
Hypoxia-Reoxygenation Impairs Autophagy-Lysosomal Machinery in Primary Human Trophoblasts Mimicking Placental Pathology of Early-Onset Preeclampsia

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
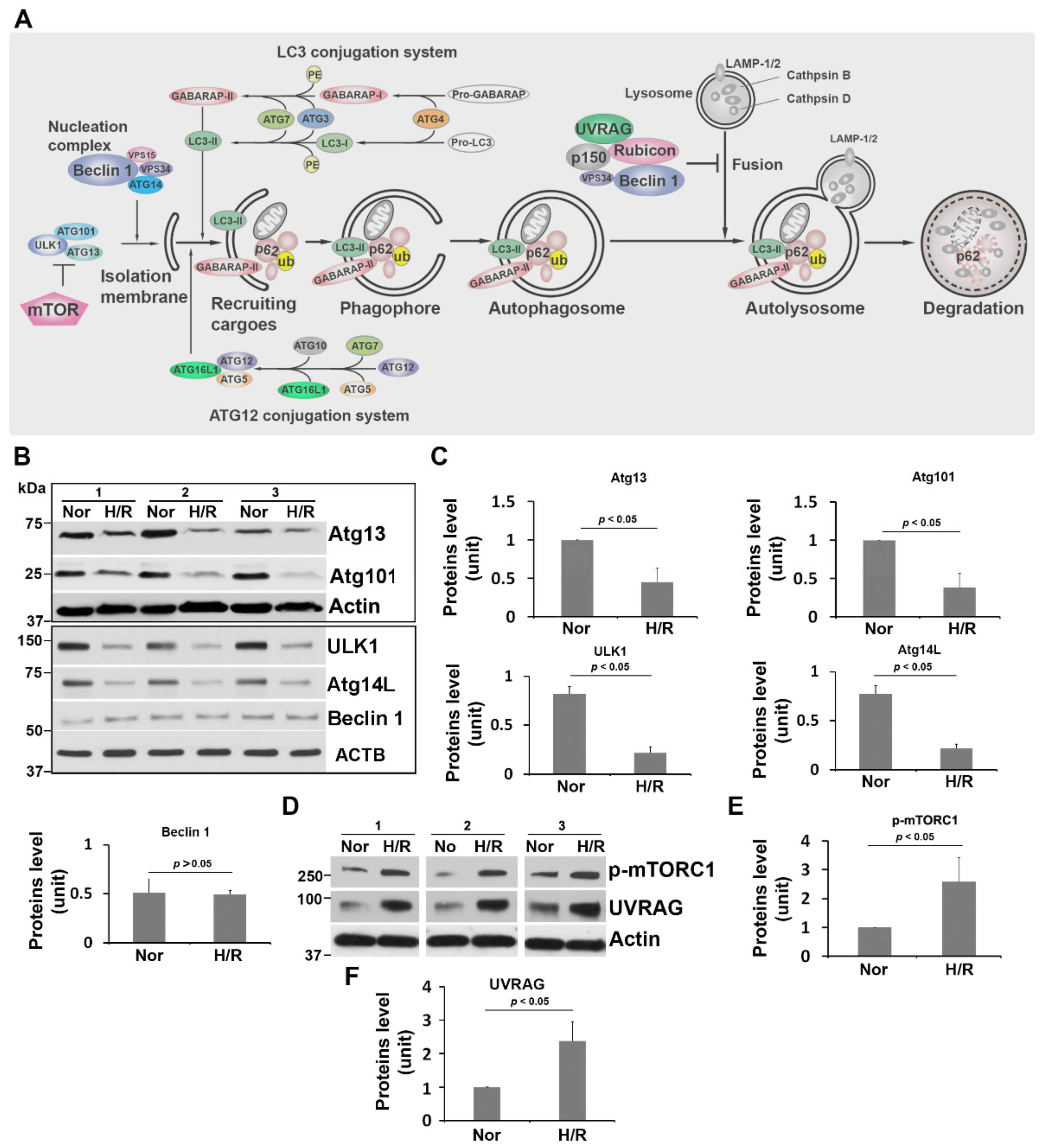
2.1. H/R Exposure Dysregulates the unc-51-like Autophagy Activating Kinase (ULK1) Regulatory Complex and the Class III Phosphatidylinositol 3-Kinase (PI3K-III) Complex in PHTs
2.2. H/R Treatment Upregulates Phosphorylated mTORC1 and UV Radiation Resistance Associated Gene Product (UVRAG) in PHTs
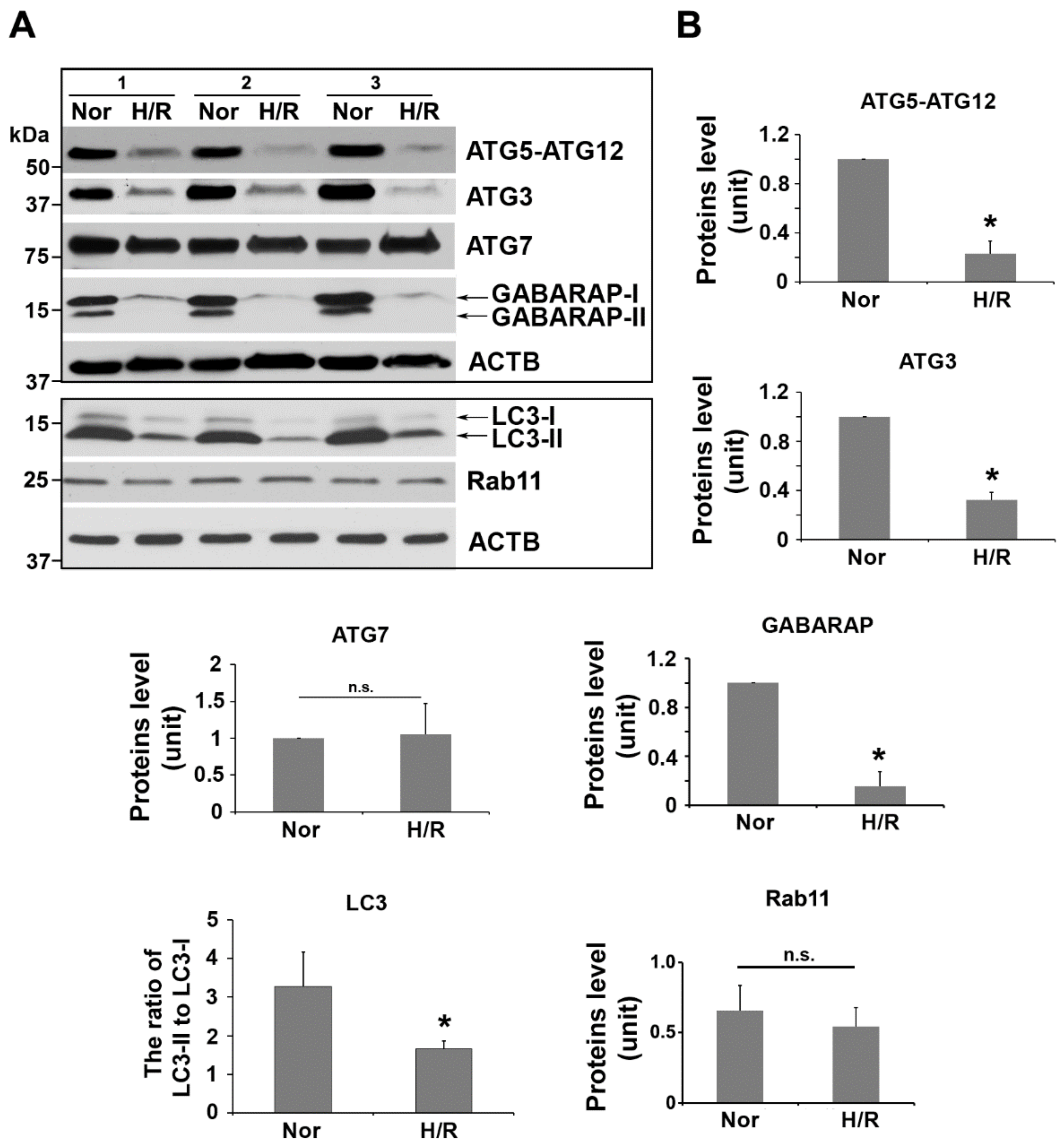
2.3. H/R Exposure Significantly Alters the Content of Several Key Autophagy Proteins, including Atg12-Atg5 Conjugation Complex, LC3, and GABARAP
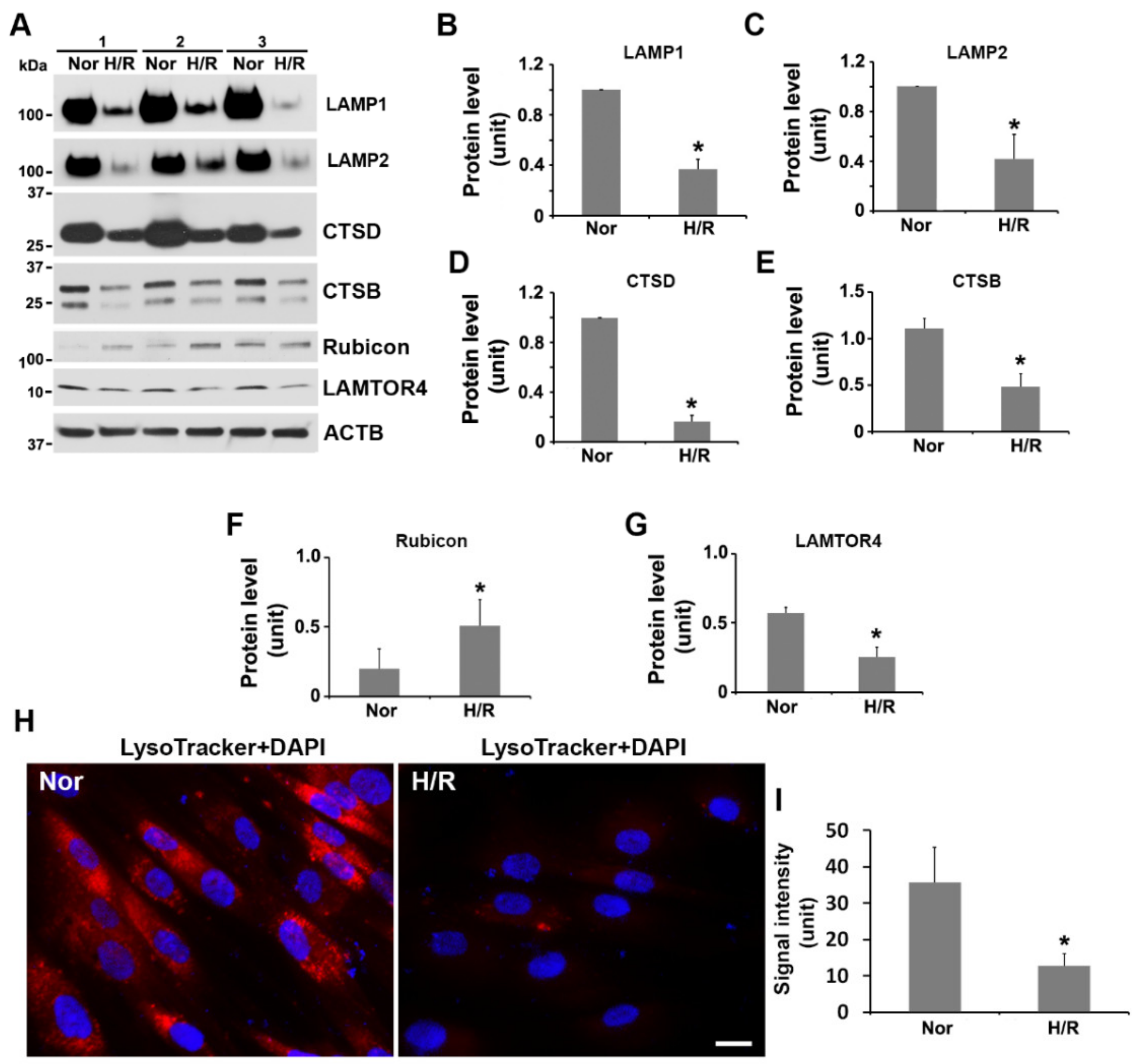
2.4. Exposure to H/R Decreases the Expression of LAMP1, LAMP2, Cathepsin B and D, and LAMTOR4, and Increases the Abundance of Rubicon and Inhibits Lysosomal Activity in PHTs
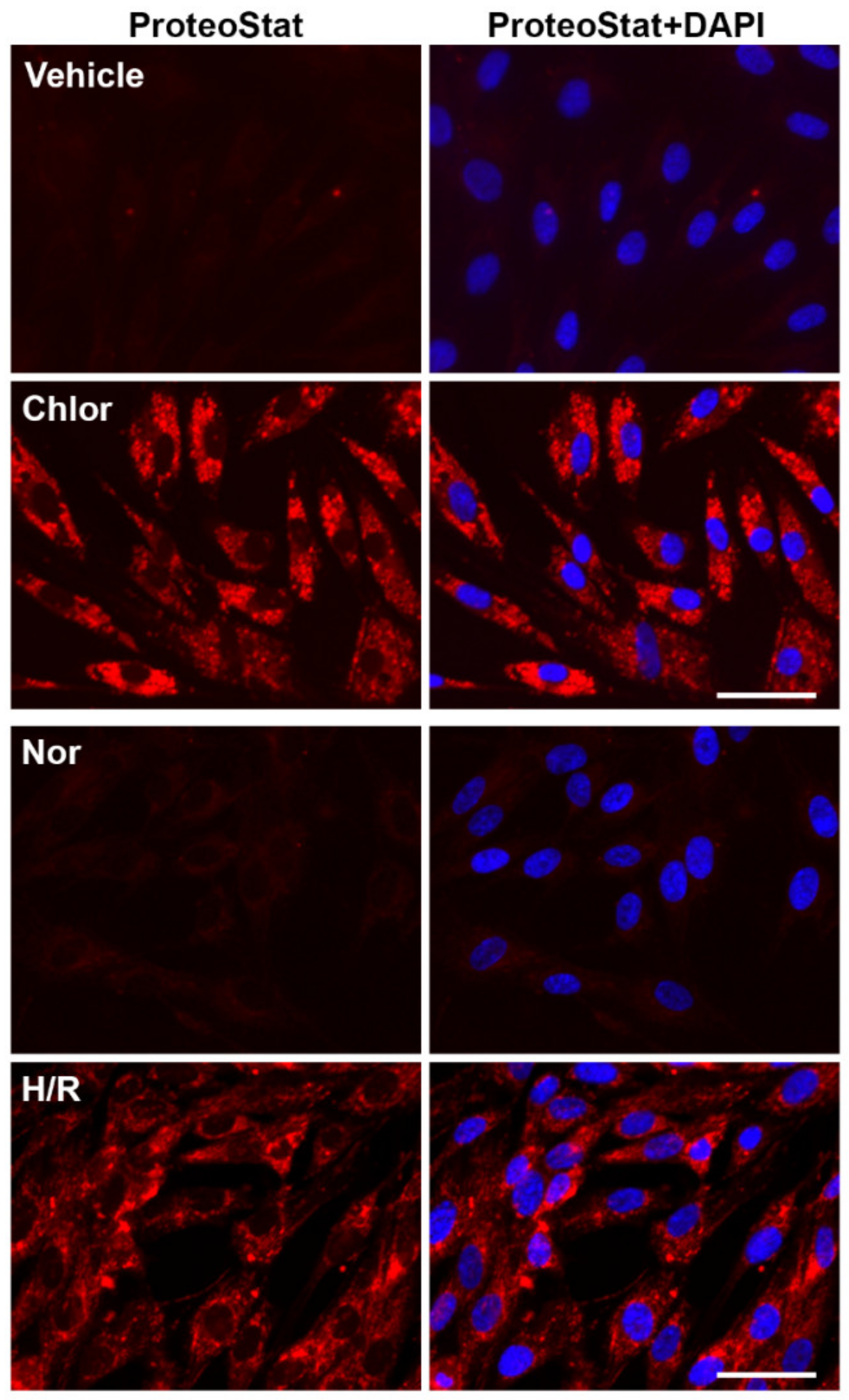
2.5. Chloroquine Treatment and H/R Exposure Induce the Accumulation of Protein Aggregates in PHTs
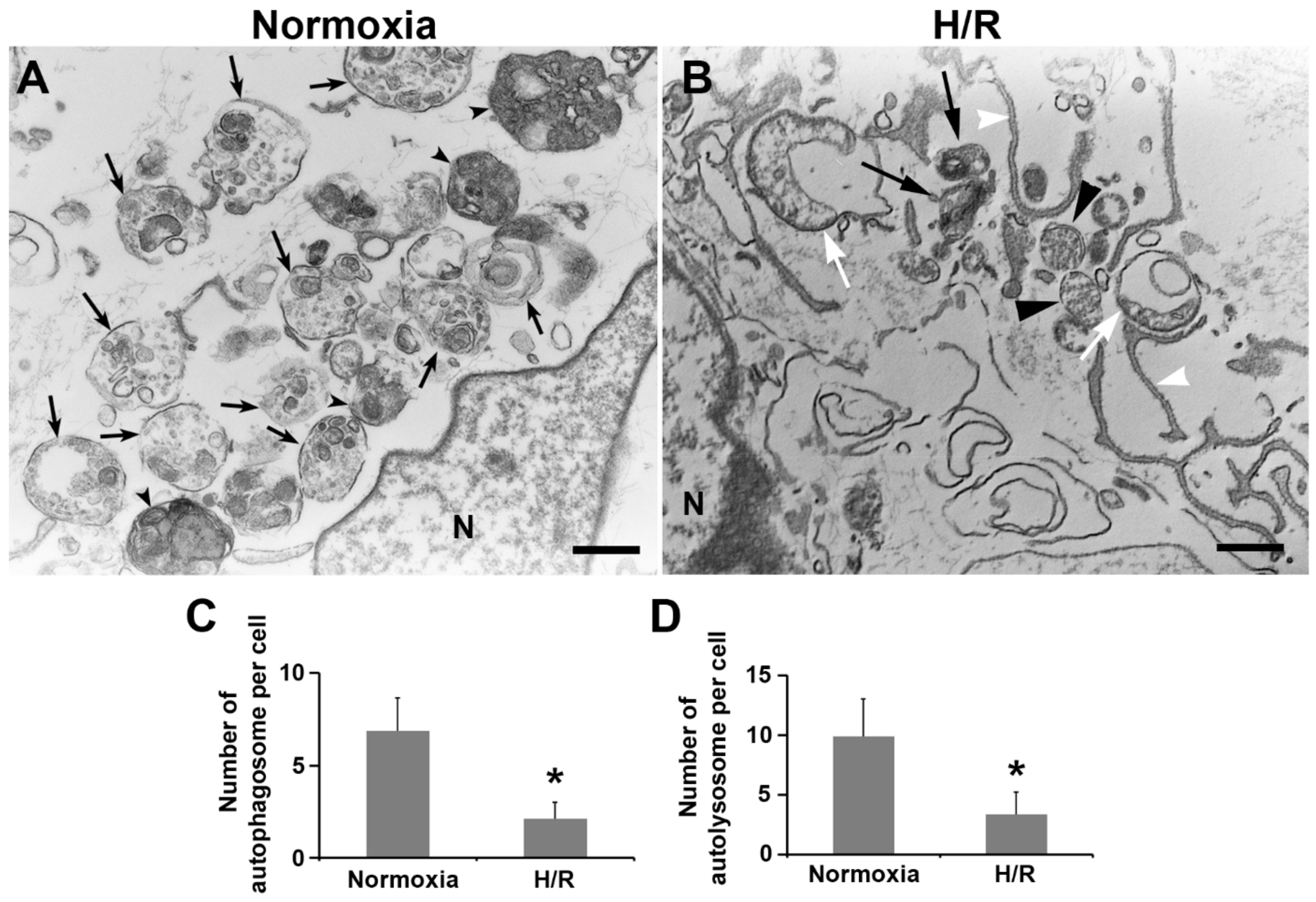
2.6. Ultrastructural Analysis of Autophagy Morphology in PHTs Exposed to H/R Condition
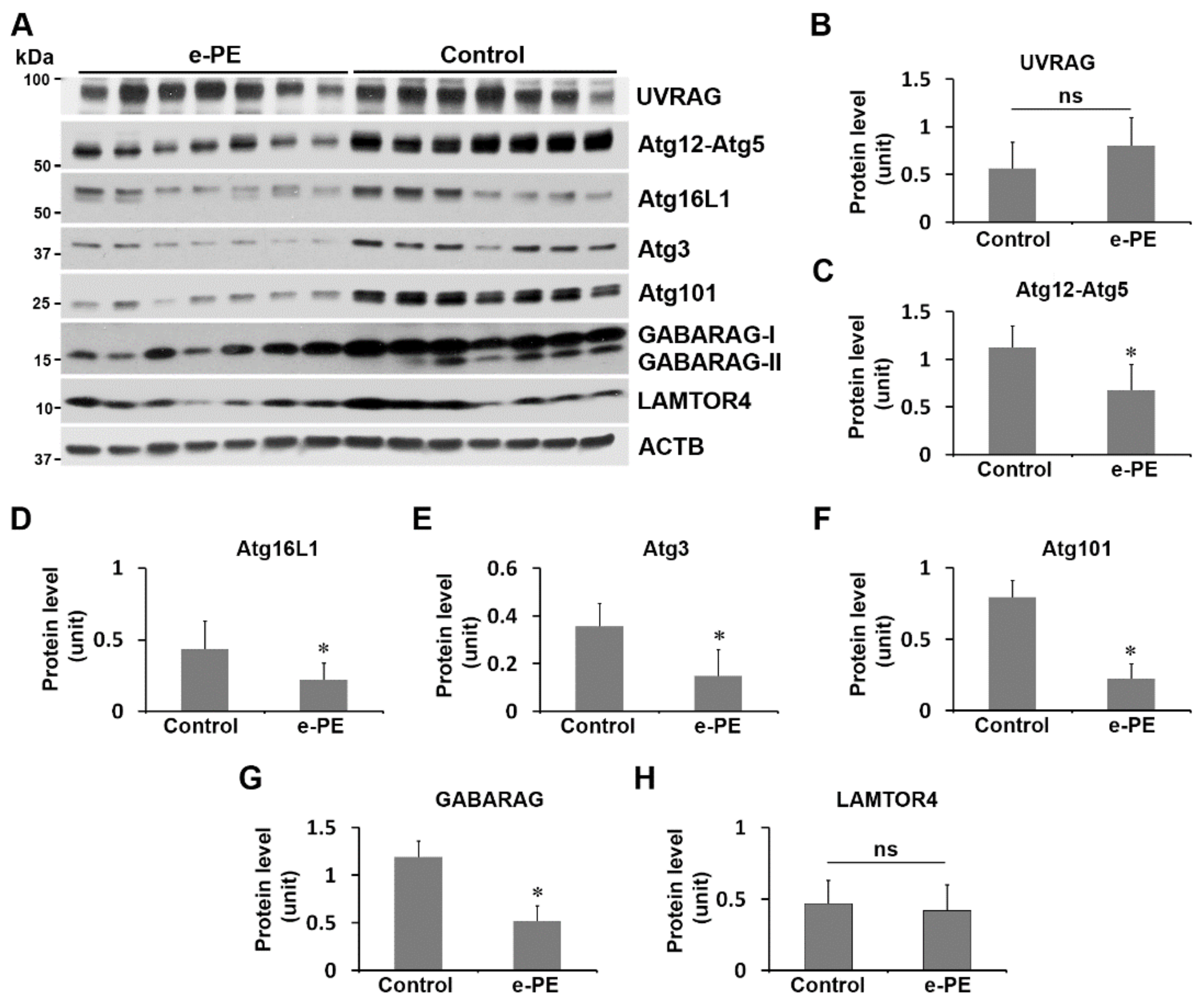
2.7. Expression of Atg101, Atg3, Atg16L1, Atg12–Atg5, and GABARAP Was Reduced in the Placenta from e-PE Deliveries
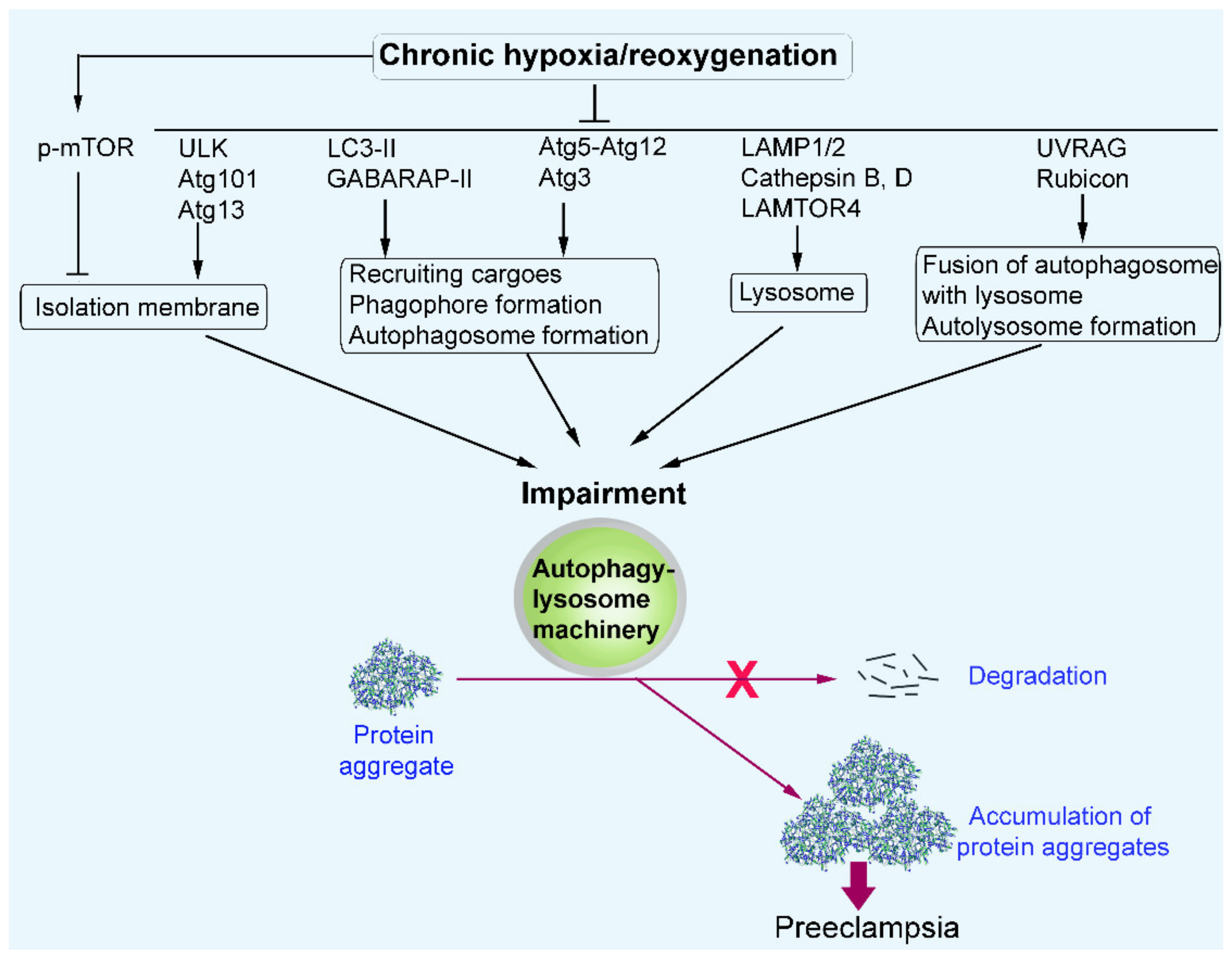
3. Discussion
4. Materials and Methods
4.1. Antibodies and Reagents
4.2. Human Subjects
4.3. Cell Culture
4.4. Hypoxia–Reoxygenation Treatment
4.5. Immunoblotting
4.6. Detection of Aggregated Proteins
4.7. LysoTracker Staining
4.8. Transmission Electron Microscopy
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yoshimori, T. Autophagy: Paying Charon’s toll. Cell 2007, 128, 833–836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuervo, A.M.; Bergamini, E.; Brunk, U.T.; Dröge, W.; Ffrench, M.; Terman, A. Autophagy and aging: The importance of maintaining “clean” cells. Autophagy 2005, 1, 131–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doherty, J.; Baehrecke, E.H. Life, death and autophagy. Nat. Cell. Biol. 2018, 20, 1110–1117. [Google Scholar] [CrossRef]
- Shimada, Y.; Klionsky, D.J. Autophagy contributes to lysosomal storage disorders. Autophagy 2012, 8, 715–716. [Google Scholar] [CrossRef] [Green Version]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [Green Version]
- Delorme-Axford, E.; Bayer, A.; Sadovsky, Y.; Coyne, C.B. Autophagy as a mechanism of antiviral defense at the maternal-fetal interface. Autophagy 2013, 9, 2173–2174. [Google Scholar] [CrossRef]
- Delorme-Axford, E.; Donker, R.B.; Mouillet, J.F.; Chu, T.; Bayer, A.; Ouyang, Y.; Wang, T.; Stolz, D.B.; Sarkar, S.N.; Morelli, A.E.; et al. Human placental trophoblasts confer viral resistance to recipient cells. Proc. Natl. Acad. Sci. USA 2013, 110, 12048–12053. [Google Scholar] [CrossRef] [Green Version]
- Cao, B.; Macones, M.; Mysorekar, I.U. ATG16L1 governs placental infection risk and preterm birth in mice and women. JCI Insight 2016, 1, e86654. [Google Scholar] [CrossRef] [Green Version]
- Hung, T.H.; Hsieh, T.T.; Chen, S.F.; Li, M.J.; Yeh, Y.L. Autophagy in the human placenta throughout gestation. PLoS ONE 2013, 8, e83475. [Google Scholar] [CrossRef]
- Nakashima, A.; Aoki, A.; Kusabiraki, T.; Cheng, S.B.; Sharma, S.; Saito, S. Autophagy regulation in preeclampsia: Pros and cons. J. Reprod. Immunol. 2017, 123, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, A.; Cheng, S.B.; Ikawa, M.; Yoshimori, T.; Huber, W.J.; Menon, R.; Huang, Z.; Fierce, J.; Padbury, J.F.; Sadovsky, Y.; et al. Evidence for lysosomal biogenesis proteome defect and impaired autophagy in preeclampsia. Autophagy 2020, 16, 1771–1785. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S. Autophagy-based diagnosis of pregnancy hypertension and pre-eclampsia. Am. J. Pathol. 2018, 188, 2457–2460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakashima, A.; Tsuda, S.; Kusabiraki, T.; Aoki, A.; Ushijima, A.; Shima, T.; Cheng, S.B.; Sharma, S.; Saito, S. Current Understanding of Autophagy in Pregnancy. Int. J. Mol. Sci. 2019, 20, 2342. [Google Scholar] [CrossRef] [Green Version]
- Nakashima, A.; Aoki, A.; Kusabiraki, T.; Shima, T.; Yoshino, O.; Cheng, S.B.; Sharma, S.; Saito, S. Role of autophagy in oocytogenesis, embryogenesis, implantation, and pathophysiology of pre-eclampsia. J. Obstet. Gynaecol. Res. 2017, 43, 633–643. [Google Scholar] [CrossRef] [Green Version]
- Saito, S.; Nakashima, A. A review of the mechanism for poor placentation in early onset preeclampsia: The role of autophagy in trophoblast invasion and vascular invasion. J. Reprod. Immunol. 2014, 101–102, 80–88. [Google Scholar] [CrossRef]
- Nakashima, A.; Yamanaka-Tatematsu, M.; Fujita, N.; Koizumi, K.; Shima, T.; Yoshida, T.; Nikaido, T.; Okamoto, A.; Yoshimori, T.; Saito, S. Impaired autophagy by soluble endoglin, under physiological hypoxia in early pregnant period, is involved in poor placentation in preeclampsia. Autophagy 2013, 9, 303–316. [Google Scholar] [CrossRef] [Green Version]
- Cheng, S.B.; Nakashima, A.; Huber, W.J.; Davis, S.; Banerjee, S.; Huang, Z.; Saito, S.; Sadovsky, Y.; Sharma, S. Pyroptosis is a critical inflammatory pathway in the placenta from early onset preeclampsia and in human trophoblasts exposed to hypoxia and endoplasmic reticulum stressors. Cell Death Dis. 2019, 10, 927. [Google Scholar] [CrossRef]
- Cheng, S.B.; Sharma, S. Preeclampsia and health risks later in life: An immunological link. Semin. Immunopathol. 2016, 38, 699–708. [Google Scholar] [CrossRef]
- Von Dadelszen, P.; Magee, L.A.; Roberts, J.M. Subclassification of preeclampsia. Hypertens Pregnancy 2003, 22, 143–148. [Google Scholar] [CrossRef]
- Sibai, B.; Dekker, G.; Kupferminc, M. Pre-eclampsia. Lancet 2005, 365, 785–789. [Google Scholar] [CrossRef]
- Roberts, J.M.; Redman, C.W. Pre-eclampsia: More than pregnancy-induced hypertension. Lancet 1993, 341, 1447–1451. [Google Scholar] [CrossRef]
- Redman, C.W.; Sargent, I.L. Latest advances in understanding preeclampsia. Science 2005, 308, 1592–1594. [Google Scholar] [CrossRef] [PubMed]
- Phipps, E.A.; Thadhani, R.; Benzing, T.; Karumanchi, S.A. Pre-eclampsia: Pathogenesis, novel diagnostics and therapies. Nat. Rev. Nephrol. 2019, 15, 275–289. [Google Scholar] [CrossRef] [PubMed]
- Staff, A.C.; Benton, S.J.; von Dadelszen, P.; Roberts, J.M.; Taylor, R.N.; Powers, R.W.; Charnock-Jones, D.S.; Redman, C.W. Redefining preeclampsia using placenta-derived biomarkers. Hypertension 2013, 61, 932–942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steegers, E.A.; von Dadelszen, P.; Duvekot, J.J.; Pijnenborg, R. Pre-eclampsia. Lancet 2010, 376, 631–644. [Google Scholar] [CrossRef]
- Vikse, B.E.; Irgens, L.M.; Leivestad, T.; Skjærven, R.; Iversen, B.M. Preeclampsia and the risk of end-stage renal disease. N. Engl. J. Med. 2008, 359, 800–809. [Google Scholar] [CrossRef]
- Saade, G.R. Pregnancy as a window to future health. Obstet. Gynecol. 2009, 114, 958–960. [Google Scholar] [CrossRef]
- Bellamy, L.; Casas, J.P.; Hingorani, A.D.; Williams, D.J. Preeclampsia and risk of cardiovascular disease and cancer in later life: A systemic review and meta-analysis. BMJ 2007, 335, 974–977. [Google Scholar] [CrossRef] [Green Version]
- Smith, G.C.; Pell, J.P.; Walsh, D. Pregnancy complications and maternal risk of ischaemic heart disease: A retrospective cohort study of 129,290 births. Lancet 2001, 357, 2002–2006. [Google Scholar] [CrossRef]
- Zhou, Y.; Gormley, M.J.; Hunkapiller, N.M.; Kapidzic, M.; Stolyarov, Y.; Feng, V.; Nishida, M.; Drake, P.M.; Bianco, K.; Wang, F.; et al. Reversal of gene dysregulation in cultured cytotrophoblasts reveals possible causes of preeclampsia. J. Clin. Investig. 2013, 123, 2862–2872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, J.M.; Hubel, C.A. Is oxidative stress the link in the two-stage model of preeclampsia? Lancet 1999, 354, 788–789. [Google Scholar] [CrossRef]
- Redman, C.W.; Sacks, G.P.; Sargent, I.L. Preeclampsia: An excessive maternal inflammatory response to pregnancy. Am. J. Obstet. Gynecol. 1999, 80, 499–506. [Google Scholar] [CrossRef]
- Burton, G.J.; Yung, H.W. Endoplasmic reticulum stress in the pathogenesis of early-onset pre-eclampsia. Pregnancy Hypertens. 2011, 1, 72–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, R.J.; Maynard, S.E.; Qian, C.; Lim, K.H.; England, L.J.; Yu, K.F.; Schisterman, E.F.; Thadhani, R.; Sachs, B.P.; Epstein, F.H.; et al. Circulating angiogenic factors and the risk of preeclampsia. N. Engl. J. Med. 2004, 350, 672–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yung, H.W.; Atkinson, D.; Campion-Smith, T.; Olovsson, M.; Charnock-Jones, D.S.; Burton, G.J. Differential activation of placental unfolded protein response pathways implies heterogeneity in causation of early- and late-onset pre-eclampsia. J. Pathol. 2014, 234, 262–276. [Google Scholar] [CrossRef] [Green Version]
- Soleymanlou, N.; Jurisica, I.; Nevo, O.; Ietta, F.; Zhang, X.; Zamudio, S.; Post, M.; Caniggia, I. Molecular evidence of placental hypoxia in preeclampsia. J. Clin. Endocrinol. Metab. 2005, 90, 4299–4308. [Google Scholar] [CrossRef] [Green Version]
- Rolfo, A.; Many, A.; Racano, A.; Tal, R.; Tagliaferro, A.; Ietta, F.; Wang, J.; Post, M.; Caniggia, I. Abnormalities in oxygen sensing define early and late onset preeclampsia as distinct pathologies. PLoS ONE 2010, 5, e13288. [Google Scholar] [CrossRef] [Green Version]
- Lai, Z.; Kalkunte, S.; Sharma, S. A critical role of interleukin-10 in modulating hypoxia-induced preeclampsia-like disease in mice. Hypertension 2011, 57, 505–514. [Google Scholar] [CrossRef] [Green Version]
- Hung, T.H.; Skepper, J.N.; Charnock-Jones, D.S.; Burton, G.J. Hypoxia-reoxygenation: A potent inducer of apoptotic changes in the human placenta and possible etiological factor in preeclampsia. Circ. Res. 2002, 90, 1274–1281. [Google Scholar] [CrossRef]
- Cheng, S.B.; Nakashima, A.; Sharma, S. Understanding Pre-Eclampsia Using Alzheimer’s Etiology: An Intriguing Viewpoint. Am. J. Reprod. Immunol. 2016, 75, 372–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, M.; Cheng, S.B.; Chen, Q.; DeSousa, J.; Stone, P.R.; James, J.L.; Chamley, L.W.; Sharma, S. Aggregated transthyretin is specifically packaged into placental nano-vesicles in preeclampsia. Sci. Rep. 2017, 7, 6694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buhimschi, I.A.; Nayeri, U.A.; Zhao, G.; Shook, L.L.; Pensalfini, A.; Funai, E.F.; Bernstein, I.M. Protein misfolding, congophilia, oligomerization, and defective amyloid processing in preeclampsia. Sci. Transl. Med. 2014, 6, 245ra92. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.Y.; Choi, S.J.; Kim, K.H.; Cho, E.Y.; Kim, J.H.; Roh, C.R. Autophagy-related proteins, LC3 and Beclin-1, in placentas from pregnancies complicated by preeclampsia. Reprod. Sci. 2008, 15, 912–920. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.Y.; Choi, S.J.; Kim, K.H.; Cho, E.Y.; Kim, J.H.; Roh, C.R. Excessive autophagy induces the failure of trophoblast invasion and vasculature: Possible relevance to the pathogenesis of preeclampsia. J. Hypertens. 2015, 33, 106–117. [Google Scholar]
- Melland-Smith, M.; Ermini, L.; Chauvin, S.; Craig-Barnes, H.; Tagliaferro, A.; Todros, T.; Post, M.; Caniggia, I. Disruption of sphingolipid metabolism augments ceramide-induced autophagy in preeclampsia. Autophagy 2015, 11, 653–669. [Google Scholar] [CrossRef]
- Hutabarat, M.; Wibowo, N.; Huppertz, B. The trophoblast survival capacity in preeclampsia. PLoS ONE 2017, 12, e0186909. [Google Scholar] [CrossRef]
- Akcora Yildiz, D.; Irtegun Kandemir, S.; Agacayak, E.; Deveci, E. Evaluation of protein levels of autophagy markers (Beclin 1 and SQSTM1/p62) and phosphorylation of cyclin E in the placenta of women with preeclampsia. Cell Mol. Biol. 2017, 63, 51–55. [Google Scholar] [CrossRef]
- Akaishi, R.; Yamada, T.; Nakabayashi, K. Autophagy in the placenta of women with hypertensive disorders in pregnancy. Placenta 2014, 35, 974–980. [Google Scholar] [CrossRef] [Green Version]
- Ozsoy, A.Z.; Cayli, S.; Sahin, C.; Ocakli, S.; Sanci, T.O.; Ilhan, D.B. Altered expression of p97/Valosin containing protein and impaired autophagy in preeclamptic human placenta. Placenta 2018, 67, 45–53. [Google Scholar] [CrossRef]
- Chen, B.; Longtine, M.S.; Nelson, D.M. Hypoxia induces autophagy in primary human trophoblasts. Endocrinology 2012, 153, 4946–4954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Hu, X.; Gao, G.; Wang, Y.; Chen, P.; Ye, Y. Autophagy protects against oxidized low density lipoprotein-mediated inflammation associated with preeclampsia. Placenta 2016, 48, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, L.; Bao, J.; Sha, X.; Cui, L.; Huang, Q.; Gu, C.; Li, X.; Liu, H. Persistent hypoxia induced autophagy leading to invasiveness of trophoblasts in placenta accreta. J. Matern. Fetal Neonatal. Med. 2019, 34, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.B.; Huang, Z.; Banerjee, S.; Jash, S.; Buxbaum, J.N.; Sharma, S. Evidence from human placenta, ER-stressed trophoblasts and transgenic mice links transthyretin proteinopathy to preeclampsia. Hypertension 2022, in press. [Google Scholar] [CrossRef]
- Wong, P.M.; Puente, C.; Ganley, I.G.; Jiang, X. The ULK1 complex: Sensing nutrient signals for autophagy activation. Autophagy 2013, 9, 124–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKnight, N.C.; Zhenyu, Y. Beclin 1, an Essential Component and Master Regulator of PI3K-III in Health and Disease. Curr. Pathobiol. Rep. 2013, 1, 231–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itakura, E.; Kishi, C.; Inoue, K.; Mizushima, N. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol. Biol. Cell. 2008, 9, 5360–5372. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.M.; Jung, C.H.; Seo, M.; Kim, E.K.; Park, J.M.; Bae, S.S.; Kim, D.H. mTORC1 phosphorylates UVRAG to negatively regulate autophagosome and endosome maturation. Mol. Cell. 2015, 57, 207–218. [Google Scholar] [CrossRef] [Green Version]
- Matsushita, M.; Suzuki, N.N.; Obara, K.; Fujioka, Y.; Ohsumi, Y.; Inagaki, F. Structure of Atg5.Atg16, a complex essential for autophagy. J. Biol. Chem. 2007, 282, 6763–6772. [Google Scholar] [CrossRef] [Green Version]
- Walczak, M.; Martens, S. Dissecting the role of the Atg12-Atg5-Atg16 complex during autophagosome formation. Autophagy 2013, 9, 424–425. [Google Scholar] [CrossRef] [Green Version]
- Tanida, I.; Mizushima, N.; Kiyooka, M.; Ohsumi, M.; Ueno, T.; Ohsumi, Y.; Kominami, E. Apg7p/Cvt2p: A novel protein-activating enzyme essential for autophagy. Mol. Biol. Cell. 1999, 10, 1367–1379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shintani, T.; Mizushima, N.; Ogawa, Y.; Matsuura, A.; Noda, T.; Ohsumi, Y. Apg10p, a novel protein-conjugating enzyme essential for autophagy in yeast. EMBO J. 1999, 18, 5234–5524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabeya, Y.; Mizushima, N.; Yamamoto, A.; Oshitani-Okamoto, S.; Ohsumi, Y.; Yoshimori, T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J. Cell Sci. 2004, 117, 2805–2812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kouroku, Y.; Fujita, E.; Tanida, I.; Ueno, T.; Isoai, A.; Kumagai, H.; Ogawa, S.; Kaufman, R.J.; Kominami, E.; Momoi, T. ER stress (PERK/eIF2alpha phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ. 2007, 14, 230–239. [Google Scholar] [CrossRef] [Green Version]
- Tanida, I.; Ueno, T.; Kominami, E. LC3 conjugation system in mammalian autophagy. Int. J. Biochem. Cell Biol. 2004, 36, 2503–2518. [Google Scholar] [CrossRef]
- Puri, C.; Vicinanza, M.; Ashkenazi, A.; Gratian, M.J.; Zhang, Q.; Bento, C.F.; Renna, M.; Menzies, F.M.; Rubinsztein, D.C. The RAB11A-Positive Compartment Is a Primary Platform for Autophagosome Assembly Mediated by WIPI2 Recognition of PI3P-RAB11A. Dev. Cell. 2018, 45, 114–131.e8. [Google Scholar] [CrossRef] [Green Version]
- Wong, S.W.; Sil, P.; Martinez, J. Rubicon: LC3-associated phagocytosis and beyond. FEBS J. 2018, 285, 1379–1388. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, S.; Hikita, H.; Tatsumi, T.; Sakamori, R.; Nozaki, Y.; Sakane, S.; Shiode, Y.; Nakabori, T.; Saito, Y.; Hiramatsu, N.; et al. Rubicon inhibits autophagy and accelerates hepatocyte apoptosis and lipid accumulation in nonalcoholic fatty liver disease in mice. Hepatology 2016, 64, 1994–2014. [Google Scholar] [CrossRef]
- Nakamura, S.; Oba, M.; Suzuki, M.; Takahashi, A.; Yamamuro, T.; Fujiwara, M.; Ikenaka, K.; Minami, S.; Tabata, N.; Yamamoto, K.; et al. Suppression of autophagic activity by Rubicon is a signature of aging. Nat. Commun. 2019, 10, 847. [Google Scholar] [CrossRef]
- Wu, S.; He, Y.; Qiu, X.; Yang, W.; Liu, W.; Li, X.; Li, Y.; Shen, H.M.; Wang, R.; Yue, Z.; et al. Targeting the potent Beclin 1-UVRAG coiled-coil interaction with designed peptides enhances autophagy and endolysosomal trafficking. Proc. Natl. Acad. Sci. USA 2018, 15, E5669–E5678. [Google Scholar] [CrossRef] [Green Version]
- Liang, C.; Lee, J.S.; Inn, K.S.; Gack, M.U.; Li, Q.; Roberts, E.A.; Vergne, I.; Deretic, V.; Feng, P.; Akazawa, C.; et al. Beclin1-binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nat. Cell Biol. 2018, 10, 776–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsunaga, K.; Saitoh, T.; Tabata, K.; Omori, H.; Satoh, T.; Kurotori, N.; Maejima, I.; Shirahama-Noda, K.; Ichimura, T.; Isobe, T.; et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat. Cell Biol. 2009, 11, 385–396. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Wang, Q.J.; Li, X.; Yan, Y.; Backer, J.M.; Chait, B.T.; Heintz, N.; Yue, Z. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nat. Cell Biol. 2009, 11, 468–476. [Google Scholar] [CrossRef] [PubMed]
- Matsunaga, K.; Noda, T.; Yoshimori, T. Binding Rubicon to cross the Rubicon. Autophagy 2009, 5, 876–877. [Google Scholar] [CrossRef] [Green Version]
- Shen, K.; Sidik, H.; Talbot, W.S. The Rag-Ragulator Complex Regulates Lysosome Function and Phagocytic Flux in Microglia. Cell Rep. 2016, 14, 547–559. [Google Scholar] [CrossRef] [Green Version]
- Cheng, S.; Banerjee, S.; Daiello, L.A.; Nakashima, A.; Jash, S.; Huang, Z.; Drake, J.D.; Ernerudh, J.; Berg, G.; Padbury, J.; et al. Novel blood test for early biomarkers of preeclampsia and Alzheimer’s disease. Sci. Rep. 2021, 11, 15934. [Google Scholar] [CrossRef]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef]
- Banerjee, S.; Huang, Z.; Wang, Z.; Nakashima, A.; Saito, S.; Sharma, S.; Cheng, S. Etiological Value of Sterile Inflammation in Preeclampsia: Is It a Non-Infectious Pregnancy Complication? Front. Cell Infect. Microbiol. 2021, 11, 694298. [Google Scholar] [CrossRef]
- Pursiheimo, J.P.; Rantanen, K.; Heikkinen, P.T.; Johansen, T.; Jaakkola, P.M. Hypoxia-activated autophagy accelerates degradation of SQSTM1/p62. Oncogene 2009, 28, 334–344. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, S.; Huang, Z.; Jash, S.; Wu, K.; Saito, S.; Nakashima, A.; Sharma, S. Hypoxia-Reoxygenation Impairs Autophagy-Lysosomal Machinery in Primary Human Trophoblasts Mimicking Placental Pathology of Early-Onset Preeclampsia. Int. J. Mol. Sci. 2022, 23, 5644. https://doi.org/10.3390/ijms23105644
Cheng S, Huang Z, Jash S, Wu K, Saito S, Nakashima A, Sharma S. Hypoxia-Reoxygenation Impairs Autophagy-Lysosomal Machinery in Primary Human Trophoblasts Mimicking Placental Pathology of Early-Onset Preeclampsia. International Journal of Molecular Sciences. 2022; 23(10):5644. https://doi.org/10.3390/ijms23105644
Chicago/Turabian StyleCheng, Shibin, Zheping Huang, Sukanta Jash, Kathleen Wu, Shigeru Saito, Akitoshi Nakashima, and Surendra Sharma. 2022. "Hypoxia-Reoxygenation Impairs Autophagy-Lysosomal Machinery in Primary Human Trophoblasts Mimicking Placental Pathology of Early-Onset Preeclampsia" International Journal of Molecular Sciences 23, no. 10: 5644. https://doi.org/10.3390/ijms23105644