
Atrial Fibrillation: Pathogenesis, Predisposing Factors, and Genetics

 , , and
, , and
Abstract
:1. Introduction
2. Fibrosis
- Interstitial fibrosis can be sub-classified into:
- (a)
- (b)
- Infiltrative interstitial fibrosis, which refers to the deposition of glycosphingolipids or insoluble proteins in the interstitial space, as seen in amyloidosis or Fabry disease respectively [12].
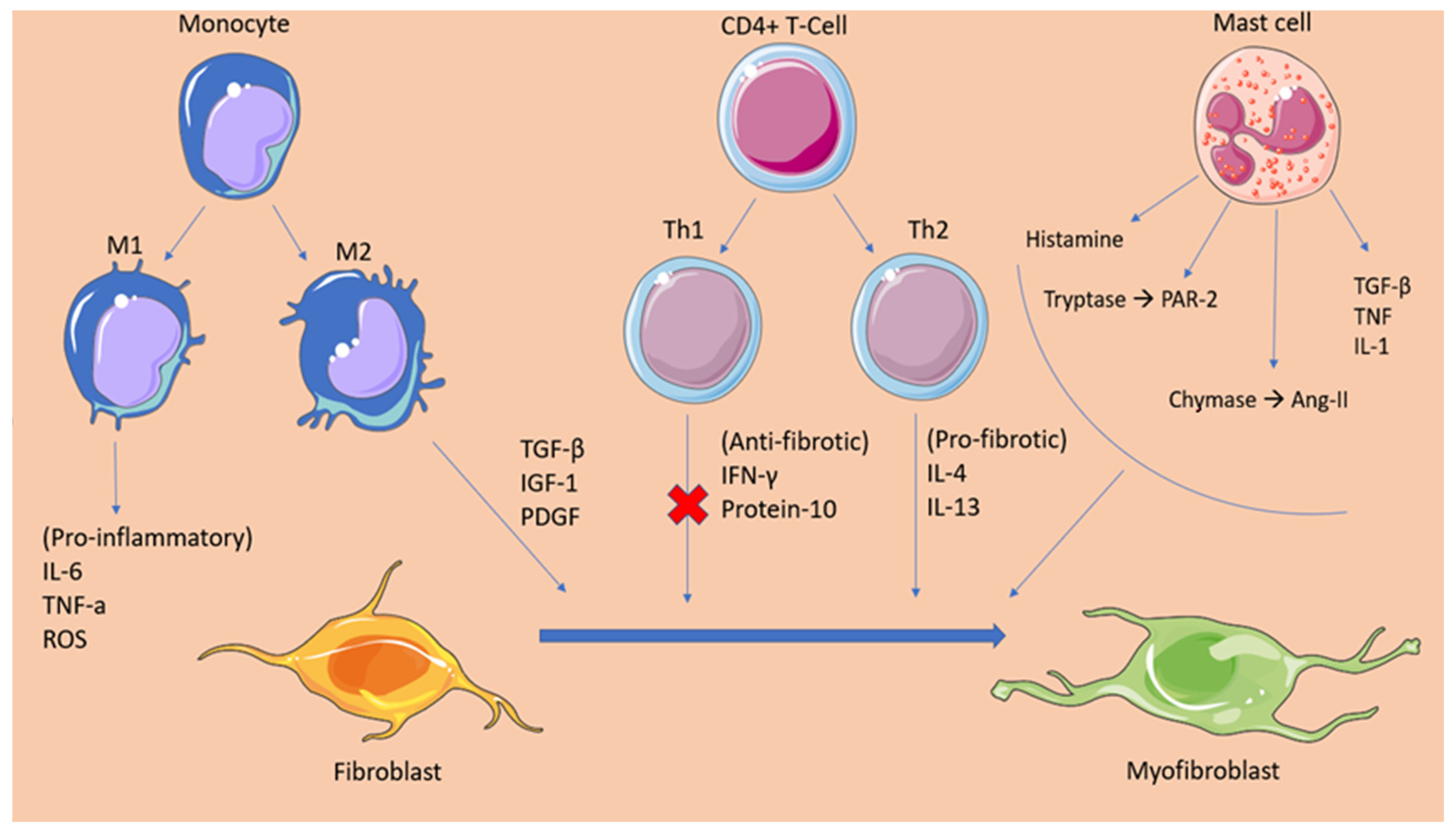
2.1. Cellular Mediators of Atrial Fibrosis
2.2. Fibrotic Mechanisms Inducing Atrial Fibrillation
3. Oxidative Stress
4. Inflammation
5. Sedentary Lifestyle
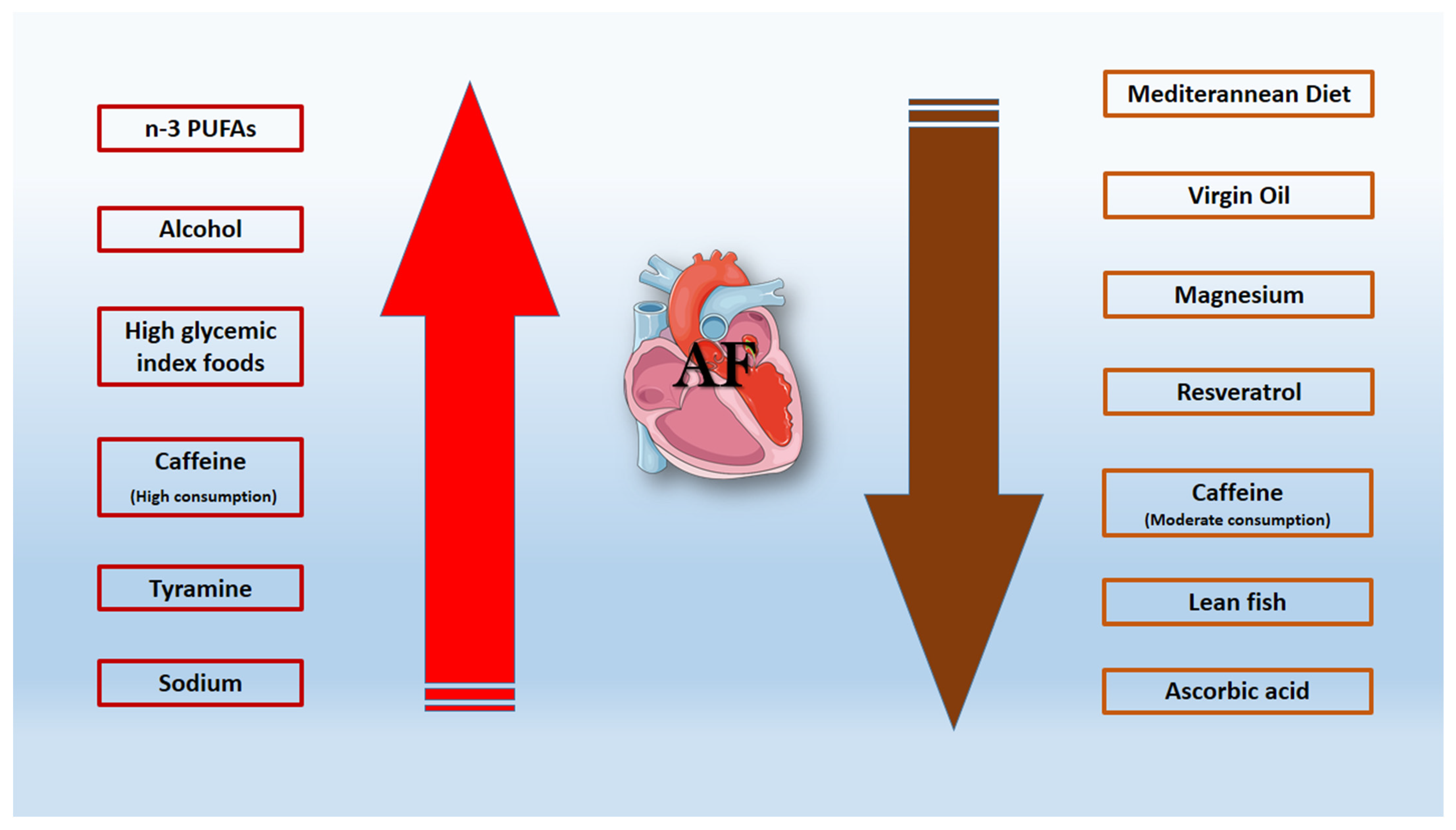
6. Dietary Habits
6.1. Alcohol-Resveratrol
6.2. Caffeine
6.3. Mediterranean Diet
6.4. Virgin Oil—Magnesium—Lean Fish
7. Genetic Factors
8. Prevention-Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lau, D.H.; Linz, D.; Sanders, P. New Findings in Atrial Fibrillation Mechanisms. Card Electrophysiol. Clin. 2019, 11, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Staerk, L.; Sherer, J.A.; Ko, D.; Benjamin, E.J.; Helm, R.H. Atrial Fibrillation: Epidemiology, Pathophysiology, and Clinical Outcomes. Circ. Res. 2017, 120, 1501–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morin, D.P.; Bernard, M.L.; Madias, C.; Rogers, P.A.; Thihalolipavan, S.; Estes, N.A., 3rd. The State of the Art: Atrial Fibrillation Epidemiology, Prevention, and Treatment. Mayo Clin. Proc. 2016, 91, 1778–1810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jalife, J.; Kaur, K. Atrial remodeling, fibrosis, and atrial fibrillation. Trends Cardiovasc. Med. 2015, 25, 475–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sagris, M.; Antonopoulos, A.S.; Theofilis, P.; Oikonomou, E.; Siasos, G.; Tsalamandris, S.; Antoniades, C.; Brilakis, E.S.; Kaski, J.C.; Tousoulis, D. Risk factors profile of young and older patients with Myocardial Infarction. Cardiovasc. Res. 2021. [Google Scholar] [CrossRef] [PubMed]
- Diavati, S.; Sagris, M.; Terentes-Printzios, D.; Vlachopoulos, C. Anticoagulation Treatment in Venous Thromboembolism: Options and Optimal Duration. Curr. Pharm. Des. 2021. [Google Scholar] [CrossRef]
- Siasos, G.; Skotsimara, G.; Oikonomou, E.; Sagris, M.; Vasiliki-Chara, M.; Bletsa, E.; Stampouloglou, P.; Theofilis, P.; Charalampous, G.; Tousoulis, D. Antithrombotic Treatment in Diabetes Mellitus: A Review of the Literature about Antiplatelet and Anticoagulation Strategies Used for Diabetic Patients in Primary and Secondary Prevention. Curr. Pharm. Des. 2020, 26, 2780–2788. [Google Scholar] [CrossRef]
- Mahmood, S.S.; Levy, D.; Vasan, R.S.; Wang, T.J. The Framingham Heart Study and the epidemiology of cardiovascular disease: A historical perspective. Lancet 2014, 383, 999–1008. [Google Scholar] [CrossRef] [Green Version]
- Nattel, S. Molecular and Cellular Mechanisms of Atrial Fibrosis in Atrial Fibrillation. JACC Clin. Electrophysiol. 2017, 3, 425–435. [Google Scholar] [CrossRef]
- Spencer, T.M.; Blumenstein, R.F.; Pryse, K.M.; Lee, S.-L.; Glaubke, D.A.; Carlson, B.E.; Elson, E.L.; Genin, G.M. Fibroblasts Slow Conduction Velocity in a Reconstituted Tissue Model of Fibrotic Cardiomyopathy. ACS Biomater. Sci. Eng. 2017, 3, 3022–3028. [Google Scholar] [CrossRef] [PubMed]
- Burstein, B.; Nattel, S. Atrial fibrosis: Mechanisms and clinical relevance in atrial fibrillation. J. Am. Coll. Cardiol. 2008, 51, 802–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinderer, S.; Schenke-Layland, K. Cardiac fibrosis—A short review of causes and therapeutic strategies. Adv. Drug Deliv. Rev. 2019, 146, 77–82. [Google Scholar] [CrossRef]
- Yao, C.; Veleva, T.; Scott, L., Jr.; Cao, S.; Li, L.; Chen, G.; Jeyabal, P.; Pan, X.; Alsina, K.M.; Abu-Taha, I.D.; et al. Enhanced Cardiomyocyte NLRP3 Inflammasome Signaling Promotes Atrial Fibrillation. Circulation 2018, 138, 2227–2242. [Google Scholar] [CrossRef]
- Nattel, S. Electrical coupling between cardiomyocytes and fibroblasts: Experimental testing of a challenging and important concept. Cardiovasc. Res. 2018, 114, 349–352. [Google Scholar] [CrossRef]
- Davis, J.; Burr, A.R.; Davis, G.F.; Birnbaumer, L.; Molkentin, J.D. A TRPC6-Dependent Pathway for Myofibroblast Transdifferentiation and Wound Healing In Vivo. Dev. Cell 2012, 23, 705–715. [Google Scholar] [CrossRef] [Green Version]
- Hoyles, R.K.; Derrett-Smith, E.C.; Khan, K.; Shiwen, X.; Howat, S.L.; Wells, A.U.; Abraham, D.J.; Denton, C.P. An Essential Role for Resident Fibroblasts in Experimental Lung Fibrosis Is Defined by Lineage-Specific Deletion of High-Affinity Type II Transforming Growth Factor β Receptor. Am. J. Respir. Crit. Care Med. 2011, 183, 249–261. [Google Scholar] [CrossRef] [PubMed]
- Leask, A. Potential Therapeutic Targets for Cardiac Fibrosis. Circ. Res. 2010, 106, 1675–1680. [Google Scholar] [CrossRef] [Green Version]
- Davis, J.; Molkentin, J.D. Myofibroblasts: Trust your heart and let fate decide. J. Mol. Cell Cardiol. 2014, 70, 9–18. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Tian, A.; Wu, J.; Yang, C.; Xing, R.; Jia, P.; Yang, L.; Zhang, Y.; Zheng, X.; Li, Z. Danshensu Inhibits β-Adrenergic Receptors-Mediated Cardiac Fibrosis by ROS/p38 MAPK Axis. Biol. Pharm. Bull. 2014, 37, 961–967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibb, A.A.; Lazaropoulos, M.P.; Elrod, J.W. Myofibroblasts and Fibrosis. Circ. Res. 2020, 127, 427–447. [Google Scholar] [CrossRef]
- Pellman, J.; Zhang, J.; Sheikh, F. Myocyte-fibroblast communication in cardiac fibrosis and arrhythmias: Mechanisms and model systems. J. Mol. Cell Cardiol. 2016, 94, 22–31. [Google Scholar] [CrossRef] [Green Version]
- Theofilis, P.; Sagris, M.; Antonopoulos, A.S.; Oikonomou, E.; Tsioufis, C.; Tousoulis, D. Inflammatory Mediators of Platelet Activation: Focus on Atherosclerosis and COVID-19. Int. J. Mol. Sci. 2021, 22, 1170. [Google Scholar] [CrossRef]
- Kim, P.; Chu, N.; Davis, J.; Kim, D.H. Mechanoregulation of Myofibroblast Fate and Cardiac Fibrosis. Adv. Biosyst 2018, 2. [Google Scholar] [CrossRef]
- Zaidi, Y.; Aguilar, E.G.; Troncoso, M.; Ilatovskaya, D.V.; DeLeon-Pennell, K.Y. Immune regulation of cardiac fibrosis post myocardial infarction. Cell Signal. 2021, 77, 109837. [Google Scholar] [CrossRef] [PubMed]
- Shiota, N.; Jin, D.; Takai, S.; Kawamura, T.; Koyama, M.; Nakamura, N.; Miyazaki, M. Chymase is activated in the hamster heart following ventricular fibrosis during the chronic stage of hypertension. FEBS Lett. 1997, 406, 301–304. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, S.; Varagic, J.; Westwood, B.M.; Chappell, M.C.; Ferrario, C.M. Uptake and Metabolism of the Novel Peptide Angiotensin-(1-12) by Neonatal Cardiac Myocytes. PLoS ONE 2011, 6, e15759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balcells, E.; Meng, Q.C.; Walter, H.; Johnson, J.; Oparil, S.; Dell’Italia, L.J. Angiotensin II formation from ACE and chymase in human and animal hearts: Methods and species considerations. Am. J. Physiol. Heart Circ. Physiol. 1997, 273, H1769–H1774. [Google Scholar] [CrossRef]
- Shimizu, M.; Tanaka, R.; Fukuyama, T.; Aoki, R.; Orito, K.; Yamane, Y. Cardiac Remodeling and Angiotensin II-Forming Enzyme Activity of the Left Ventricle in Hamsters with Chronic Pressure Overload Induced by Ascending Aortic Stenosis. J. Vet. Med Sci. 2006, 68, 271–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLarty, J.L.; Meléndez, G.C.; Brower, G.L.; Janicki, J.S.; Levick, S.P. Tryptase/Protease-Activated Receptor 2 Interactions Induce Selective Mitogen-Activated Protein Kinase Signaling and Collagen Synthesis by Cardiac Fibroblasts. Hypertension 2011, 58, 264–270. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Shen, L.; Li, X.; Luo, T.; Wei, X.; Zhang, J.; Cao, S.; Huang, X.; Fukushima, Y.; Bin, J.; et al. Disruption of histamine H2 receptor slows heart failure progression through reducing myocardial apoptosis and fibrosis. Clin. Sci. 2014, 127, 435–448. [Google Scholar] [CrossRef] [PubMed]
- Morgan, L.G.; Levick, S.P.; Voloshenyuk, T.G.; Murray, D.B.; Forman, M.F.; Brower, G.L.; Janicki, J.S. A novel technique for isolating functional mast cells from the heart. Inflamm Res. 2008, 57, 241–246. [Google Scholar] [CrossRef]
- Nattel, S. How does fibrosis promote atrial fibrillation persistence: In silico findings, clinical observations, and experimental data. Cardiovasc. Res. 2016, 110, 295–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allessie, M.A.; de Groot, N.M.; Houben, R.P.; Schotten, U.; Boersma, E.; Smeets, J.L.; Crijns, H.J. Electropathological substrate of long-standing persistent atrial fibrillation in patients with structural heart disease: Longitudinal dissociation. Circ. Arrhythm Electrophysiol. 2010, 3, 606–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krul, S.P.; Berger, W.R.; Smit, N.W.; van Amersfoorth, S.C.; Driessen, A.H.; van Boven, W.J.; Fiolet, J.W.; van Ginneken, A.C.; van der Wal, A.C.; de Bakker, J.M.; et al. Atrial fibrosis and conduction slowing in the left atrial appendage of patients undergoing thoracoscopic surgical pulmonary vein isolation for atrial fibrillation. Circ. Arrhythm Electrophysiol. 2015, 8, 288–295. [Google Scholar] [CrossRef] [Green Version]
- Hansen, B.J.; Zhao, J.; Csepe, T.A.; Moore, B.T.; Li, N.; Jayne, L.A.; Kalyanasundaram, A.; Lim, P.; Bratasz, A.; Powell, K.A.; et al. Atrial fibrillation driven by micro-anatomic intramural re-entry revealed by simultaneous sub-epicardial and sub-endocardial optical mapping in explanted human hearts. Eur. Heart J. 2015, 36, 2390–2401. [Google Scholar] [CrossRef] [Green Version]
- Samman Tahhan, A.; Sandesara, P.B.; Hayek, S.S.; Alkhoder, A.; Chivukula, K.; Hammadah, M.; Mohamed-Kelli, H.; O’Neal, W.T.; Topel, M.; Ghasemzadeh, N.; et al. Association between oxidative stress and atrial fibrillation. Heart Rhythm. 2017, 14, 1849–1855. [Google Scholar] [CrossRef]
- Sovari, A.A.; Dudley, S.C., Jr. Reactive oxygen species-targeted therapeutic interventions for atrial fibrillation. Front. Physiol. 2012, 3, 311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, W.; Santulli, G.; Reiken, S.R.; Yuan, Q.; Osborne, B.W.; Chen, B.-X.; Marks, A.R. Mitochondrial oxidative stress promotes atrial fibrillation. Sci. Rep. 2015, 5, 11427. [Google Scholar] [CrossRef] [Green Version]
- Yoo, S.; Aistrup, G.; Shiferaw, Y.; Ng, J.; Mohler, P.J.; Hund, T.J.; Waugh, T.; Browne, S.; Gussak, G.; Gilani, M.; et al. Oxidative stress creates a unique, CaMKII-mediated substrate for atrial fibrillation in heart failure. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shan, J.; Xie, W.; Betzenhauser, M.; Reiken, S.; Chen, B.X.; Wronska, A.; Marks, A.R. Calcium leak through ryanodine receptors leads to atrial fibrillation in 3 mouse models of catecholaminergic polymorphic ventricular tachycardia. Circ. Res. 2012, 111, 708–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babusikova, E.; Kaplan, P.; Lehotsky, J.; Jesenak, M.; Dobrota, D. Oxidative modification of rat cardiac mitochondrial membranes and myofibrils by hydroxyl radicals. Gen. Physiol. Biophys. 2004, 23, 327–335. [Google Scholar]
- Conen, D.; Ridker, P.M.; Everett, B.M.; Tedrow, U.B.; Rose, L.; Cook, N.R.; Buring, J.E.; Albert, C.M. A multimarker approach to assess the influence of inflammation on the incidence of atrial fibrillation in women. Eur. Heart J. 2010, 31, 1730–1736. [Google Scholar] [CrossRef] [PubMed]
- Kallergis, E.M.; Manios, E.G.; Kanoupakis, E.M.; Mavrakis, H.E.; Kolyvaki, S.G.; Lyrarakis, G.M.; Chlouverakis, G.I.; Vardas, P.E. The role of the post-cardioversion time course of hs-CRP levels in clarifying the relationship between inflammation and persistence of atrial fibrillation. Heart 2008, 94, 200–204. [Google Scholar] [CrossRef]
- Rotter, M.; Jaïs, P.; Vergnes, M.-C.; Nurden, P.; Takahashi, Y.; Sanders, P.; Rostock, T.; Hocini, M.; Sacher, F.; Haïssaguerre, M. Decline in C-Reactive Protein After Successful Ablation of Long-Lasting Persistent Atrial Fibrillation. J. Am. Coll. Cardiol. 2006, 47, 1231–1233. [Google Scholar] [CrossRef]
- Mouselimis, D.; Tsarouchas, A.S.; Pagourelias, E.D.; Bakogiannis, C.; Theofilogiannakos, E.K.; Loutradis, C.; Fragakis, N.; Vassilikos, V.P.; Papadopoulos, C.E. Left atrial strain, intervendor variability, and atrial fibrillation recurrence after catheter ablation: A systematic review and meta-analysis. Hellenic J. Cardiol. 2020, 61, 154–164. [Google Scholar] [CrossRef]
- Gao, G.; Dudley, S.C., Jr. Redox regulation, NF-kappaB, and atrial fibrillation. Antioxid Redox Signal. 2009, 11, 2265–2277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theofilis, P.; Sagris, M.; Oikonomou, E.; Antonopoulos, A.S.; Siasos, G.; Tsioufis, C.; Tousoulis, D. Inflammatory Mechanisms Contributing to Endothelial Dysfunction. Biomedicines 2021, 9, 781. [Google Scholar] [CrossRef] [PubMed]
- Satou, R.; Penrose, H.; Navar, L.G. Inflammation as a Regulator of the Renin-Angiotensin System and Blood Pressure. Curr. Hypertens. Rep. 2018, 20, 100. [Google Scholar] [CrossRef]
- Liang, F.; Wang, Y. Coronary heart disease and atrial fibrillation: A vicious cycle. Am. J. Physiol. Heart Circ. Physiol. 2021, 320, H1–H12. [Google Scholar] [CrossRef] [PubMed]
- Aronson, D.; Boulos, M.; Suleiman, A.; Bidoosi, S.; Agmon, Y.; Kapeliovich, M.; Beyar, R.; Markiewicz, W.; Hammerman, H.; Suleiman, M. Relation of C-reactive protein and new-onset atrial fibrillation in patients with acute myocardial infarction. Am. J. Cardiol. 2007, 100, 753–757. [Google Scholar] [CrossRef]
- Marcus, G.M.; Whooley, M.A.; Glidden, D.V.; Pawlikowska, L.; Zaroff, J.G.; Olgin, J.E. Interleukin-6 and atrial fibrillation in patients with coronary artery disease: Data from the Heart and Soul Study. Am. Heart J. 2008, 155, 303–309. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Shao, L.; Ma, J. Toll-Like Receptors 2 and 4 Predict New-Onset Atrial Fibrillation in Acute Myocardial Infarction Patients. Int. Heart J. 2018, 59, 64–70. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Sharma, D.; Du, F.; Liu, Y. The role of Toll-like receptor 2 and hypoxia-induced transcription factor-1α in the atrial structural remodeling of non-valvular atrial fibrillation. Int. J. Cardiol. 2013, 168, 2940–2941. [Google Scholar] [CrossRef] [PubMed]
- Maehama, T.; Okura, H.; Imai, K.; Saito, K.; Yamada, R.; Koyama, T.; Hayashida, A.; Neishi, Y.; Kawamoto, T.; Yoshida, K. Systemic inflammation and left atrial thrombus in patients with non-rheumatic atrial fibrillation. J. Cardiol. 2010, 56, 118–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaski, J.C.; Arrebola-Moreno, A.L. Inflamación y trombosis en la fibrilación auricular. Rev. Esp. Cardiol. 2011, 64, 551–553. [Google Scholar] [CrossRef]
- Shantsila, E.; Lip, G.Y. The role of monocytes in thrombotic disorders. Insights from tissue factor, monocyte-platelet aggregates and novel mechanisms. Thromb. Haemost. 2009, 102, 916–924. [Google Scholar] [CrossRef] [PubMed]
- Nair, G.M.; Nery, P.B.; Redpath, C.J.; Birnie, D.H. The Role Of Renin Angiotensin System In Atrial Fibrillation. J. Atr. Fibrillation 2014, 6, 972. [Google Scholar] [CrossRef] [PubMed]
- Sagris, M.; Theofilis, P.; Antonopoulos, A.S.; Tsioufis, C.; Oikonomou, E.; Antoniades, C.; Crea, F.; Kaski, J.C.; Tousoulis, D. Inflammatory Mechanisms in COVID-19 and Atherosclerosis: Current Pharmaceutical Perspectives. Int. J. Mol. Sci. 2021, 22, 6607. [Google Scholar] [CrossRef]
- Liu, K.; Daviglus, M.L.; Loria, C.M.; Colangelo, L.A.; Spring, B.; Moller, A.C.; Lloyd-Jones, D.M. Healthy lifestyle through young adulthood and the presence of low cardiovascular disease risk profile in middle age: The Coronary Artery Risk Development in (Young) Adults (CARDIA) study. Circulation 2012, 125, 996–1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaugai, S.; Meng, W.Y.; Ali Sepehry, A. Effects of RAAS Blockers on Atrial Fibrillation Prophylaxis: An Updated Systematic Review and Meta-Analysis of Randomized Controlled Trials. J. Cardiovasc. Pharmacol. Ther. 2016, 21, 388–404. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, F.I.; Castro, A. Obesity, adiposopathy, and quantitative imaging biomarkers. Radiol. Bras. 2017, 50, VII–VIII. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogunmoroti, O.; Michos, E.D.; Aronis, K.N.; Salami, J.A.; Blankstein, R.; Virani, S.S.; Spatz, E.S.; Allen, N.B.; Rana, J.S.; Blumenthal, R.S.; et al. Life’s Simple 7 and the risk of atrial fibrillation: The Multi-Ethnic Study of Atherosclerosis. Atherosclerosis 2018, 275, 174–181. [Google Scholar] [CrossRef]
- Sagris, M.; Kokkinidis, D.G.; Lempesis, I.G.; Giannopoulos, S.; Rallidis, L.; Mena-Hurtado, C.; Bakoyiannis, C. Nutrition, dietary habits, and weight management to prevent and treat patients with peripheral artery disease. Rev. Cardiovasc. Med. 2020, 21, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Carter, S.; Hartman, Y.; Holder, S.; Thijssen, D.H.; Hopkins, N.D. Sedentary Behavior and Cardiovascular Disease Risk: Mediating Mechanisms. Exerc. Sport Sci. Rev. 2017, 45, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Andresen, B.T.; Zhang, C. Inflammation and reactive oxygen species in cardiovascular disease. World J. Cardiol. 2010, 2, 408–410. [Google Scholar] [CrossRef]
- Abed, H.S.; Wittert, G.A.; Leong, D.P.; Shirazi, M.G.; Bahrami, B.; Middeldorp, M.E.; Lorimer, M.F.; Lau, D.H.; Antic, N.A.; Brooks, A.G.; et al. Effect of weight reduction and cardiometabolic risk factor management on symptom burden and severity in patients with atrial fibrillation: A randomized clinical trial. JAMA 2013, 310, 2050–2060. [Google Scholar] [CrossRef]
- Sagris, M.; Giannopoulos, S.; Giannopoulos, S.; Tzoumas, A.; Texakalidis, P.; Charisis, N.; Kokkinidis, D.G.; Malgor, R.D.; Mouawad, N.J.; Bakoyiannis, C. Transcervical carotid artery revascularization: A systematic review and meta-analysis of outcomes. J. Vasc. Surg. 2021, 74, 657–665.e12. [Google Scholar] [CrossRef]
- Lavie, C.J.; Pandey, A.; Lau, D.H.; Alpert, M.A.; Sanders, P. Obesity and Atrial Fibrillation Prevalence, Pathogenesis, and Prognosis: Effects of Weight Loss and Exercise. J. Am. Coll. Cardiol. 2017, 70, 2022–2035. [Google Scholar] [CrossRef]
- Davi, G.; Falco, A. Oxidant stress, inflammation and atherogenesis. Lupus 2005, 14, 760–764. [Google Scholar] [CrossRef]
- Larsson, S.C.; Drca, N.; Wolk, A. Alcohol consumption and risk of atrial fibrillation: A prospective study and dose-response meta-analysis. J. Am. Coll. Cardiol. 2014, 64, 281–289. [Google Scholar] [CrossRef]
- Ariansen, I.; Reims, H.M.; Gjesdal, K.; Olsen, M.H.; Ibsen, H.; Devereux, R.B.; Okin, P.M.; Kjeldsen, S.E.; Dahlof, B.; Wachtell, K. Impact of alcohol habits and smoking on the risk of new-onset atrial fibrillation in hypertensive patients with ECG left ventricular hypertrophy: The LIFE study. Blood Press 2012, 21, 6–11. [Google Scholar] [CrossRef]
- Di Castelnuovo, A.; Costanzo, S.; Bonaccio, M.; Rago, L.; De Curtis, A.; Persichillo, M.; Bracone, F.; Olivieri, M.; Cerletti, C.; Donati, M.B.; et al. Moderate Alcohol Consumption Is Associated With Lower Risk for Heart Failure But Not Atrial Fibrillation. JACC Heart Fail. 2017, 5, 837–844. [Google Scholar] [CrossRef] [PubMed]
- Baczko, I.; Light, P.E. Resveratrol and derivatives for the treatment of atrial fibrillation. Ann. N. Y. Acad. Sci. 2015, 1348, 68–74. [Google Scholar] [CrossRef] [Green Version]
- Stephan, L.S.; Almeida, E.D.; Markoski, M.M.; Garavaglia, J.; Marcadenti, A. Red Wine, Resveratrol and Atrial Fibrillation. Nutrients 2017, 9, 1190. [Google Scholar] [CrossRef] [Green Version]
- Kawada, T. Caffeine Consumption and Atrial Fibrillation: A Risk Assessment. Cardiology 2019, 142, 194. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Fan, W.; Budoff, M.J.; Heckbert, S.R.; Amsterdam, E.A.; Alonso, A.; Wong, N.D. Intermittent Nonhabitual Coffee Consumption and Risk of Atrial Fibrillation: The Multi-Ethnic Study of Atherosclerosis. J. Atr. Fibrillation 2019, 12, 2205. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.; Hu, Z.; Lu, X.; Huang, J.; Gu, D. Caffeine intake and atrial fibrillation incidence: Dose response meta-analysis of prospective cohort studies. Can. J. Cardiol. 2014, 30, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Bodar, V.; Chen, J.; Gaziano, J.M.; Albert, C.; Djousse, L. Coffee Consumption and Risk of Atrial Fibrillation in the Physicians’ Health Study. J. Am. Heart Assoc. 2019, 8, e011346. [Google Scholar] [CrossRef] [Green Version]
- Casiglia, E.; Tikhonoff, V.; Albertini, F.; Gasparotti, F.; Mazza, A.; Montagnana, M.; Danese, E.; Benati, M.; Spinella, P.; Palatini, P. Caffeine intake reduces incident atrial fibrillation at a population level. Eur. J. Prev. Cardiol. 2018, 25, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- Abdelfattah, R.; Kamran, H.; Lazar, J.; Kassotis, J. Does Caffeine Consumption Increase the Risk of New-Onset Atrial Fibrillation? Cardiology 2018, 140, 106–114. [Google Scholar] [CrossRef]
- Pastori, D.; Carnevale, R.; Bartimoccia, S.; Nocella, C.; Tanzilli, G.; Cangemi, R.; Vicario, T.; Catena, M.; Violi, F.; Pignatelli, P. Does Mediterranean Diet Reduce Cardiovascular Events and Oxidative Stress in Atrial Fibrillation? Antioxid Redox Signal. 2015, 23, 682–687. [Google Scholar] [CrossRef]
- Huang, W.L.; Yang, J.; Yang, J.; Wang, H.B.; Yang, C.J.; Yang, Y. Vitamin D and new-onset atrial fibrillation: A meta-analysis of randomized controlled trials. Hellenic J. Cardiol. 2018, 59, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Pastori, D.; Carnevale, R.; Menichelli, D.; Nocella, C.; Bartimoccia, S.; Novo, M.; Leo, I.; Violi, F.; Pignatelli, P. Is There an Interplay Between Adherence to Mediterranean Diet, Antioxidant Status, and Vascular Disease in Atrial Fibrillation Patients? Antioxid Redox Signal. 2016, 25, 751–755. [Google Scholar] [CrossRef]
- Pignatelli, P.; Pastori, D.; Farcomeni, A.; Nocella, C.; Bartimoccia, S.; Vicario, T.; Bucci, T.; Carnevale, R.; Violi, F. Mediterranean diet reduces thromboxane A2 production in atrial fibrillation patients. Clin. Nutr. 2015, 34, 899–903. [Google Scholar] [CrossRef]
- Pastori, D.; Carnevale, R.; Nocella, C.; Novo, M.; Santulli, M.; Cammisotto, V.; Menichelli, D.; Pignatelli, P.; Violi, F. Gut-Derived Serum Lipopolysaccharide is Associated With Enhanced Risk of Major Adverse Cardiovascular Events in Atrial Fibrillation: Effect of Adherence to Mediterranean Diet. J. Am. Heart Assoc. 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Larsson, S.C.; Drca, N.; Michaelsson, K. Serum Magnesium and Calcium Levels and Risk of Atrial Fibrillation. Circ. Genom. Precis. Med. 2019, 12, e002349. [Google Scholar] [CrossRef]
- Storz, M.A.; Helle, P. Atrial fibrillation risk factor management with a plant-based diet: A review. J. Arrhythm. 2019, 35, 781–788. [Google Scholar] [CrossRef] [Green Version]
- Dinesen, P.T.; Joensen, A.M.; Rix, T.A.; Tjonneland, A.; Schmidt, E.B.; Lundbye-Christensen, S.; Overvad, K. Effect of Dietary Intake of Saturated Fatty Acids on the Development of Atrial Fibrillation and the Effect of Replacement of Saturated With Monounsaturated and Polyunsaturated Fatty Acids. Am. J. Cardiol. 2017, 120, 1129–1132. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, L.M.; Lundbye-Christensen, S.; Schmidt, E.B.; Calder, P.C.; Schierup, M.H.; Tjonneland, A.; Parner, E.T.; Overvad, K. Long-chain n-3 and n-6 polyunsaturated fatty acids and risk of atrial fibrillation: Results from a Danish cohort study. PLoS ONE 2017, 12, e0190262. [Google Scholar] [CrossRef] [Green Version]
- Larsson, S.C.; Wolk, A. Fish, long-chain omega-3 polyunsaturated fatty acid intake and incidence of atrial fibrillation: A pooled analysis of two prospective studies. Clin. Nutr. 2017, 36, 537–541. [Google Scholar] [CrossRef]
- Li, F.R.; Chen, G.C.; Qin, J.; Wu, X. DietaryFish and Long-Chain n-3 Polyunsaturated Fatty Acids Intake and Risk of Atrial Fibrillation: A Meta-Analysis. Nutrients 2017, 9, 955. [Google Scholar] [CrossRef] [Green Version]
- Nattel, S.; Dobrev, D. Electrophysiological and molecular mechanisms of paroxysmal atrial fibrillation. Nat. Rev. Cardiol. 2016, 13, 575–590. [Google Scholar] [CrossRef] [PubMed]
- Levin, M.G.; Judy, R.; Gill, D.; Vujkovic, M.; Verma, S.S.; Bradford, Y.; Regeneron Genetics, C.; Ritchie, M.D.; Hyman, M.C.; Nazarian, S.; et al. Genetics of height and risk of atrial fibrillation: A Mendelian randomization study. PLoS Med. 2020, 17, e1003288. [Google Scholar] [CrossRef]
- Ebana, Y.; Furukawa, T. Networking analysis on superior vena cava arrhythmogenicity in atrial fibrillation. Int. J. Cardiol. Heart Vasc. 2019, 22, 150–153. [Google Scholar] [CrossRef]
- Ebana, Y.; Nitta, J.; Takahashi, Y.; Miyazaki, S.; Suzuki, M.; Liu, L.; Hirao, K.; Kanda, E.; Isobe, M.; Furukawa, T. Association of the Clinical and Genetic Factors With Superior Vena Cava Arrhythmogenicity in Atrial Fibrillation. Circ. J. 2017, 82, 71–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Low, S.K.; Takahashi, A.; Ebana, Y.; Ozaki, K.; Christophersen, I.E.; Ellinor, P.T.; Consortium, A.F.; Ogishima, S.; Yamamoto, M.; Satoh, M.; et al. Identification of six new genetic loci associated with atrial fibrillation in the Japanese population. Nat. Genet. 2017, 49, 953–958. [Google Scholar] [CrossRef]
- Lee, J.Y.; Kim, T.H.; Yang, P.S.; Lim, H.E.; Choi, E.K.; Shim, J.; Shin, E.; Uhm, J.S.; Kim, J.S.; Joung, B.; et al. Korean atrial fibrillation network genome-wide association study for early-onset atrial fibrillation identifies novel susceptibility loci. Eur. Heart J. 2017, 38, 2586–2594. [Google Scholar] [CrossRef] [Green Version]
- Mechakra, A.; Footz, T.; Walter, M.; Aranega, A.; Hernandez-Torres, F.; Morel, E.; Millat, G.; Yang, Y.Q.; Chahine, M.; Chevalier, P.; et al. A Novel PITX2c Gain-of-Function Mutation, p.Met207Val, in Patients With Familial Atrial Fibrillation. Am. J. Cardiol. 2019, 123, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Zhang, M.; Li, L.; Bai, Y.; Zhou, Y.; Moon, A.M.; Kaminski, H.J.; Martin, J.F. Pitx2, an atrial fibrillation predisposition gene, directly regulates ion transport and intercalated disc genes. Circ. Cardiovasc. Genet. 2014, 7, 23–32. [Google Scholar] [CrossRef] [Green Version]
- Syeda, F.; Kirchhof, P.; Fabritz, L. PITX2-dependent gene regulation in atrial fibrillation and rhythm control. J. Physiol. 2017, 595, 4019–4026. [Google Scholar] [CrossRef] [Green Version]
- Adam, O.; Lohfelm, B.; Thum, T.; Gupta, S.K.; Puhl, S.L.; Schafers, H.J.; Bohm, M.; Laufs, U. Role of miR-21 in the pathogenesis of atrial fibrosis. Basic Res. Cardiol. 2012, 107, 278. [Google Scholar] [CrossRef]
- Mase, M.; Grasso, M.; Avogaro, L.; Nicolussi Giacomaz, M.; D’Amato, E.; Tessarolo, F.; Graffigna, A.; Denti, M.A.; Ravelli, F. Upregulation of miR-133b and miR-328 in Patients With Atrial Dilatation: Implications for Stretch-Induced Atrial Fibrillation. Front. Physiol. 2019, 10, 1133. [Google Scholar] [CrossRef] [PubMed]
- Shen, N.N.; Zhang, C.; Li, Z.; Kong, L.C.; Wang, X.H.; Gu, Z.C.; Wang, J.L. MicroRNA expression signatures of atrial fibrillation: The critical systematic review and bioinformatics analysis. Exp. Biol. Med. 2020, 245, 42–53. [Google Scholar] [CrossRef] [Green Version]
- Zhelankin, A.V.; Vasiliev, S.V.; Stonogina, D.A.; Babalyan, K.A.; Sharova, E.I.; Doludin, Y.V.; Shchekochikhin, D.Y.; Generozov, E.V.; Akselrod, A.S. Elevated Plasma Levels of Circulating Extracellular miR-320a-3p in Patients with Paroxysmal Atrial Fibrillation. Int. J. Mol. Sci. 2020, 21, 3485. [Google Scholar] [CrossRef]
- Alonso, A.; Krijthe, B.P.; Aspelund, T.; Stepas, K.A.; Pencina, M.J.; Moser, C.B.; Sinner, M.F.; Sotoodehnia, N.; Fontes, J.D.; Janssens, A.C.; et al. Simple risk model predicts incidence of atrial fibrillation in a racially and geographically diverse population: The CHARGE-AF consortium. J. Am. Heart Assoc. 2013, 2, e000102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hindricks, G.; Potpara, T.; Dagres, N.; Arbelo, E.; Bax, J.J.; Blomstrom-Lundqvist, C.; Boriani, G.; Castella, M.; Dan, G.A.; Dilaveris, P.E.; et al. 2020 ESC Guidelines for the diagnosis and management of atrial fibrillation developed in collaboration with the European Association for Cardio-Thoracic Surgery (EACTS): The Task Force for the diagnosis and management of atrial fibrillation of the European Society of Cardiology (ESC) Developed with the special contribution of the European Heart Rhythm Association (EHRA) of the ESC. Eur. Heart J. 2021, 42, 373–498. [Google Scholar] [CrossRef] [PubMed]
- Schnabel, R.B.; Sullivan, L.M.; Levy, D.; Pencina, M.J.; Massaro, J.M.; D’Agostino, R.B., Sr.; Newton-Cheh, C.; Yamamoto, J.F.; Magnani, J.W.; Tadros, T.M.; et al. Development of a risk score for atrial fibrillation (Framingham Heart Study): A community-based cohort study. Lancet 2009, 373, 739–745. [Google Scholar] [CrossRef] [Green Version]
- Khatib, R.; Joseph, P.; Briel, M.; Yusuf, S.; Healey, J. Blockade of the renin-angiotensin-aldosterone system (RAAS) for primary prevention of non-valvular atrial fibrillation: A systematic review and meta analysis of randomized controlled trials. Int. J. Cardiol. 2013, 165, 17–24. [Google Scholar] [CrossRef]
- Kumagai, K.; Nakashima, H.; Urata, H.; Gondo, N.; Arakawa, K.; Saku, K. Effects of angiotensin II type 1 receptor antagonist on electrical and structural remodeling in atrial fibrillation. J. Am. Coll. Cardiol. 2003, 41, 2197–2204. [Google Scholar] [CrossRef] [Green Version]
- Bonora, B.M.; Raschi, E.; Avogaro, A.; Fadini, G.P. SGLT-2 inhibitors and atrial fibrillation in the Food and Drug Administration adverse event reporting system. Cardiovasc. Diabetol. 2021, 20, 39. [Google Scholar] [CrossRef] [PubMed]
- Dobrev, D.; Potpara, T.S. Smart device-based detection of atrial fibrillation: Opportunities and challenges in the emerging world of digital health. Int. J. Cardiol. 2020, 302, 108–109. [Google Scholar] [CrossRef] [PubMed]
- Seshadri, D.R.; Bittel, B.; Browsky, D.; Houghtaling, P.; Drummond, C.K.; Desai, M.Y.; Gillinov, A.M. Accuracy of Apple Watch for Detection of Atrial Fibrillation. Circulation 2020, 141, 702–703. [Google Scholar] [CrossRef] [PubMed]
- Roselli, C.; Rienstra, M.; Ellinor, P.T. Genetics of Atrial Fibrillation in 2020: GWAS, Genome Sequencing, Polygenic Risk, and Beyond. Circ. Res. 2020, 127, 21–33. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Protein | Protein Serum Levels Difference | Atrial Tissue Levels Difference | Predictor for AF |
|---|---|---|---|
| CRP | NA | NA | Yes |
| MCP-1 | + | + | No |
| MPO | NA | + | No |
| TGF-β | NA | + | No |
| TNF | NA | + | No |
| HSP-27 | + | + | NA |
| HSP-70 | - | - | NA |
| IL-1 | NA | NA | NA |
| IL-6 | NA | + | No |
| IL-8 | + | + | NA |
| IL-10 | NA | + | NA |
| Gene of | Polymorphism-Mutation | Action |
|---|---|---|
| ABCC9 (I KATP) KCNA5 (I Kur) HCN4 (I f) KCND3 (I Ks) KCNE1 (IKs) KCNE2 (IKs) KCNE3 (IKs) KCNE4 (IKs) KCNE5 (IKs) KCNH2 (IKr) KCNJ2 (I K1) KCNJ5 (I KAch) KCNJ8 (I KATP) KCNN3 (IAHP) KCNQ1 (IKs) | Potassium (K+) channel genes | The increased K+ current abbreviates refractoriness and promotes re-entry, while tending to reduce automaticity |
| SCN1B SCN2B SCN3B SCN4B SCN5A SCN10A | Sodium (Na+) channel genes | Delay repolarization and promote Ca+2 mediated after depolarization |
| GJA5 | Mutations in the gap junctional protein | Re-entry mechanism |
| NUP155 | Nuclear pore complex (nucleoporin) Nup155 | Re-entry mechanism |
| E169K | Junctophilin mutation | Delay repolarization and promote Ca+2 mediated after depolarization enhancing RyR2 Ca+2 leak |
| CASR | rs1801725 | Delay repolarization and promote Ca+2 mediated after depolarization |
| PITX2 | rs2200733 rs10033464 rs2634073 | PITX2 deficiency results in electrical and structural remodelling |
| NURL1 | rs6584555 rs6584555 | Undefined |
| PRRX1 | rs593479 | Undefined |
| CAV1 | rs1177384 | Undefined |
| CUX2 | rs649002 | Undefined |
| ZFHX3 | rs12932445 | Undefined |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sagris, M.; Vardas, E.P.; Theofilis, P.; Antonopoulos, A.S.; Oikonomou, E.; Tousoulis, D. Atrial Fibrillation: Pathogenesis, Predisposing Factors, and Genetics. Int. J. Mol. Sci. 2022, 23, 6. https://doi.org/10.3390/ijms23010006
Sagris M, Vardas EP, Theofilis P, Antonopoulos AS, Oikonomou E, Tousoulis D. Atrial Fibrillation: Pathogenesis, Predisposing Factors, and Genetics. International Journal of Molecular Sciences. 2022; 23(1):6. https://doi.org/10.3390/ijms23010006
Chicago/Turabian StyleSagris, Marios, Emmanouil P. Vardas, Panagiotis Theofilis, Alexios S. Antonopoulos, Evangelos Oikonomou, and Dimitris Tousoulis. 2022. "Atrial Fibrillation: Pathogenesis, Predisposing Factors, and Genetics" International Journal of Molecular Sciences 23, no. 1: 6. https://doi.org/10.3390/ijms23010006