
Loss of SDHB Induces a Metabolic Switch in the hPheo1 Cell Line toward Enhanced OXPHOS

 , ,
, ,
Abstract
:1. Introduction
2. Results
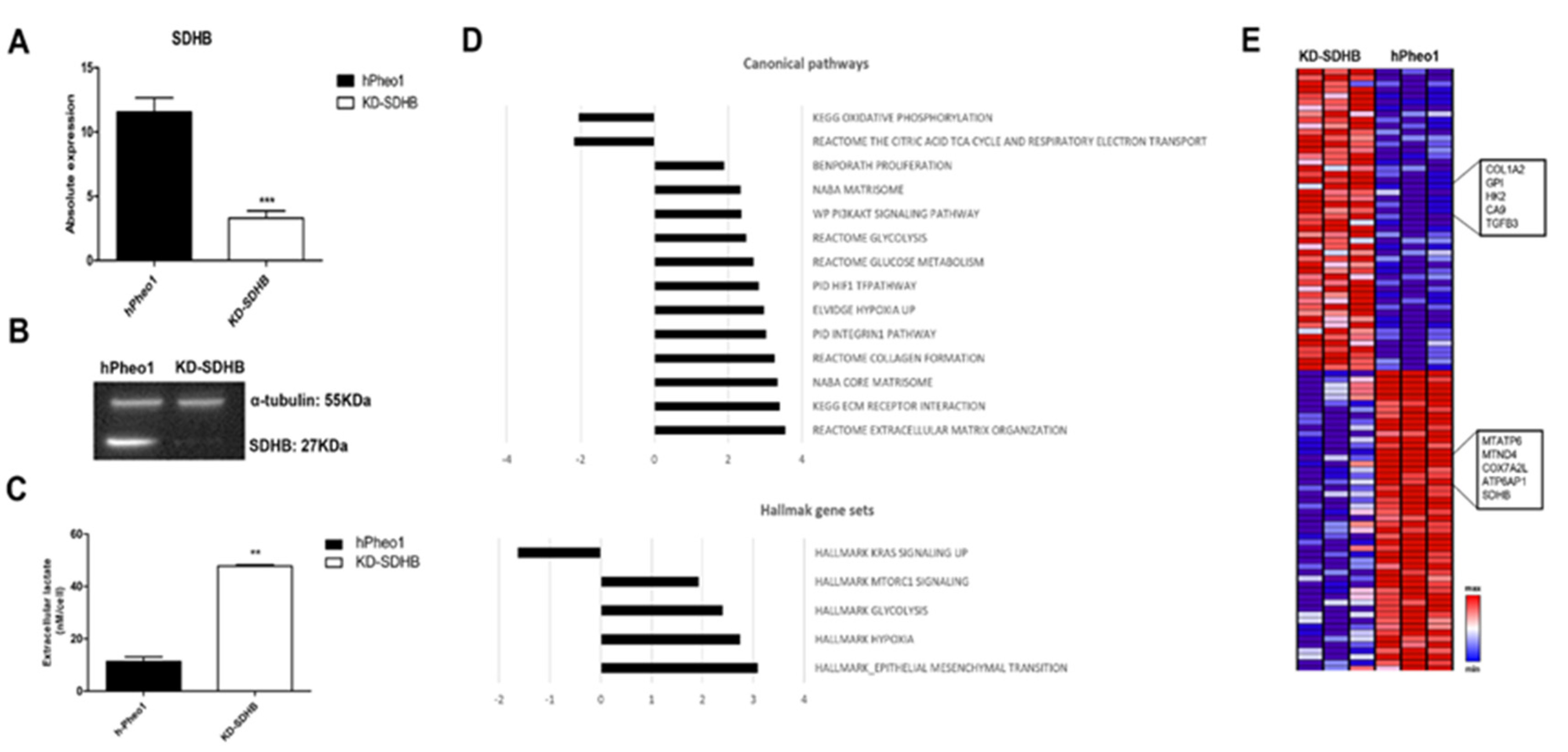
2.1. Downregulating SDHB in hPheo1 Cell
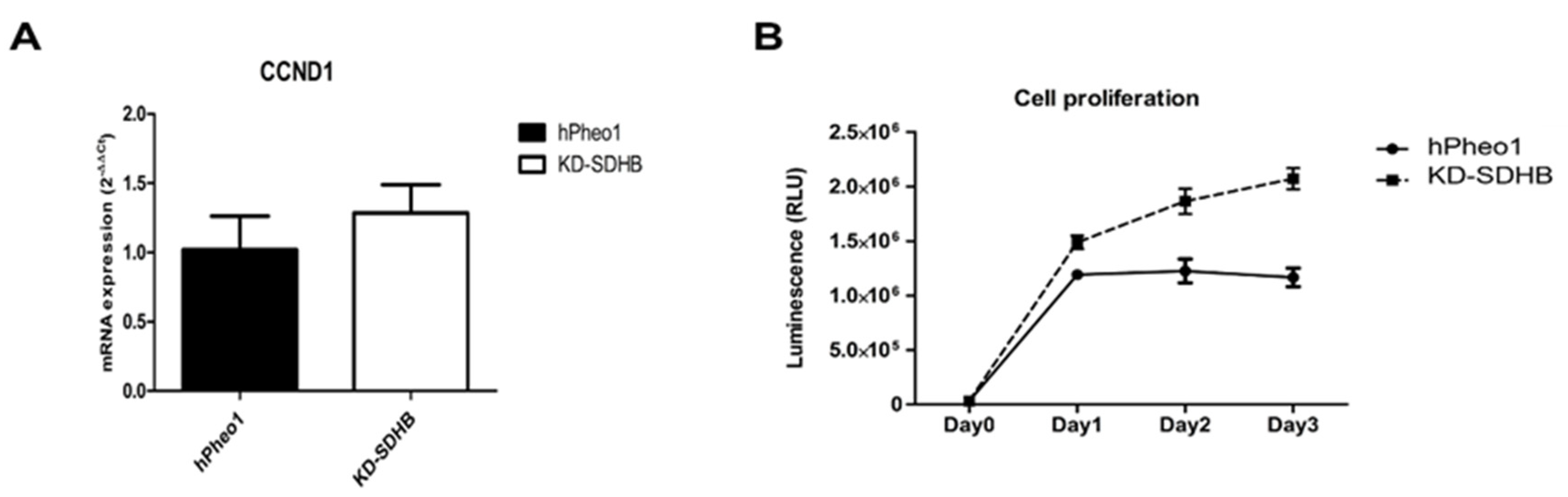
2.2. Effects in Cell Proliferation
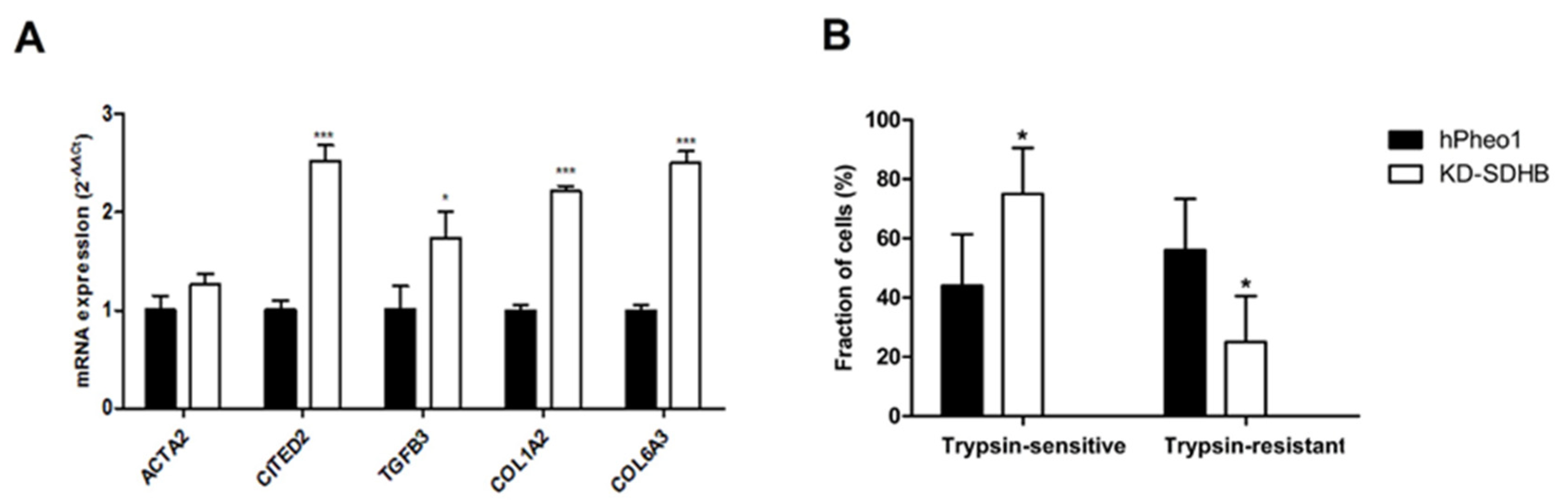
2.3. Effects on Cellular Adhesion
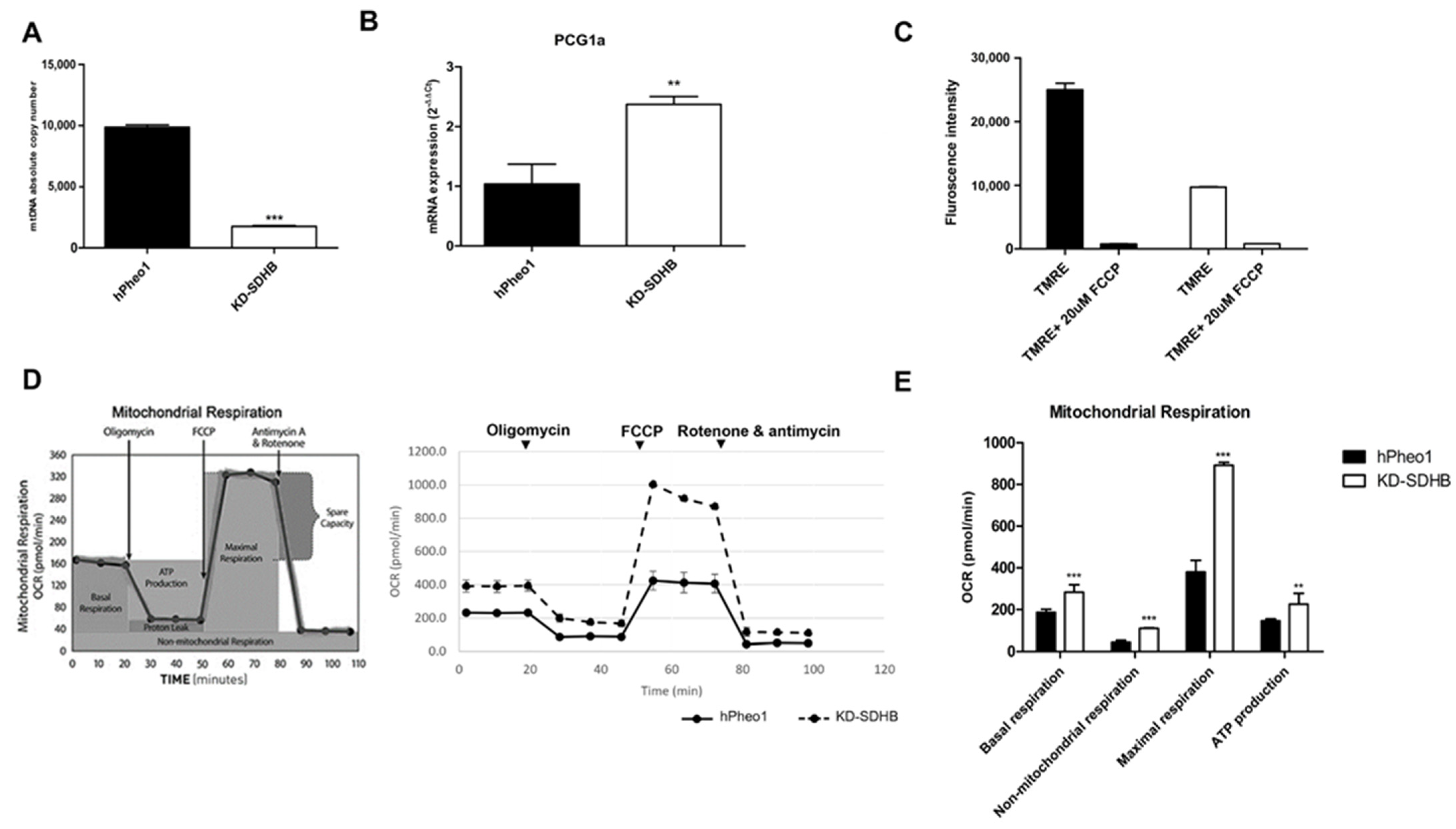
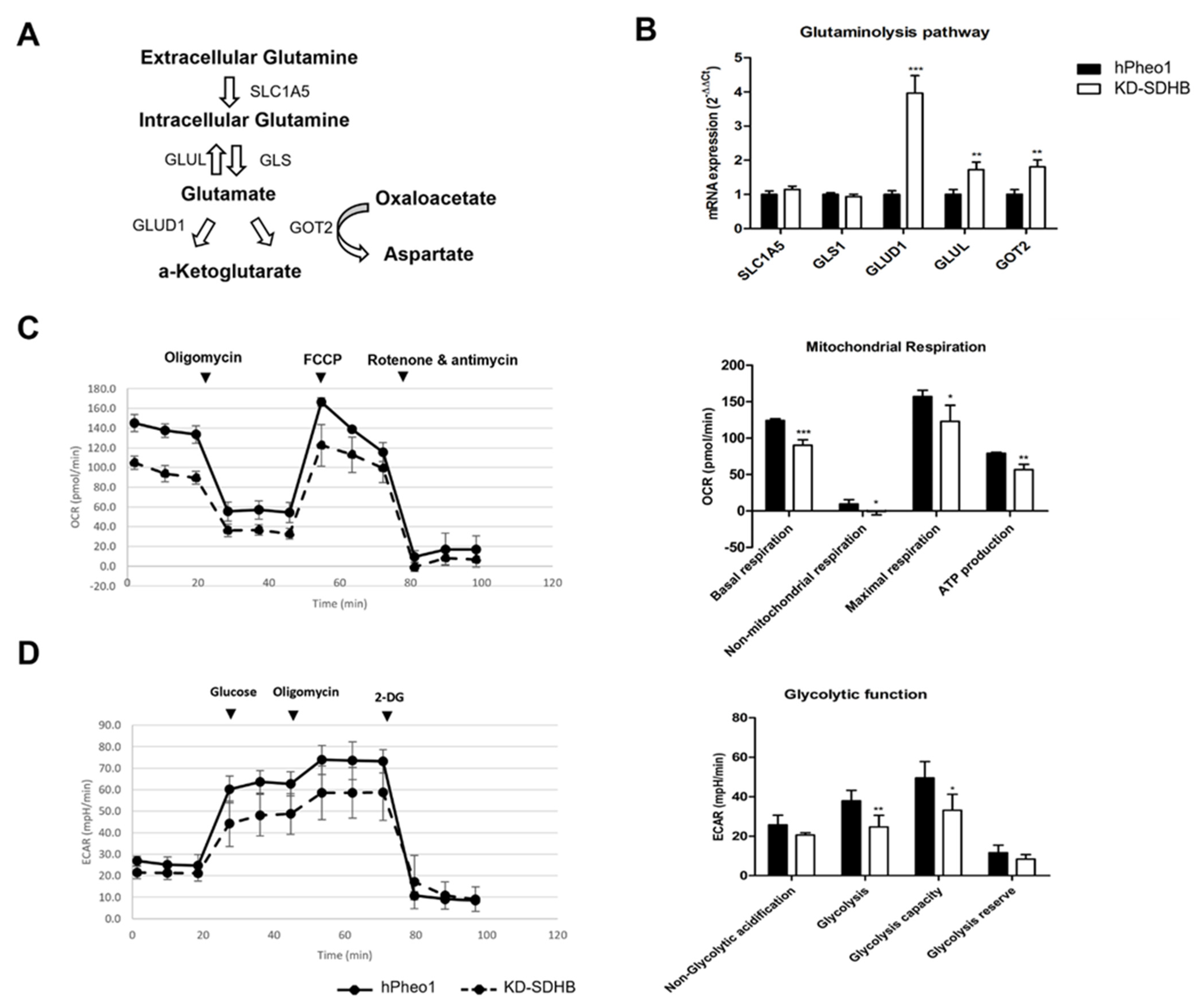
2.4. Effects in Mitochondrial Respiration
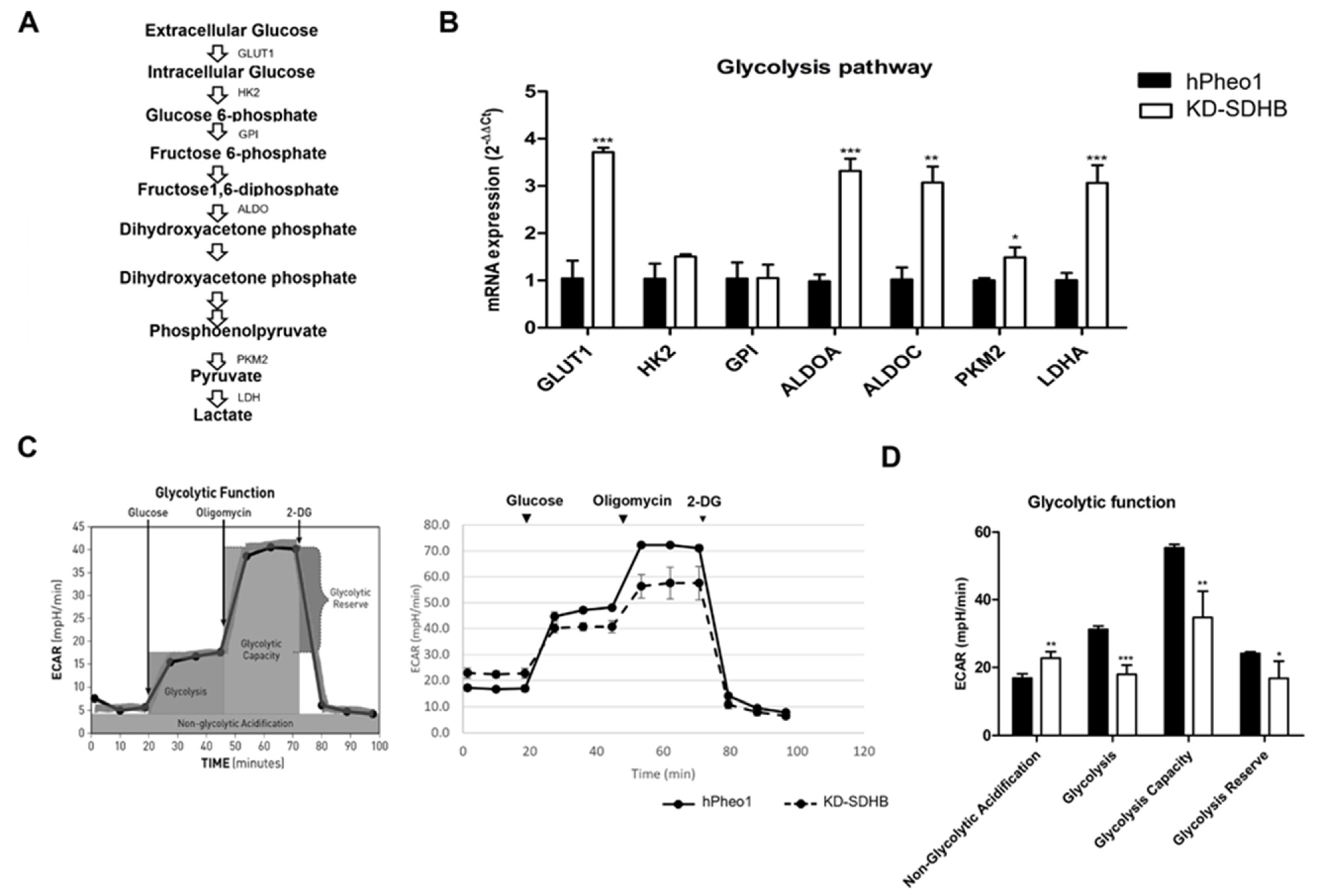
2.5. Effects in Glycolysis
2.6. Effects in Glutaminolysis
2.7. GLUD1 and SDHB Gene Expression in PCC/PGL
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. SDHB Knockdown with CRISPRcas9
4.3. DNA Extraction and Sequencing Confirmation
4.4. Spheroid Formation
4.5. RNA Extraction
4.6. SDHB Gene Expression
4.7. Western Blotting
4.8. Biochemical Analysis
4.9. Microarray Analysis
4.10. RT-qPCR
4.11. Cell Proliferation
4.12. Clonogenic Assay
4.13. Differential Trypsinization
4.14. Mitochondrial Content Measurement
4.15. Mitochondrial Membrane Potential (ψm)
4.16. Measurements of Glycolytic and OXPHOS Activities
4.17. PCC/PGL TCGA Cohort Analysis
4.18. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- van Nederveen, F.H.; Gaal, J.; Favier, J.; Korpershoek, E.; Oldenburg, R.A.; de Bruyn, E.M.C.A.; Sleddens, H.F.B.M.; Derkx, P.; Rivière, J.; Dannenberg, H.; et al. An Immunohistochemical Procedure to Detect Patients with Paraganglioma and Phaeochromocytoma with Germline SDHB, SDHC, or SDHD Gene Mutations: A Retrospective and Prospective Analysis. Lancet Oncol. 2009, 10, 764–771. [Google Scholar] [CrossRef] [Green Version]
- Williamson, S.R.; Eble, J.N.; Amin, M.B.; Gupta, N.S.; Smith, S.C.; Sholl, L.M.; Montironi, R.; Hirsch, M.S.; Hornick, J.L. Succinate Dehydrogenase-Deficient Renal Cell Carcinoma: Detailed Characterization of 11 Tumors Defining a Unique Subtype of Renal Cell Carcinoma. Mod. Pathol. 2015, 28, 80–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Wang, W.; Xiang, B.; Li, N.; Huang, S.; Zhou, W.; Sun, Y.; Wang, X.; Ma, J.; Li, G.; et al. Reduced Succinate Dehydrogenase B Expression Is Associated with Growth and De-Differentiation of Colorectal Cancer Cells. Tumour Biol. 2013, 34, 2337–2347. [Google Scholar] [CrossRef] [PubMed]
- Dalla Pozza, E.; Dando, I.; Pacchiana, R.; Liboi, E.; Scupoli, M.T.; Donadelli, M.; Palmieri, M. Regulation of Succinate Dehydrogenase and Role of Succinate in Cancer. Semin. Cell Dev. Biol. 2020, 98, 4–14. [Google Scholar] [CrossRef]
- Kitazawa, S.; Ebara, S.; Ando, A.; Baba, Y.; Satomi, Y.; Soga, T.; Hara, T. Succinate Dehydrogenase B-Deficient Cancer Cells Are Highly Sensitive to Bromodomain and Extra-Terminal Inhibitors. Oncotarget 2017, 8, 28922–28938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkadi, B.; Meszaros, K.; Krencz, I.; Canu, L.; Krokker, L.; Zakarias, S.; Barna, G.; Sebestyen, A.; Papay, J.; Hujber, Z.; et al. Glutaminases as a Novel Target for SDHB-Associated Pheochromocytomas/Paragangliomas. Cancers 2020, 12, 599. [Google Scholar] [CrossRef] [Green Version]
- Amar, L.; Baudin, E.; Burnichon, N.; Peyrard, S.; Silvera, S.; Bertherat, J.; Bertagna, X.; Schlumberger, M.; Jeunemaitre, X.; Gimenez-Roqueplo, A.-P.; et al. Succinate Dehydrogenase B Gene Mutations Predict Survival in Patients with Malignant Pheochromocytomas or Paragangliomas. J. Clin. Endocrinol. Metab. 2007, 92, 3822–3828. [Google Scholar] [CrossRef]
- Lussey-Lepoutre, C.; Hollinshead, K.E.R.; Ludwig, C.; Menara, M.; Morin, A.; Castro-Vega, L.-J.; Parker, S.J.; Janin, M.; Martinelli, C.; Ottolenghi, C.; et al. Loss of Succinate Dehydrogenase Activity Results in Dependency on Pyruvate Carboxylation for Cellular Anabolism. Nat. Commun. 2015, 6, 8784. [Google Scholar] [CrossRef]
- Pollard, P.J.; Brière, J.J.; Alam, N.A.; Barwell, J.; Barclay, E.; Wortham, N.C.; Hunt, T.; Mitchell, M.; Olpin, S.; Moat, S.J.; et al. Accumulation of Krebs Cycle Intermediates and Over-Expression of HIF1alpha in Tumours Which Result from Germline FH and SDH Mutations. Hum. Mol. Genet. 2005, 14, 2231–2239. [Google Scholar] [CrossRef]
- Selak, M.A.; Armour, S.M.; MacKenzie, E.D.; Boulahbel, H.; Watson, D.G.; Mansfield, K.D.; Pan, Y.; Simon, M.C.; Thompson, C.B.; Gottlieb, E. Succinate Links TCA Cycle Dysfunction to Oncogenesis by Inhibiting HIF-Alpha Prolyl Hydroxylase. Cancer Cell 2005, 7, 77–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gimenez-Roqueplo, A.P.; Favier, J.; Rustin, P.; Mourad, J.J.; Plouin, P.F.; Corvol, P.; Rötig, A.; Jeunemaitre, X. The R22X Mutation of the SDHD Gene in Hereditary Paraganglioma Abolishes the Enzymatic Activity of Complex II in the Mitochondrial Respiratory Chain and Activates the Hypoxia Pathway. Am. J. Hum. Genet. 2001, 69, 1186–1197. [Google Scholar] [CrossRef] [Green Version]
- Gimenez-Roqueplo, A.-P.; Favier, J.; Rustin, P.; Rieubland, C.; Kerlan, V.; Plouin, P.-F.; Rötig, A.; Jeunemaitre, X. Functional Consequences of a SDHB Gene Mutation in an Apparently Sporadic Pheochromocytoma. J. Clin. Endocrinol. Metab. 2002, 87, 4771–4774. [Google Scholar] [CrossRef] [Green Version]
- Cervera, A.M.; Apostolova, N.; Crespo, F.L.; Mata, M.; McCreath, K.J. Cells Silenced for SDHB Expression Display Characteristic Features of the Tumor Phenotype. Cancer Res. 2008, 68, 4058–4067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Cui, H. Targeting Glutamine Induces Apoptosis: A Cancer Therapy Approach. Int. J. Mol. Sci. 2015, 16, 22830–22855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villar, V.H.; Merhi, F.; Djavaheri-Mergny, M.; Durán, R.V. Glutaminolysis and Autophagy in Cancer. Autophagy 2015, 11, 1198–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghayee, H.K.; Bhagwandin, V.J.; Stastny, V.; Click, A.; Ding, L.-H.; Mizrachi, D.; Zou, Y.S.; Chari, R.; Lam, W.L.; Bachoo, R.M.; et al. Progenitor Cell Line (HPheo1) Derived from a Human Pheochromocytoma Tumor. PLoS ONE 2013, 8, e65624. [Google Scholar] [CrossRef] [Green Version]
- Errichiello, E.; Venesio, T. Mitochondrial DNA Variations in Tumors: Drivers or Passengers? Mitochondrial DNA-New Insights 2018. [Google Scholar] [CrossRef] [Green Version]
- Ohshima, K.; Morii, E. Metabolic Reprogramming of Cancer Cells during Tumor Progression and Metastasis. Metabolites 2021, 11, 28. [Google Scholar] [CrossRef]
- Saxena, N.; Maio, N.; Crooks, D.R.; Ricketts, C.J.; Yang, Y.; Wei, M.-H.; Fan, T.W.-M.; Lane, A.N.; Sourbier, C.; Singh, A.; et al. SDHB-Deficient Cancers: The Role of Mutations That Impair Iron Sulfur Cluster Delivery. J. Natl. Cancer Inst. 2015, 108, djv287. [Google Scholar] [CrossRef] [Green Version]
- Hujber, Z.; Horváth, G.; Petővári, G.; Krencz, I.; Dankó, T.; Mészáros, K.; Rajnai, H.; Szoboszlai, N.; Leenders, W.P.J.; Jeney, A.; et al. GABA, Glutamine, Glutamate Oxidation and Succinic Semialdehyde Dehydrogenase Expression in Human Gliomas. J. Exp. Clin. Cancer Res. 2018, 37, 271. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Chen, H.; Xue, L.; He, W.; Shu, W.; Wu, H.; Wang, Z. High Throughput Proteomic and Metabolic Profiling Identified Target Correction of Metabolic Abnormalities as a Novel Therapeutic Approach in Head and Neck Paraganglioma. Transl. Oncol. 2021, 14, 101146. [Google Scholar] [CrossRef]
- Favier, J.; Brière, J.-J.; Burnichon, N.; Rivière, J.; Vescovo, L.; Benit, P.; Giscos-Douriez, I.; Reyniès, A.D.; Bertherat, J.; Badoual, C.; et al. The Warburg Effect Is Genetically Determined in Inherited Pheochromocytomas. PLoS ONE 2009, 4, e7094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fliedner, S.M.J.; Kaludercic, N.; Jiang, X.-S.; Hansikova, H.; Hajkova, Z.; Sladkova, J.; Limpuangthip, A.; Backlund, P.S.; Wesley, R.; Martiniova, L.; et al. Warburg Effect’s Manifestation in Aggressive Pheochromocytomas and Paragangliomas: Insights from a Mouse Cell Model Applied to Human Tumor Tissue. PLoS ONE 2012, 7, e40949. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.C.; Yu, Y.C.; Sung, Y.; Han, J.M. Glutamine Reliance in Cell Metabolism. Exp. Mol. Med. 2020, 52, 1496–1516. [Google Scholar] [CrossRef]
- Yoo, H.C.; Park, S.J.; Nam, M.; Kang, J.; Kim, K.; Yeo, J.H.; Kim, J.-K.; Heo, Y.; Lee, H.S.; Lee, M.Y.; et al. A Variant of SLC1A5 Is a Mitochondrial Glutamine Transporter for Metabolic Reprogramming in Cancer Cells. Cell Metab. 2020, 31, 267–283.e12. [Google Scholar] [CrossRef]
- Durán, R.V.; Oppliger, W.; Robitaille, A.M.; Heiserich, L.; Skendaj, R.; Gottlieb, E.; Hall, M.N. Glutaminolysis Activates Rag-MTORC1 Signaling. Mol. Cell 2012, 47, 349–358. [Google Scholar] [CrossRef] [Green Version]
- LeBleu, V.S.; O’Connell, J.T.; Gonzalez Herrera, K.N.; Wikman, H.; Pantel, K.; Haigis, M.C.; de Carvalho, F.M.; Damascena, A.; Domingos Chinen, L.T.; Rocha, R.M.; et al. PGC-1α Mediates Mitochondrial Biogenesis and Oxidative Phosphorylation in Cancer Cells to Promote Metastasis. Nat. Cell Biol. 2014, 16, 992–1003. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, T.; Kishikawa, T.; Sato, T.; Takeda, N.; Sugiura, Y.; Seimiya, T.; Sekiba, K.; Ohno, M.; Iwata, T.; Ishibashi, R.; et al. Mutant KRAS Drives Metabolic Reprogramming and Autophagic Flux in Premalignant Pancreatic Cells. Cancer Gene Ther. 2021, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Csibi, A.; Fendt, S.-M.; Li, C.; Poulogiannis, G.; Choo, A.Y.; Chapski, D.J.; Jeong, S.M.; Dempsey, J.; Parkhitko, A.; Morrison, T.; et al. The MTORC1 Pathway Stimulates Glutamine Metabolism and Cell Proliferation by Repressing SIRT4. Cell 2013, 153, 840–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.; Zhu, J.; Yu, M.; Cai, C.; Zhou, Y.; Yu, M.; Fu, Z.; Gong, Y.; Yang, B.; Li, Y.; et al. Glutamate Dehydrogenase Is a Novel Prognostic Marker and Predicts Metastases in Colorectal Cancer Patients. J. Transl. Med. 2015, 13, 144. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Li, C.; Allen, A.; Stanley, C.A.; Smith, T.J. The Structure and Allosteric Regulation of Glutamate Dehydrogenase. Neurochem. Int. 2011, 59, 445–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, L.; Li, D.; Alesi, G.N.; Fan, J.; Kang, H.-B.; Lu, Z.; Boggon, T.J.; Jin, P.; Yi, H.; Wright, E.R.; et al. Glutamate Dehydrogenase 1 Signals through Antioxidant Glutathione Peroxidase 1 to Regulate Redox Homeostasis and Tumor Growth. Cancer Cell 2015, 27, 257–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walia, V.; Elble, R.C. Enrichment for Breast Cancer Cells with Stem/Progenitor Properties by Differential Adhesion. Stem Cells Dev. 2010, 19, 1175–1182. [Google Scholar] [CrossRef] [PubMed]
- D’Antongiovanni, V.; Martinelli, S.; Richter, S.; Canu, L.; Guasti, D.; Mello, T.; Romagnoli, P.; Pacak, K.; Eisenhofer, G.; Mannelli, M.; et al. The Microenvironment Induces Collective Migration in SDHB-Silenced Mouse Pheochromocytoma Spheroids. Endocr. Relat. Cancer 2017, 24, 555–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loriot, C.; Domingues, M.; Berger, A.; Menara, M.; Ruel, M.; Morin, A.; Castro-Vega, L.-J.; Letouzé, É.; Martinelli, C.; Bemelmans, A.-P.; et al. Deciphering the Molecular Basis of Invasiveness in Sdhb-Deficient Cells. Oncotarget 2015, 6, 32955–32965. [Google Scholar] [CrossRef] [Green Version]
- Henke, E.; Nandigama, R.; Ergün, S. Extracellular Matrix in the Tumor Microenvironment and Its Impact on Cancer Therapy. Front. Mol. Biosci. 2020, 6, 160. [Google Scholar] [CrossRef] [Green Version]
- Koenig, A.; Mueller, C.; Hasel, C.; Adler, G.; Menke, A. Collagen Type I Induces Disruption of E-Cadherin-Mediated Cell-Cell Contacts and Promotes Proliferation of Pancreatic Carcinoma Cells. Cancer Res. 2006, 66, 4662–4671. [Google Scholar] [CrossRef] [Green Version]
- Tzanakakis, G.; Kavasi, R.-M.; Voudouri, K.; Berdiaki, A.; Spyridaki, I.; Tsatsakis, A.; Nikitovic, D. Role of the Extracellular Matrix in Cancer-Associated Epithelial to Mesenchymal Transition Phenomenon. Dev. Dyn. 2018, 247, 368–381. [Google Scholar] [CrossRef] [Green Version]
- Liao, J.; Suen, H.C.; Luk, A.C.S.; Lee, A.W.T.; Ng, J.K.W.; Chan, T.H.T.; Cheung, M.Y.; Chan, D.Y.L.; Qi, H.; Chan, W.Y.; et al. ScATAC-Seq Reveals Epigenetic Heterogeneity Associated with an EMT-like Process in Male Germline Stem Cells and Its Regulation by G9a. bioRxiv 2020. [Google Scholar] [CrossRef]
- Lee, J.M.; Dedhar, S.; Kalluri, R.; Thompson, E.W. The Epithelial-Mesenchymal Transition: New Insights in Signaling, Development, and Disease. J. Cell Biol. 2006, 172, 973–981. [Google Scholar] [CrossRef]
- Shintani, Y.; Maeda, M.; Chaika, N.; Johnson, K.R.; Wheelock, M.J. Collagen I Promotes Epithelial-to-Mesenchymal Transition in Lung Cancer Cells via Transforming Growth Factor–β Signaling. Am. J. Respir Cell Mol. Biol. 2008, 38, 95–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vignais, M.; Fafet, P. TGFβ-Dependent Epithelial-Mesenchymal Transition. In Madame Curie Bioscience Database [Internet]; Landes Bioscience: Austin, TX, USA, 2000–2013. Available online: https://www.ncbi.nlm.nih.gov/books/NBK6525/ (accessed on 6 December 2021).
- Mammoto, T.; Jiang, A.; Jiang, E.; Panigrahy, D.; Kieran, M.W.; Mammoto, A. Role of Collagen Matrix in Tumor Angiogenesis and Glioblastoma Multiforme Progression. Am. J. Pathol. 2013, 183, 1293–1305. [Google Scholar] [CrossRef] [Green Version]
- Provenzano, P.P.; Inman, D.R.; Eliceiri, K.W.; Knittel, J.G.; Yan, L.; Rueden, C.T.; White, J.G.; Keely, P.J. Collagen Density Promotes Mammary Tumor Initiation and Progression. BMC Med. 2008, 6, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kauppila, S.; Stenbäck, F.; Risteli, J.; Jukkola, A.; Risteli, L. Aberrant Type I and Type III Collagen Gene Expression in Human Breast Cancer In Vivo. J. Pathol. 1998, 186, 262–268. [Google Scholar] [CrossRef]
- Liang, Y.; Lv, Z.; Huang, G.; Qin, J.; Li, H.; Nong, F.; Wen, B. Prognostic Significance of Abnormal Matrix Collagen Remodeling in Colorectal Cancer Based on Histologic and Bioinformatics Analysis. Oncol. Rep. 2020, 44, 1671–1685. [Google Scholar] [CrossRef]
- Ungefroren, H. Autocrine TGF-β in Cancer: Review of the Literature and Caveats in Experimental Analysis. Int. J. Mol. Sci. 2021, 22, 977. [Google Scholar] [CrossRef]
- Irizarry, R.A.; Hobbs, B.; Collin, F.; Beazer-Barclay, Y.D.; Antonellis, K.J.; Scherf, U.; Speed, T.P. Exploration, Normalization, and Summaries of High Density Oligonucleotide Array Probe Level Data. Biostatistics 2003, 4, 249–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franken, N.A.P.; Rodermond, H.M.; Stap, J.; Haveman, J.; van Bree, C. Clonogenic Assay of Cells in Vitro. Nat. Protoc. 2006, 1, 2315–2319. [Google Scholar] [CrossRef]
- Rossitti, H.M.; Dutta, R.K.; Larsson, C.; Ghayee, H.K.; Söderkvist, P.; Gimm, O. Activation of RAS Signalling Is Associated with Altered Cell Adhesion in Phaeochromocytoma. Int. J. Mol. Sci. 2020, 21, 8072. [Google Scholar] [CrossRef]
- Memon, A.A.; Zöller, B.; Hedelius, A.; Wang, X.; Stenman, E.; Sundquist, J.; Sundquist, K. Quantification of Mitochondrial DNA Copy Number in Suspected Cancer Patients by a Well Optimized DdPCR Method. Biomol. Detect. Quantif. 2017, 13, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Fishbein, L.; Leshchiner, I.; Walter, V.; Danilova, L.; Robertson, A.G.; Johnson, A.R.; Lichtenberg, T.M.; Murray, B.A.; Ghayee, H.K.; Else, T.; et al. Comprehensive Molecular Characterization of Pheochromocytoma and Paraganglioma. Cancer Cell 2017, 31, 181–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koivunen, P.; Hirsilä, M.; Remes, A.M.; Hassinen, I.E.; Kivirikko, K.I.; Myllyharju, J. Inhibition of Hypoxia-Inducible Factor (HIF) Hydroxylases by Citric Acid Cycle Intermediates: Possible Links Between Cell Metabolism and Stabilization Of Hif. J. Biol. Chem. 2007, 282, 4524–4532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roh, T.H.; Yim, H.; Roh, J.; Lee, K.B.; Park, S.H.; Jeong, S.-Y.; Kim, S.-H.; Kim, J.-H. The Loss of Succinate Dehydrogenase B Expression Is Frequently Identified in Hemangioblastoma of the Central Nervous System. Sci. Rep. 2019, 9, 5873. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Sequence (5′→3′) | Amplicon Length (bp) |
|---|---|---|
| CCND1 | F: GAGGAGCTGCTGCAAATGG R: CGGCCAGGTTCCACTTGA | 58 (G) |
| ACTA2 | F: CGGGAGAAAATGACTCAAATTATGTT R: CATACATGGCTGGGACATTGAA | 62 (G) |
| PCG1-α | F: TGAGAGGGCCAAGCAAAG R: ATAAATCACACGGCGCTCTT | 64 (G) |
| TGFB3 | F: TATGTGATTGCCATCTTTGCC R: TGGACCTGCAGTGGCTAAACA | 54 (G) |
| COL1A2 | F: ACCACAGGGTGTTCAAGGTG R: CAGGACCAGGGAGACCAAAC | 149 (G) |
| COL6A3 | F: CCTAACCACATATGTTAGTGGAGGT R: GAATGTCTCGCTTGCTCTCTG | 71 (G) |
| GLUT1 | F: TTGTGGGCATGTGCTTCCAG R: ATCGAAGGTCCGGCCTTTAG | 134 (G) |
| HK2 | F: GGTGGACAGGATACGAGAAAACR: ACATCACATTTCGGAGCCAG | 141 (G) |
| GPI | F: AAATCGCCCAACCAACTC R: ATGATGCCCTGAACGAAG | 102 (G) |
| ALDOA | F: TTGTGGGCATCAAGGTAG R: TAGTCTCGCCATTTGTCC | 60 (G) |
| ALDOC | F: ATCGTCGTGGGCATCAAGG R: TTGGGCACAGCGTTCTGAG | 105 (G) |
| PKM2 | F: ATCGTCCTCACCAAGTCTGG R: GAAGATGCCACGGTACAGGT | 126 (A) |
| LDHA | F: GAG GTG ATC AAA CTC AAA GGC T R: CAT GGT GGA AAC TGG GTG C | 111 (G) |
| SLC1A5 | F: CTCGATTCGTTCCTGGATCTT R: GTTCCGGTGATATTCCTCTCTTC | 107 (A) |
| GLS1 | F: TCTACAGGATTGCGAACGTCT R: CTTTGTCTAGCATGACACCATCT | 100 (A) |
| GLUD1 | F: GGTCATCGAAGGCTACCG R: TCAGTGCTGTAACGGATACCTC | 75 (A) |
| GLUL | F: CCTGCTTGTATGCTGGAGTC R: GATCTCCCATGCTGATTCC | 105 (A) |
| GOT2 | F: GACCAAATTGGCATGTTCTGT R: CGGCCATCTTTTGTCATGTA | 95 (A) |
| ACTB | F: CTCTTCCAGCCTTCCTTCCT R: AGCACTGTGTTGGCGTACAG | 116 |
| HPRT1 | F: ATGGACTAATTATGGACAGGACTGAA R: CTCCCATCTCCTTCATCACATCT | 60 |
| GUSB | F: CAAGACAGTGGGCTGGTGAATTA R: CTTGAACAGGTTACTGCCCTTGAC | 55 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tabebi, M.; Kumar Dutta, R.; Skoglund, C.; Söderkvist, P.; Gimm, O. Loss of SDHB Induces a Metabolic Switch in the hPheo1 Cell Line toward Enhanced OXPHOS. Int. J. Mol. Sci. 2022, 23, 560. https://doi.org/10.3390/ijms23010560
Tabebi M, Kumar Dutta R, Skoglund C, Söderkvist P, Gimm O. Loss of SDHB Induces a Metabolic Switch in the hPheo1 Cell Line toward Enhanced OXPHOS. International Journal of Molecular Sciences. 2022; 23(1):560. https://doi.org/10.3390/ijms23010560
Chicago/Turabian StyleTabebi, Mouna, Ravi Kumar Dutta, Camilla Skoglund, Peter Söderkvist, and Oliver Gimm. 2022. "Loss of SDHB Induces a Metabolic Switch in the hPheo1 Cell Line toward Enhanced OXPHOS" International Journal of Molecular Sciences 23, no. 1: 560. https://doi.org/10.3390/ijms23010560