
Immune Signaling Kinases in Amyotrophic Lateral Sclerosis (ALS) and Frontotemporal Dementia (FTD)

Abstract
:1. Pathological and Molecular Features of ALS and FTD
2. Importance of Immune-Related Kinases in ALS and FTD
3. Kinases and Immune Signaling in ALS and FTD Pathogenesis
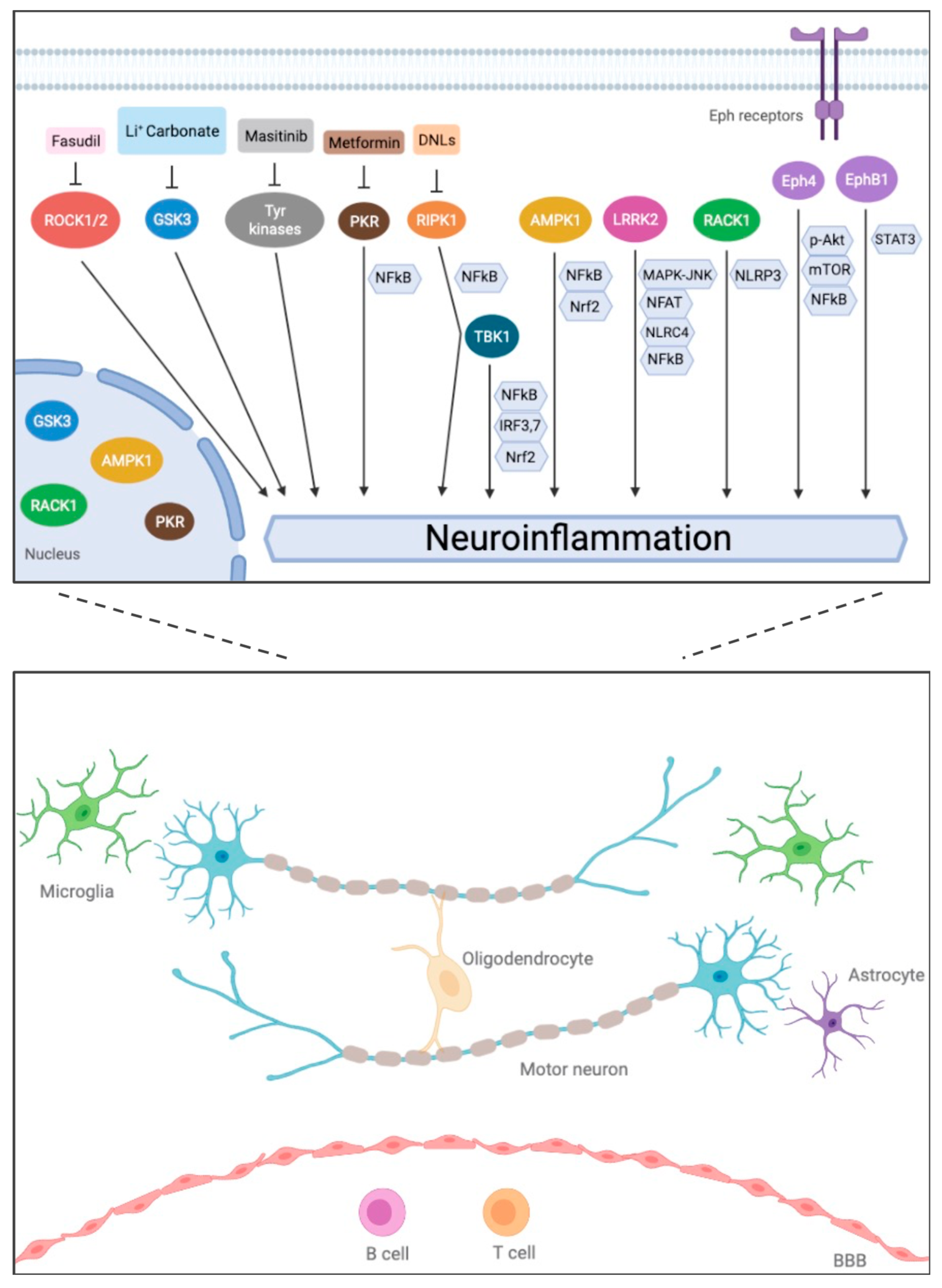
3.1. TBK1 (TANK-Binding Kinase 1)
3.2. RIPK1 (Receptor-Interacting Kinase 1)
3.3. Eph Receptors (Ephrin Receptor Family)
3.4. RACK1 (Receptor of Activated Protein C Kinase 1)
3.5. AMPK (AMP-Activated Protein Kinase) and LRRK2 (Leucine-Rich-Repeat Kinase 2), New Immune Signaling Kinases in ALS/FTD
4. Other Emerging Signaling Kinases in ALS/FTD
5. Immune Kinase Modulation for ALS and FTD in Disease Managing and in Clinical Trials
5.1. Masitinib: A Receptor Tyr Kinase Inhibitor
5.2. Fasudil: A ROCK1/2 (Rho-Kinase 1/2) Inhibitor
5.3. DNL Compounds: RIPK1 Inhibitors
5.4. Lithium Carbonate: A GSK3α/β (Glycogen Synthase Kinase 3) Inhibitor
6. Immune Kinase Modulators in the Pipeline towards ALS/FTD Therapy
6.1. Metformin: A PKR Inhibitor
6.2. C4: An Epha4 Ligand
7. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Caballero-Hernandez, D.; Toscano, M.G.; Cejudo-Guillen, M.; Garcia-Martin, M.L.; Lopez, S.; Franco, J.M.; Quintana, F.J.; Roodveldt, C.; Pozo, D. The “Omics” of Amyotrophic Lateral Sclerosis. Trends Mol. Med. 2016, 22, 53–67. [Google Scholar] [CrossRef]
- Jimenez-Pacheco, A.; Franco, J.M.; Lopez, S.; Gomez-Zumaquero, J.M.; Magdalena Leal-Lasarte, M.; Caballero-Hernandez, D.E.; Cejudo-Guillén, M.; Pozo, D. Epigenetic Mechanisms of Gene Regulation in Amyotrophic Lateral Sclerosis. Adv. Exp. Med. Biol. 2017, 978, 255–275. [Google Scholar] [CrossRef] [PubMed]
- McCauley, M.E.; Baloh, R.H. Inflammation in ALS/FTD pathogenesis. Acta Neuropathol. 2019, 137, 715–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beers, D.R.; Appel, S.H. Immune dysregulation in amyotrophic lateral sclerosis: Mechanisms and emerging therapies. Lancet Neurol. 2019, 18, 211–220. [Google Scholar] [CrossRef]
- Mejzini, R.; Flynn, L.L.; Pitout, I.L.; Fletcher, S.; Wilton, S.D.; Akkari, P.A. ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now? Front. Neurosci. 2019, 13, 1310. [Google Scholar] [CrossRef] [Green Version]
- Wood, A.; Gurfinkel, Y.; Polain, N.; Lamont, W.; Lyn Rea, S. Molecular Mechanisms Underlying TDP-43 Pathology in Cellular and Animal Models of ALS and FTLD. Int. J. Mol. Sci. 2021, 22, 4705. [Google Scholar] [CrossRef]
- Ransohoff, R.M. How neuroinflammation contributes to neurodegeneration. Science 2016, 353, 777–783. [Google Scholar] [CrossRef]
- Lomen-Hoerth, C.; Anderson, T.; Miller, B. The overlap of amyotrophic lateral sclerosis and frontotemporal dementia. Neurology 2002, 59, 1077–1079. [Google Scholar] [CrossRef]
- Strong, M.J.; Grace, G.M.; Freedman, M.; Lomen-Hoerth, C.; Woolley, S.; Goldstein, L.H.; Murphy, J.; Shoesmith, C.; Rosenfeld, J.; Leigh, P.N.; et al. Consensus criteria for the diagnosis of frontotemporal cognitive and behavioural syndromes in amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 2009, 10, 131–146. [Google Scholar] [CrossRef] [PubMed]
- Abramzon, Y.A.; Fratta, P.; Traynor, B.J.; Chia, R. The Overlapping Genetics of Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Front. Neurosci. 2020, 14, 42. [Google Scholar] [CrossRef] [Green Version]
- Mannini, B.; Vecchi, G.; Labrador-Garrido, A.; Fabre, B.; Fani, G.; Franco, J.M.; Lilley, K.; Pozo, D.; Vendruscolo, M.; Chiti, F.; et al. Differential Interactome and Innate Immune Response Activation of Two Structurally Distinct Misfolded Protein Oligomers. ACS Chem. Neurosci. 2019, 10, 3464–3478. [Google Scholar] [CrossRef] [PubMed]
- Leal-Lasarte, M.; Mannini, B.; Chiti, F.; Vendruscolo, M.; Dobson, C.M.; Roodveldt, C.; Pozo, D. Distinct responses of human peripheral blood cells to different misfolded protein oligomers. Immunology 2021, 64, 358–371. [Google Scholar] [CrossRef] [PubMed]
- Haukedal, H.; Freude, K. Implications of Microglia in Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. J. Mol. Biol. 2019, 431, 1818–1829. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Vandoorne, T.; Steyaert, J.; Staats, K.A.; Van Den Bosch, L. The multifaceted role of kinases in amyotrophic lateral sclerosis: Genetic, pathological and therapeutic implications. Brain 2020, 143, 1651–1673. [Google Scholar] [CrossRef] [PubMed]
- Cirulli, E.T.; Lasseigne, B.N.; Petrovski, S.; Sapp, P.C.; Dion, P.A.; Leblond, C.S.; Couthouis, J.; Lu, Y.F.; Wang, Q.; Krueger, B.J.; et al. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 2015, 347, 1436–1441. [Google Scholar] [CrossRef] [Green Version]
- Freischmidt, A.; Wieland, T.; Richter, B.; Ruf, W.; Schaeffer, V.; Müller, K.; Marroquin, N.; Nordin, F.; Hübers, A.; Weydt, P.; et al. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat. Neurosci. 2015, 18, 631–636. [Google Scholar] [CrossRef]
- Oakes, J.A.; Davies, M.C.; Collins, M.O. TBK1: A new player in ALS linking autophagy and neuroinflammation. Mol. Brain 2017, 10, 5. [Google Scholar] [CrossRef] [Green Version]
- Gijselinck, I.; Van Mossevelde, S.; van der Zee, J.; Sieben, A.; Philtjens, S.; Heeman, B.; Engelborghs, S.; Vandenbulcke, M.; De Baets, G.; Bäumer, V.; et al. Loss of TBK1 is a frequent cause of frontotemporal dementia in a Belgian cohort. Neurology 2015, 85, 2116–2125. [Google Scholar] [CrossRef] [Green Version]
- Pottier, C.; Bieniek, K.F.; Finch, N.; van de Vorst, M.; Baker, M.; Perkersen, R.; Brown, P.; Ravenscroft, T.; van Blitterswijk, M.; Nicholson, A.M.; et al. Whole-genome sequencing reveals important role for TBK1 and OPTN mutations in frontotemporal lobar degeneration without motor neuron disease. Acta Neuropathol. 2015, 130, 77–92. [Google Scholar] [CrossRef]
- de Majo, M.; Topp, S.D.; Smith, B.N.; Nishimura, A.L.; Chen, H.J.; Gkazi, A.S.; Miller, J.; Wong, C.H.; Vance, C.; Baas, F.; et al. ALS-associated missense and nonsense TBK1 mutations can both cause loss of kinase function. Neurobiol. Aging 2018, 71, 266.e1–266.e10. [Google Scholar] [CrossRef]
- Duan, W.; Guo, M.; Yi, L.; Zhang, J.; Bi, Y.; Liu, Y.; Li, Y.; Li, Z.; Ma, Y.; Zhang, G.; et al. Deletion of Tbk1 disrupts autophagy and reproduces behavioral and locomotor symptoms of FTD-ALS in mice. Aging 2019, 11, 2457–2476. [Google Scholar] [CrossRef]
- van der Zee, J.; Gijselinck, I.; Van Mossevelde, S.; Perrone, F.; Dillen, L.; Heeman, B.; Bäumer, V.; Engelborghs, S.; De Bleecker, J.; Baets, J.; et al. TBK1 Mutation Spectrum in an Extended European Patient Cohort with Frontotemporal Dementia and Amyotrophic Lateral Sclerosis. Hum. Mutat. 2017, 38, 297–309. [Google Scholar] [CrossRef] [Green Version]
- Foster, A.D.; Downing, P.; Figredo, E.; Polain, N.; Stott, A.; Layfield, R.; Rea, S.L. ALS-associated TBK1 variant p.G175S is defective in phosphorylation of p62 and impacts TBK1-mediated signalling and TDP-43 autophagic degradation. Mol. Cell. Neurosci. 2020, 108, 103539. [Google Scholar] [CrossRef]
- Moore, A.S.; Holzbaur, E.L. Dynamic recruitment and activation of ALS-associated TBK1 with its target optineurin are required for efficient mitophagy. Proc. Natl. Acad. Sci. USA 2016, 113, E3349–E3358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, L.; Zhang, S.Y.; Casanova, J.L.; Sancho-Shimizu, V. Human TBK1: A Gatekeeper of Neuroinflammation. Trends Mol. Med. 2016, 22, 511–527. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Cheung, J.; Gerbino, V.; Ahlsén, G.; Zimanyi, C.; Hirsh, D.; Maniatis, T. Effects of ALS-associated TANK binding kinase 1 mutations on protein-protein interactions and kinase activity. Proc. Natl. Acad. Sci. USA 2019, 116, 24517–24526. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Jin, T.; Zhu, H.; Chen, H.; Ofengeim, D.; Zou, C.; Mifflin, L.; Pan, L.; Amin, P.; Li, W.; et al. TBK1 Suppresses RIPK1-Driven Apoptosis and Inflammation during Development and in Aging. Cell 2018, 174, 1477–1491.e19. [Google Scholar] [CrossRef] [Green Version]
- Brenner, D.; Sieverding, K.; Bruno, C.; Lüningschrör, P.; Buck, E.; Mungwa, S.; Fischer, L.; Brockmann, S.J.; Ulmer, J.; Bliederhäuser, C.; et al. Heterozygous Tbk1 loss has opposing effects in early and late stages of ALS in mice. J. Exp. Med. 2019, 216, 267–278. [Google Scholar] [CrossRef] [Green Version]
- Gerbino, V.; Kaunga, E.; Ye, J.; Canzio, D.; O’Keeffe, S.; Rudnick, N.D.; Guarnieri, P.; Lutz, C.M.; Maniatis, T. The Loss of TBK1 Kinase Activity in Motor Neurons or in All Cell Types Differentially Impacts ALS Disease Progression in SOD1 Mice. Neuron 2020, 106, 789–805.e5. [Google Scholar] [CrossRef]
- Yuan, J.; Amin, P.; Ofengeim, D. Necroptosis and RIPK1-mediated neuroinflammation in CNS diseases. Nat. Rev. Neurosci. 2019, 20, 19–33. [Google Scholar] [CrossRef]
- Humphries, F.; Yang, S.; Wang, B.; Moynagh, P.N. RIP kinases: Key decision makers in cell death and innate immunity. Cell Death Differ. 2015, 22, 225–236. [Google Scholar] [CrossRef] [Green Version]
- Christofferson, D.E.; Li, Y.; Hitomi, J.; Zhou, W.; Upperman, C.; Zhu, H.; Gerber, S.A.; Gygi, S.; Yuan, J. A novel role for RIP1 kinase in mediating TNFα production. Cell Death Dis. 2012, 3, e320. [Google Scholar] [CrossRef]
- Ito, Y.; Ofengeim, D.; Najafov, A.; Das, S.; Saberi, S.; Li, Y.; Hitomi, J.; Zhu, H.; Chen, H.; Mayo, L.; et al. RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Science 2016, 353, 603–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mifflin, L.; Hu, Z.; Dufort, C.; Hession, C.C.; Walker, A.J.; Niu, K.; Zhu, H.; Liu, N.; Liu, J.S.; Levin, J.Z.; et al. A RIPK1-regulated inflammatory microglial state in amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2021, 118, e2025102118. [Google Scholar] [CrossRef]
- Kania, A.; Klein, R. Mechanisms of ephrin-Eph signalling in development, physiology and disease. Nat. Rev. Mol. Cell Biol. 2016, 17, 240–256. [Google Scholar] [CrossRef] [PubMed]
- Van Hoecke, A.; Schoonaert, L.; Lemmens, R.; Timmers, M.; Staats, K.A.; Laird, A.S.; Peeters, E.; Philips, T.; Goris, A.; Dubois, B.; et al. EPHA4 is a disease modifier of amyotrophic lateral sclerosis in animal models and in humans. Nat. Med. 2012, 18, 1418–1422. [Google Scholar] [CrossRef] [PubMed]
- Goldshmit, Y.; Galea, M.P.; Wise, G.; Bartlett, P.F.; Turnley, A.M. Axonal regeneration and lack of astrocytic gliosis in EphA4-deficient mice. J. Neurosci. 2004, 24, 10064–10073. [Google Scholar] [CrossRef] [Green Version]
- Kowalski, E.A.; Chen, J.; Hazy, A.; Fritsch, L.E.; Gudenschwager-Basso, E.K.; Chen, M.; Wang, X.; Qian, Y.; Zhou, M.; Byerly, M.; et al. Peripheral loss of EphA4 ameliorates TBI-induced neuroinflammation and tissue damage. J. Neuroinflamm. 2019, 16, 210. [Google Scholar] [CrossRef]
- Schoonaert, L.; Rué, L.; Roucourt, B.; Timmers, M.; Little, S.; Chávez-Gutiérrez, L.; Dewilde, M.; Joyce, P.; Curnock, A.; Weber, P.; et al. Identification and characterization of Nanobodies targeting the EphA4 receptor. J. Biol. Chem. 2017, 292, 11452–11465. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Cooper, L.T.; Boyd, A.W.; Bartlett, P.F. Decreased signalling of EphA4 improves functional performance and motor neuron survival in the SOD1 G93A ALS mouse model. Sci. Rep. 2018, 8, 11393. [Google Scholar] [CrossRef] [Green Version]
- Wu, B.; De, S.K.; Kulinich, A.; Salem, A.F.; Koeppen; Wang, R.; Barile, E.; Wang, S.; Zhang, D.; Ethell, I.; et al. Potent and Selective EphA4 Agonists for the Treatment of ALS. Cell Chem. Biol. 2017, 24, 293–305. [Google Scholar] [CrossRef] [Green Version]
- Ling, K.K.; Jackson, M.; Alkam, D.; Liu, D.; Allaire, N.; Sun, C.; Kiaei, M.; McCampbell, A.; Rigo, F. Antisense-mediated reduction of EphA4 in the adult CNS does not improve the function of mice with amyotrophic lateral sclerosis. Neurobiol. Dis. 2018, 114, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Rué, L.; Timmers, M.; Lenaerts, A.; Smolders, S.; Poppe, L.; de Boer, A.; Van Den Bosch, L.; Van Damme, P.; Robberecht, W.; Lemmens, R. Reducing EphA4 before disease onset does not affect survival in a mouse model of Amyotrophic Lateral Sclerosis. Sci. Rep. 2019, 9, 14112. [Google Scholar] [CrossRef] [Green Version]
- Tyzack, G.E.; Hall, C.E.; Sibley; Cymes, T.; Forostyak, S.; Carlino, G.; Meyer, I.F.; Schiavo, G.; Zhang, S.C.; Gibbons, G.M.; et al. A neuroprotective astrocyte state is induced by neuronal signal EphB1 but fails in ALS models. Nat. Commun. 2017, 8, 1164. [Google Scholar] [CrossRef] [PubMed]
- Russo, A.; Scardigli, R.; La Regina, F.; Murray, M.E.; Romano, N.; Dickson, D.W.; Wolozin, B.; Cattaneo, A.; Ceci, M. Increased cytoplasmic TDP-43 reduces global protein synthesis by interacting with RACK1 on polyribosomes. Hum. Mol. Genet. 2017, 26, 1407–1418. [Google Scholar] [CrossRef] [Green Version]
- Yin, H.; Song, S.; Pan, X. Knockdown of miR-155 protects microglia against LPS-induced inflammatory injury via targeting RACK1: A novel research for intracranial infection. J. Inflamm. 2017, 14, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, Y.; Zhang, L.; Angosto-Bazarra, D.; Pelegrín, P.; Núñez, G.; He, Y. RACK1 Mediates NLRP3 Inflammasome Activation by Promoting NLRP3 Active Conformation and Inflammasome Assembly. Cell Rep. 2020, 33, 108405. [Google Scholar] [CrossRef]
- Perera, N.D.; Turner, B.J. AMPK Signalling and Defective Energy Metabolism in Amyotrophic Lateral Sclerosis. Neurochem. Res. 2016, 41, 544–553. [Google Scholar] [CrossRef]
- Beckers, J.; Tharkeshwar, A.K.; Van Damme, P. C9orf72 ALS-FTD: Recent evidence for dysregulation of the autophagy-lysosome pathway at multiple levels. Autophagy 2021, 17, 3306–3322. [Google Scholar] [CrossRef]
- Root, J.; Merino, P.; Nuckols, A.; Johnson, M.; Kukar, T. Lysosome dysfunction as a cause of neurodegenerative diseases: Lessons from frontotemporal dementia and amyotrophic lateral sclerosis. Neurobiol. Dis. 2021, 154, 105360. [Google Scholar] [CrossRef]
- Saito, M.; Saito, M.; Das, B.C. Involvement of AMP-activated protein kinase in neuroinflammation and neurodegeneration in the adult and developing brain. Int. J. Dev. Neurosci. 2019, 77, 48–59. [Google Scholar] [CrossRef]
- Broce, I.; Karch, C.M.; Wen, N.; Fan, C.C.; Wang, Y.; Tan, C.H.; Kouri, N.; Ross, O.A.; Höglinger, G.U.; Muller, U.; et al. Immune-related genetic enrichment in frontotemporal dementia: An analysis of genome-wide association studies. PLoS Med. 2018, 15, e1002487. [Google Scholar] [CrossRef]
- Dächsel, J.C.; Ross, O.A.; Mata, I.F.; Kachergus, J.; Toft, M.; Cannon, A.; Baker, M.; Adamson, J.; Hutton, M.; Dickson, D.W.; et al. Lrrk2 G2019S substitution in frontotemporal lobar degeneration with ubiquitin-immunoreactive neuronal inclusions. Acta Neuropathol. 2007, 113, 601–606. [Google Scholar] [CrossRef]
- Ferrari, R.; Wang, Y.; Vandrovcova, J.; Guelfi, S.; Witeolar, A.; Karch, C.M.; Schork, A.J.; Fan, C.C.; Brewer, J.B.; Momeni, P.; et al. Genetic architecture of sporadic frontotemporal dementia and overlap with Alzheimer’s and Parkinson’s diseases. J. Neurol. Neurosurg. Psychiatry 2017, 88, 152–164. [Google Scholar] [CrossRef]
- Ciani, M.; Bonvicini, C.; Scassellati, C.; Carrara, M.; Maj, C.; Fostinelli, S.; Binetti, G.; Ghidoni, R.; Benussi, L. The Missing Heritability of Sporadic Frontotemporal Dementia: New Insights from Rare Variants in Neurodegenerative Candidate Genes. Int. J. Mol. Sci. 2019, 20, 3903. [Google Scholar] [CrossRef] [Green Version]
- Ahmadi Rastegar, D.; Dzamko, N. Leucine Rich Repeat Kinase 2 and Innate Immunity. Front. Neurosci. 2020, 14, 193. [Google Scholar] [CrossRef] [Green Version]
- Wallings, R.L.; Tansey, M.G. LRRK2 regulation of immune-pathways and inflammatory disease. Biochem. Soc. Trans. 2019, 47, 1581–1595. [Google Scholar] [CrossRef] [PubMed]
- Gloeckner, C.J.; Schumacher, A.; Boldt, K.; Ueffing, M. The Parkinson disease-associated protein kinase LRRK2 exhibits MAPKKK activity and phosphorylates MKK3/6 and MKK4/7, in vitro. J. Neurochem. 2009, 109, 959–968. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.H.; Chan, D.; Wolozin, B. LRRK2 and the stress response: Interaction with MKKs and JNK-interacting proteins. Neurodegener. Dis. 2010, 7, 68–75. [Google Scholar] [CrossRef]
- Chen, C.Y.; Weng, Y.H.; Chien, K.Y.; Lin, K.J.; Yeh, T.H.; Cheng, Y.P.; Lu, C.S.; Wang, H.L. (G2019S) LRRK2 activates MKK4-JNK pathway and causes degeneration of SN dopaminergic neurons in a transgenic mouse model of PD. Cell Death Differ. 2012, 19, 1623–1633. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Lee, J.; Krummey, S.; Lu, W.; Cai, H.; Lenardo, M.J. The kinase LRRK2 is a regulator of the transcription factor NFAT that modulates the severity of inflammatory bowel disease. Nat. Immunol. 2011, 12, 1063–1070. [Google Scholar] [CrossRef] [Green Version]
- Panagiotakopoulou, V.; Ivanyuk, D.; De Cicco, S.; Haq, W.; Arsić, A.; Yu, C.; Messelodi, D.; Oldrati, M.; Schöndorf, D.C.; Perez, M.J.; et al. Interferon-γ signaling synergizes with LRRK2 in neurons and microglia derived from human induced pluripotent stem cells. Nat. Commun. 2020, 11, 5163. [Google Scholar] [CrossRef] [PubMed]
- Takagawa, T.; Kitani, A.; Fuss, I.; Levine, B.; Brant, S.R.; Peter, I.; Tajima, M.; Nakamura, S.; Strober, W. An increase in LRRK2 suppresses autophagy and enhances Dectin-1-induced immunity in a mouse model of colitis. Sci. Transl. Med. 2018, 10, eaan8162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Liu, X.; Li, Y.; Zhao, J.; Liu, Z.; Hu, Z.; Wang, Y.; Yao, Y.; Miller, A.W.; Su, B.; et al. LRRK2 promotes the activation of NLRC4 inflammasome during Salmonella Typhimurium infection. J. Exp. Med. 2017, 214, 3051–3066. [Google Scholar] [CrossRef] [PubMed]
- Russo, I.; Berti, G.; Plotegher, N.; Bernardo, G.; Filograna, R.; Bubacco, L.; Greggio, E. Leucine-rich repeat kinase 2 positively regulates inflammation and down-regulates NF-κB p50 signaling in cultured microglia cells. J. Neuroinflamm. 2015, 12, 230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russo, I.; Di Benedetto, G.; Kaganovich, A.; Ding, J.; Mercatelli, D.; Morari, M.; Cookson, M.R.; Bubacco, L.; Greggio, E. Leucine-rich repeat kinase 2 controls protein kinase A activation state through phosphodiesterase 4. J. Neuroinflamm. 2018, 15, 297. [Google Scholar] [CrossRef]
- Ho, C.C.-Y.; Rideout, H.J.; Ribe, E.; Troy, C.M.; Dauer, W.T. The Parkinson Disease Protein Leucine-Rich Repeat Kinase 2 Transduces Death Signals via Fas-Associated Protein with Death Domain and Caspase-8 in a Cellular Model of Neurodegeneration. J. Neurosci. 2009, 29, 1011. [Google Scholar] [CrossRef]
- Amin, P.; Florez, M.; Najafov, A.; Pan, H.; Geng, J.; Ofengeim, D.; Dziedzic, S.A.; Wang, H.; Barrett, V.J.; Ito, Y.; et al. Regulation of a distinct activated RIPK1 intermediate bridging complex I and complex II in TNFα-mediated apoptosis. Proc. Natl. Acad. Sci. USA 2018, 115, E5944. [Google Scholar] [CrossRef] [Green Version]
- Balachandran, S.; Roberts, P.C.; Brown, L.E.; Truong, H.; Pattnaik, A.K.; Archer, D.R.; Barber, G.N. Essential role for the dsRNA-dependent protein kinase PKR in innate immunity to viral infection. Immunity 2000, 13, 129–141. [Google Scholar] [CrossRef] [Green Version]
- Prudencio, M.; Gonzales, P.K.; Cook, C.N.; Gendron, T.F.; Daughrity, L.M.; Song, Y.; Ebbert, M.T.W.; van Blitterswijk, M.; Zhang, Y.J.; Jansen-West, K.; et al. Repetitive element transcripts are elevated in the brain of C9orf72 ALS/FTLD patients. Hum. Mol. Genet. 2017, 26, 3421–3431. [Google Scholar] [CrossRef]
- Li, W.; Jin, Y.; Prazak, L.; Hammell, M.; Dubnau, J. Transposable elements in TDP-43-mediated neurodegenerative disorders. PLoS ONE 2012, 7, e44099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LaRocca, T.J.; Mariani, A.; Watkins, L.R.; Link, C.D. TDP-43 knockdown causes innate immune activation via protein kinase R in astrocytes. Neurobiol. Dis. 2019, 132, 104514. [Google Scholar] [CrossRef]
- Hu, J.H.; Zhang, H.; Wagey, R.; Krieger, C.; Pelech, S.L. Protein kinase and protein phosphatase expression in amyotrophic lateral sclerosis spinal cord. J. Neurochem. 2003, 85, 432–442. [Google Scholar] [CrossRef] [PubMed]
- Zu, T.; Guo, S.; Bardhi, O.; Ryskamp, D.A.; Li, J.; Khoramian Tusi, S.; Engelbrecht, A.; Klippel, K.; Chakrabarty, P.; Nguyen, L.; et al. Metformin inhibits RAN translation through PKR pathway and mitigates disease in C9orf72 ALS/FTD mice. Proc. Natl. Acad. Sci. USA 2020, 117, 18591–18599. [Google Scholar] [CrossRef]
- Ash, P.E.; Bieniek, K.F.; Gendron, T.F.; Caulfield, T.; Lin, W.L.; Dejesus-Hernandez, M.; van Blitterswijk, M.M.; Jansen-West, K.; Paul, J.W.; Rademakers, R.; et al. Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron 2013, 77, 639–646. [Google Scholar] [CrossRef] [Green Version]
- Mori, K.; Weng, S.M.; Arzberger, T.; May, S.; Rentzsch, K.; Kremmer, E.; Schmid, B.; Kretzschmar, H.A.; Cruts, M.; Van Broeckhoven, C.; et al. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 2013, 339, 1335–1338. [Google Scholar] [CrossRef]
- Zu, T.; Liu, Y.; Bañez-Coronel, M.; Reid, T.; Pletnikova, O.; Lewis, J.; Miller, T.M.; Harms, M.B.; Falchook, A.E.; Subramony, S.H.; et al. RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc. Natl. Acad. Sci. USA 2013, 110, E4968–E4977. [Google Scholar] [CrossRef] [Green Version]
- Leal-Lasarte, M.M.; Franco, J.M.; Labrador-Garrido, A.; Pozo, D.; Roodveldt, C. Extracellular TDP-43 aggregates target MAPK/MAK/MRK overlapping kinase (MOK) and trigger caspase-3/IL-18 signaling in microglia. FASEB J. 2017, 31, 2797–2816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyata, Y.; Akashi, M.; Nishida, E. Molecular cloning and characterization of a novel member of the MAP kinase superfamily. Genes Cells 1999, 4, 299–309. [Google Scholar] [CrossRef]
- Fu, Z. Regulations and Functions of ICK/MAK/MOK—A Novel MAPK-Related Kinase Family Linked to Human Diseases. In Protein Kinases; InTech: London, UK, 2012. [Google Scholar]
- Panza, F.; Lozupone, M.; Seripa, D.; Daniele, A.; Watling, M.; Giannelli, G.; Imbimbo, B.P. Development of disease-modifying drugs for frontotemporal dementia spectrum disorders. Nat. Rev. Neurol. 2020, 16, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Jaiswalv, M.K. Riluzole and edaravone: A tale of two amyotrophic lateral sclerosis drugs. Med. Res. Rev. 2019, 39, 733–748. [Google Scholar] [CrossRef] [PubMed]
- Cheah, B.C.; Vucic, S.; Krishnan, A.V.; Kiernan, K. Riluzole, neuroprotection and amyotrophic lateral sclerosis. Curr. Med. Chem. 2010, 17, 1942–1959. [Google Scholar] [CrossRef]
- Noh, K.M.; Hwang, J.Y.; Shin, H.C.; Koh, J.Y. A novel neuroprotective mechanism of riluzole: Direct inhibition of protein kinase C. Neurobiol. Dis. 2000, 7, 375–383. [Google Scholar] [CrossRef] [Green Version]
- Altman, A.; Kong, K.F. Protein Kinase C Enzymes in the Hematopoietic and Immune Systems. Annu. Rev. Immunol. 2016, 34, 511–538. [Google Scholar] [CrossRef] [PubMed]
- Dubreuil, P.; Letard, S.; Ciufolini, M.; Gros, L.; Humbert, M.; Castéran, N.; Borge, L.; Hajem, B.; Lermet, A.; Sippl, W.; et al. Masitinib (AB1010), a potent and selective tyrosine kinase inhibitor targeting KIT. PLoS ONE 2009, 4, e7258. [Google Scholar] [CrossRef]
- Trias, E.; Ibarburu, S.; Barreto-Núñez, R.; Babdor, J.; Maciel, T.T.; Guillo, M.; Gros, L.; Dubreuil, P.; Díaz-Amarilla, P.; Cassina, P.; et al. Post-paralysis tyrosine kinase inhibition with masitinib abrogates neuroinflammation and slows disease progression in inherited amyotrophic lateral sclerosis. J. Neuroinflamm. 2016, 13, 177. [Google Scholar] [CrossRef]
- Palomo, V.; Nozal, N.; Rojas-Prats, E.; Gil, C.; Martinez, A. Protein kinase inhibitors for amyotrophic lateral sclerosis therapy. Br. J. Pharmacol. 2021, 178, 1316–1335. [Google Scholar] [CrossRef] [PubMed]
- Liscic, R.M.; Alberici, A.; Cairns, N.J.; Romano, M.; Buratti, E. From basic research to the clinic: Innovative therapies for ALS and FTD in the pipeline. Mol. Neurodegener. 2020, 15, 31. [Google Scholar] [CrossRef]
- Trias, E.; Ibarburu, S.; Barreto-Núñez, R.; Varela, V.; Moura, I.C.; Dubreuil, P.; Hermine, O.; Beckman, J.S.; Barbeito, L. Evidence for mast cells contributing to neuromuscular pathology in an inherited model of ALS. JCI Insight 2017, 2, e95934. [Google Scholar] [CrossRef] [Green Version]
- Trias, E.; King, P.H.; Si, Y.; Kwon, Y.; Varela, V.; Ibarburu, S.; Kovacs, M.; Moura, I.C.; Beckman, J.S.; Hermine, O.; et al. Mast cells and neutrophils mediate peripheral motor pathway degeneration in ALS. JCI Insight 2018, 3, 3249. [Google Scholar] [CrossRef]
- Mora, J.S.; Genge, A.; Chio, A.; Estol, C.J.; Chaverri, D.; Hernández, M.; Marín, S.; Mascias, J.; Rodriguez, G.E.; Povedano, M.; et al. Masitinib as an add-on therapy to riluzole in patients with amyotrophic lateral sclerosis: A randomized clinical trial. Amyotroph. Lateral Scler. Frontotemporal. Degener. 2020, 21, 5–14. [Google Scholar] [CrossRef] [Green Version]
- Roser, A.E.; Tönges, L.; Lingor, P. Modulation of Microglial Activity by Rho-Kinase (ROCK) Inhibition as Therapeutic Strategy in Parkinson’s Disease and Amyotrophic Lateral Sclerosis. Front. Aging Neurosci. 2017, 9, 94. [Google Scholar] [CrossRef]
- Hensel, N.; Rademacher, S.; Claus, P. Chatting with the neighbors: Crosstalk between Rho-kinase (ROCK) and other signaling pathways for treatment of neurological disorders. Front. Neurosci. 2015, 9, 198. [Google Scholar] [CrossRef] [Green Version]
- Yan, J.; Zhou, X.; Guo, J.J.; Mao, L.; Wang, Y.J.; Sun, J.; Sun, L.X.; Zhang, L.Y.; Zhou, X.F.; Liao, H. Nogo-66 inhibits adhesion and migration of microglia via GTPase Rho pathway in vitro. J. Neurochem. 2012, 120, 721–731. [Google Scholar] [CrossRef]
- Borrajo, A.; Rodriguez-Perez, A.I.; Villar-Cheda, B.; Guerra, M.J.; Labandeira-Garcia, J.L. Inhibition of the microglial response is essential for the neuroprotective effects of Rho-kinase inhibitors on MPTP-induced dopaminergic cell death. Neuropharmacology 2014, 85, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Conti, A.; Riva, N.; Pesca, M.; Iannaccone, S.; Cannistraci, C.V.; Corbo, M.; Previtali, S.C.; Quattrini, A.; Alessio, M. Increased expression of Myosin binding protein H in the skeletal muscle of amyotrophic lateral sclerosis patients. Biochim. Biophys. Acta 2014, 1842, 99–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tönges, L.; Günther, R.; Suhr, M.; Jansen, J.; Balck, A.; Saal, K.A.; Barski, E.; Nientied, T.; Götz, A.A.; Koch, J.C.; et al. Rho kinase inhibition modulates microglia activation and improves survival in a model of amyotrophic lateral sclerosis. Glia 2014, 62, 217–232. [Google Scholar] [CrossRef]
- Ding, J.; Li, Q.Y.; Wang, X.; Sun, C.H.; Lu, C.Z.; Xiao, B.G. Fasudil protects hippocampal neurons against hypoxia-reoxygenation injury by suppressing microglial inflammatory responses in mice. J. Neurochem. 2010, 114, 1619–1629. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Li, Y.; Yu, J.; Guo, M.; Meng, J.; Liu, C.; Xie, Y.; Feng, L.; Xiao, B.; Ma, C. Rho kinase inhibitor fasudil regulates microglia polarization and function. Neuroimmunomodulation 2013, 20, 313–322. [Google Scholar] [CrossRef]
- Günther, R.; Balckv, A.; Koch, J.C.; Nientiedt, T.; Sereda, M.; Bähr, M.; Lingor, P.; Tönges, L. Rho Kinase Inhibition with Fasudil in the SOD1 G93A Mouse Model of Amyotrophic Lateral Sclerosis-Symptomatic Treatment Potential after Disease Onset. Front. Pharmacol. 2017, 8, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stankiewicz, T.R.; Pena, C.; Bouchard, R.; Linseman, D.A. Dysregulation of Rac or Rho elicits death of motor neurons and activation of these GTPases is altered in the G93A mutant hSOD1 mouse model of amyotrophic lateral sclerosis. Neurobiol. Dis. 2020, 136, 104743. [Google Scholar] [CrossRef]
- Joshi, A.R.; Muke, I.; Bobylev, I.; Lehmann, H.C. ROCK inhibition improves axonal regeneration in a preclinical model of amyotrophic lateral sclerosis. J. Comp. Neurol. 2019, 527, 2334–2340. [Google Scholar] [CrossRef] [PubMed]
- Harris, P.A.; Berger, S.B.; Jeong, J.U.; Nagilla, R.; Bandyopadhyay, D.; Campobasso, N.; Capriotti, C.A.; Cox, J.A.; Dare, L.; Dong, X.; et al. Discovery of a First-in-Class Receptor Interacting Protein 1 (RIP1) Kinase Specific Clinical Candidate (GSK2982772) for the Treatment of Inflammatory Diseases. J. Med. Chem. 2017, 60, 1247–1261. [Google Scholar] [CrossRef]
- Mifflin, L.; Ofengeim, D.; Yuan, J. Receptor-interacting protein kinase 1 (RIPK1) as a therapeutic target. Nat. Rev. Drug Discov. 2020, 19, 553–571. [Google Scholar] [CrossRef] [PubMed]
- Globe Newswire by Notified. Available online: https://www.globenewswire.com/news-release/2020/06/09/2045940/0/en/Denali-Therapeutics-Provides-Broad-Update-on-Its-RIPK1-Program-Partnered-With-Sanofi.html (accessed on 20 September 2021).
- ALS New Today. Available online: https://alsnewstoday.com/news-posts/2020/06/15/denali-sanofi-pause-dnl747-studies-favor-advancing-dnl788/ (accessed on 20 October 2021).
- Beurel, E. Regulation by glycogen synthase kinase-3 of inflammation and T cells in CNS diseases. Front. Mol. Neurosci. 2011, 4, 18. [Google Scholar] [CrossRef] [Green Version]
- Hoffmeister, L.; Diekmann, M.; Brand, K.; Huber, R. GSK3: A Kinase Balancing Promotion and Resolution of Inflammation. Cells 2020, 9, 820. [Google Scholar] [CrossRef] [Green Version]
- Hong, M.; Chen, D.C.; Klein, P.S.; Lee, V.M. Lithium reduces tau phosphorylation by inhibition of glycogen synthase kinase-3. J. Biol. Chem. 1997, 272, 25326–25332. [Google Scholar] [CrossRef] [Green Version]
- Noble, W.; Planel, E.; Zehr, C.; Olm, V.; Meyerson, J.; Suleman, F.; Gaynor, K.; Wang, L.; LaFrancois, J.; Feinstein, B.; et al. Inhibition of glycogen synthase kinase-3 by lithium correlates with reduced tauopathy and degeneration in vivo. Proc. Natl. Acad. Sci. USA 2005, 102, 6990–6995. [Google Scholar] [CrossRef] [Green Version]
- Puglisi-Allegra, S.; Ruggieri, S.; Fornai, F. Translational evidence for lithium-induced brain plasticity and neuroprotection in the treatment of neuropsychiatric disorders. Transl. Psychiatry 2021, 11, 366. [Google Scholar] [CrossRef]
- NIH US Library of Medicine. Available online: https://clinicaltrials.gov/ct2/show/NCT02862210 (accessed on 20 October 2021).
- Lv, Z.; Guo, Y. Metformin and Its Benefits for Various Diseases. Front. Endocrinol. 2020, 11, 191. [Google Scholar] [CrossRef] [PubMed]
- Kaneb, H.M.; Sharp, P.S.; Rahmani-Kondori, N.; Wells, D.J. Metformin treatment has no beneficial effect in a dose-response survival study in the SOD1(G93A) mouse model of ALS and is harmful in female mice. PLoS ONE 2011, 6, e24189. [Google Scholar] [CrossRef]
- Peters, O.M.; Ghasemi, M.; Brown, R.H., Jr. Emerging mechanisms of molecular pathology in ALS. J. Clin. Investig. 2015, 125, 2548. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Drug | Kinase Target | Study Phase | Year Launched 1 | Code Number | Disease |
|---|---|---|---|---|---|
| Masitinib | Tyrosine kinase receptors | III | 2020–2022 | NCT03127267 | ALS |
| Fasudil | ROCK | II | 2020 | NCT03792490 | ALS |
| DNL747 | RIPK1 | Ib (paused) | 2020 | NCT03757351 | ALS |
| DNL788/ SAR443820 | RIPK1 | II (announced) | Q1 2022 | N/A | ALS |
| Lithium carbonate | GSK3 | II (recruiting) | 2021 | NCT02862210 | FTD |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-García, R.; Martín-Herrero, L.; Blanca-Pariente, L.; Pérez-Cabello, J.; Roodveldt, C. Immune Signaling Kinases in Amyotrophic Lateral Sclerosis (ALS) and Frontotemporal Dementia (FTD). Int. J. Mol. Sci. 2021, 22, 13280. https://doi.org/10.3390/ijms222413280
García-García R, Martín-Herrero L, Blanca-Pariente L, Pérez-Cabello J, Roodveldt C. Immune Signaling Kinases in Amyotrophic Lateral Sclerosis (ALS) and Frontotemporal Dementia (FTD). International Journal of Molecular Sciences. 2021; 22(24):13280. https://doi.org/10.3390/ijms222413280
Chicago/Turabian StyleGarcía-García, Raquel, Laura Martín-Herrero, Laura Blanca-Pariente, Jesús Pérez-Cabello, and Cintia Roodveldt. 2021. "Immune Signaling Kinases in Amyotrophic Lateral Sclerosis (ALS) and Frontotemporal Dementia (FTD)" International Journal of Molecular Sciences 22, no. 24: 13280. https://doi.org/10.3390/ijms222413280