
The Thousand Polish Genomes—A Database of Polish Variant Allele Frequencies

 , , , , , , , , ,
, , , , , , , , ,  , , , , , , , and add
Show full author list
, , , , , , , and add
Show full author list
Abstract
:1. Introduction
2. Results
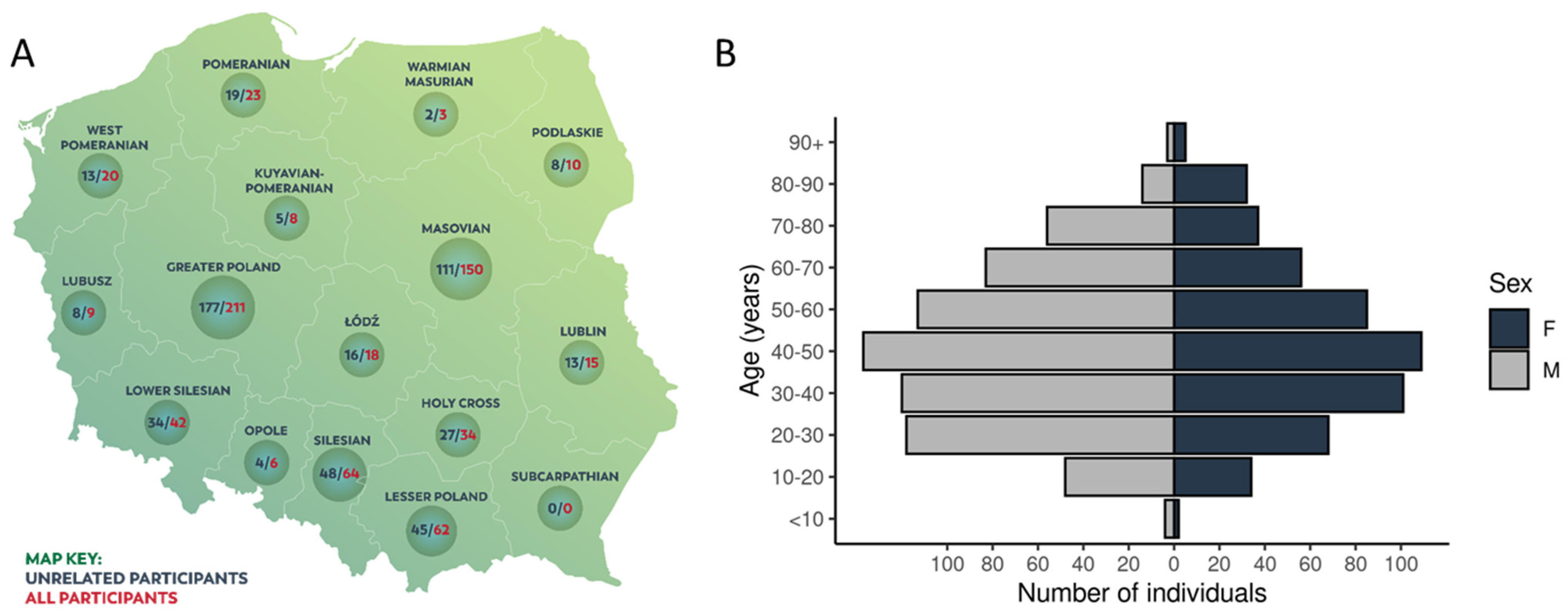
2.1. Characteristics of the Cohort
2.2. Genetic Variation in the Polish Population
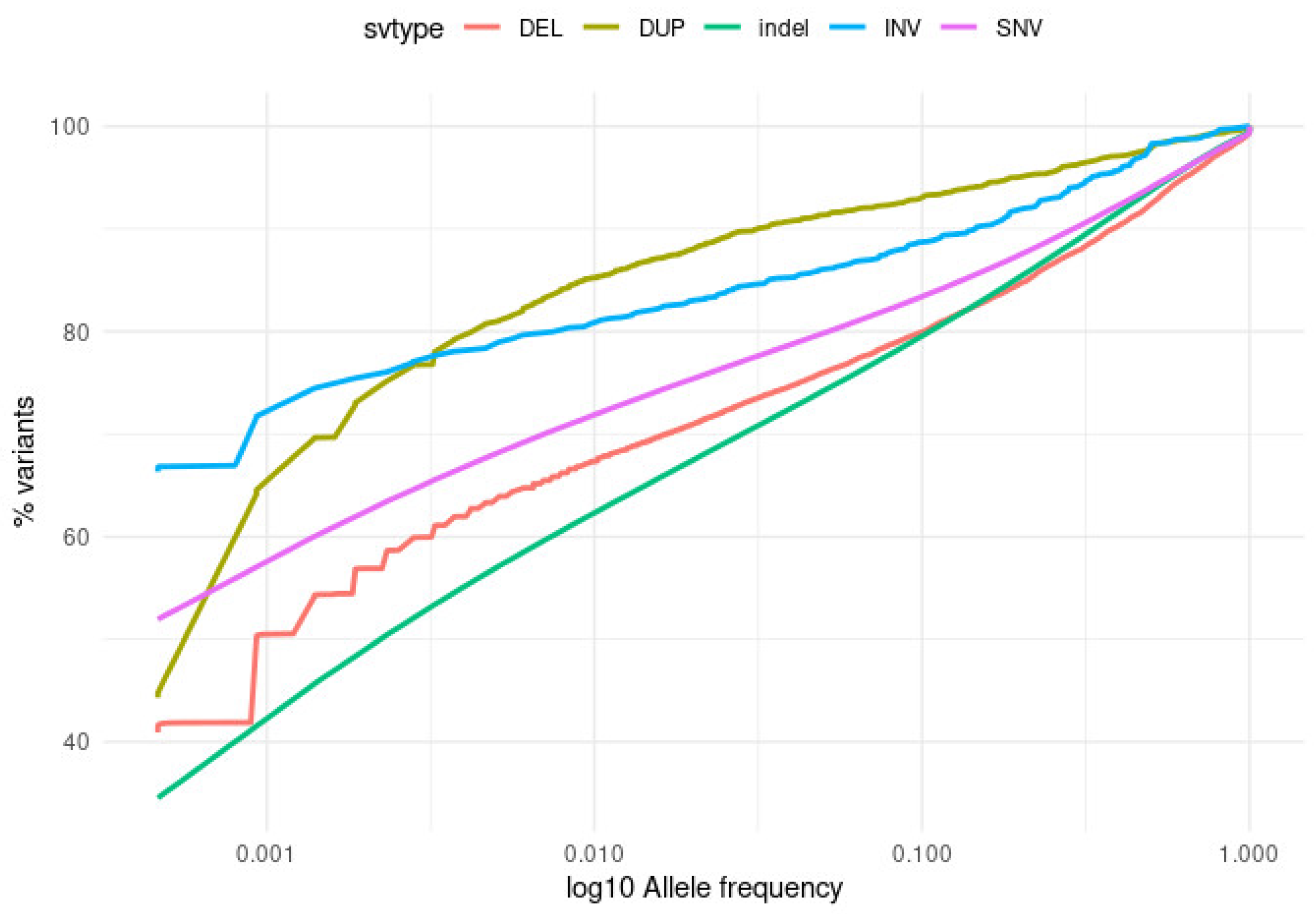
2.2.1. Small and Structural Variants
2.2.2. Distribution of Allele-Frequencies
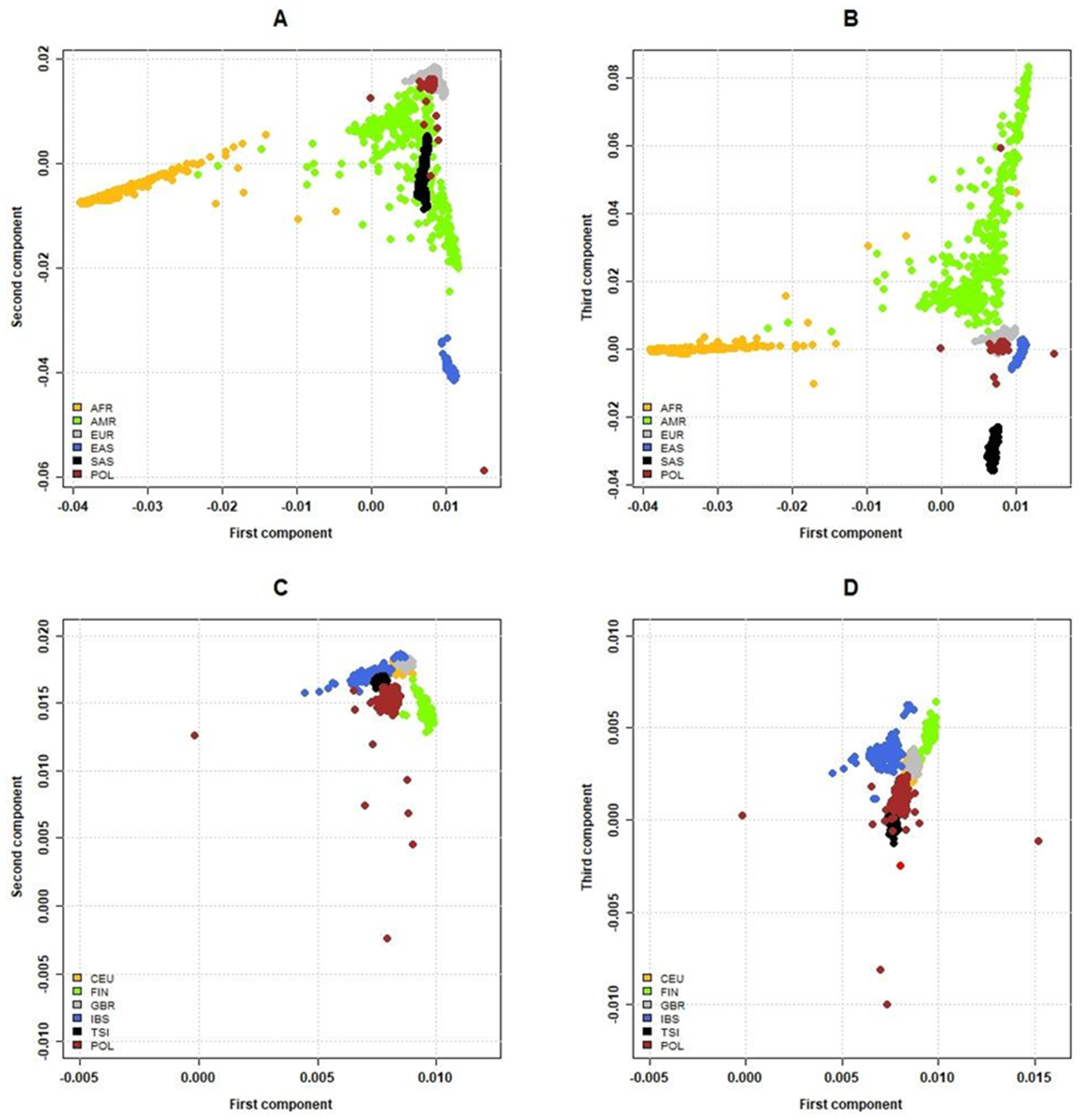
2.3. Relationship of the Polish and Global Populations
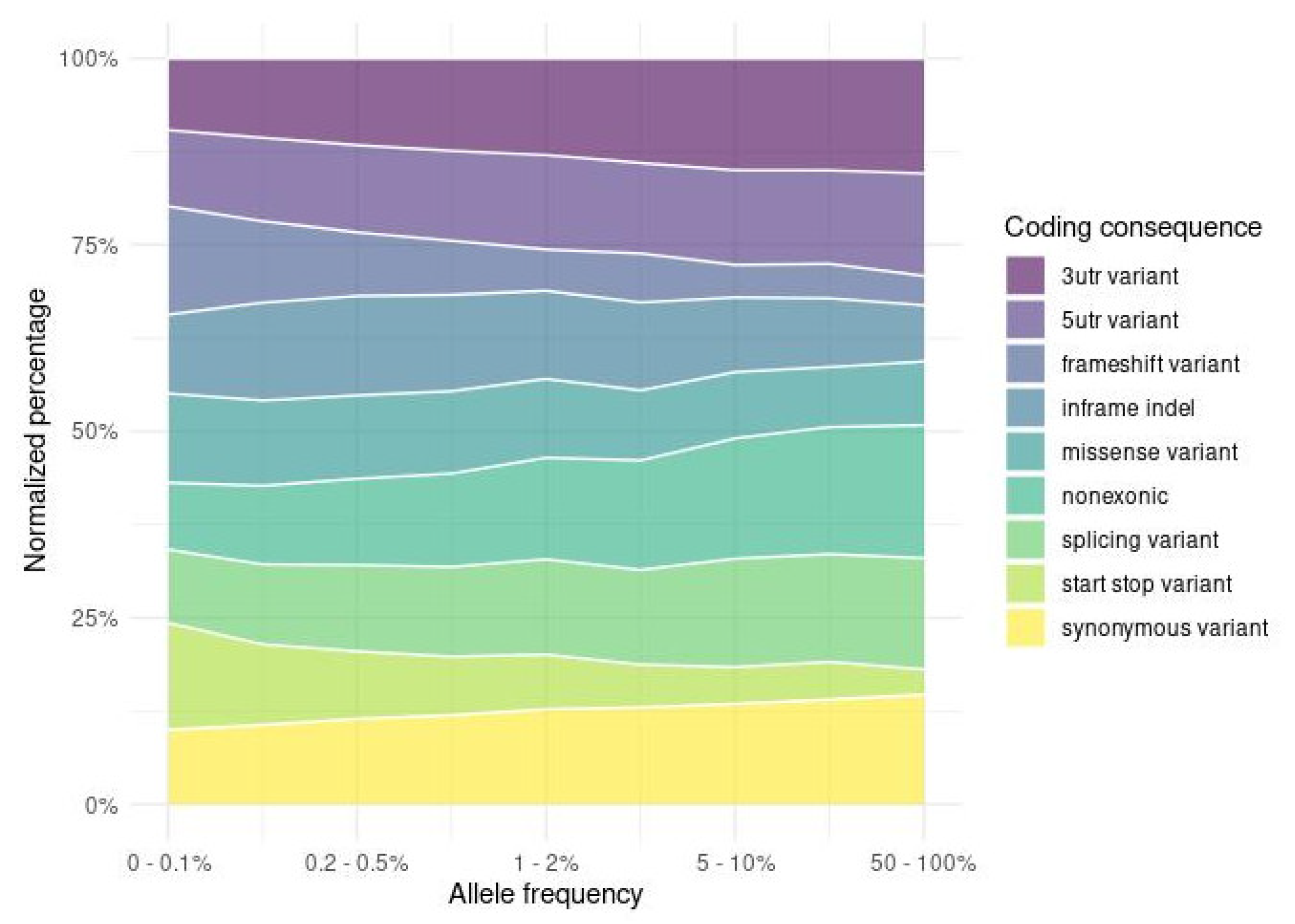
2.4. Functional Impact of the Variants
2.4.1. Impacts of Small Variants on Protein Function
2.4.2. Disease Causing Variation
2.4.3. Pathogenic Allele Burden
3. Discussion
4. Materials and Methods
4.1. Donors’ Characteristics and Sample Collection
4.2. Compliance with Ethical Standards
4.3. Total Quality Management Utilised in the Study
4.4. Sample Collection and Whole-Genome Sequencing
4.5. WGS Data Processing
4.6. Variant Analysis
4.7. Population Analysis
4.8. Pathogenic Allele Burden
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- The 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [Green Version]
- Frazer, K.A.; Murray, S.S.; Schork, N.J.; Topol, E.J. Human genetic variation and its contribution to complex traits. Nat. Rev. Genet. 2009, 10, 241–251. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2017, 46, D1062–D1067. [Google Scholar] [CrossRef] [Green Version]
- The International HapMap Consortium. The International HapMap Project. Nature 2003, 426, 789–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alfoldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Taliun, D.; Harris, D.N.; Kessler, M.D.; Carlson, J.; Szpiech, Z.A.; Torres, R.; Taliun, S.A.G.; Corvelo, A.; Gogarten, S.M.; NHLBI Trans-Omics for Precision Medicine (TOPMed) Consortium; et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature 2021, 590, 290–299. [Google Scholar] [CrossRef] [PubMed]
- Manolio, T.A.; Collins, F.S. The HapMap and Genome-Wide Association Studies in Diagnosis and Therapy. Annu. Rev. Med. 2009, 60, 443–456. [Google Scholar] [CrossRef] [Green Version]
- Bycroft, C.; Freeman, C.; Petkova, D.; Band, G.; Elliott, L.T.; Sharp, K.; Motyer, A.; Vukcevic, D.; Delaneau, O.; O’Connell, J.; et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 2018, 562, 203–209. [Google Scholar] [CrossRef] [Green Version]
- Zerbino, D.R.; Achuthan, P.; Akanni, W.; Amode, M.R.; Barrell, D.; Bhai, J.; Billis, K.; Cummins, C.; Gall, A.; Girón, C.G.; et al. Ensembl 2018. Nucleic Acids Res. 2018, 46, D754–D761. [Google Scholar] [CrossRef]
- The Severe COVID-19 GWAS Group. Genomewide Association Study of Severe COVID-19 with Respiratory Failure. N. Engl. J. Med. 2020, 383, 1522–1534. [Google Scholar] [CrossRef]
- Popejoy, A.B.; Fullerton, S.M. Genomics is failing on diversity. Nature 2016, 538, 161–164. [Google Scholar] [CrossRef] [Green Version]
- Mallick, S.; Li, H.; Lipson, M.; Mathieson, I.; Gymrek, M.; Racimo, F.; Zhao, M.; Chennagiri, N.; Nordenfelt, S.; Tandon, A.; et al. The Simons Genome Diversity Project: 300 genomes from 142 diverse populations. Nature 2016, 538, 201–206. [Google Scholar] [CrossRef]
- Bergström, A.; McCarthy, S.A.; Hui, R.; Almarri, M.A.; Ayub, Q.; Danecek, P.; Chen, Y.; Felkel, S.; Hallast, P.; Kamm, J.; et al. Insights into human genetic variation and population history from 929 diverse genomes. Science 2020, 367, eaay5012. [Google Scholar] [CrossRef]
- Kars, M.E.; Başak, A.N.; Onat, O.E.; Bilguvar, K.; Choi, J.; Itan, Y.; Çağlar, C.; Palvadeau, R.; Casanova, J.-L.; Cooper, D.N.; et al. The genetic structure of the Turkish population reveals high levels of variation and admixture. Proc. Natl. Acad. Sci. USA 2021, 118, e2026076118. [Google Scholar] [CrossRef]
- The Genome of the Netherlands Consortium. Whole-genome sequence variation, population structure and demographic history of the Dutch population. Nat. Genet. 2014, 46, 818–825. [Google Scholar] [CrossRef]
- Boomsma, D.; Wijmenga, C.; Slagboom, P.; Swertz, M.; Karssen, L.C.; Abdellaoui, M.; Ye, K.; Guryev, V.; Vermaat, J.; van Dijk, F.; et al. The Genome of the Netherlands: Design, and project goals. Eur. J. Hum. Genet. 2013, 22, 221–227. [Google Scholar] [CrossRef] [Green Version]
- Sidore, C.; Busonero, F.; Maschio, A.; Porcu, E.; Naitza, S.; Zoledziewska, M.; Mulas, A.; Pistis, G.; Steri, A.M.; Danjou, F.; et al. Genome sequencing elucidates Sardinian genetic architecture and augments association analyses for lipid and blood inflammatory markers. Nat. Genet. 2015, 47, 1272–1281. [Google Scholar] [CrossRef] [Green Version]
- Wohlers, I.; Künstner, A.; Munz, M.; Olbrich, M.; Fähnrich, A.; Calonga-Solís, V.; Ma, C.; Hirose, M.; El-Mosallamy, S.; Salama, M.; et al. An integrated personal and population-based Egyptian genome reference. Nat. Commun. 2020, 11, 1–10. [Google Scholar] [CrossRef]
- Jeon, S.; Bhak, Y.; Choi, Y.; Jeon, Y.; Kim, S.; Jang, J.; Jang, J.; Blazyte, A.; Kim, C.; Kim, Y.; et al. Korean Genome Project: 1094 Korean personal genomes with clinical information. Sci. Adv. 2020, 6, eaaz7835. [Google Scholar] [CrossRef]
- Puechmaille, S.J. Data from: The program STRUCTURE does not reliably recover the correct population structure when sampling is uneven: Sub-sampling and new estimators alleviate the problem. Mol. Ecol. Resour. 2016, 16, 608–627. [Google Scholar] [CrossRef] [PubMed]
- Jankowski, M.; Daca-Roszak, P.; Trautmann, A.; Milovanova, A.; Balasz-Chmielewska, I.; Lipska-Ziętkiewicz, B. Clinical delineation of the NUP93 glomerulopathy prevalent in Central and Eastern Europe. Pediatric Nephrol. 2021, 36, 3306. [Google Scholar]
- Szczecinska, W.; Nesteruk, D.; Wertheim-Tysarowska, K.; Greenblatt, D.; Baty, D.; Browne, F.; Liu, L.; Ozoemena, L.; Terron-Kwiatkowski, A.; McGrath, J.; et al. Under-recognition of acral peeling skin syndrome: 59 new cases with 15 novel mutations. Br. J. Dermatol. 2014, 171, 1206–1210. [Google Scholar] [CrossRef] [PubMed]
- Hartig, M.B.; Iuso, A.; Haack, T.; Kmiec, T.; Jurkiewicz, E.; Heim, K.; Roeber, S.; Tarabin, V.; Dusi, S.; Krajewska-Walasek, M.; et al. Absence of an Orphan Mitochondrial Protein, C19orf12, Causes a Distinct Clinical Subtype of Neurodegeneration with Brain Iron Accumulation. Am. J. Hum. Genet. 2011, 89, 543–550. [Google Scholar] [CrossRef] [Green Version]
- Dusatkova, P.; Pfäffle, R.; Brown, M.R.; Akulevich, N.; Arnhold, I.J.; A Kalina, M.; Kot, K.; Kržišnik, C.; Lemos, M.C.; Malikova, J.; et al. Genesis of two most prevalent PROP1 gene variants causing combined pituitary hormone deficiency in 21 populations. Eur. J. Hum. Genet. 2015, 24, 415–420. [Google Scholar] [CrossRef] [Green Version]
- Gudbjartsson, D.F.; Helgason, H.; Gudjonsson, S.A.; Zink, F.; Oddsson, A.; Gylfason, A.; Besenbacher, S.; Magnusson, G.; Halldórsson, B.; Hjartarson, E.; et al. Large-scale whole-genome sequencing of the Icelandic population. Nat. Genet. 2015, 47, 435–444. [Google Scholar] [CrossRef]
- Lazaridis, I. The evolutionary history of human populations in Europe. Curr. Opin. Genet. Dev. 2018, 53, 21–27. [Google Scholar] [CrossRef] [Green Version]
- Veltman, J.A.; Brunner, H.G. De novo mutations in human genetic disease. Nat. Rev. Genet. 2012, 13, 565–575. [Google Scholar] [CrossRef]
- Gravel, S.; Henn, B.M.; Gutenkunst, R.N.; Indap, A.; Marth, G.T.; Clark, A.G.; Yu, F.; Gibbs, R.A.; Bustamante, C.D.; The 1000 Genomes Project. Demographic history and rare allele sharing among human populations. Proc. Natl. Acad. Sci. USA 2011, 108, 11983–11988. [Google Scholar] [CrossRef] [Green Version]
- Mathieson, I.; McVean, G. Differential confounding of rare and common variants in spatially structured populations. Nat. Genet. 2012, 44, 243–246. [Google Scholar] [CrossRef] [Green Version]
- Ceballos, F.C.; Joshi, P.K.; Clark, D.W.; Ramsay, M.; Wilson, J.F. Runs of homozygosity: Windows into population history and trait architecture. Nat. Rev. Genet. 2018, 19, 220–234. [Google Scholar] [CrossRef]
- Pemberton, T.J.; Absher, D.; Feldman, M.W.; Myers, R.M.; Rosenberg, N.A.; Li, J.Z. Genomic Patterns of Homozygosity in Worldwide Human Populations. Am. J. Hum. Genet. 2012, 91, 275–292. [Google Scholar] [CrossRef] [Green Version]
- Witsch-Baumgartner, M.; Ciara, E.; Löffler, J.; Menzel, H.J.; Seedorf, U.; Burn, J.; Gillessen-Kaesbach, G.; Hoffmann, G.F.; Fitzky, B.U.; Mundy, H.; et al. Frequency gradients of DHCR7 mutations in patients with Smith-Lemli-Opitz syndrome in Europe: Evidence for different origins of common mutations. Eur. J. Hum. Genet. 2001, 9, 45–50. [Google Scholar] [CrossRef]
- Matsuura, S.; Tauchi, H.; Nakamura, A.; Kondo, N.; Sakamoto, S.; Endo, S.; Smeets, D.; Solder, B.; Belohradsky, B.H.; der Kaloustian, V.M.; et al. Positional cloning of the gene for Nijmegen breakage syndrome. Nat. Genet. 1998, 19, 179–181. [Google Scholar] [CrossRef]
- Koczkowska, M.; Zuk, M.; Gorczynski, A.; Ratajska, M.; Lewandowska, M.; Biernat, W.; Limon, J.; Wasag, B. Detection of somatic BRCA1/2 mutations in ovarian cancer—Next-generation sequencing analysis of 100 cases. Cancer Med. 2016, 5, 1640–1646. [Google Scholar] [CrossRef] [Green Version]
- Metcalfe, K.A.; Poll, A.; Royer, R.; Llacuachaqui, M.; Tulman, A.; Sun, P.; Narod, S.A. Screening for Founder Mutations in BRCA1 and BRCA2 in Unselected Jewish Women. J. Clin. Oncol. 2010, 28, 387–391. [Google Scholar] [CrossRef]
- Moslehi, R.; Chu, W.; Karlan, B.; Fishman, D.; Risch, H.; Fields, A.; Smotkin, D.; Ben-David, Y.; Rosenblatt, J.; Russo, D.; et al. BRCA1 and BRCA2 Mutation Analysis of 208 Ashkenazi Jewish Women with Ovarian Cancer. Am. J. Hum. Genet. 2000, 66, 1259–1272. [Google Scholar] [CrossRef] [Green Version]
- Sudmant, P.H.; Rausch, T.; Gardner, E.J.; Handsaker, R.E.; Abyzov, A.; Huddleston, J.; Zhang, Y.; Ye, K.; Jun, G.; Fritz, M.H.-Y.; et al. An integrated map of structural variation in 2,504 human genomes. Nature 2015, 526, 75–81. [Google Scholar] [CrossRef] [Green Version]
- Sztromwasser, P.; Michalak, A.; Małachowska, B.; Młudzik, P.; Antosik, K.; Hogendorf, A.; Zmysłowska, A.; Borowiec, M.; Młynarski, W.; Fendler, W. A cross-sectional study of patients referred for HNF1B-MODY genetic testing due to cystic kidneys and diabetes. Pediatr. Diabetes 2020, 21, 422–430. [Google Scholar] [CrossRef] [Green Version]
- Cameron, D.L.; di Stefano, L.; Papenfuss, A.T. Comprehensive evaluation and characterisation of short read general-purpose structural variant calling software. Nat. Commun. 2019, 10, 324. [Google Scholar] [CrossRef]
- Chiang, C.; Scott, A.J.; Davis, J.R.; Tsang, E.K.; Li, X.; Kim, Y.; Hadzic, T.; Damani, F.N.; Ganel, L.; GTEx Consortium; et al. The impact of structural variation on human gene expression. Nat. Genet. 2017, 49, 692–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ACMG Secondary Findings Working Group; Miller, D.T.; Lee, K.; Chung, W.K.; Gordon, A.S.; Herman, G.E.; Klein, T.E.; Stewart, D.R.; Amendola, L.M.; Adelman, K.; et al. ACMG SF v3.0 list for reporting of secondary findings in clinical exome and genome sequencing: A policy statement of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2021, 23, 1381–1390. [Google Scholar] [CrossRef] [PubMed]
- Hehir-Kwa, J.Y.; Marschall, T.; Kloosterman, W.P.; Francioli, L.C.; Baaijens, J.; Dijkstra, L.J.; Abdellaoui, A.; Koval, V.; Thung, D.T.; The Genome of the Netherlands Consortium; et al. A high-quality human reference panel reveals the complexity and distribution of genomic structural variants. Nat. Commun. 2016, 7, 12989. [Google Scholar] [CrossRef]
- Collins, R.L.; Brand, H.; Karczewski, K.J.; Zhao, X.; Alföldi, J.; Francioli, L.C.; Khera, A.V.; Lowther, C.; Gauthier, L.D.; Wang, H.; et al. A structural variation reference for medical and population genetics. Nature 2020, 581, 444–451. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.; Layer, R.M.; Faust, G.G.; Lindberg, M.R.; Rose, D.B.; Garrison, E.P.; Marth, G.T.; Quinlan, A.R.; Hall, I.M. SpeedSeq: Ultra-fast personal genome analysis and interpretation. Nat. Methods 2015, 12, 966–968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. ArXiv preprint 2013, arXiv:1303.3997. [Google Scholar]
- Faust, G.G.; Hall, I.M. SAMBLASTER: Fast duplicate marking and structural variant read extraction. Bioinformatics 2014, 30, 2503–2505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedersen, B.; Quinlan, A. Mosdepth: Quick coverage calculation for genomes and exomes. Bioinformatics 2018, 34, 867–868. [Google Scholar] [CrossRef]
- Poplin, R.; Chang, P.-C.; Alexander, D.; Schwartz, S.; Colthurst, T.; Ku, A.; Newburger, D.; Dijamco, J.; Nguyen, N.; Afshar, P.T.; et al. A universal SNP and small-indel variant caller using deep neural networks. Nat. Biotechnol. 2018, 36, 983–987. [Google Scholar] [CrossRef]
- Yun, T.; Li, H.; Chang, P.-C.; Lin, M.F.; Carroll, A.; McLean, C.Y. Accurate, scalable cohort variant calls using DeepVariant and GLnexus. Bioinformatics 2020, 36, 5582–5589. [Google Scholar] [CrossRef]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; O Pollard, M.; Whitwham, A.; Keane, T.; A McCarthy, S.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. GigaScience 2021, 10, giab008. [Google Scholar] [CrossRef]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [Green Version]
- Sim, N.-L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef]
- Quang, D.; Chen, Y.; Xie, X. DANN: A deep learning approach for annotating the pathogenicity of genetic variants. Bioinformatics 2014, 31, 761–763. [Google Scholar] [CrossRef] [Green Version]
- Goodstadt, L. Ruffus: A lightweight Python library for computational pipelines. Bioinformatics 2010, 26, 2778–2779. [Google Scholar] [CrossRef]
- Tange, O. GNU Parallel: The Command-Line Power Tool. USENIX Mag. 2011, 36, 42–47. [Google Scholar]
- Harrison, S.M.; Riggs, E.R.; Maglott, D.R.; Lee, J.M.; Azzariti, D.R.; Niehaus, A.; Ramos, E.M.; Martin, C.L.; Landrum, M.J.; Rehm, H.L. Using ClinVar as a Resource to Support Variant Interpretation. Curr. Protoc. Hum. Genet. 2016, 89, 8.16.1–8.16.23. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Wickham, H.; Averick, M.; Bryan, J.; Chang, W.; McGowan, L.D.A.; François, R.; Grolemund, G.; Hayes, A.; Henry, L.; Hester, J.; et al. Welcome to the Tidyverse. J. Open Source Softw. 2019, 4, 1686. [Google Scholar] [CrossRef]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Liu, J.-X. StructureSelector: A web-based software to select and visualize the optimal number of clusters using multiple methods. Mol. Ecol. Resour. 2017, 18, 176–177. [Google Scholar] [CrossRef] [PubMed]
- Weir, B.S.; Cockerham, C.C. Estimating F-Statistics for the Analysis of Population Structure. Evolution 1984, 38, 1358–1370. [Google Scholar] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IMPACT | |||||
|---|---|---|---|---|---|
| VARIANT_CLASS | AF | HIGH | MODERATE | LOW | MODIFIER |
| deletion | >0.5% | 412 | 603 | 855 | 1,208,322 |
| insertion | 260 | 573 | 977 | 1,380,654 | |
| SNV | 1109 | 35,717 | 41,402 | 10,877,171 | |
| deletion | 0.1–0.5% | 392 | 492 | 316 | 433,985 |
| insertion | 197 | 345 | 376 | 529,654 | |
| SNV | 852 | 23,682 | 18,675 | 4,375,036 | |
| deletion | <0.1% | 2849 | 1988 | 1003 | 1,295,678 |
| insertion | 1382 | 1144 | 826 | 1,037,730 | |
| SNV | 5432 | 119,843 | 80,467 | 17,817,903 | |
| TOTAL | 12,885 | 184,387 | 144,897 | 38,956,133 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaja, E.; Lejman, A.; Sielski, D.; Sypniewski, M.; Gambin, T.; Dawidziuk, M.; Suchocki, T.; Golik, P.; Wojtaszewska, M.; Mroczek, M.; et al. The Thousand Polish Genomes—A Database of Polish Variant Allele Frequencies. Int. J. Mol. Sci. 2022, 23, 4532. https://doi.org/10.3390/ijms23094532
Kaja E, Lejman A, Sielski D, Sypniewski M, Gambin T, Dawidziuk M, Suchocki T, Golik P, Wojtaszewska M, Mroczek M, et al. The Thousand Polish Genomes—A Database of Polish Variant Allele Frequencies. International Journal of Molecular Sciences. 2022; 23(9):4532. https://doi.org/10.3390/ijms23094532
Chicago/Turabian StyleKaja, Elżbieta, Adrian Lejman, Dawid Sielski, Mateusz Sypniewski, Tomasz Gambin, Mateusz Dawidziuk, Tomasz Suchocki, Paweł Golik, Marzena Wojtaszewska, Magdalena Mroczek, and et al. 2022. "The Thousand Polish Genomes—A Database of Polish Variant Allele Frequencies" International Journal of Molecular Sciences 23, no. 9: 4532. https://doi.org/10.3390/ijms23094532