
AXL Knock-Out in SNU475 Hepatocellular Carcinoma Cells Provides Evidence for Lethal Effect Associated with G2 Arrest and Polyploidization

 , , , and
, , , and
Abstract
:1. Introduction
2. Results
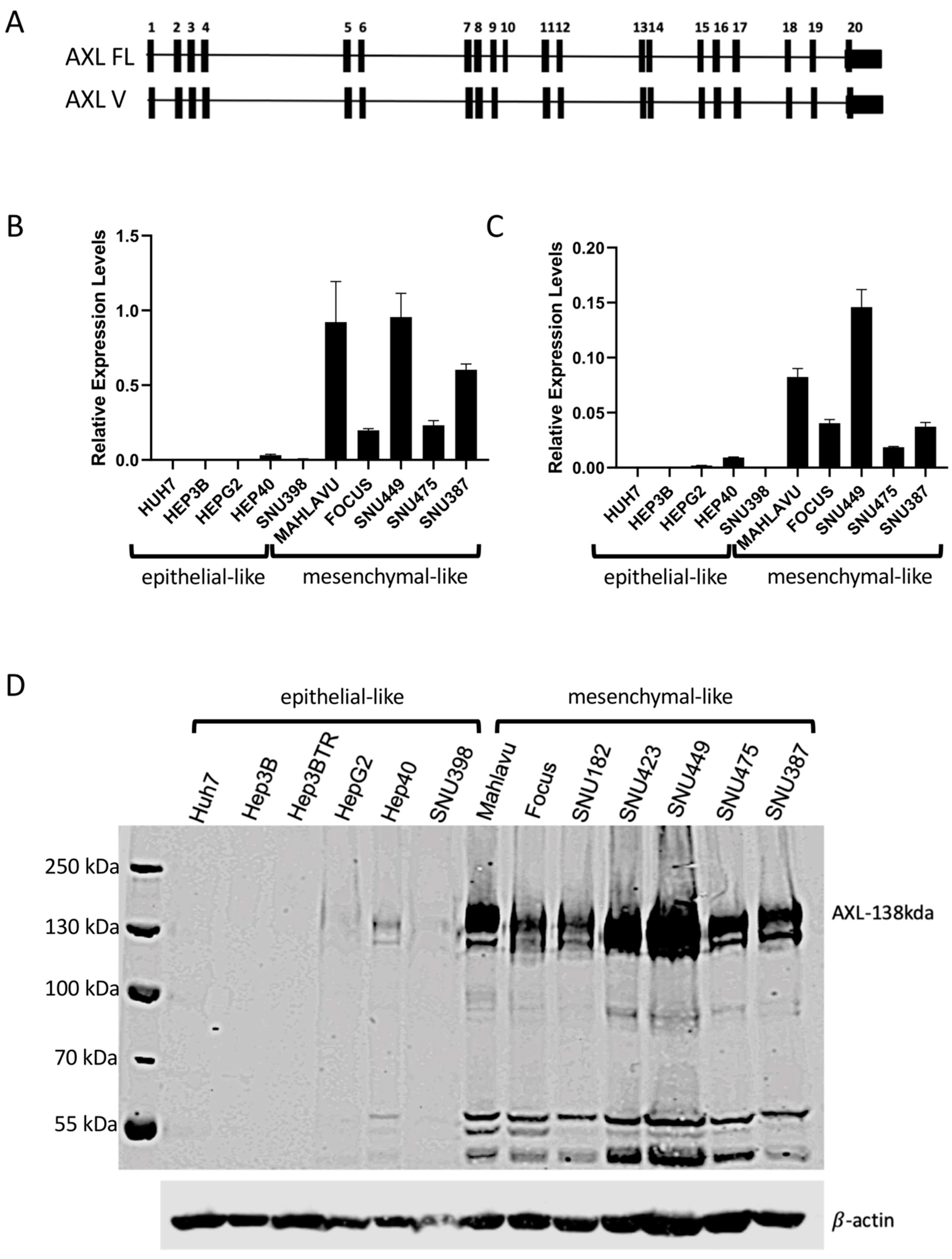
2.1. AXL Has Elevated Expression in Mesenchymal-like HCC Cell Lines
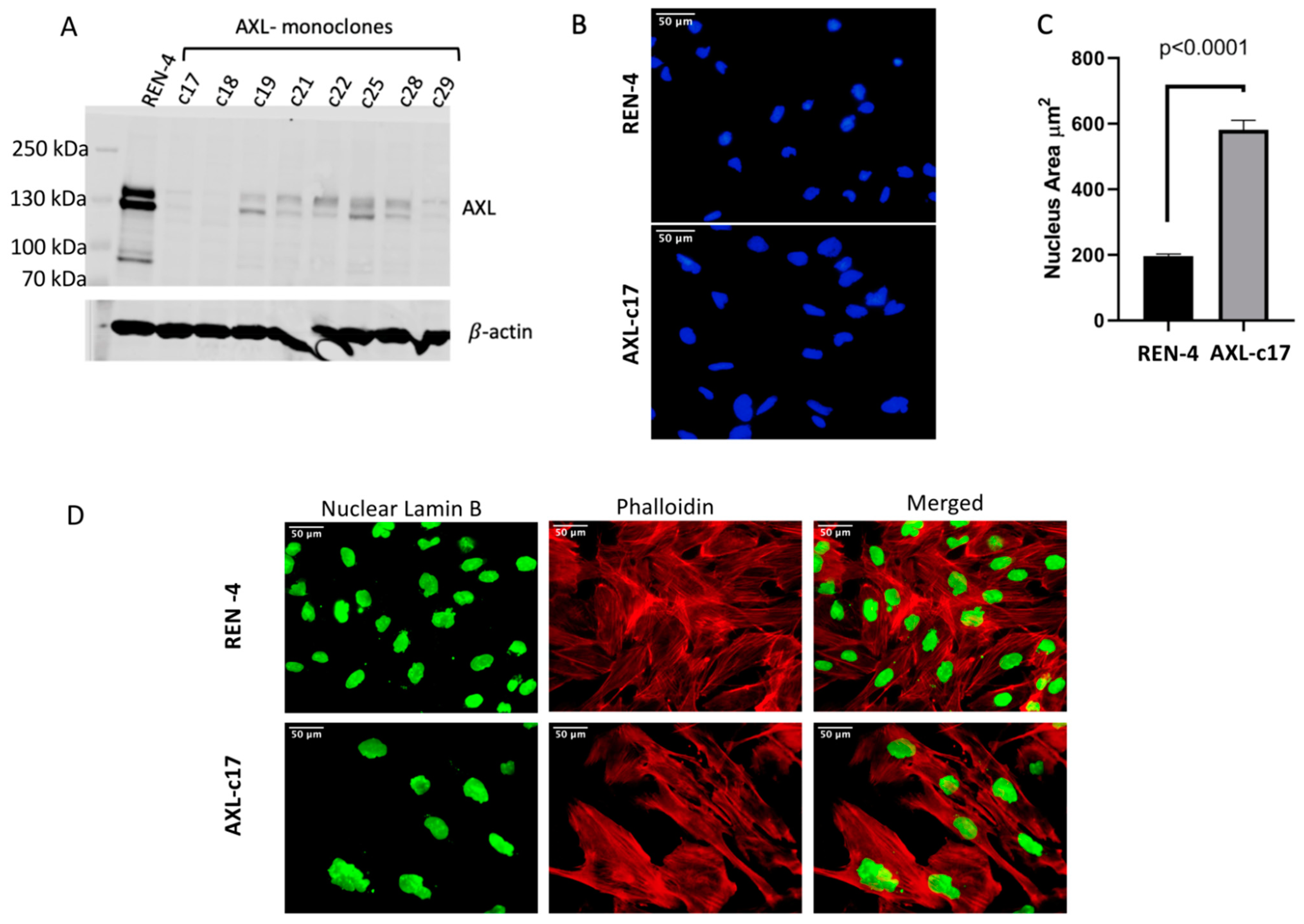
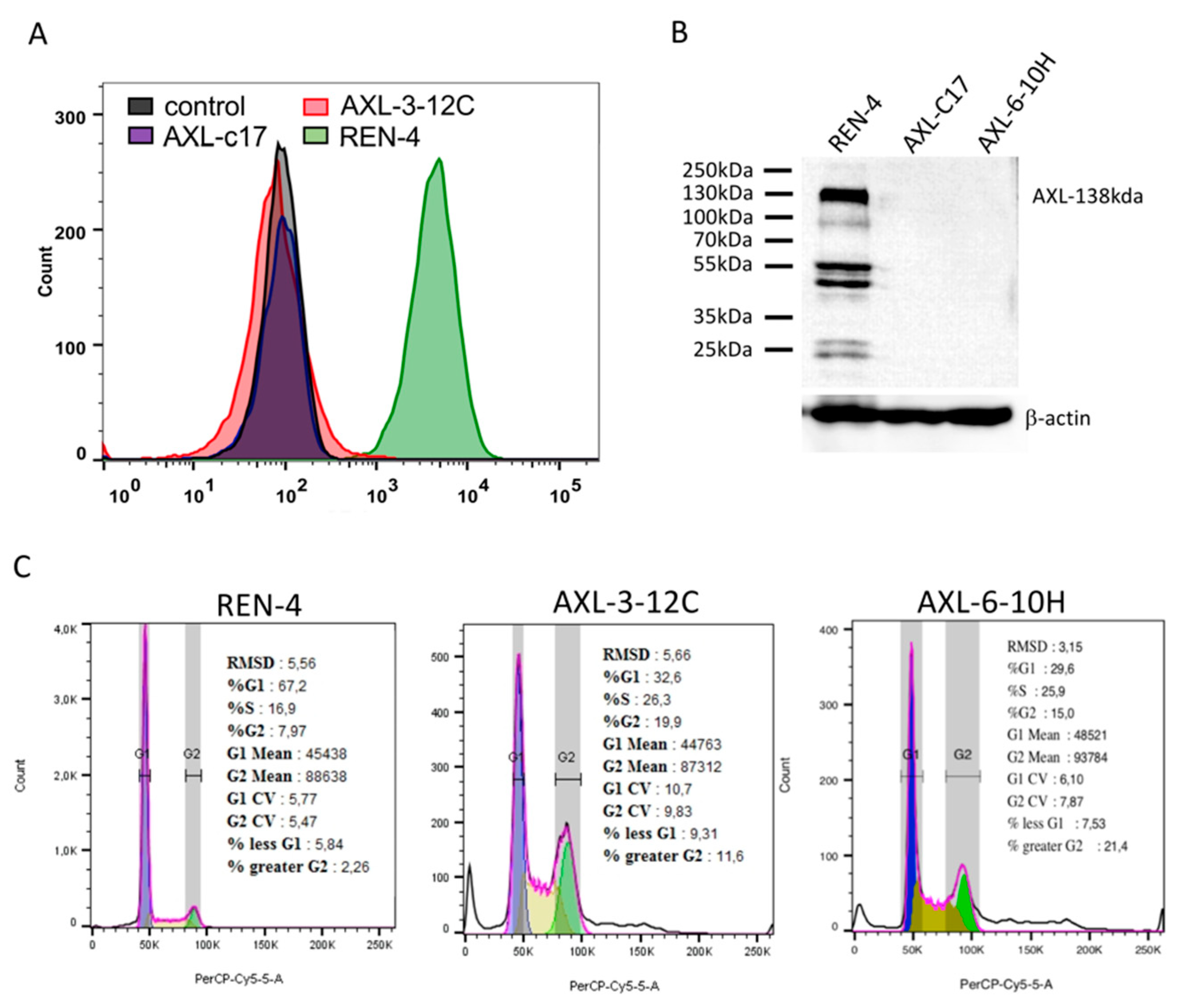
2.2. Successful Knockout of AXL Gene in SNU475 HCC Cell Line
2.3. Augmented Nucleus Size and Different Cellular Morphology of AXL Knock-Out SNU449 Clone AXL-c17
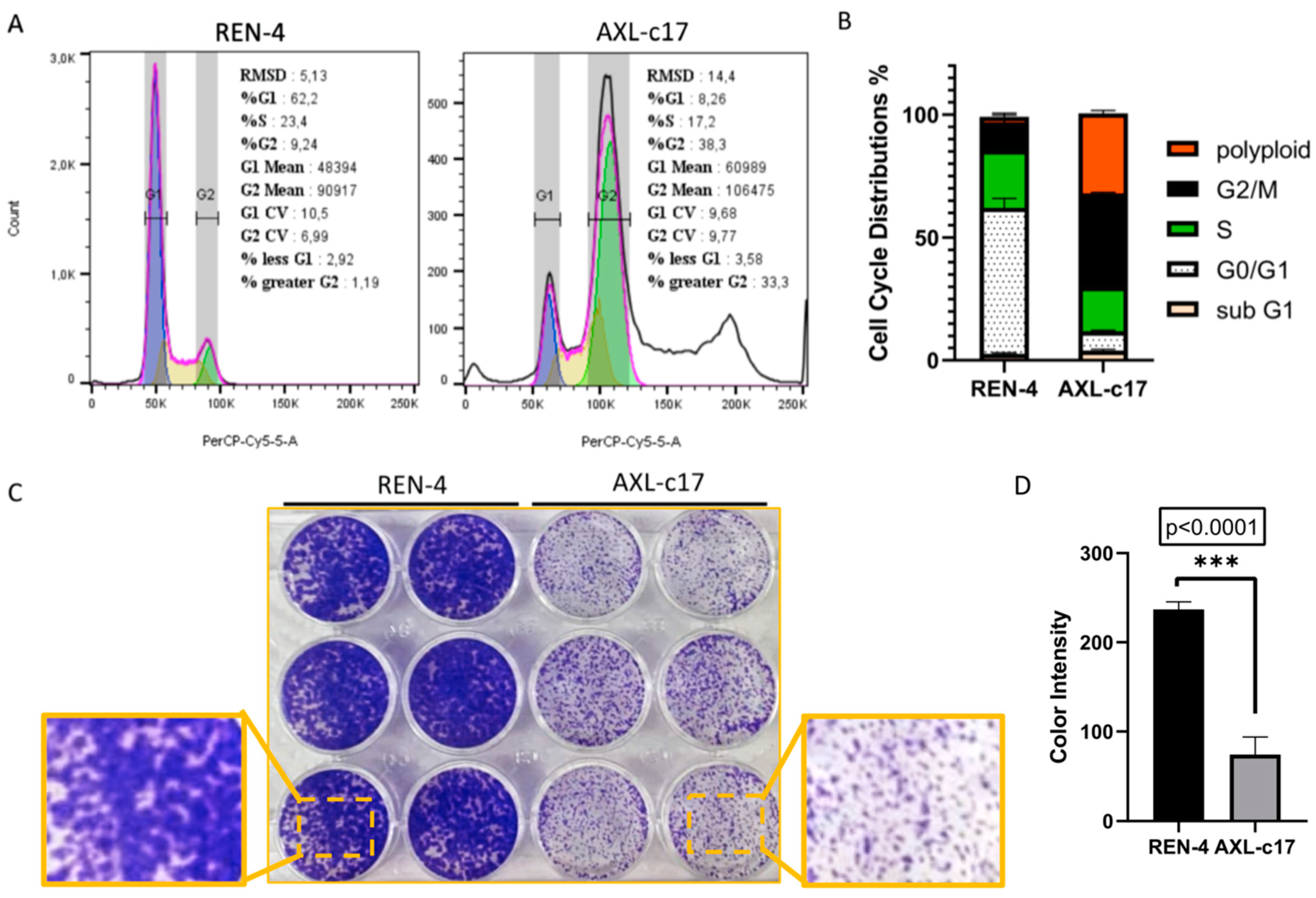
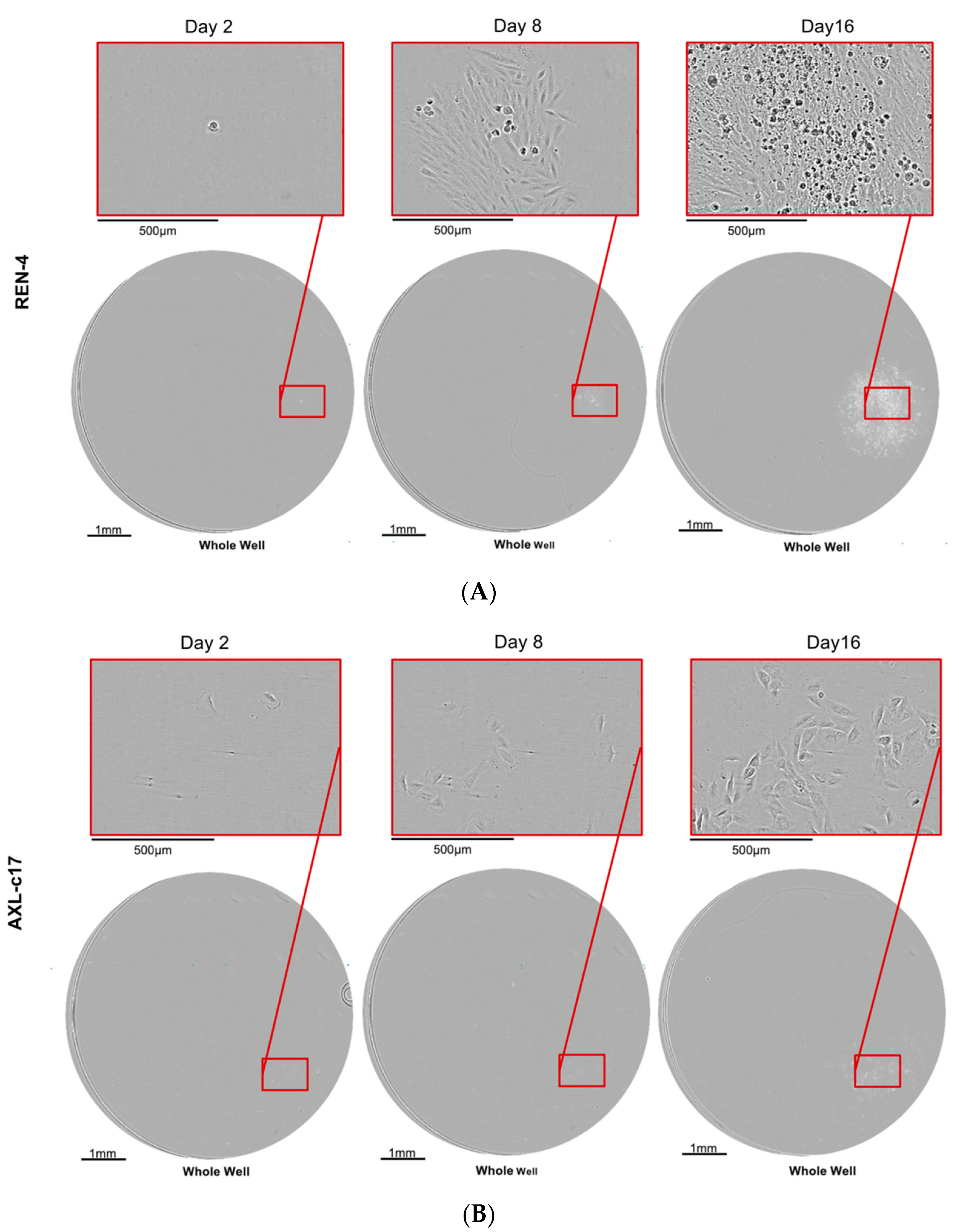
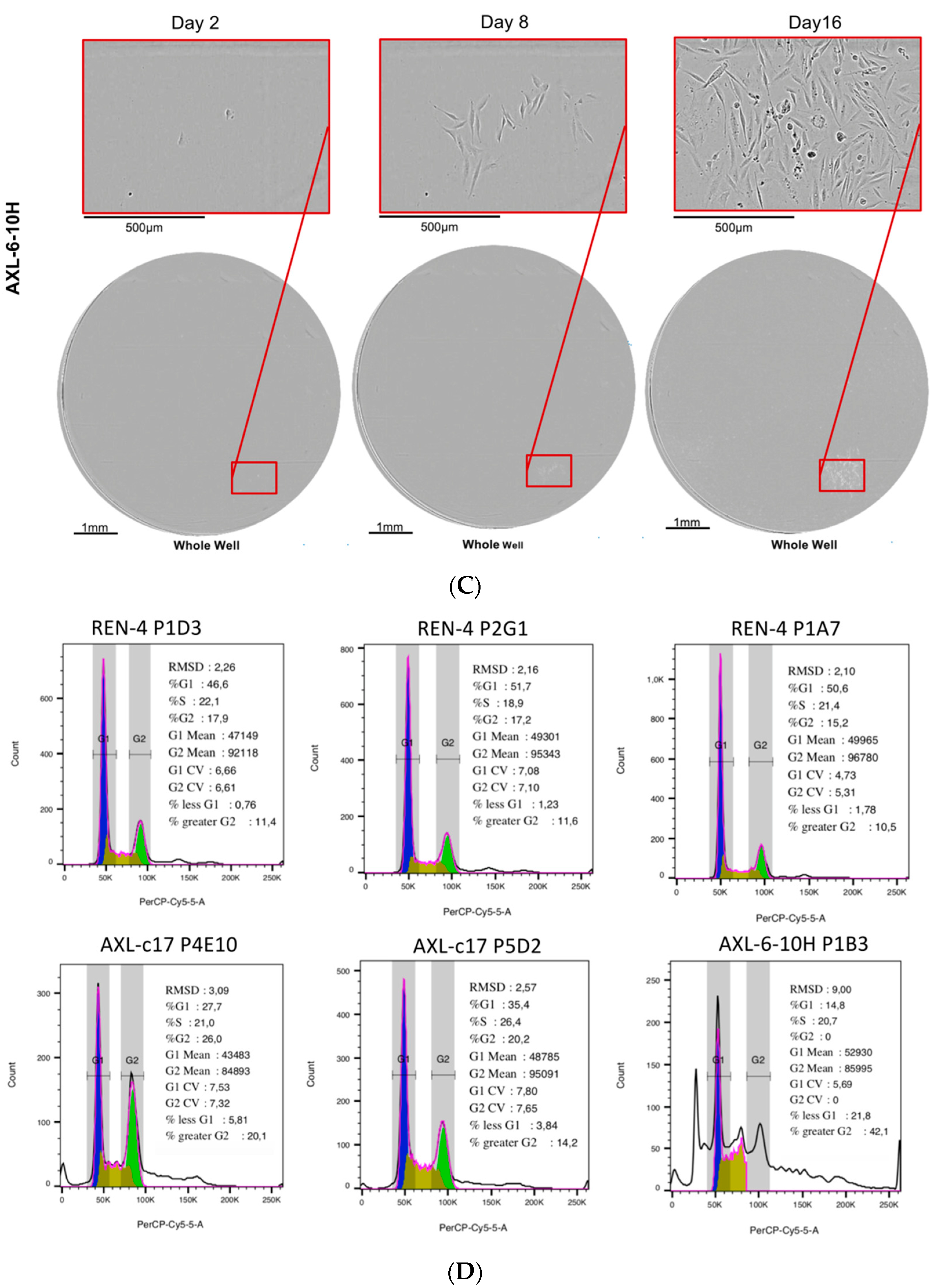
2.4. AXL Knock-Out Cells Exhibit Cell Cycle Aberrations, Polyploidy, and Low Clonogenicity
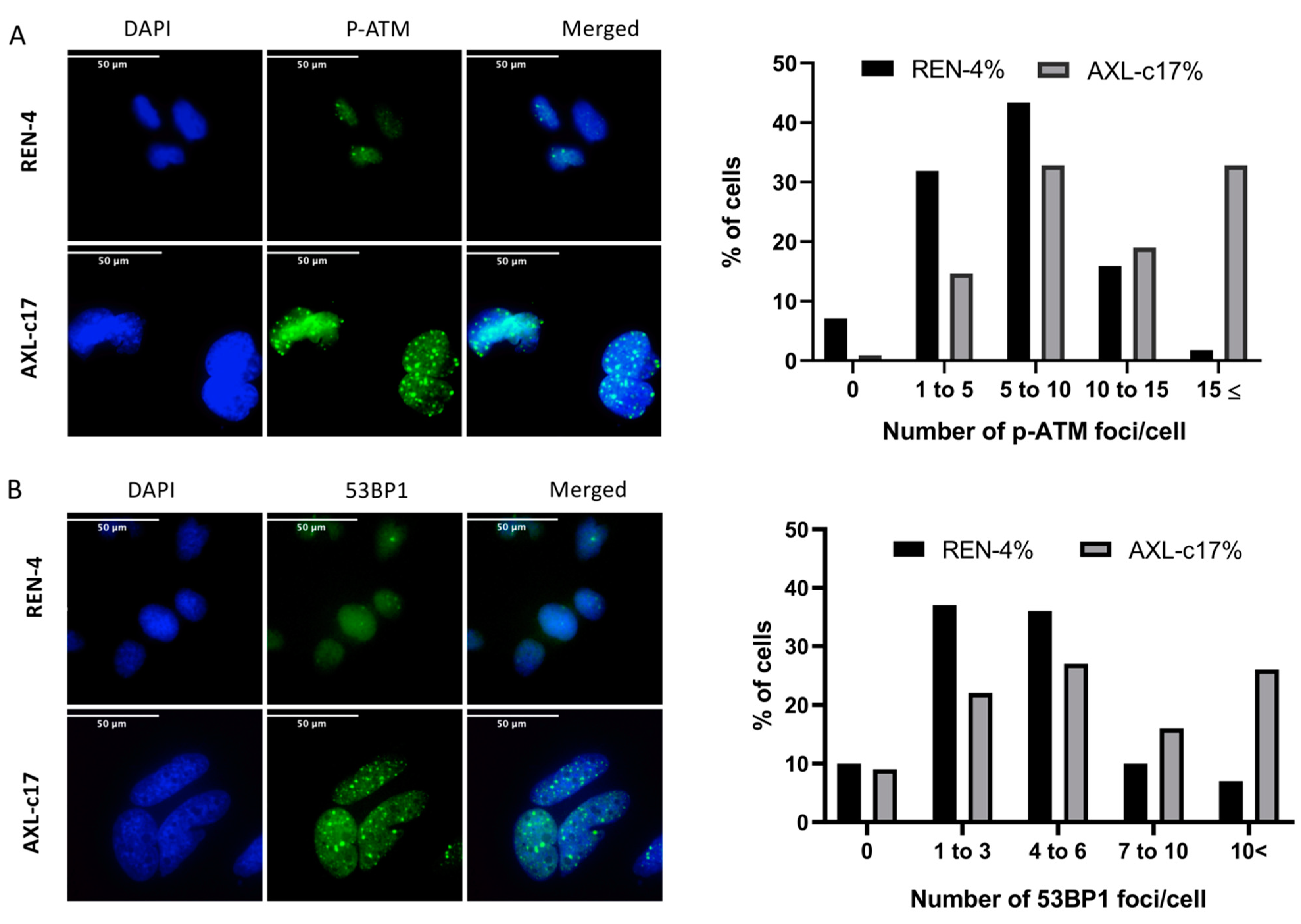
2.5. G2 Arrest and Polyploidization of AXL Knock-Out Cells Is Associated with Features of Unresolved DNA Damage Response
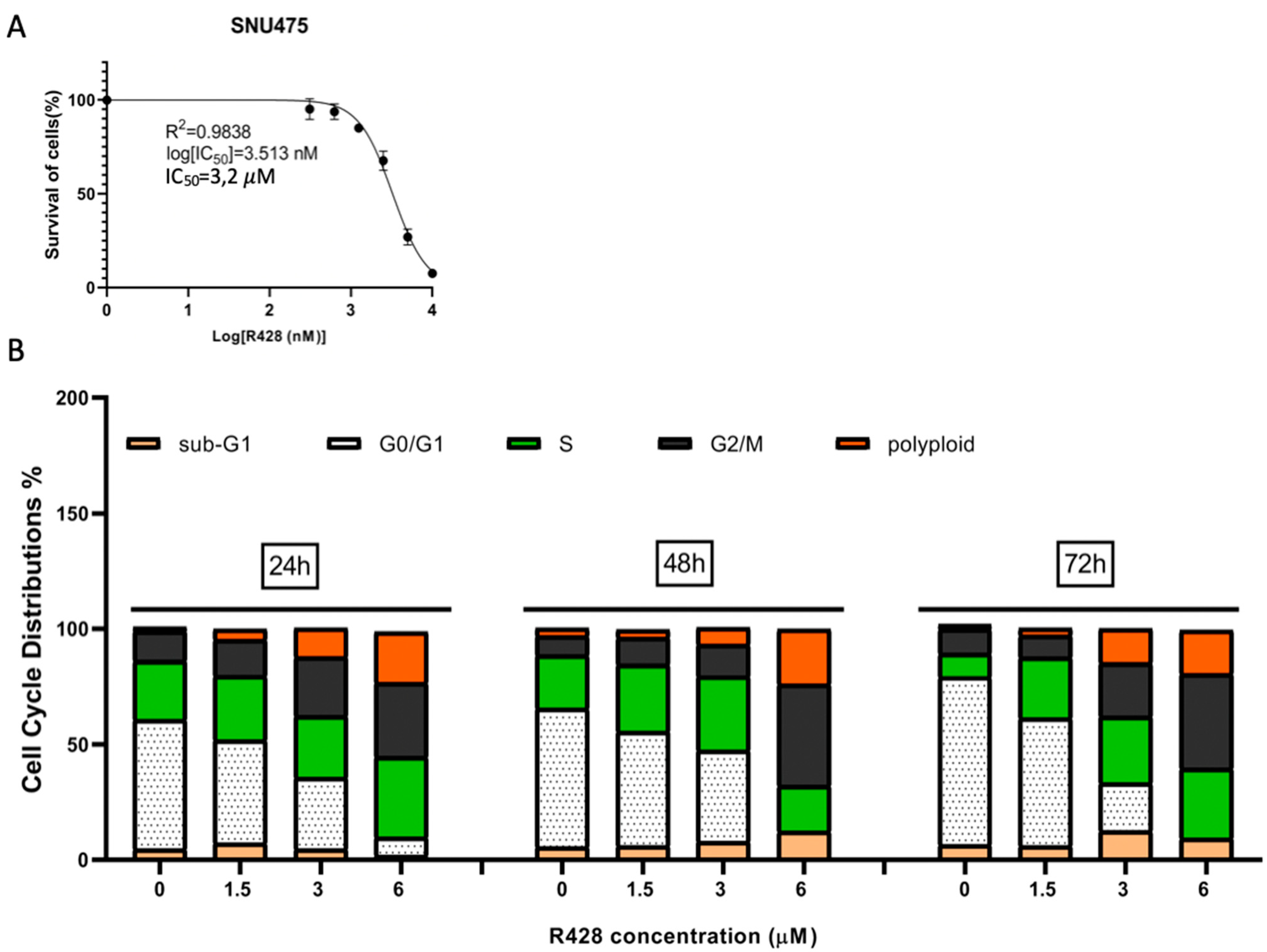
2.6. Induction of G2 Arrest and Polyploidization by Pharmacological Inhibition of AXL in SNU475 Cells
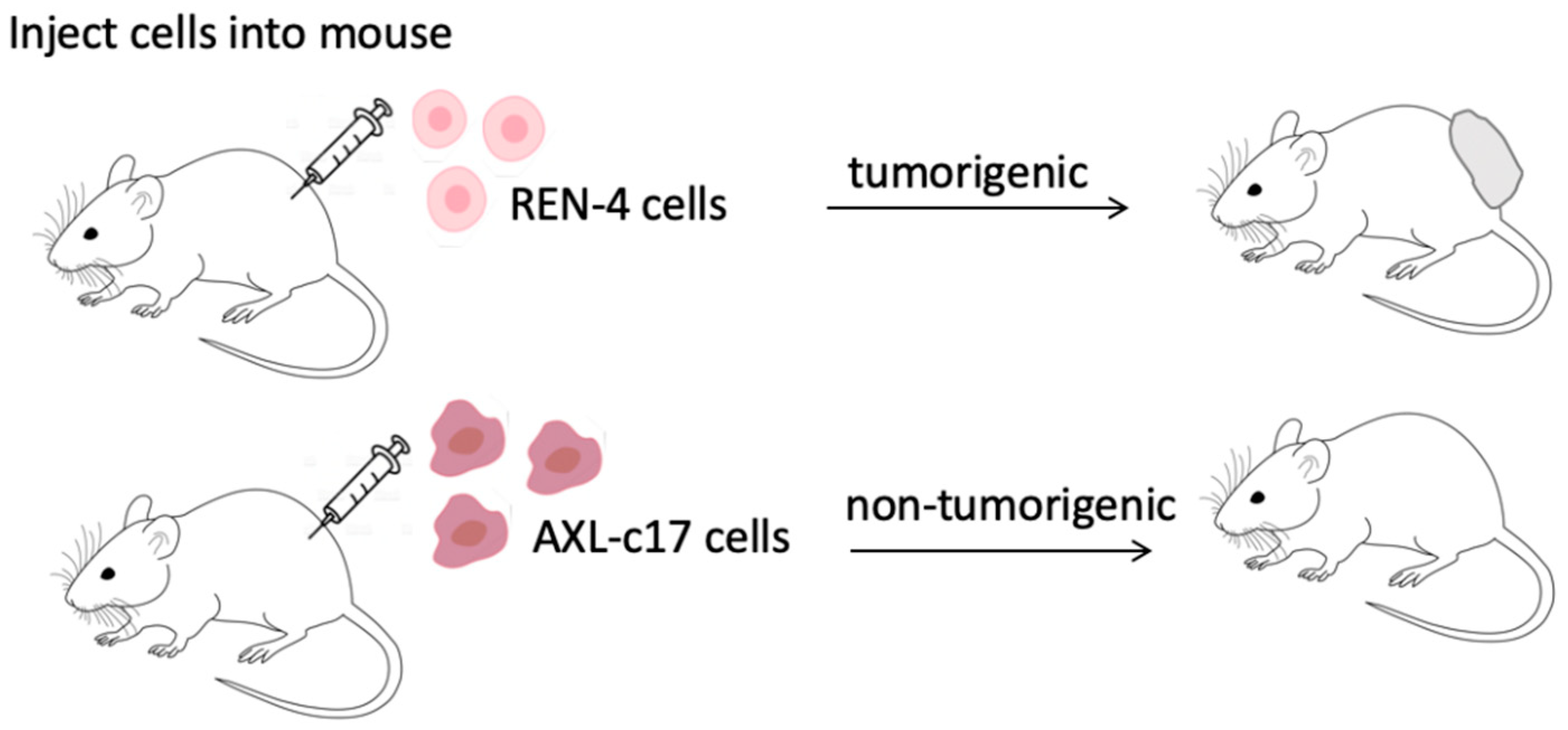
2.7. AXL Knock-Out Cells Lost Their Tumorigenic Ability
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Antibodies
4.3. Cloning
4.4. Viral Packaging and Transduction
4.5. Obtaining Monoclones
4.6. Western Blot
4.7. Immunofluorescence
4.8. Colony Formation
4.9. Cell Cycle
4.10. Flow Cytometry
4.11. Quantitative Real-Time PCR (qPCR)
4.12. Clonality-Proliferation Assay
4.13. Sulforhodamine B Assay
4.14. Animal Studies
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Kim, D.W.; Talati, C.; Kim, R. Hepatocellular carcinoma (HCC): Beyond sorafenib—Chemotherapy. J. Gastrointest. Oncol. 2017, 8, 256–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zucman-Rossi, J.; Villanueva, A.; Nault, J.-C.; Llovet, J.M. Genetic Landscape and Biomarkers of Hepatocellular Carcinoma. Gastroenterology 2015, 149, 1226–1239.e4. [Google Scholar] [CrossRef] [Green Version]
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 2021, 7, 6. [Google Scholar] [CrossRef]
- El-Khoueiry, A.B.; Hanna, D.L.; Llovet, J.; Kelley, R.K. Cabozantinib: An evolving therapy for hepatocellular carcinoma. Cancer Treat. Rev. 2021, 98, 102221. [Google Scholar] [CrossRef] [PubMed]
- Lemke, G. Biology of the TAM receptors. Cold Spring Harb. Perspect. Biol. 2013, 5, a009076. [Google Scholar] [CrossRef] [PubMed]
- Lemke, G. How macrophages deal with death. Nat. Rev. Immunol. 2019, 19, 539–549. [Google Scholar] [CrossRef] [PubMed]
- Golkowski, M.; Lau, H.-T.; Chan, M.; Kenerson, H.; Vidadala, V.N.; Shoemaker, A.; Maly, D.J.; Yeung, R.S.; Gujral, T.S.; Ong, S.-E. Pharmacoproteomics Identifies Kinase Pathways that Drive the Epithelial-Mesenchymal Transition and Drug Resistance in Hepatocellular Carcinoma. Cell Syst. 2020, 11, 196–207.e7. [Google Scholar] [CrossRef]
- Gay, C.M.; Balaji, K.; Byers, L.A. Giving AXL the axe: Targeting AXL in human malignancy. Br. J. Cancer 2017, 116, 415–423. [Google Scholar] [CrossRef]
- Liu, J.; Wang, K.; Yan, Z.; Xia, Y.; Li, J.; Shi, L.; Zou, Q.; Wan, X.; Jiao, B.; Wang, H.; et al. Axl expression stratifies patients with poor prognosis after hepatectomy for hepatocellular carcinoma. PLoS ONE 2016, 11, e0154767. [Google Scholar] [CrossRef]
- Lee, H.J.; Jeng, Y.M.; Chen, Y.L.; Chung, L.; Yuan, R.H. Gas6/Axl pathway promotes tumor invasion through the transcriptional activation of slug in hepatocellular carcinoma. Carcinogenesis 2014, 35, 769–775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Jia, L.; Ma, H.; Li, Y.; Ma, Z.; Zhao, Y. Axl gene knockdown inhibits the metastasis properties of hepatocellular carcinoma via PI3K/Akt-PAK1 signal pathway. Tumor Biol. 2014, 35, 3809–3817. [Google Scholar] [CrossRef]
- Goyette, M.-A.; Elkholi, I.E.; Apcher, C.; Kuasne, H.; Rothlin, C.V.; Muller, W.J.; Richard, D.E.; Park, M.; Gratton, J.-P.; Côté, J.-F. Targeting Axl favors an antitumorigenic microenvironment that enhances immunotherapy responses by decreasing Hif-1α levels. Proc. Natl. Acad. Sci. USA 2021, 118, e2023868118. [Google Scholar] [CrossRef] [PubMed]
- Balaji, K.; Vijayaraghavan, S.; Diao, L.; Tong, P.; Fan, Y.; Carey, J.P.W.; Bui, T.N.; Warner, S.; Heymach, J.V.; Hunt, K.K.; et al. AXL inhibition suppresses the DNA damage response and sensitizes cells to PARP inhibition in multiple cancers. Mol. Cancer Res. 2017, 15, 45–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flem-Karlsen, K.; McFadden, E.; Omar, N.; Haugen, M.H.; Øy, G.F.; Ryder, T.; Gullestad, H.P.; Hermann, R.; Mælandsmo, G.M.; Flørenes, V.A. Targeting AXL and the DNA Damage Response Pathway as a Novel Therapeutic Strategy in Melanoma. Mol. Cancer Ther. 2020, 19, 895–905. [Google Scholar] [CrossRef] [Green Version]
- Goyette, M.-A.; Duhamel, S.; Aubert, L.; Pelletier, A.; Savage, P.; Thibault, M.-P.; Johnson, R.M.; Carmeliet, P.; Basik, M.; Gaboury, L.; et al. The Receptor Tyrosine Kinase AXL Is Required at Multiple Steps of the Metastatic Cascade during HER2-Positive Breast Cancer Progression. Cell Rep. 2018, 23, 1476–1490. [Google Scholar] [CrossRef] [PubMed]
- Scharf, I.; Bierbaumer, L.; Huber, H.; Wittmann, P.; Haider, C.; Pirker, C.; Berger, W.; Mikulits, W. Dynamics of CRISPR/CAS9-mediated genomic editing of the AXL locus in hepatocellular carcinoma cells. Oncol. Lett. 2018, 15, 2441–2450. [Google Scholar] [CrossRef]
- Reichl, P.; Dengler, M.; van Zijl, F.; Huber, H.; Führlinger, G.; Reichel, C.; Sieghart, W.; Peck-Radosavljevic, M.; Grubinger, M.; Mikulits, W. Axl activates autocrine transforming growth factor-β signaling in hepatocellular carcinoma. Hepatology 2015, 61, 930–941. [Google Scholar] [CrossRef] [Green Version]
- He, L.; Zhang, J.; Jiang, L.; Jin, C.; Zhao, Y.; Yang, G.; Jia, L. Differential expression of Axl in hepatocellular carcinoma and correlation with tumor lymphatic metastasis. Mol. Carcinog. 2010, 49, 882–891. [Google Scholar] [CrossRef]
- Caruso, S.; Calatayud, A.-L.; Pilet, J.; La Bella, T.; Rekik, S.; Imbeaud, S.; Letouzé, E.; Meunier, L.; Bayard, Q.; Rohr-Udilova, N.; et al. Analysis of Liver Cancer Cell Lines Identifies Agents with Likely Efficacy Against Hepatocellular Carcinoma and Markers of Response. Gastroenterology 2019, 157, 760–776. [Google Scholar] [CrossRef]
- O’Bryan, J.P.; Frye, R.A.; Cogswell, P.C.; Neubauer, A.; Kitch, B.; Prokop, C.; Espinosa, R.; Le Beau, M.M.; Earp, H.S.; Liu, E.T. Axl, a Transforming Gene Isolated From Primary Human Myeloid Leukemia Cells, Encodes a Novel Receptor Tyrosine Kinase. Mol. Cell. Biol. 1991, 11, 5016–5031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, S.; Almouzni, G. Chromatin dynamics during the cell cycle at centromeres. Nat. Rev. Genet. 2017, 18, 192–208. [Google Scholar] [CrossRef]
- Kuerbitz, S.J.; Plunkett, B.S.; Walsh, W.V.; Kastan, M.B. Wild-type p53 is a cell cycle checkpoint determinant following irradiation. Proc. Natl. Acad. Sci. USA 1992, 89, 7491–7495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowe, S.W.; Bodis, S.; McClatchey, A.; Remington, L.; Ruley, H.E.; Fisher, D.E.; Housman, D.E.; Jacks, T. p53 Status and the Efficacy of Cancer Therapy in Vivo. Science 1994, 266, 807–810. [Google Scholar] [CrossRef]
- Illidge, T.M.; Cragg, M.S.; Fringes, B.; Olive, P.; Erenpreisa, J.A. Polyploid giant cells provide a survival mechanism for p53 mutant cells after DNA damage. Cell Biol. Int. 2000, 24, 621–633. [Google Scholar] [CrossRef] [Green Version]
- Hwang, S.-J.; Lozano, G.; Amos, C.I.; Strong, L.C. Germline p53 Mutations in a Cohort with Childhood Sarcoma: Sex Differences in Cancer Risk. Am. J. Hum. Genet. 2003, 72, 975–983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sancar, A.; Lindsey-Boltz, L.A.; Ünsal-Kaçmaz, K.; Linn, S. Molecular Mechanisms of Mammalian DNA Repair and the DNA Damage Checkpoints. Annu. Rev. Biochem. 2004, 73, 39–85. [Google Scholar] [CrossRef] [Green Version]
- Gire, V.; Dulic, V. Senescence from G2 arrest, revisited. Cell Cycle 2015, 14, 297–304. [Google Scholar] [CrossRef] [Green Version]
- Holland, S.J.; Pan, A.; Franci, C.; Hu, Y.; Chang, B.; Li, W.; Duan, M.; Torneros, A.; Yu, J.; Heckrodt, T.J.; et al. R428, a Selective Small Molecule Inhibitor of Axl Kinase, Blocks Tumor Spread and Prolongs Survival in Models of Metastatic Breast Cancer. Cancer Res. 2010, 70, 1544–1554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Z.Y.; Chuang, W.L. Genes responsible for the characteristics of primary cultured invasive phenotype hepatocellular carcinoma cells. Biomed. Pharmacother. 2012, 66, 454–458. [Google Scholar] [CrossRef]
- Pinato, D.J.; Brown, M.W.; Trousil, S.; Aboagye, E.O.; Beaumont, J.; Zhang, H.; Coley, H.M.; Mauri, F.A.; Sharma, R. Integrated analysis of multiple receptor tyrosine kinases identifies Axl as a therapeutic target and mediator of resistance to sorafenib in hepatocellular carcinoma. Br. J. Cancer 2019, 120, 512–521. [Google Scholar] [CrossRef] [Green Version]
- Hsu, C.; Hsieh, P.; Chen, Y.; Lo, G.; Lin, H.; Dai, C.; Huang, J.; Chuang, W.; Chen, Y.; Yu, M.; et al. Axl and autophagy LC3 expression in tumors is strongly associated with clinical prognosis of hepatocellular carcinoma patients after curative resection. Cancer Med. 2019, 8, 3453–3463. [Google Scholar] [CrossRef]
- Staufer, K.; Dengler, M.; Huber, H.; Marculescu, R.; Stauber, R.; Lackner, C.; Dienes, H.-P.; Kivaranovic, D.; Schachner, C.; Zeitlinger, M.; et al. The non-invasive serum biomarker soluble Axl accurately detects advanced liver fibrosis and cirrhosis. Cell Death Dis. 2017, 8, e3135. [Google Scholar] [CrossRef] [Green Version]
- Dengler, M.; Staufer, K.; Huber, H.; Stauber, R.; Bantel, H.; Weiss, K.H.; Starlinger, P.; Pock, H.; Plachky, P.K.; Gotthardt, D.N.; et al. Soluble Axl is an accurate biomarker of cirrhosis and hepatocellular carcinoma development: Results from a large scale multicenter analysis. Oncotarget 2017, 8, 46234–46248. [Google Scholar] [CrossRef]
- Song, X.; Wu, A.; Ding, Z.; Liang, S.; Zhang, C. Soluble Axl Is a Novel Diagnostic Biomarker of Hepatocellular Carcinoma in Chinese Patients with Chronic Hepatitis B Virus Infection. Cancer Res. Treat. 2020, 52, 789–797. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.Z.; Chan, S.W.; Liu, A.M.; Wong, K.F.; Fan, S.T.; Chen, J.; Poon, R.T.; Zender, L.; Lowe, S.W.; Hong, W.; et al. AXL receptor kinase is a mediator of YAP-dependent oncogenic functions in hepatocellular carcinoma. Oncogene 2011, 30, 1229–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, W.; Jiang, X.; Huang, L. RNA interference technology. In Comprehensive Biotechnology; Elsevier: Amsterdam, The Netherlands, 2019; ISBN 9780444640475. [Google Scholar]
- Ramkumar, K.; Stewart, C.A.; Cargill, K.R.; Della Corte, C.M.; Wang, Q.; Shen, L.; Diao, L.; Cardnell, R.J.; Peng, D.H.; Rodriguez, B.L.; et al. AXL Inhibition Induces DNA Damage and Replication Stress in Non–Small Cell Lung Cancer Cells and Promotes Sensitivity to ATR Inhibitors. Mol. Cancer Res. 2021, 19, 485–497. [Google Scholar] [CrossRef] [PubMed]
- Kariolis, M.S.; Miao, Y.R.; Diep, A.; Nash, S.E.; Olcina, M.M.; Jiang, D.; Ii, D.S.J.; Kapur, S.; Mathews, I.I.; Koong, A.C.; et al. Inhibition of the GAS6/AXL pathway augments the efficacy of chemotherapies. J. Clin. Investig. 2017, 127, 183–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Middleton, A.; Suman, R.; O’Toole, P.; Akopyan, K.; Lindqvist, A. p53-dependent polyploidisation after DNA damage in G2 phase. bioRxiv 2020. [Google Scholar] [CrossRef]
- Oz, O.; Iscan, E.; Batur, T.; Ozturk, M. 3d organoid modelling of hepatoblast-like and mesenchymal-like hepatocellular carcinoma cell lines. Hepatoma Res. 2021, 7, 60. [Google Scholar] [CrossRef]
- Iscan, E.; Ekin, U.; Yildiz, G.; Oz, O.; Keles, U.; Suner, A.; Cakan-Akdogan, G.; Ozhan, G.; Nekulova, M.; Vojtesek, B.; et al. TAp73β Can Promote Hepatocellular Carcinoma Dedifferentiation. Cancers 2021, 13, 783. [Google Scholar] [CrossRef] [PubMed]
- Senturk, S.; Shirole, N.H.; Nowak, D.G.; Corbo, V.; Pal, D.; Vaughan, A.; Tuveson, D.A.; Trotman, L.C.; Kinney, J.B.; Sordella, R. Rapid and tunable method to temporally control gene editing based on conditional Cas9 stabilization. Nat. Commun. 2017, 8, 14370. [Google Scholar] [CrossRef] [PubMed]
- Shalem, O.; Sanjana, N.E.; Hartenian, E.; Shi, X.; Scott, D.A.; Mikkelsen, T.S.; Heckl, D.; Ebert, B.L.; Root, D.E.; Doench, J.G.; et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 2014, 343, 84–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Injected Cell Number | Number of Animals with Tumors REN-4 | Number of Animals with Tumors AXL-c17 |
|---|---|---|
| 1 × 106 | 1/1 | 0/1 |
| 2 × 105 | 4/4 | 0/3 |
| Total | 5/5 | 0/4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Batur, T.; Argundogan, A.; Keles, U.; Mutlu, Z.; Alotaibi, H.; Senturk, S.; Ozturk, M. AXL Knock-Out in SNU475 Hepatocellular Carcinoma Cells Provides Evidence for Lethal Effect Associated with G2 Arrest and Polyploidization. Int. J. Mol. Sci. 2021, 22, 13247. https://doi.org/10.3390/ijms222413247
Batur T, Argundogan A, Keles U, Mutlu Z, Alotaibi H, Senturk S, Ozturk M. AXL Knock-Out in SNU475 Hepatocellular Carcinoma Cells Provides Evidence for Lethal Effect Associated with G2 Arrest and Polyploidization. International Journal of Molecular Sciences. 2021; 22(24):13247. https://doi.org/10.3390/ijms222413247
Chicago/Turabian StyleBatur, Tugce, Ayse Argundogan, Umur Keles, Zeynep Mutlu, Hani Alotaibi, Serif Senturk, and Mehmet Ozturk. 2021. "AXL Knock-Out in SNU475 Hepatocellular Carcinoma Cells Provides Evidence for Lethal Effect Associated with G2 Arrest and Polyploidization" International Journal of Molecular Sciences 22, no. 24: 13247. https://doi.org/10.3390/ijms222413247