
Transcriptomic Profiling Reveals AKR1C1 and AKR1C3 Mediate Cisplatin Resistance in Signet Ring Cell Gastric Carcinoma via Autophagic Cell Death


 ,
,  and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Long-Term Exposure to Cisplatin Treatment Can Promote Drug Resistance in SRCGC
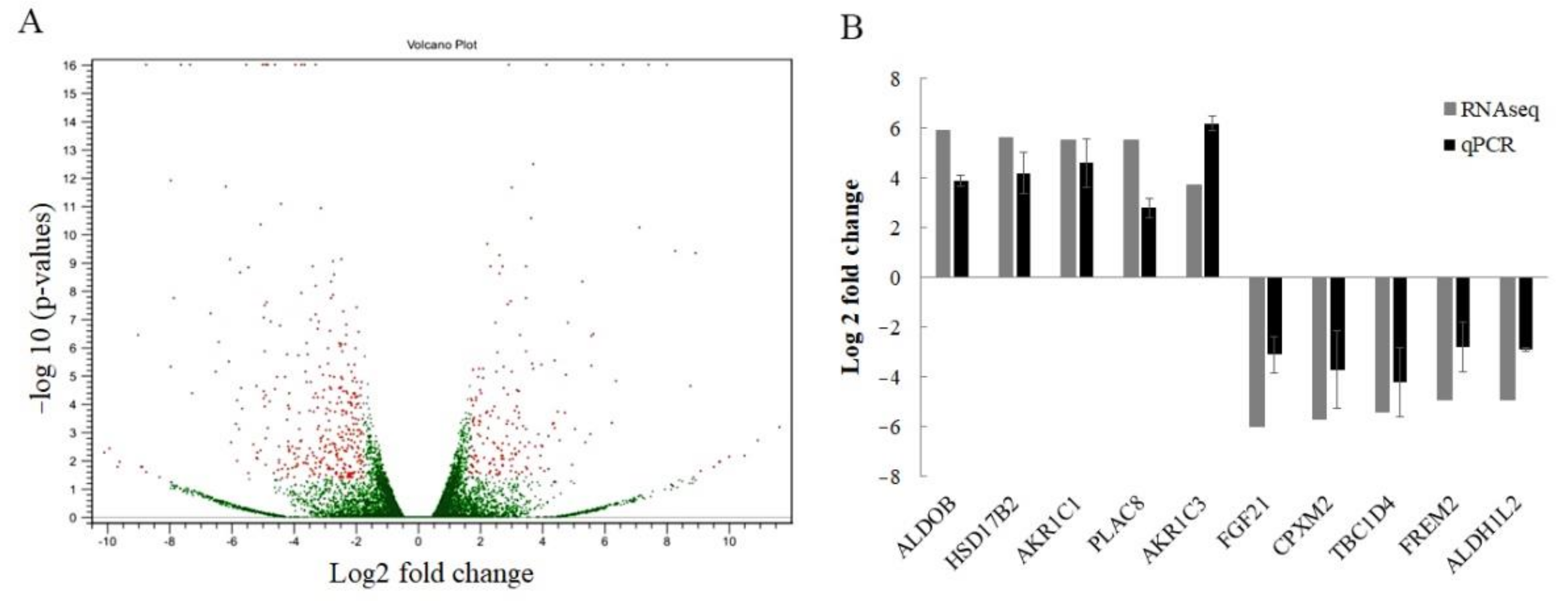
2.2. Identification of Candidate Genes to Promote Cisplatin Resistance in SRCGC Using Transcriptomic Sequencing
2.3. Verification of Cisplatin Resistance Related Genes in KATO/DDP Cell Line
2.4. Inhibition of AKR1C1 and AKR1C3 in Resensitizing the Cisplatin Cytotoxicity
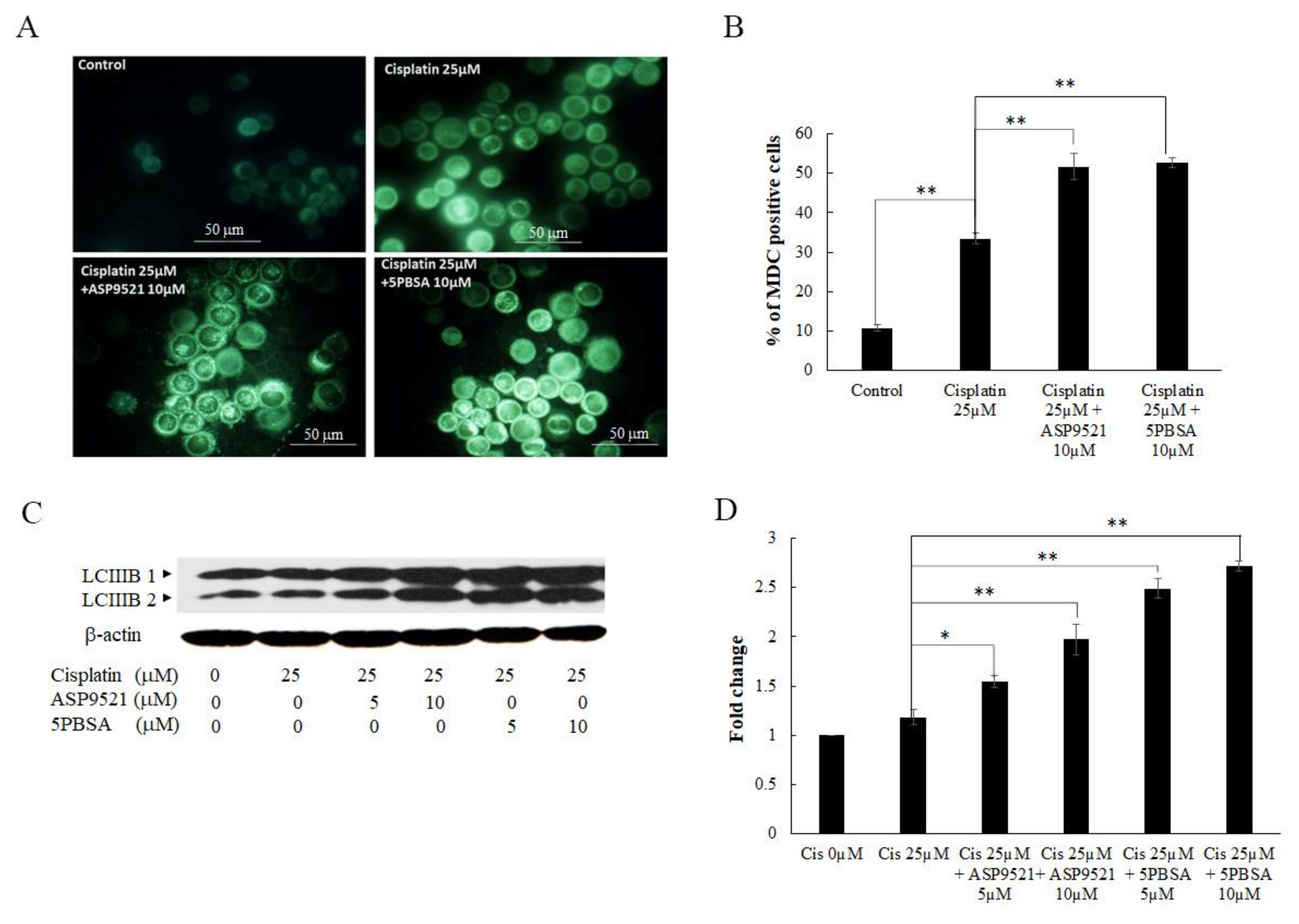
2.5. Inhibition of AKR1C1- and AKR1C3-Induced Cell Death via Autophagy
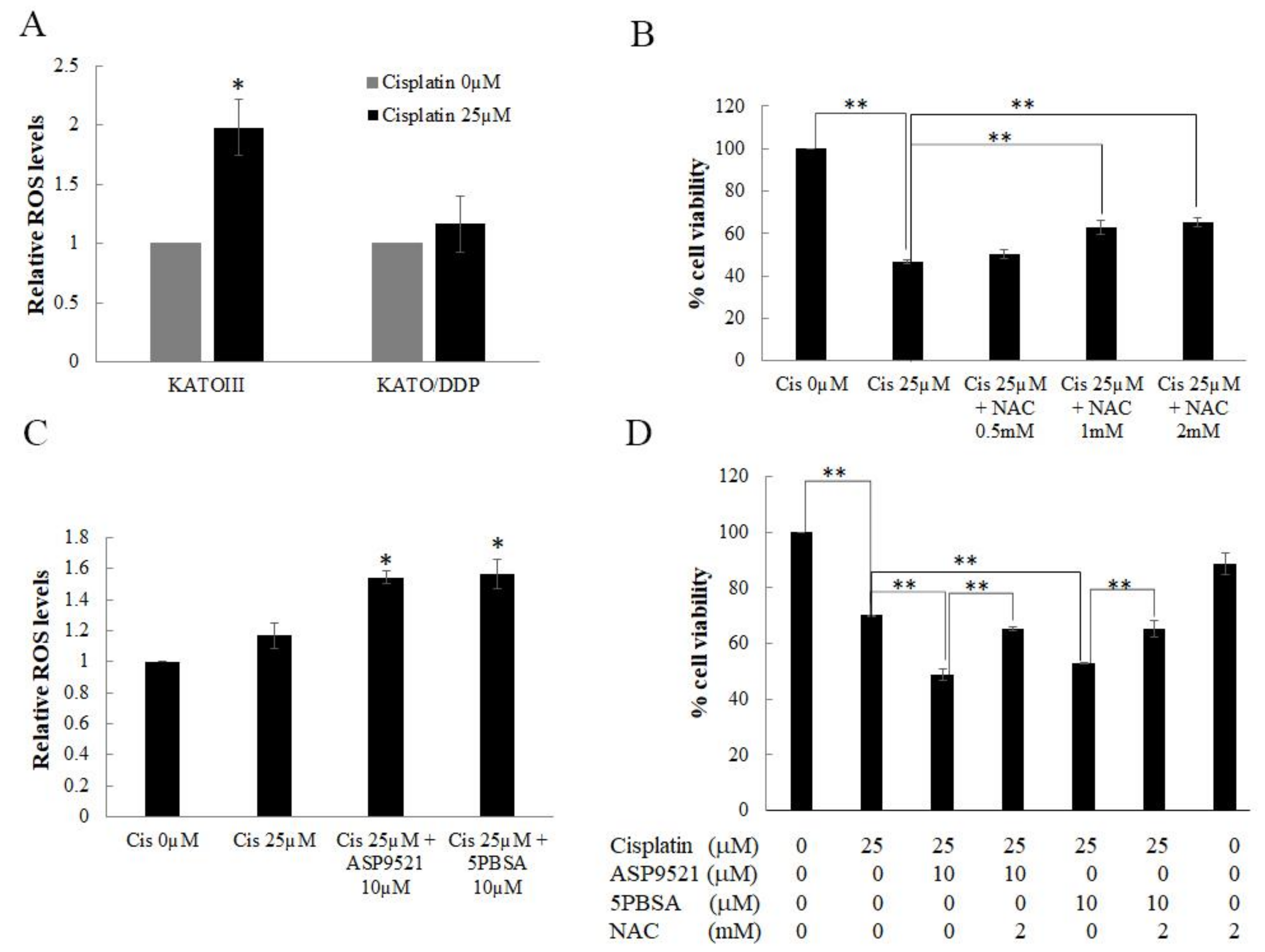
2.6. AKR1C1 and 1C3 Mediated Chemo-Resistance in KATO/DPP by Regulating Redox Homeostasis
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Cell Line and Culture Condition
4.3. Establishment of Cisplatin Resistance Gastric Cancer Cell Line
4.4. Cell Viability Test
4.5. RNA Isolation, Transcriptomic Sequencing, and Analysis
4.6. RNA Isolation, Reverse Transcription, and Quantitative PCR
4.7. Western Blotting Analysis
4.8. Intracellular ROS Measurement
4.9. Apoptosis Assay
4.10. Monodensylcadaverine Staining
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 3-MA | 3-Methyladenine |
| 5PBSA | 3-Bromo-5-phenylsalicylic acid |
| AKR | Aldoketoreductase |
| AKR1C1 | Aldoketoreductase1C1 |
| AKR1C3 | Aldoketoreductase1C3 |
| CYPs | Microsomal cytochrome P450 enzymes |
| DCF-DA | 2’,7’-Dichlorofluorescin diacetate |
| DEGs | Diffrentially expressed genes |
| DMEM | Dulbecco’s Modified Eagle Medium |
| FBS | Fetal bovine serum |
| GO | Gene ontology |
| HNE | 4-Hydroxynonenal |
| MDC | Monodansylcadaverine |
| NAC | N acetyl cysteine |
| NCBI | National center for biotechnology information |
| Nrf2 | Nuclear factor erythroid related factor 2 |
| PBS | Phosphate-buffered saline |
| QC | Quality Control |
| ROS | Reactive oxygen species |
| SRA | Sequence read archive |
| SRCGC | Signet ring cell gastric carcinoma |
References
- Rawla, P.; Barsouk, A. Epidemiology of gastric cancer: Global trends, risk factors and prevention. Gastroenterol. Rev. 2019, 14, 26–38. [Google Scholar] [CrossRef] [PubMed]
- Bamboat, Z.M.; Tang, L.H.; Vinuela, E.; Kuk, D.; Gonen, M.; Shah, M.A.; Brennan, M.F.; Coit, D.G.; Strong, V.E. Stage-stratified prognosis of signet ring cell histology in patients undergoing curative resection for gastric adenocarcinoma. Ann. Surg. Oncol. 2014, 21, 1678–1685. [Google Scholar] [CrossRef] [PubMed]
- Ajani, J.; Buyse, M.; Lichinitser, M.; Gorbunova, V.; Bodoky, G.; Douillard, J.; Cascinu, S.; Heinemann, V.; Zaucha, R.; Carrato, A. Combination of cisplatin/S-1 in the treatment of patients with advanced gastric or gastroesophageal adenocarcinoma: Results of noninferiority and safety analyses compared with cisplatin/5-fluorouracil in the First-Line Advanced Gastric Cancer Study. Eur. J. 2013, 49, 3616–3624. [Google Scholar] [CrossRef]
- Fichtinger-Schepman, A.M.; van der Veer, J.L.; den Hartog, J.H.; Lohman, P.H.; Reedijk, J. Adducts of the antitumor drug cis-diamminedichloroplatinum(II) with DNA: Formation, identification, and quantitation. Biochemistry 1985, 24, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Kleih, M.; Bopple, K.; Dong, M.; Gaissler, A.; Heine, S.; Olayioye, M.A.; Aulitzky, W.E.; Essmann, F. Direct impact of cisplatin on mitochondria induces ROS production that dictates cell fate of ovarian cancer cells. Cell Death Dis. 2019, 10, 851. [Google Scholar] [CrossRef] [Green Version]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makovec, T. Cisplatin and beyond: Molecular mechanisms of action and drug resistance development in cancer chemotherapy. Radiol. Oncol. 2019, 53, 148–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsunaga, T.; Yamaguchi, A.; Morikawa, Y.; Kezuka, C.; Takazawa, H.; Endo, S.; El-Kabbani, O.; Tajima, K.; Ikari, A.; Hara, A. Induction of aldo-keto reductases (AKR1C1 and AKR1C3) abolishes the efficacy of daunorubicin chemotherapy for leukemic U937 cells. Anti-Cancer Drugs 2014, 25, 868–877. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Hong, X.; Gao, X.; Gu, X.; Xiong, W.; Zhao, J.; Yu, H.; Cui, M.; Xie, M.; Bai, Y.; et al. Methyl jasmonate enhances the radiation sensitivity of esophageal carcinoma cells by inhibiting the 11-ketoprostaglandin reductase activity of AKR1C3. Cancer Manag. Res. 2018, 10, 3149–3158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Luca, A.; Parker, L.J.; Ang, W.H.; Rodolfo, C.; Gabbarini, V.; Hancock, N.C.; Palone, F.; Mazzetti, A.P.; Menin, L.; Morton, C.J. A structure-based mechanism of cisplatin resistance mediated by glutathione transferase P1-1. Proc. Natl. Acad. Sci. USA 2019, 116, 13943–13951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Q.; Ding, Y.; Song, L.; Xu, J. Correlation of UGT1A1 and ERCC1 gene polymorphisms with the outcome of combined irinotecan plus cisplatin treatment in recurrent ovarian cancer. Genet. Mol. Res. 2015, 14, 7241–7247. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Feng, M.; Li, X.; Zhong, P.; Guo, A.; Chen, G.; Xu, Q.; Ye, Y. Transcriptome profiling of cancer and normal tissues from cervical squamous cancer patients by deep sequencing. Mol. Med. Rep. 2017, 16, 2075–2088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, C.-M.; Chang, L.-L.; Ying, M.-D.; Cao, J.; He, Q.-J.; Zhu, H.; Yang, B. Aldo–keto reductase AKR1C1–AKR1C4: Functions, regulation, and intervention for anti-cancer therapy. Front. Pharmacol. 2017, 8, 119. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.F.; Wang, S.G.; Zhao, Z.Y.; Li, W.L. AKR1C1-3, notably AKR1C3, are distinct biomarkers for liver cancer diagnosis and prognosis: Database mining in malignancies. Oncol. Lett. 2019, 18, 4515–4522. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Xu, Z.; Sun, J.; Lv, H.; Wang, Y.; Ni, Y.; Chen, S.; Hu, C.; Wang, L.; Chen, W. Cisplatin resistance in gastric cancer cells is involved with GPR30-mediated epithelial-mesenchymal transition. J. Cell Mol. Med. 2020, 24, 3625–3633. [Google Scholar] [CrossRef] [PubMed]
- Wongsirisin, P.; Yamada, S.L.; Yodkeeree, S.; Punfa, W.; Limtrakul, P. Association of DNA repair and drug transporter in relation to chemosensitivity in primary culture of thai gastric cancer patients. Biol. Pharm. Bull. 2018, 41, 360–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebert, B.; Kisiela, M.; Malátková, P.; El-Hawari, Y.; Maser, E. Regulation of human carbonyl reductase 3 (CBR3; SDR21C2) expression by Nrf2 in cultured cancer cells. Biochemistry 2010, 49, 8499–8511. [Google Scholar] [CrossRef]
- Hu, Y.; Cui, J.; Jin, L.; Su, Y.; Zhang, X. LRPPRC contributes to the cisplatin resistance of lung cancer cells by regulating MDR1 expression. Oncol. Rep. 2021, 45, 1. [Google Scholar] [CrossRef]
- Melguizo, C.; Prados, J.; Luque, R.; Ortiz, R.; Caba, O.; Álvarez, P.J.; Gonzalez, B.; Aranega, A. Modulation of MDR1 and MRP3 gene expression in lung cancer cells after paclitaxel and carboplatin exposure. Int. J. Mol. Sci. 2012, 13, 16624–16635. [Google Scholar] [CrossRef] [PubMed]
- Slyskova, J.; Sabatella, M.; Ribeiro-Silva, C.; Stok, C.; Theil, A.F.; Vermeulen, W.; Lans, H. Base and nucleotide excision repair facilitate resolution of platinum drugs-induced transcription blockage. Nucleic Acids Res. 2018, 46, 9537–9549. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.-C.; Chu, C.-B.; Liu, K.-J.; Huang, C.-Y.F.; Chang, J.-Y.; Pan, W.-Y.; Chen, H.-H.; Cheng, Y.-H.; Lee, K.-D.; Chen, M.-F. Gene expression profiling for analysis acquired oxaliplatin resistant factors in human gastric carcinoma TSGH-S3 cells: The role of IL-6 signaling and Nrf2/AKR1C axis identification. Biochem. Pharmacol. 2013, 86, 872–887. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, R.; Tsuda, M.; Yoshida, K.; Tanino, M.; Kimura, T.; Nishihara, H.; Abe, T.; Shinohara, N.; Nonomura, K.; Tanaka, S. Aldo-keto reductase 1C1 induced by interleukin-1β mediates the invasive potential and drug resistance of metastatic bladder cancer cells. Sci. Rep. 2016, 6, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Huebbers, C.U.; Verhees, F.; Poluschkin, L.; Olthof, N.C.; Kolligs, J.; Siefer, O.G.; Henfling, M.; Ramaekers, F.C.; Preuss, S.F.; Beutner, D. Upregulation of AKR1C1 and AKR1C3 expression in OPSCC with integrated HPV16 and HPV-negative tumors is an indicator of poor prognosis. Int. J. Cancer 2019, 144, 2465–2477. [Google Scholar] [CrossRef]
- Zhong, T.; Xu, F.; Xu, J.; Liu, L.; Chen, Y. Aldo-keto reductase 1C3 (AKR1C3) is associated with the doxorubicin resistance in human breast cancer via PTEN loss. Biomed. Pharmacother. 2015, 69, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Matsunaga, T.; Hojo, A.; Yamane, Y.; Endo, S.; El-Kabbani, O.; Hara, A. Pathophysiological roles of aldo–keto reductases (AKR1C1 and AKR1C3) in development of cisplatin resistance in human colon cancers. Chem. Biol. Interact. 2013, 202, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.; Krishnamurthy, S. Cellular responses to Cisplatin-induced DNA damage. J. Nucleic Acids 2010, 2010, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Zhong, X.; Yuan, H.; Guo, Y.; Song, D.; Qi, F.; Zhu, Z.; Wang, X.; Guo, Z. Interfering in apoptosis and DNA repair of cancer cells to conquer cisplatin resistance by platinum (IV) prodrugs. Chem. Sci. 2020, 11, 3829–3835. [Google Scholar] [CrossRef]
- Zhou, C.; Wang, Z.; Li, J.; Wu, X.; Fan, N.; Li, D.; Liu, F.; Plum, P.S.; Hoppe, S.; Hillmer, A.M. Aldo-Keto Reductase 1C3 Mediates Chemotherapy Resistance in Esophageal Adenocarcinoma via ROS Detoxification. Cancer 2021, 13, 2403. [Google Scholar] [CrossRef] [PubMed]
- Matsunaga, T.; Kawabata, S.; Yanagihara, Y.; Kezuka, C.; Kato, M.; Morikawa, Y.; Endo, S.; Chen, H.; Iguchi, K.; Ikari, A. Pathophysiological roles of autophagy and aldo-keto reductases in development of doxorubicin resistance in gastrointestinal cancer cells. Chem. Biol. Interact. 2019, 314, 108839. [Google Scholar] [CrossRef] [PubMed]
- Magnano, S.; Barroeta, P.H.; Duffy, R.; O’Sullivan, J.; Zisterer, D.M. Cisplatin induces autophagy-associated apoptosis in human oral squamous cell carcinoma (OSCC) mediated in part through reactive oxygen species. Toxicol. Appl. Pharmacol. 2021, 427, 115646. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.-J.; Zhang, Y.-M.; Zhang, L.; Huang, B.; Tao, F.-F.; Chen, W.; Guo, Z.-J.; Xu, Q.; Sun, Y. Novel monofunctional platinum (II) complex Mono-Pt induces apoptosis-independent autophagic cell death in human ovarian carcinoma cells, distinct from cisplatin. Autophagy 2013, 9, 996–1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray, S.; Roy, K. Cisplatin-induced lipid peroxidation and its inhibition with ascorbic acid. Indian J. Pharm. Sci. 2006, 68, 199–204. [Google Scholar]
- Matsunaga, T.; Yamaji, Y.; Tomokuni, T.; Morita, H.; Morikawa, Y.; Suzuki, A.; Yonezawa, A.; Endo, S.; Ikari, A.; Iguchi, K. Nitric oxide confers cisplatin resistance in human lung cancer cells through upregulation of aldo-keto reductase 1B10 and proteasome. Free Radic. Res. 2014, 48, 1371–1385. [Google Scholar] [CrossRef]
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid. Med. Cell. Longev. 2014, 2014, 60438. [Google Scholar] [CrossRef] [PubMed]
- Burczynski, M.E.; Sridhar, G.R.; Palackal, N.T.; Penning, T.M. The reactive oxygen species-and Michael acceptor-inducible human aldo-keto reductase AKR1C1 reduces the α, β-unsaturated aldehyde 4-hydroxy-2-nonenal to 1, 4-dihydroxy-2-nonene. J. Biol. Chem. 2001, 276, 2890–2897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antelmann, H.; Helmann, J.D. Thiol-based redox switches and gene regulation. Antioxid. Redox Signal. 2011, 14, 1049–1063. [Google Scholar] [CrossRef]
- Matsui, Y.; Takagi, H.; Qu, X.; Abdellatif, M.; Sakoda, H.; Asano, T.; Levine, B.; Sadoshima, J. Distinct roles of autophagy in the heart during ischemia and reperfusion: Roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ. Res. 2007, 100, 914–922. [Google Scholar] [CrossRef]
- Chen, Y.; McMillan-Ward, E.; Kong, J.; Israels, S.; Gibson, S. Oxidative stress induces autophagic cell death independent of apoptosis in transformed and cancer cells. Cell Death Differ. 2008, 15, 171–182. [Google Scholar] [CrossRef]
- Lee, Y.J.; Kim, N.-Y.; Suh, Y.-A.; Lee, C. Involvement of ROS in curcumin-induced autophagic cell death. Korean J. Physiol. Pharmacol. 2011, 15, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Phoo, N.L.L.; Dejkriengkraikul, P.; Khaw-On, P.; Yodkeeree, S. Transcriptomic Profiling Reveals AKR1C1 and AKR1C3 Mediate Cisplatin Resistance in Signet Ring Cell Gastric Carcinoma via Autophagic Cell Death. Int. J. Mol. Sci. 2021, 22, 12512. https://doi.org/10.3390/ijms222212512
Phoo NLL, Dejkriengkraikul P, Khaw-On P, Yodkeeree S. Transcriptomic Profiling Reveals AKR1C1 and AKR1C3 Mediate Cisplatin Resistance in Signet Ring Cell Gastric Carcinoma via Autophagic Cell Death. International Journal of Molecular Sciences. 2021; 22(22):12512. https://doi.org/10.3390/ijms222212512
Chicago/Turabian StylePhoo, Nang Lae Lae, Pornngarm Dejkriengkraikul, Patompong Khaw-On, and Supachai Yodkeeree. 2021. "Transcriptomic Profiling Reveals AKR1C1 and AKR1C3 Mediate Cisplatin Resistance in Signet Ring Cell Gastric Carcinoma via Autophagic Cell Death" International Journal of Molecular Sciences 22, no. 22: 12512. https://doi.org/10.3390/ijms222212512