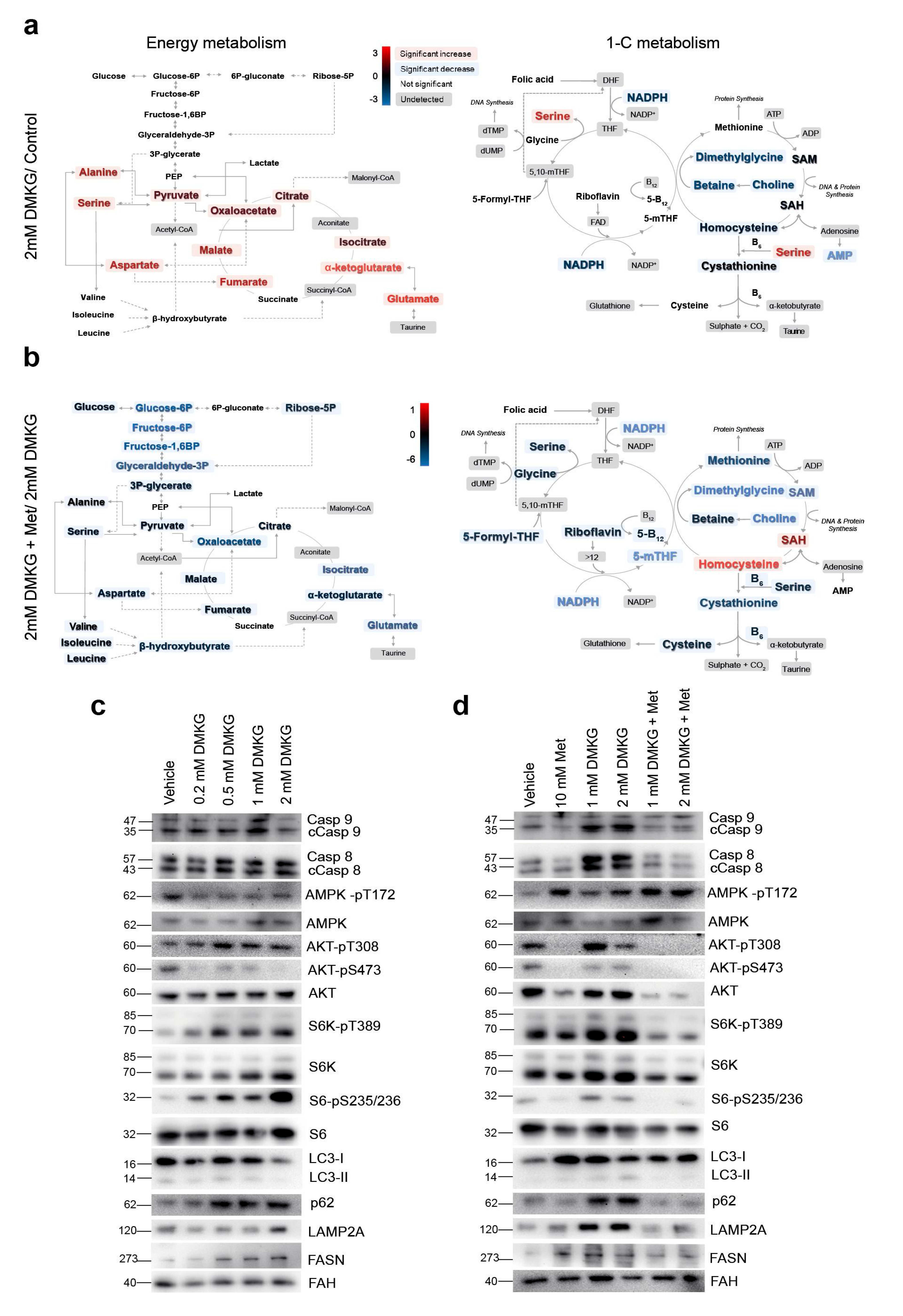
2.4. Metabolomics Revealed the Role of Glutaminolysis Activation in NASH Metabolic Rewiring
2.5. Increased α-Ketoglutarate Altered Mitochondrial Metabolism and Apoptosis in Cultured Hepatocytes
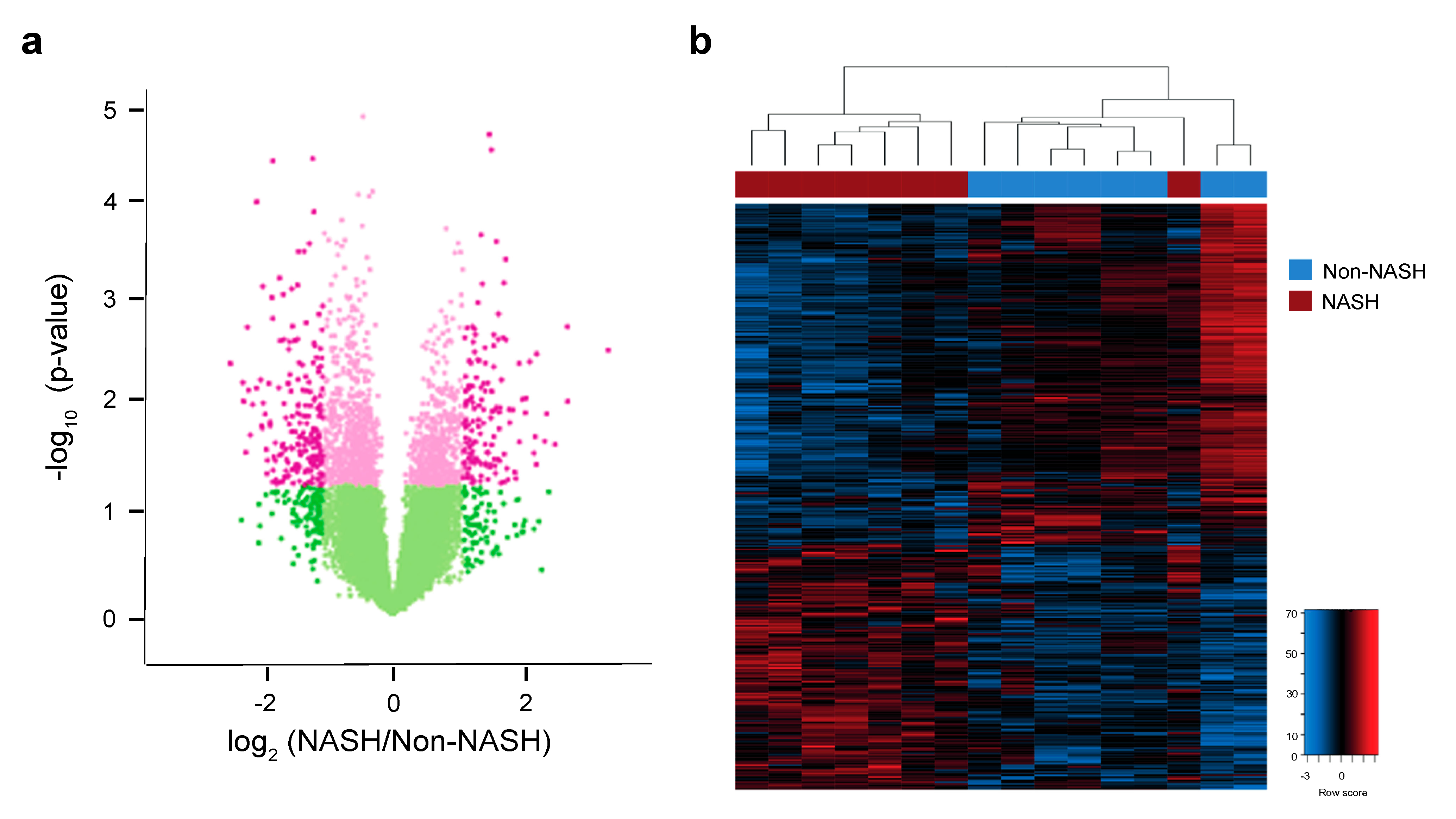
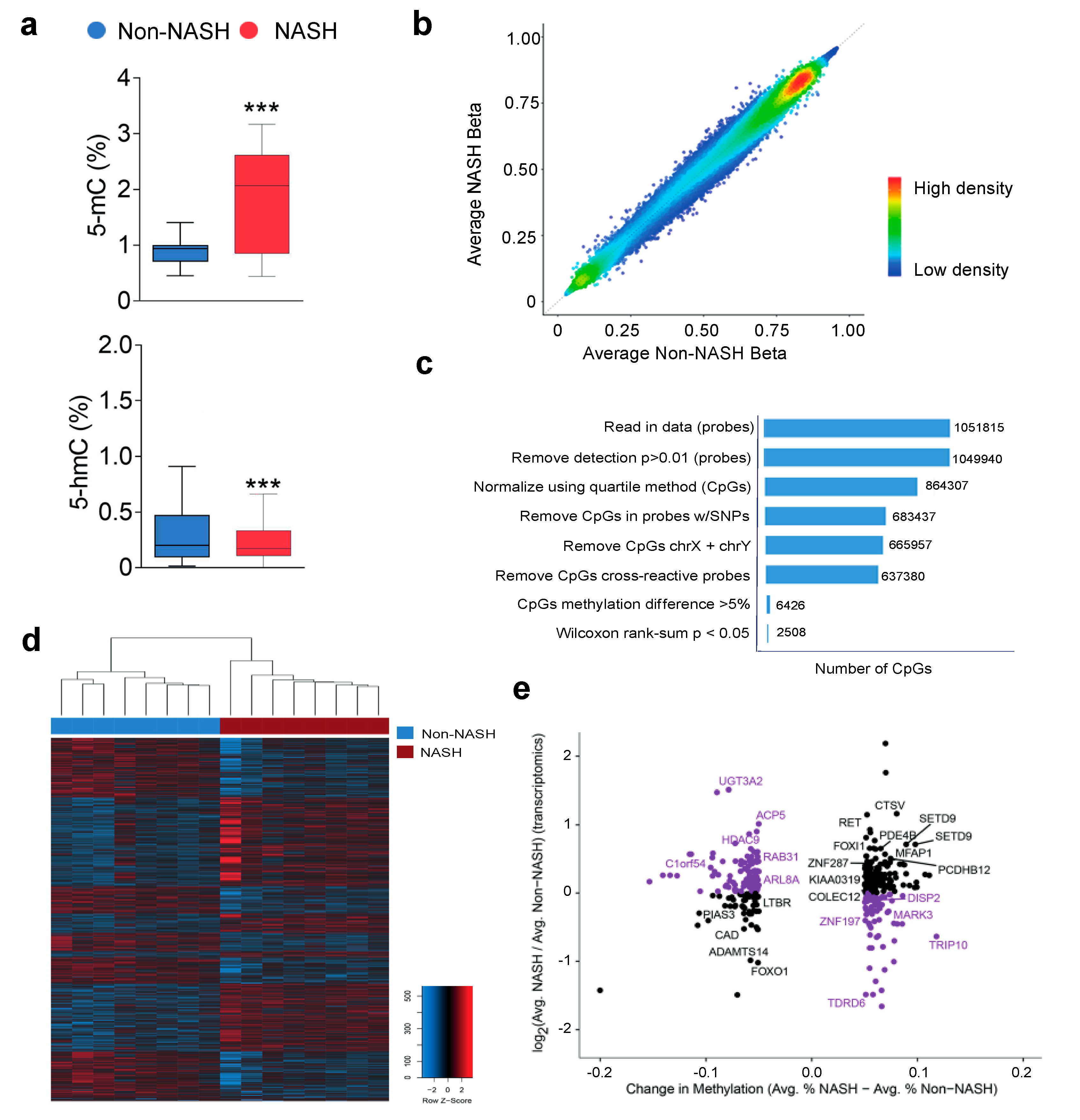
2.6. Analysis of Hepatic Transcriptome Signatures and Differentially Methylated Genomic Regions
3. Discussion
4. Materials and Methods
4.1. Participants
4.2. Analytical Methods
4.3. Statistical Analysis
4.4. Ethics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Polyzos, S.A.; Kountouras, J.; Mantzoros, C.S. Obesity and nonalcoholic fatty liver disease: From pathophysiology to therapeutics. Metabolism 2019, 92, 82–97. [Google Scholar] [CrossRef]
- Koliaki, C.; Szendroedi, J.; Kaul, K.; Jelenik, T.; Nowotny, P.; Jankowiak, F.; Herder, C.; Carstensen, M.; Krausch, M.; Knoefel, W.T.; et al. Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell. Metab. 2015, 21, 739–746. [Google Scholar] [CrossRef] [Green Version]
- Hernández-Alvarez, M.I.; Sebastián, D.; Vives, S.; Ivanova, S.; Bartoccioni, P.; Kakimoto, P.; Plana, N.; Veiga, S.R.; Hernández, V.; Vasconcelos, N.; et al. Deficient endoplasmic reticulum-mitochondrial phosphatidylserine transfer causes liver disease. Cell 2019, 177, 881–895. [Google Scholar] [CrossRef] [PubMed]
- Cuyàs, E.; Corominas-Faja, B.; Joven, J.; Menendez, J.A. Cell cycle regulation by the nutrient-sensing mammalian target of rapamycin (mTOR) pathway. Methods Mol. Biol. 2014, 1170, 113–144. [Google Scholar] [CrossRef] [PubMed]
- Calvo, N.; Beltrán-Debón, R.; Rodríguez-Gallego, E.; Hernández-Aguilera, A.; Guirro, M.; Mariné-Casadó, R.; Millá, L.; Alegret, J.M.; Sabench, F.; del Castillo, D.; et al. Liver fat deposition and mitochondrial dysfunction in morbid obesity: An approach combining metabolomics with liver imaging and histology. World J. Gastroenterol. 2015, 21, 7529–7544. [Google Scholar] [CrossRef] [PubMed]
- Cabré, N.; Luciano-Mateo, F.; Baiges-Gayà, G.; Fernández-Arroyo, S.; Rodríguez-Tomàs, E.; Hernández-Aguilera, A.; París, M.; Sabench, F.; Del Castillo, D.; López-Miranda, J.; et al. Plasma metabolic alterations in patients with severe obesity and non-alcoholic steatohepatitis. Aliment. Pharmacol. Ther. 2020, 51, 374–387. [Google Scholar] [CrossRef] [PubMed]
- Riera-Borrull, M.; Rodríguez-Gallego, E.; Hernández-Aguilera, A.; Luciano, F.; Ras, R.; Cuyàs, E.; Camps, J.; Segura-Carretero, A.; Menendez, J.A.; Joven, J.; et al. Exploring the process of energy generation in pathophysiology by trgeted metabolomics: Performance of a simple and quantitative method. J. Am. Soc. Mass Spectrom. 2016, 27, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Mansouri, A.; Gattolliat, C.H.; Asselah, T. Mitochondrial dysfunction and signaling in chronic liver diseases. Gastroenterology 2018, 155, 629–647. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, L.B.; Gui, D.Y.; Vander Heiden, M.G. Altered metabolite levels in cancer: Implications for tumour biology and cancer therapy. Nat. Rev. Cancer 2016, 16, 680–693. [Google Scholar] [CrossRef]
- Eslam, M.; Valenti, L.; Romeo, S. Genetics and epigenetics of NAFLD and NASH: Clinical impact. J. Hepatol. 2018, 68, 268–279. [Google Scholar] [CrossRef]
- Guzy, R.D.; Sharma, B.; Bell, E.; Chandel, N.S.; Schumacker, P.T. Loss of the SdhB, but Not the SdhA, subunit of complex II triggers reactive oxygen species-dependent hypoxia-inducible factor activation and tumorigenesis. Mol. Cell. Biol. 2008, 28, 718–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villar, V.H.; Durán, R.V. Glutamoptosis: A new cell death mechanism inhibited by autophagy during nutritional imbalance. Autophagy 2017, 13, 1078–1079. [Google Scholar] [CrossRef] [Green Version]
- Menendez, J.A.; Cufí, S.; Oliveras-Ferraros, C.; Vellon, L.; Joven, J.; Vazquez-Martin, A. Gerosuppressant metformin: Less is more. Aging 2011, 3, 348–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mauro, C.; Leow, S.C.; Anso, E.; Rocha, S.; Thotakura, A.K.; Tornatore, L.; Moretti, M.; De Smaele, E.; Beg, A.A.; Tergaonkar, V.; et al. NF-κB controls energy homeostasis and metabolic adaptation by upregulating mitochondrial respiration. Nat. Cell. Biol. 2011, 13, 1272–1279. [Google Scholar] [CrossRef] [PubMed]
- Schrepfer, E.; Scorrano, L. Mitofusins, from mitochondria to metabolism. Mol. Cell 2016, 61, 683–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabré, N.; Luciano-Mateo, F.; Fernández-Arroyo, S.; Baiges-Gayà, G.; Hernández-Aguilera, A.; Fibla, M.; Fernández-Julià, R.; París, M.; Sabench, F.; Castillo, D.D.; et al. Laparoscopic sleeve gastrectomy reverses non-alcoholic fatty liver disease modulating oxidative stress and inflammation. Metabolism 2019, 99, 81–89. [Google Scholar] [CrossRef]
- Canivet, C.M.; David, N.; Pailhoriès, H.; Briand, M.; Guy, C.D.; Bouchez, O.; Hunault, G.; Fizanne, L.; Lannes, A.; Oberti, F.; et al. Cross-linkage between bacterial taxonomy and gene functions: A study of metagenome-assembled genomes of gut microbiota in adult non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2021, 53, 722–732. [Google Scholar] [CrossRef]
- Parthasarathy, G.; Revelo, X.; Malhi, H. Pathogenesis of nonalcoholic steatohepatitis: An overview. Hepatol. Commun. 2020, 4, 478–492. [Google Scholar] [CrossRef] [Green Version]
- Luedde, T.; Kaplowitz, N.; Schwabe, R. Cell death and cell death responses in liver disease: Mechanisms and clinical relevance. Gastroenterology 2014, 147, 765–783. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Gallego, E.; Guirro, M.; Riera-Borrull, M.; Hernández-Aguilera, A.; Mariné-Casadó, R.; Fernández-Arroyo, S.; Beltrán-Debón, R.; Sabench, F.; Hernández, M.; del Castillo, D.; et al. Mapping of the circulating metabolome reveals α-ketoglutarate as a predictor of morbid obesity-associated non-alcoholic fatty liver disease. Int. J. Obes. 2015, 39, 279–287. [Google Scholar] [CrossRef]
- Dai, W.; Xu, L.; Yu, X.; Zhang, G.; Guo, H.; Liu, H.; Song, G.; Weng, S.; Dong, L.; Zhu, J.; et al. OGDHL silencing promotes hepatocellular carcinoma by reprogramming glutamine metabolism. J. Hepatol. 2019, 72, 909–923. [Google Scholar] [CrossRef] [PubMed]
- Simon, J.; Nuñez-García, M.; Fernández-Tussy, P.; Barbier-Torres, L.; Fernández-Ramos, D.; Gómez-Santos, B.; Buqué, X.; Lopitz-Otsoa, F.; Goikoetxea-Usandizaga, N.; Serrano-Macia, M.; et al. Targeting hepatic glutaminase 1 ameliorates non-alcoholic steatohepatitis by restoring very-low-density lipoprotein triglyceride assembly. Cell. Metab. 2020, 31, 605–622. [Google Scholar] [CrossRef] [PubMed]
- Carey, B.W.; Finley, L.W.; Cross, J.R.; Allis, C.D.; Thompson, C.B. Intracellular α-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature 2015, 518, 413–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, B.; Karin, M. Obesity, inflammation, and liver cancer. J. Hepatol. 2012, 56, 704–713. [Google Scholar] [CrossRef] [Green Version]
- Zhao, P.; Sun, X.; Chaggan, C.; Liao, Z.; In Wong, K.; He, F.; Singh, S.; Loomba, R.; Karin, M.; Witztum, J.L.; et al. An AMPK-caspase-6 axis controls liver damage in nonalcoholic steatohepatitis. Science 2020, 367, 652–660. [Google Scholar] [CrossRef]
- Vilar-Gomez, E.; Calzadilla-Bertot, L.; Wong, V.W.; Castellanos, M.; Aller-de la Fuente, R.; Eslam, M.; Wong, G.L.; George, J.; Romero-Gomez, M.; Adams, L.A. Type 2 diabetes and metformin use associate with outcomes of patients with nonalcoholic steatohepatitis-related, Child-Pugh A cirrhosis. Clin. Gastroenterol. Hepatol. 2021, 19, 136–145. [Google Scholar] [CrossRef]
- Ahrens, M.; Ammerpohl, O.; von Schönfels, W.; Kolarova, J.; Bens, S.; Itzel, T.; Teufel, A.; Herrmann, A.; Brosch, M.; Hinrichsen, H.; et al. DNA methylation analysis in nonalcoholic fatty liver disease suggests distinct disease-specific and remodeling signatures after bariatric surgery. Cell. Metab. 2013, 18, 296–302. [Google Scholar] [CrossRef] [Green Version]
- Suppli, M.P.; Rigbolt, K.T.G.; Veidal, S.S.; Heebøll, S.; Eriksen, P.L.; Demant, M.; Bagger, J.I.; Nielsen, J.C.; Oró, D.; Thrane, S.W.; et al. Hepatic transcriptome signatures in patients with varying degrees of nonalcoholic fatty liver disease compared with healthy normal-weight individuals. Am. J. Physiol. Gastrointest. Liver Physiol. 2019, 316, G462–G472. [Google Scholar] [CrossRef]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Diehl, A.M.; Brunt, E.M.; Cusi, K.; Charlton, M.; Sanyal, A.J. The diagnosis and management of non-alcoholic fatty liver disease: Practice Guideline by the American Association for the Study of Liver Diseases, American College of Gastroenterology, and the American Gastroenterological Association. Hepatology 2012, 55, 2005–2023. [Google Scholar] [CrossRef]
- Kleiner, D.E.; Brunt, E.M.; Van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.C.; Torbenson, M.S.; Unalp-Arida, A.; et al. Nonalcoholic Steatohepatitis Clinical Research Network. Design and validation of a histological scoring system for nonalcoholic atty liver disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef]
- Rodríguez-Gallego, E.; Riera-Borrull, M.; Hernández-Aguilera, A.; Mariné-Casadó, R.; Rull, A.; Beltrán-Debón, R.; Luciano-Mateo, F.; Menendez, J.A.; Vazquez-Martin, A.; Sirvent, J.J.; et al. Ubiquitous transgenic overexpression of C-C chemokine ligand 2: A model to assess the combined effect of high energy intake and continuous low-grade inflammation. Mediat. Inflamm. 2013, 2013, 953841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luciano-Mateo, F.; Cabré, N.; Fernández-Arroyo, S.; Baiges-Gaya, G.; Hernández-Aguilera, A.; Rodríguez-Tomàs, E.; Mercado-Gómez, M.; Menendez, J.A.; Camps, J.; Joven, J. Chemokine (C-C motif) ligand 2 gene ablation protects low-density lipoprotein and paraoxonase-1 double deficient mice from liver injury, oxidative stress and inflammation. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1555–1566. [Google Scholar] [CrossRef] [PubMed]
- Cuyàs, E.; Fernández-Arroyo, S.; Verdura, S.; García, R.Á.; Stursa, J.; Werner, L.; Blanco-González, E.; Montes-Bayón, M.; Joven, J.; Viollet, B.; et al. Metformin regulates global DNA methylation via mitochondrial one-carbon metabolism. Oncogene 2018, 37, 963–970. [Google Scholar] [CrossRef] [PubMed]
- Akalin, A.; Franke, V.; Vlahoviček, K.; Mason, C.E.; Schübeler, D. Genomation: A toolkit to summarize, annotate and visualize genomic intervals. Bioinformatics 2015, 31, 1127–1129. [Google Scholar] [CrossRef]
- Gu, Z.; Gu, L.; Eils, R.; Schlesner, M.; Brors, B. Circlize implements and enhances circular visualization in R. Bioinformatics 2014, 30, 2811–2812. [Google Scholar] [CrossRef] [Green Version]
- Rosa-Garrido, M.; Chapski, D.J.; Schmitt, A.D.; Kimball, T.H.; Karbassi, E.; Monte, E.; Balderas, E.; Pellegrini, M.; Shih, T.T.; Soehalim, E.; et al. High-resolution mapping of chromatin conformation in cardiac myocytes reveals structural remodeling of the epigenome in heart failure. Circulation 2017, 136, 1613–1625. [Google Scholar] [CrossRef]
- Aranda, N.; Viteri, F.E.; Montserrat, C.; Arija, V. Effects of C282Y, H63D, and S65C HFE gene mutations, diet, and life-style factors on iron status in a general Mediterranean population from Tarragona, Spain. Ann. Hematol. 2010, 89, 767–773. [Google Scholar] [CrossRef] [Green Version]
- Mechanick, J.I.; Apovian, C.; Brethauer, S.; Garvey, W.T.; Joffe, A.M.; Kim, J. Clinical practice guidelines for the perioperative nutrition, metabolic, and nonsurgical support of patients undergoing bariatric procedures. Surg. Obes. Relat. Dis. 2020, 16, 175–247. [Google Scholar] [CrossRef]
- Vives, M.; Molina, A.; Danús, M.; Rebenaque, E.; Blanco, S.; París, M. Analysis of gastric physiology after laparoscopic sleeve gastrectomy (LSG) with or without antral preservation in relation to metabolic response: A randomised study. Obes. Surg. 2017, 27, 2836–2844. [Google Scholar] [CrossRef]
- Zhang, W.; Sargis, R.M.; Volden, P.A.; Carmean, C.M.; Sun, X.J.; Brady, M.J. PCB 126 and other dioxin-like PCBs specifically suppress hepatic PEPCK expression via the aryl hydrocarbon receptor. PLoS ONE 2012, 7, e37103. [Google Scholar] [CrossRef] [Green Version]
- Qiao, A.; Jin, X.; Pang, J.; Moskophidis, D.; Mivechi, N.F. The transcriptional regulator of the chaperone response HSF1 controls hepatic bioenergetics and protein homeostasis. J. Cell Biol. 2017, 216, 723–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cabré, N.; Luciano-Mateo, F.; Chapski, D.J.; Baiges-Gaya, G.; Fernández-Arroyo, S.; Hernández-Aguilera, A.; Castañé, H.; Rodríguez-Tomàs, E.; París, M.; Sabench, F.; et al. Laparoscopic Sleeve Gastrectomy in Patients with Severe Obesity Restores Adaptive Responses Leading to Nonalcoholic Steatohepatitis. Int. J. Mol. Sci. 2022, 23, 7830. https://doi.org/10.3390/ijms23147830
Cabré N, Luciano-Mateo F, Chapski DJ, Baiges-Gaya G, Fernández-Arroyo S, Hernández-Aguilera A, Castañé H, Rodríguez-Tomàs E, París M, Sabench F, et al. Laparoscopic Sleeve Gastrectomy in Patients with Severe Obesity Restores Adaptive Responses Leading to Nonalcoholic Steatohepatitis. International Journal of Molecular Sciences. 2022; 23(14):7830. https://doi.org/10.3390/ijms23147830
Chicago/Turabian StyleCabré, Noemí, Fedra Luciano-Mateo, Douglas J. Chapski, Gerard Baiges-Gaya, Salvador Fernández-Arroyo, Anna Hernández-Aguilera, Helena Castañé, Elisabet Rodríguez-Tomàs, Marta París, Fàtima Sabench, and et al. 2022. "Laparoscopic Sleeve Gastrectomy in Patients with Severe Obesity Restores Adaptive Responses Leading to Nonalcoholic Steatohepatitis" International Journal of Molecular Sciences 23, no. 14: 7830. https://doi.org/10.3390/ijms23147830