
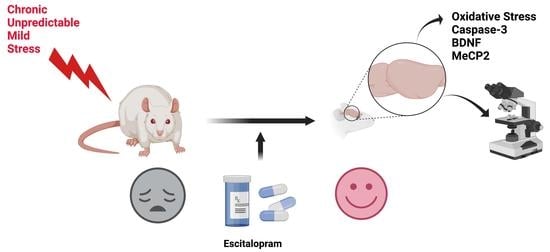
Escitalopram Targets Oxidative Stress, Caspase-3, BDNF and MeCP2 in the Hippocampus and Frontal Cortex of a Rat Model of Depression Induced by Chronic Unpredictable Mild Stress

 , ,
, ,  , ,
, ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
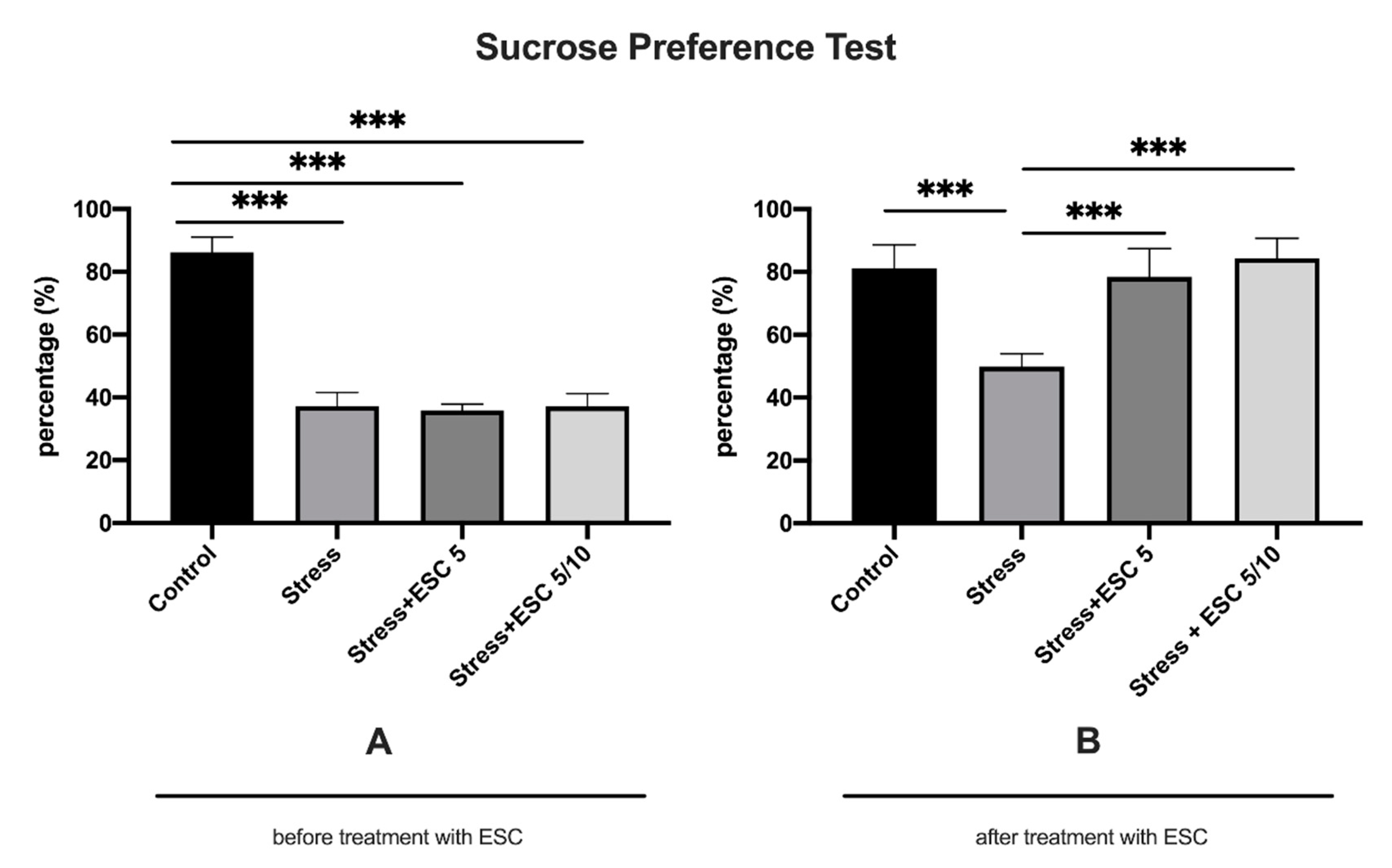
2.1. The Effect of CUMS Procedure and Escitalopram Treatment on Sucrose Preference
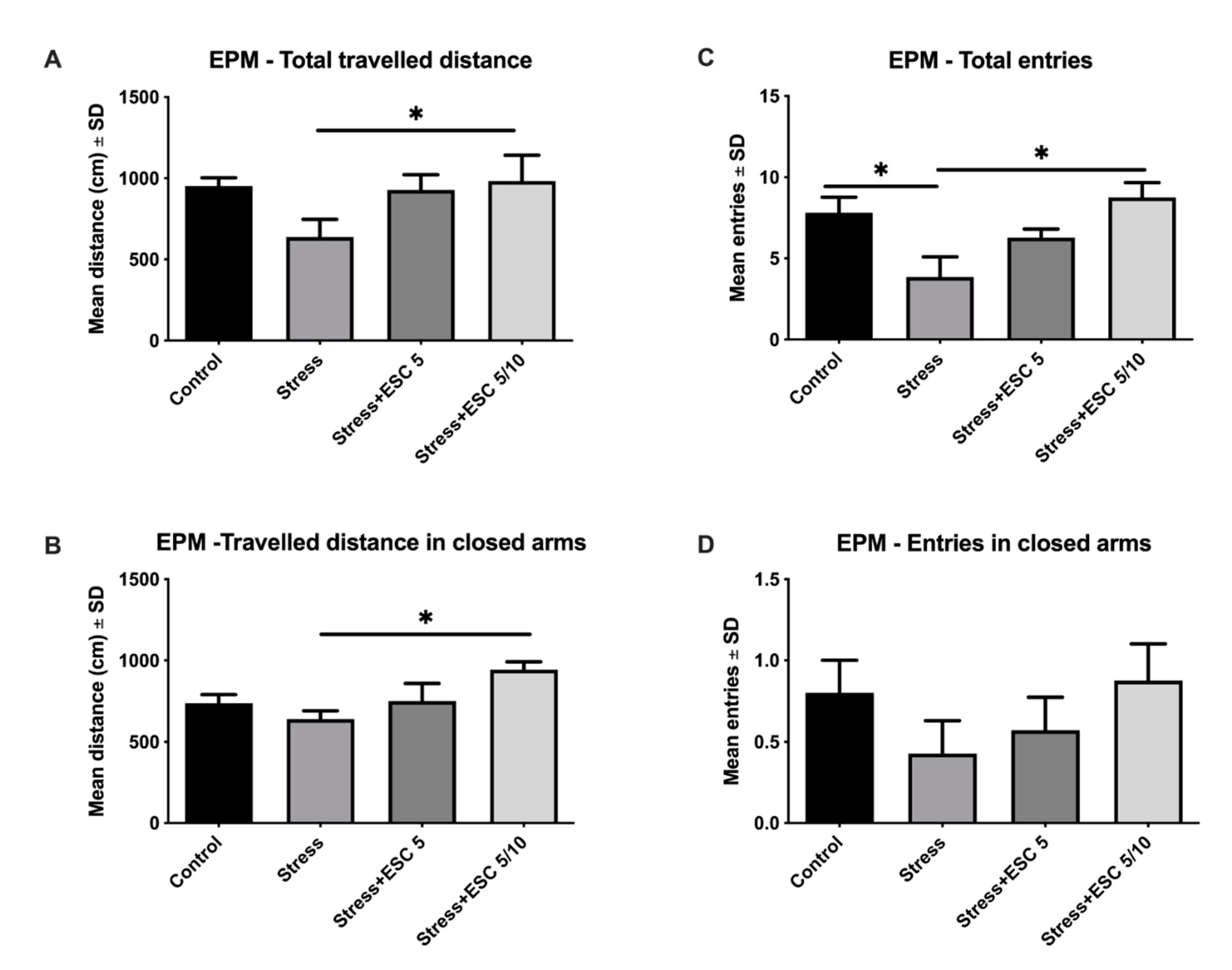
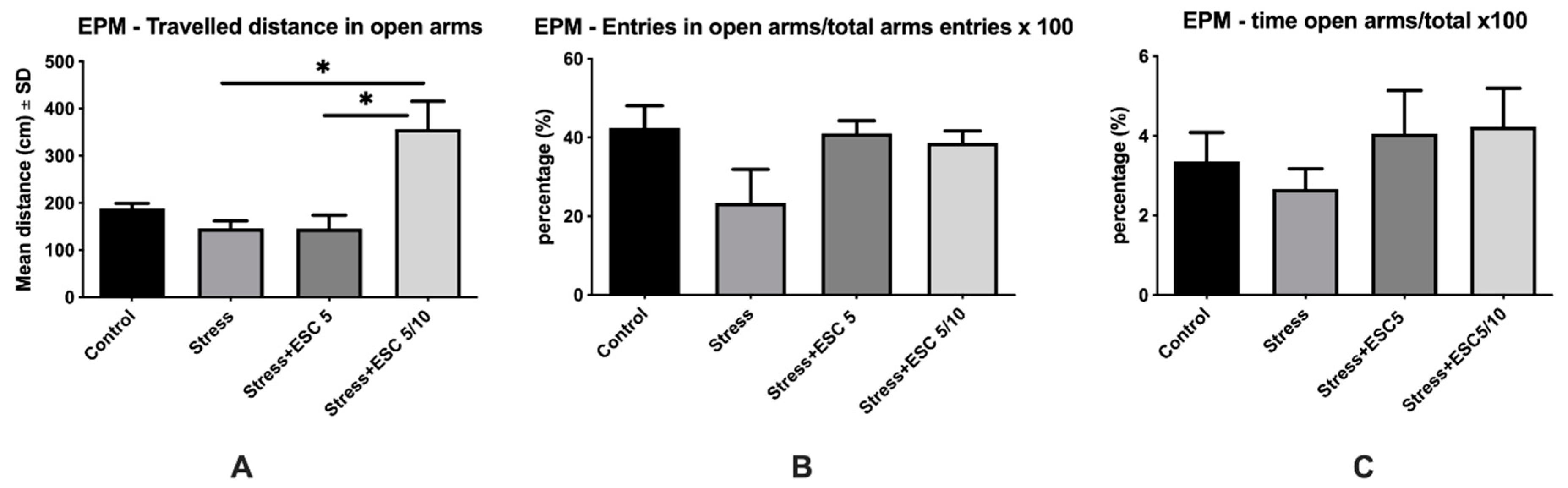
2.2. EPM Evaluation
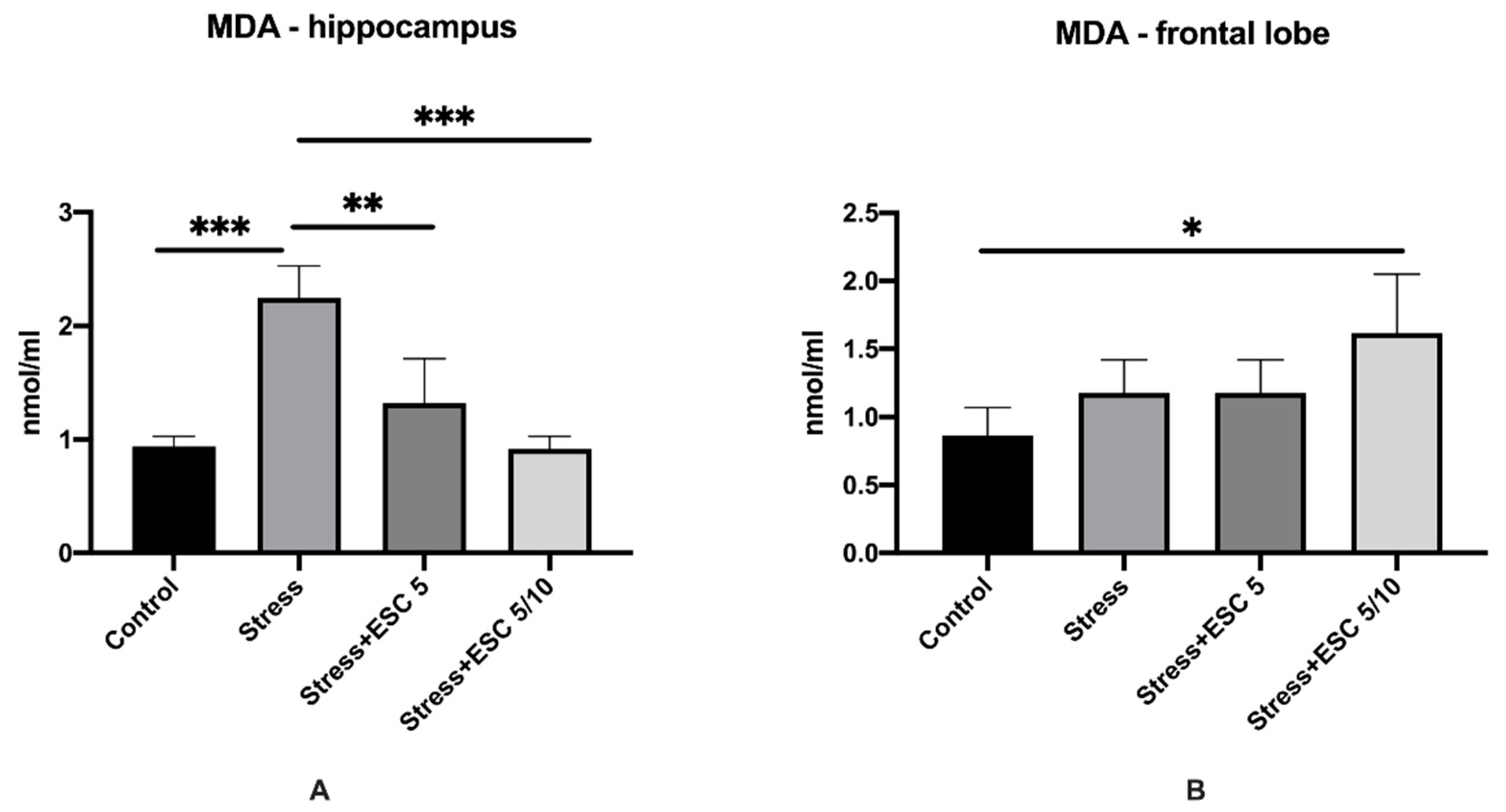
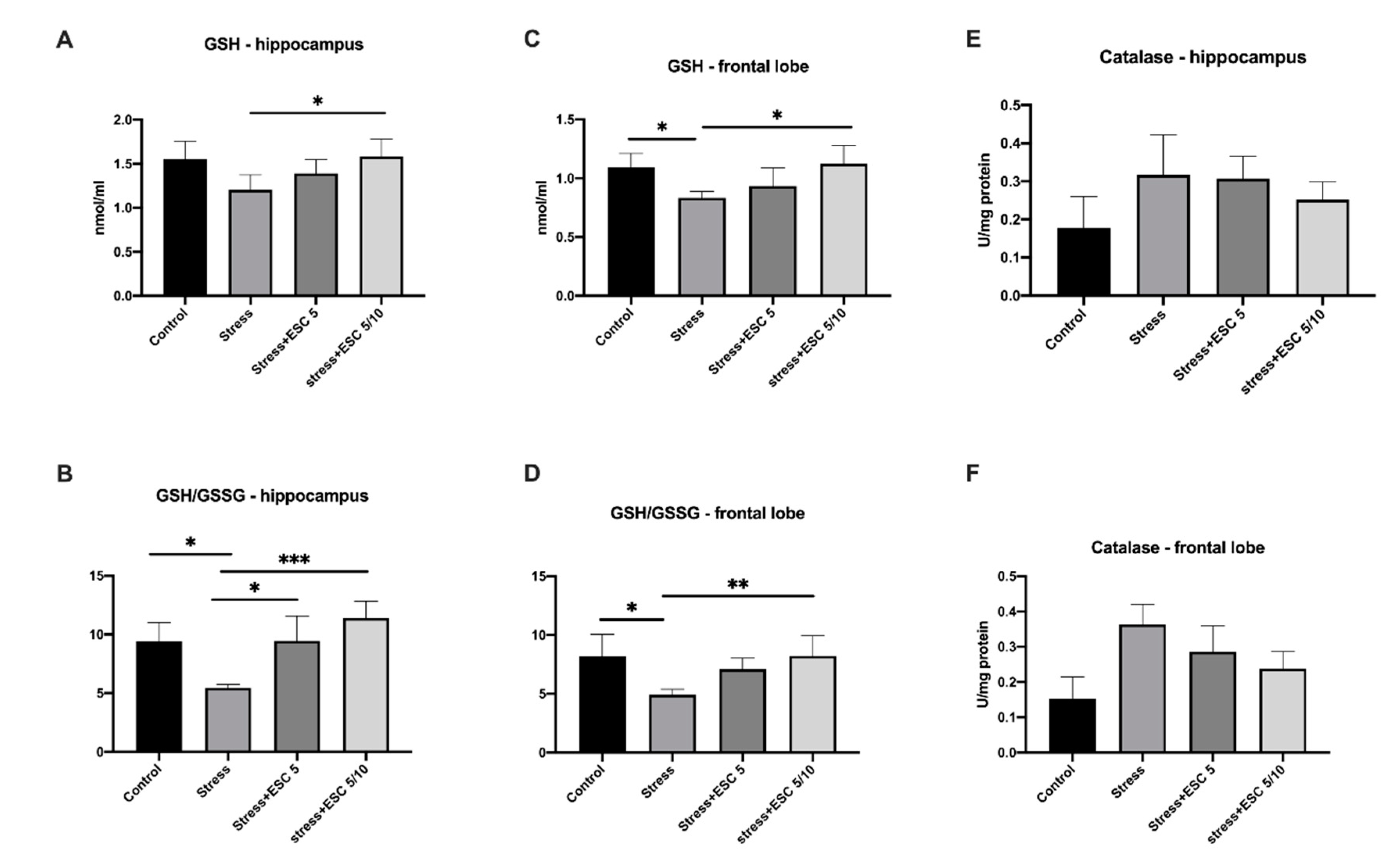
2.3. Oxidative Stress Assessment in the Hippocampus and Frontal Cortex
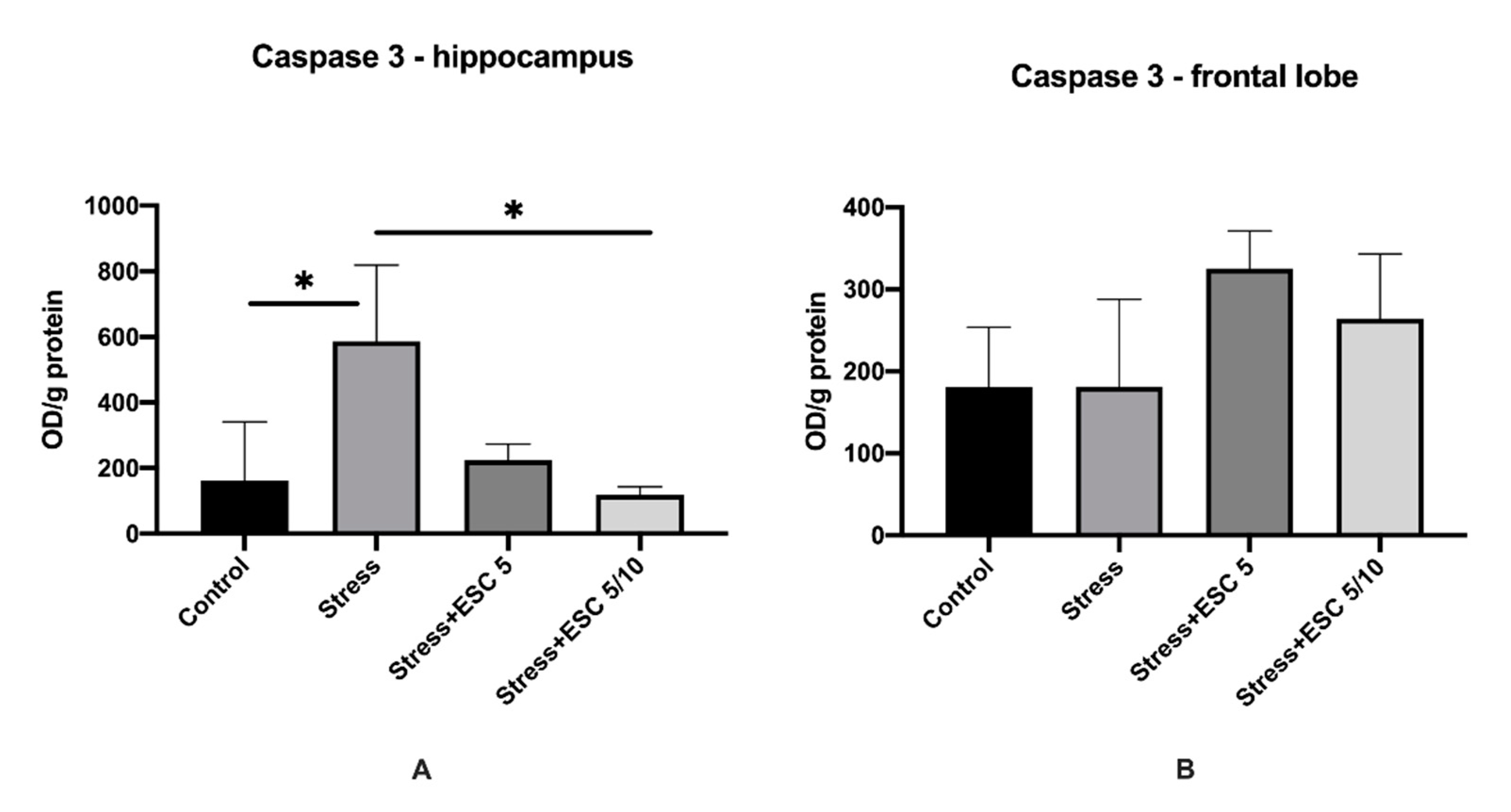
2.4. Caspase-3 Activity in the Hippocampus and Frontal Cortex
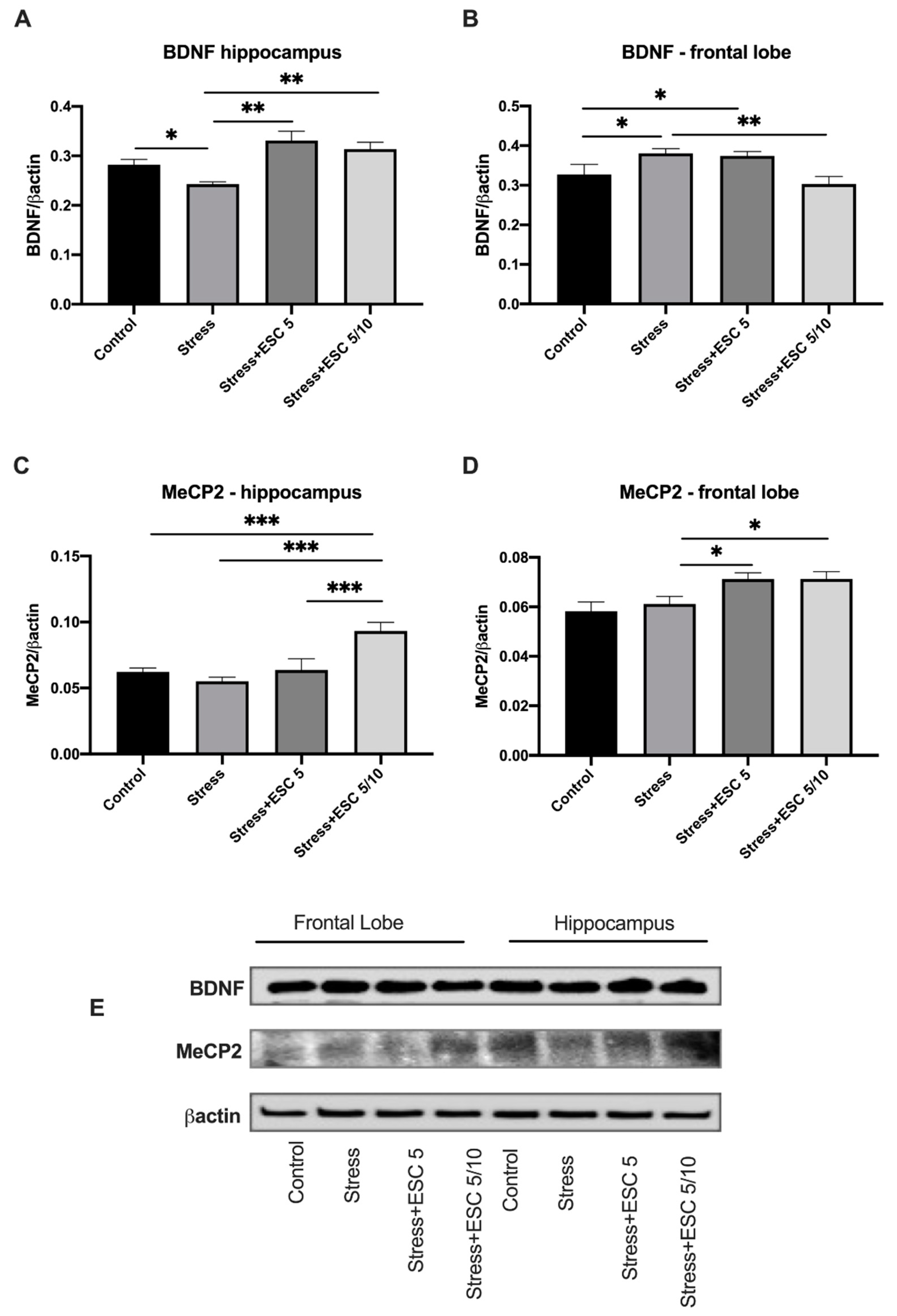
2.5. Quantitative Estimation of BDNF and MeCP2 Expressions
2.6. Histological Examination of Hippocampus and Frontal Cortex Tissues
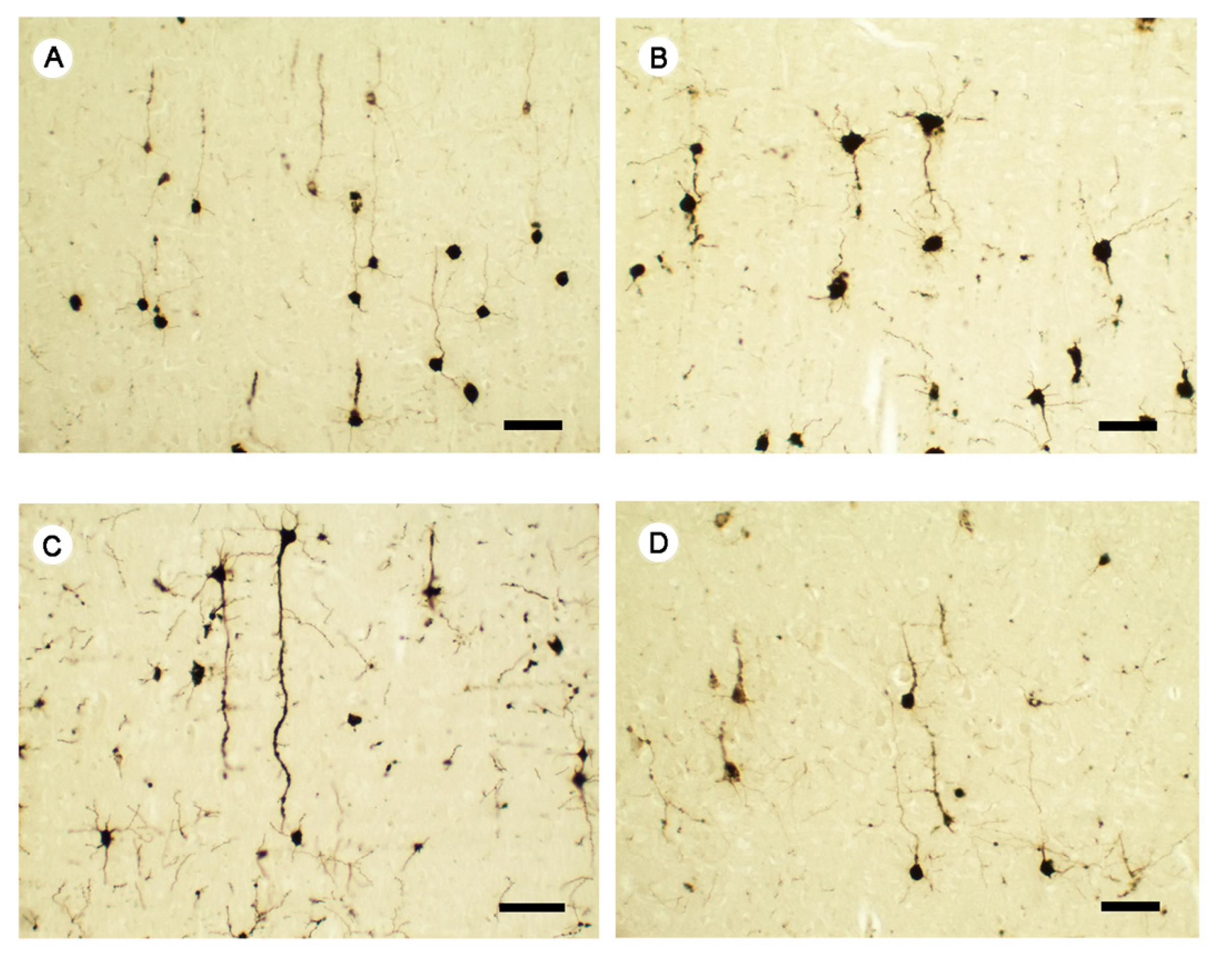
2.6.1. Golgi-Cox
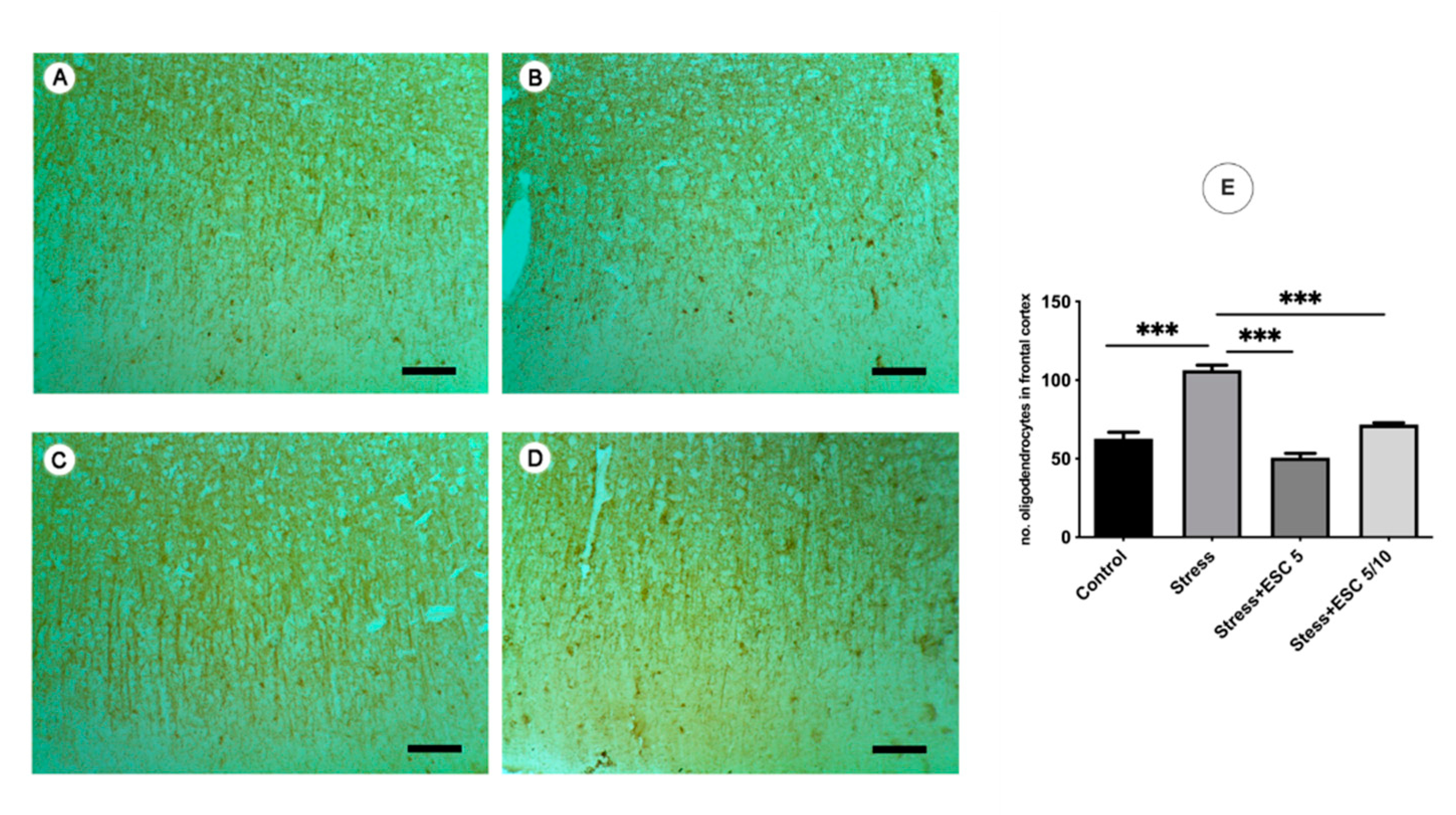
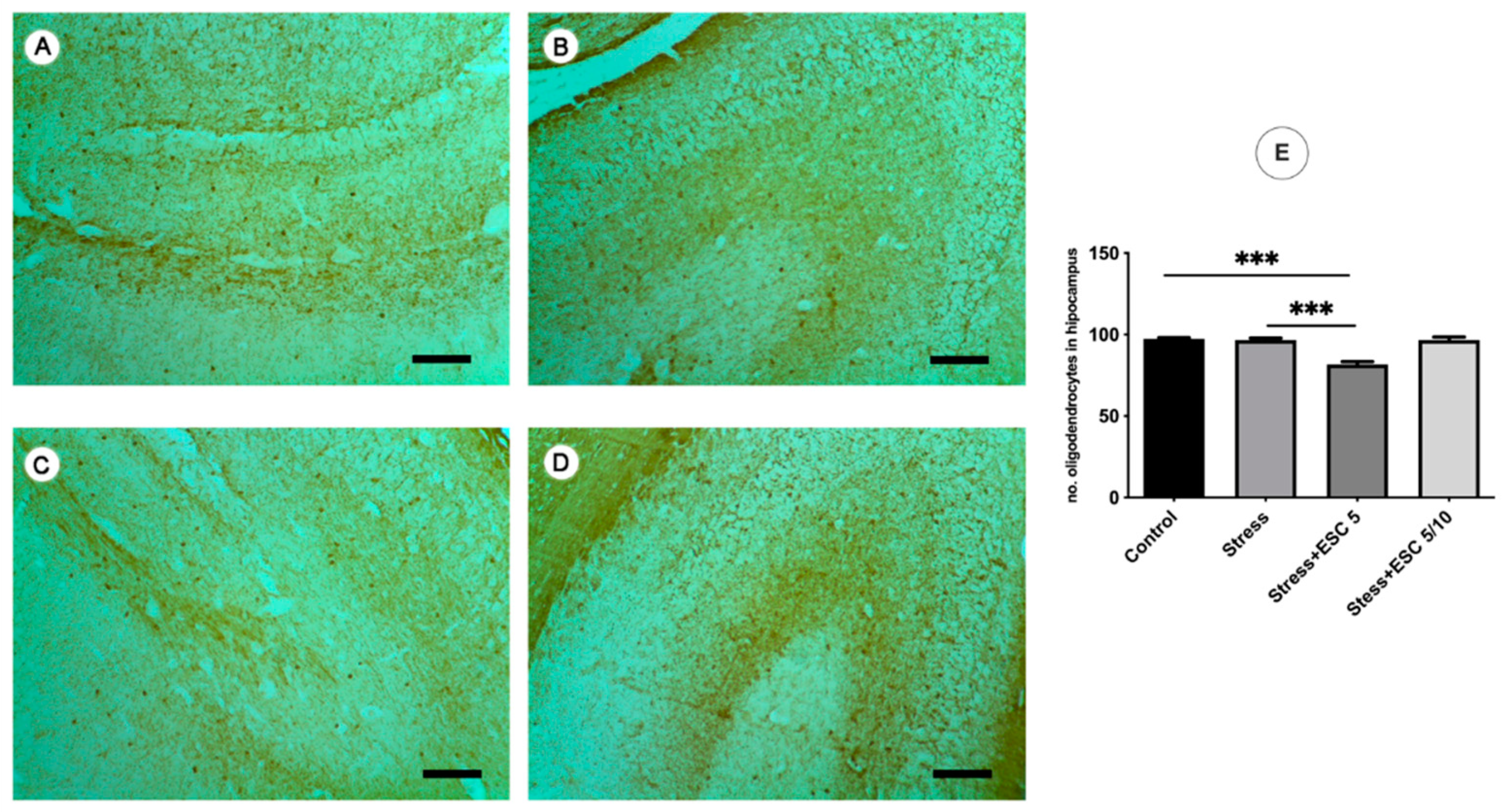
2.6.2. 2′,3′-Cyclic-nucleotide 3′-phosphodiesterase (CNPase)
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Animals and Housing
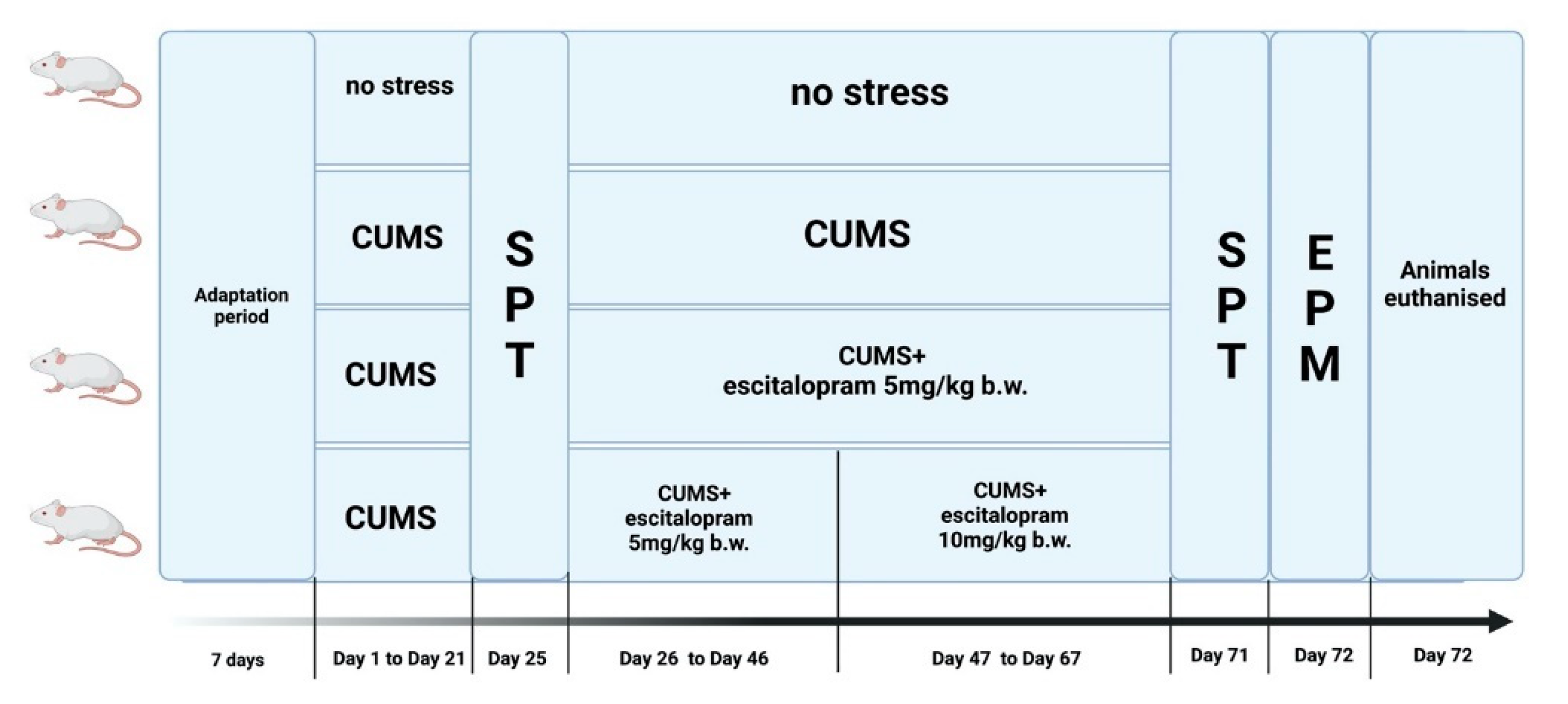
4.3. Experimental Design
4.4. Chronic Unpredictable Mild Stress Protocol
4.5. Behavioural Testing
4.5.1. Sucrose Preference Test
4.5.2. Elevated Plus Maze
4.6. Oxidative Stress Markers
4.7. Quantitative Estimation of BDNF and MeCP2 Expressions
4.8. Histological and Immunohistochemical Investigation of Hippocampus and Frontal Lobe
4.8.1. Immunohistochemistry
4.8.2. Golgi-Cox Impregnation
4.9. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO. Depression and Other Common Mental Disorders: Global Health Estimates; Licence: CC BY-NC-SA 3.0 IGO; World Health Organization: Geneva, Switzerland, 2017. [Google Scholar]
- Ferrari, A.; Somerville, A.; Baxter, A.; Norman, R.; Patten, S.; Vos, T.; Whiteford, H. Global variation in the prevalence and incidence of major depressive disorder: A systematic review of the epidemiological literature. Psychol. Med. 2012, 43, 471–481. [Google Scholar] [CrossRef]
- Al-Harbi, K.S. Treatment-resistant depression: Therapeutic trends, challenges, and future directions. Patient Pref. Adherence 2012, 6, 369–388. [Google Scholar] [CrossRef] [Green Version]
- Reuter, S.; Gupta, S.; Chaturvedi, M.; Aggarwal, B. Oxidative stress, inflammation, and cancer: How are they linked? Free Radic. Biol. Med. 2010, 49, 1603–1616. [Google Scholar] [CrossRef] [Green Version]
- Pandya, C.; Howell, K.; Pillai, A. Antioxidants as potential therapeutics for neuropsychiatric disorders. Prog. Neuropsychopharmacol. Biol. Psychiatry 2013, 46, 214–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maes, M.; Galecki, P.; Chang, Y.; Berk, M. A review on the oxidative and nitrosative stress (O&NS) pathways in major depression and their possible contribution to the (neuro)degenerative processes in that illness. Prog. Neuropsychopharmacol. Biol. Psychiatry 2011, 35, 676–692. [Google Scholar]
- Dean, J.; Keshavan, M. The neurobiology of depression: An integrated view. Asian J. Psychiatry 2017, 27, 101–111. [Google Scholar] [CrossRef]
- Maydych, V. The interplay between stress, inflammation, and emotional attention: Relevance for depression. Front. Neurosci 2019, 13, 384. [Google Scholar] [CrossRef]
- Cruz-Pereira, J.; Rea, K.; Nolan, Y.; O’Leary, O.; Dinan, T.; Cryan, J. Depression’s unholy trinity: Dysregulated stress, immunity, and the microbiome. Annu. Rev. Psychol. 2020, 71, 49–78. [Google Scholar] [CrossRef]
- Willner, P. The chronic mild stress (CMS) model of depression: History, evaluation and usage. Neurobiol Stress 2017, 6, 78–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scotton, E.; Colombo, R.; Reis, J.; Possebon, G.; Hizo, G.; Valiati, F.; Géa, L.; Bristot, G.; Salvador, M.; Silva, T.; et al. BDNF prevents central oxidative damage in a chronic unpredictable mild stress model: The possible role of prdx-1 in anhedonic behavior. Behav. Brain Res. 2020, 378, 112245. [Google Scholar] [CrossRef] [PubMed]
- You, J.; Yun, S.; Nam, K.; Kang, C.; Won, R.; Lee, E. Mechanism of glucocorticoid-induced oxidative stress in rat hippocampal slice cultures. Can. J. Physiol. Pharmacol. 2009, 87, 440–447. [Google Scholar] [CrossRef]
- Lee, Y.; Kim, H.; Lee, C.; Hyun, S.; Ko, M.; Lee, B.; Hwang, D.; Ka, M. 2-Phenylethylamine (PEA) Ameliorates Corticosterone-Induced Depression-Like Phenotype via the BDNF/Trkb/CREB Signaling Pathway. Int. J. Mol. Sci. 2020, 21, 9103. [Google Scholar] [CrossRef]
- Kubera, M.; Obuchowicz, E.; Goehler, L.; Brzeszcz, J.; Maes, M. In Animal Models, Psychosocial Stress-Induced (Neuro)Inflammation, Apoptosis and Reduced Neurogenesis Are Associated to the Onset of Depression. Prog. Neuropsychopharmacol. Biol. Psychiatry 2011, 35, 744–759. [Google Scholar] [CrossRef] [PubMed]
- Dionisie, V.; Filip, G.A.; Manea, M.C.; Manea, M.; Riga, S. The anti-inflammatory role of SSRI and SNRI in the treatment of depression: A review of human and rodent research studies. Inflammopharmacology 2021, 29, 75–90. [Google Scholar] [CrossRef] [PubMed]
- Bakunina, N.; Pariante, C.; Zunszain, P. Immune Mechanisms Linked to Depression via Oxidative Stress and Neuroprogression. Immunology 2015, 144, 365–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, W.; Wilson-Delfosse, A.; Mieyal, J. Dysregulation of Glutathione Homeostasis in Neurodegenerative Diseases. Nutrients 2012, 4, 1399–1440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Ma, S.; Qu, R. SCLM, Total Saponins Extracted Fromchaihu-Jia-Longgu-Muli-Tang, Reduces Chronic Mild Stress-Induced Apoptosis in the Hippocampus in Mice. Pharm. Biol. 2010, 48, 840–848. [Google Scholar] [CrossRef]
- Wigner, P.; Synowiec, E.; Jóźwiak, P.; Czarny, P.; Bijak, M.; Białek, K.; Szemraj, J.; Gruca, P.; Papp, M.; Śliwiński, T. The Effect of Chronic Mild Stress and Escitalopram on the Expression and Methylation Levels of Genes Involved in the Oxidative and Nitrosative Stresses As Well As Tryptophan Catabolites Pathway in the Blood and Brain Structures. Int. J. Mol. Sci. 2020, 22, 10. [Google Scholar] [CrossRef]
- McEwen, B.; Nasca, C.; Gray, J. Stress Effects on Neuronal Structure: Hippocampus, Amygdala, and Prefrontal Cortex. Neuropsychopharmacology 2015, 41, 3–23. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Hu, W.; Zhang, W.; Yang, L.; Li, Y.; Xiao, Z.; Zhang, M.; He, Z. Paeoniflorin Attenuates Impairment of Spatial Learning and Hippocampal Long-Term Potentiation in Mice Subjected to Chronic Unpredictable Mild Stress. Psychopharmacology 2019, 236, 2823–2834. [Google Scholar] [CrossRef]
- Dwivedi, Y. Brain-Derived Neurotrophic Factor: Role in Depression and Suicide. Neuropsychiatr. Dis. Treat. 2009, 5, 433–449. [Google Scholar] [CrossRef] [Green Version]
- Lima Giacobbo, B.; Doorduin, J.; Klein, H.; Dierckx, R.; Bromberg, E.; de Vries, E. Brain-Derived Neurotrophic Factor in Brain Disorders: Focus on Neuroinflammation. Mol. Neurobiol. 2018, 56, 3295–3312. [Google Scholar] [CrossRef] [Green Version]
- Miao, Z.; Wang, Y.; Sun, Z. The Relationships between Stress, Mental Disorders, and Epigenetic Regulation of BDNF. Int. J. Mol. Sci. 2020, 21, 1375. [Google Scholar] [CrossRef] [Green Version]
- Taliaz, D.; Stall, N.; Dar, D.; Zangen, A. Knockdown of Brain-Derived Neurotrophic Factor in Specific Brain Sites Precipitates Behaviors Associated with Depression and Reduces Neurogenesis. Mol. Psychiatry 2009, 15, 80–92. [Google Scholar] [CrossRef] [Green Version]
- Autry, A.; Monteggia, L. Brain-Derived Neurotrophic Factor and Neuropsychiatric Disorders. Pharmacol. Rev. 2012, 64, 238–258. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Chen, Z. The Role of BDNF in Depression on the Basis of Its Location in the Neural Circuitry. Acta Pharmacol. Sin. 2010, 32, 3–11. [Google Scholar] [CrossRef] [Green Version]
- Bouvier, E.; Brouillard, F.; Molet, J.; Claverie, D.; Cabungcal, J.; Cresto, N.; Doligez, N.; Rivat, C.; Do, K.; Bernard, C.; et al. Nrf2-Dependent Persistent Oxidative Stress Results in Stress-Induced Vulnerability to Depression. Mol. Psychiatry 2017, 22, 1701–1713. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.; Cha, M.; Lee, B. Neuroprotective Effect of Antioxidants in the Brain. Int. J. Mol. Sci. 2020, 21, 7152. [Google Scholar] [CrossRef]
- Hashimoto, K. Essential Role of Keap1-Nrf2 Signaling in Mood Disorders: Overview and Future Perspective. Front. Pharmacol. 2018, 9, 1182. [Google Scholar] [CrossRef]
- Martín-Hernández, D.; Bris, Á.; MacDowell, K.; García-Bueno, B.; Madrigal, J.; Leza, J.; Caso, J. Modulation of the Antioxidant Nuclear Factor (Erythroid 2-Derived)-Like 2 Pathway by Antidepressants in Rats. Neuropharmacology 2016, 103, 79–91. [Google Scholar] [CrossRef]
- Martín-Hernández, D.; Caso, J.; Javier Meana, J.; Callado, L.; Madrigal, J.; García-Bueno, B.; Leza, J. Intracellular Inflammatory and Antioxidant Pathways in Postmortem Frontal Cortex of Subjects with Major Depression: Effect of Antidepressants. J. Neuroinflamm. 2018, 15, 251. [Google Scholar] [CrossRef]
- Yao, W.; Zhang, J.; Ishima, T.; Dong, C.; Yang, C.; Ren, Q.; Ma, M.; Han, M.; Wu, J.; Suganuma, H.; et al. Role of Keap1-Nrf2 Signaling in Depression and Dietary Intake of Glucoraphanin Confers Stress Resilience in Mice. Sci. Rep. 2016, 6, 30659. [Google Scholar] [CrossRef] [Green Version]
- Pejhan, S.; Rastegar, M. Role of DNA Methyl-Cpg-Binding Protein Mecp2 in Rett Syndrome Pathobiology and Mechanism of Disease. Biomolecules 2021, 11, 75. [Google Scholar] [CrossRef]
- Su, M.; Hong, J.; Zhao, Y.; Liu, S.; Xue, X. Mecp2 Controls Hippocampal Brain-Derived Neurotrophic Factor Expression via Homeostatic Interactions with Microrna-132 in Rats with Depression. Mol. Med. Rep. 2015, 12, 5399–5406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutchinson, A.; Deng, J.; Cohen, S.; West, A. Phosphorylation of Mecp2 at Ser421 Contributes to Chronic Antidepressant Action. J. Neurosci. 2012, 32, 14355–14363. [Google Scholar] [CrossRef] [PubMed]
- Filosa, S.; Pecorelli, A.; D’Esposito, M.; Valacchi, G.; Hajek, J. Exploring the Possible Link between Mecp2 and Oxidative Stress in Rett Syndrome. Free Radic. Biol. Med. 2015, 88, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Raison, C.; Miller, A. The Neuroimmunology of Stress and Depression. Semin. Clin. Neuropsychiatry 2001, 6, 277–295. [Google Scholar] [CrossRef] [PubMed]
- Hammen, C. Stress and Depression. Annu. Rev. Clin. Psychol. 2005, 1, 293–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duman, R.; Aghajanian, G.; Sanacora, G.; Krystal, J. Synaptic Plasticity and Depression: New Insights from Stress and Rapid-Acting Antidepressants. Nat. Med. 2016, 22, 238–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bondi, C.; Rodriguez, G.; Gould, G.; Frazer, A.; Morilak, D. Chronic Unpredictable Stress Induces A Cognitive Deficit and Anxiety-Like Behavior in Rats That Is Prevented by Chronic Antidepressant Drug Treatment. Neuropsychopharmacology 2007, 33, 320–331. [Google Scholar] [CrossRef] [Green Version]
- Willner, P. Chronic Mild Stress (CMS) Revisited: Consistency and Behavioural-Neurobiological Concordance in the Effects of CMS. Neuropsychobiology 2005, 52, 90–110. [Google Scholar] [CrossRef] [PubMed]
- Vollmayr, B.; Henn, F. Stress Models of Depression. Clin. Neurosci. Res. 2003, 3, 245–251. [Google Scholar] [CrossRef]
- Kennedy, S.; Lam, R.; McIntyre, R.; Tourjman, S.; Bhat, V.; Blier, P.; Hasnain, M.; Jollant, F.; Levitt, A.; MacQueen, G.; et al. Canadian Network For Mood and Anxiety Treatments (CANMAT) 2016 Clinical Guidelines For the Management of Adults with Major Depressive Disorder. Can. J. Psychiatry 2016, 61, 540–560. [Google Scholar] [CrossRef] [PubMed]
- Haduch, A.; Rysz, M.; Papp, M.; Daniel, W. The Activity of Brain and Liver Cytochrome P450 2D (CYP2D) Is Differently Affected by Antidepressants in the Chronic Mild Stress (CMS) Model of Depression in the Rat. Biochem. Pharmacol. 2018, 156, 398–405. [Google Scholar] [CrossRef] [PubMed]
- Jayatissa, M.; Bisgaard, C.; West, M.; Wiborg, O. The Number of Granule Cells in Rat Hippocampus Is Reduced After Chronic Mild Stress and Re-Established After Chronic Escitalopram Treatment. Neuropharmacology 2008, 54, 530–541. [Google Scholar] [CrossRef] [PubMed]
- Mnie-Filali, O.; El Mansari, M.; Scarna, H.; Zimmer, L.; Sánchez, C.; Haddjeri, N. Escitalopram: A selective inhibitor and allosteric modulator of the serotonin transporter. Encephale 2007, 33, 965–972. [Google Scholar] [CrossRef] [PubMed]
- Kaminska, K.; Rogoz, Z. The Antidepressant- and Anxiolytic-Like Effects Following Co-Treatment with Escitalopram and Risperidone in Rats. J. Physiol. Pharmacol. 2016, 67, 471–480. [Google Scholar]
- Kurhe, Y.; Mahesh, R.; Gupta, D.; Thangaraj, D. Effect of (4A) A Novel 5-HT3 Receptor Antagonist on Chronic Unpredictable Mild Stress Induced Depressive-Like Behavior in Mice: An Approach Using Behavioral Tests Battery. J. Basic Clin. Physiol. Pharmacol. 2015, 26, 25–33. [Google Scholar] [CrossRef]
- Näslund, J.; Studer, E.; Pettersson, R.; Hagsäter, M.; Nilsson, S.; Nissbrandt, H.; Eriksson, E. Differences in Anxiety-Like Behavior within A Batch of Wistar Rats Are Associated with Differences in Serotonergic Transmission, Enhanced by Acute SRI Administration, and Abolished by Serotonin Depletion. Int. J. Neuropsychopharmacol. 2015, 18, pyv018. [Google Scholar] [CrossRef] [Green Version]
- Doron, R.; Lotan, D.; Versano, Z.; Benatav, L.; Franko, M.; Armoza, S.; Kately, N.; Rehavi, M. Escitalopram Or Novel Herbal Mixture Treatments During Or Following Exposure to Stress Reduce Anxiety-Like Behavior Through Corticosterone and BDNF Modifications. PLoS ONE 2014, 9, e91455. [Google Scholar] [CrossRef]
- Scheggi, S.; De Montis, M.; Gambarana, C. Making Sense of Rodent Models of Anhedonia. Int. J. Neuropsychopharmacol. 2018, 21, 1049–1065. [Google Scholar] [CrossRef]
- Yankelevitch-Yahav, R.; Franko, M.; Huly, A.; Doron, R. The Forced Swim Test as a Model of Depressive-Like Behavior. J. Vis. Exp. 2015, 97, e52587. [Google Scholar] [CrossRef] [PubMed]
- Rai, A.; Gill, M.; Kinra, M.; Shetty, R.; Krishnadas, N.; Rao, C.; Sumalatha, S.; Kumar, N. Catechin Ameliorates Depressive Symptoms in Sprague Dawley Rats Subjected to Chronic Unpredictable Mild Stress by Decreasing Oxidative Stress. Biomed. Rep. 2019, 11, 78–84. [Google Scholar] [CrossRef] [Green Version]
- Gill, M.; Kinra, M.; Rai, A.; Chamallamudi, M.; Kumar, N. Evaluation of Antidepressant Activity of Methanolic Extract of Saraca Asoca Bark in a Chronic Unpredictable Mild Stress Model. Neuroreport 2018, 29, 134–140. [Google Scholar] [CrossRef]
- Alboni, S.; Benatti, C.; Capone, G.; Corsini, D.; Caggia, F.; Tascedda, F.; Mendlewicz, J.; Brunello, N. Time-Dependent Effects of Escitalopram on Brain Derived Neurotrophic Factor (BDNF) and Neuroplasticity Related Targets in the Central Nervous System of Rats. Eur. J. Pharmacol. 2010, 643, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Seo, M.; Lee, J.; Park, S. Effects of Escitalopram and Ibuprofen on a Depression-Like Phenotype Induced by Chronic Stress in Rats. Neurosci. Lett. 2019, 696, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Xiong, F.; Zhang, L. Role of the Hypothalamic–Pituitary–Adrenal Axis in Developmental Programming of Health and Disease. Front. Neuroendocrinol. 2013, 34, 27–46. [Google Scholar] [CrossRef] [Green Version]
- Frodl, T.; O’Keane, V. How Does the Brain Deal with Cumulative Stress? A Review with Focus on Developmental Stress, HPA Axis Function and Hippocampal Structure in Humans. Neurobiol. Dis. 2013, 52, 24–37. [Google Scholar] [CrossRef]
- Spiers, J.; Chen, H.; Sernia, C.; Lavidis, N. Activation of the Hypothalamic-Pituitary-Adrenal Stress Axis Induces Cellular Oxidative Stress. Front. Neurosci. 2015, 8, 456. [Google Scholar] [CrossRef] [Green Version]
- Popa-Wagner, A.; Mitran, S.; Sivanesan, S.; Chang, E.; Buga, A. ROS and Brain Diseases: The Good, the Bad, and the Ugly. Oxid. Med. Cell. Longev. 2013, 2013, 963520. [Google Scholar] [CrossRef]
- Cobley, J.; Fiorello, M.; Bailey, D. 13 Reasons Why the Brain Is Susceptible to Oxidative Stress. Redox Biol. 2018, 15, 490–503. [Google Scholar] [CrossRef]
- Che, Y.; Zhou, Z.; Shu, Y.; Zhai, C.; Zhu, Y.; Gong, S.; Cui, Y.; Wang, J. Chronic Unpredictable Stress Impairs Endogenous Antioxidant Defense in Rat Brain. Neurosci. Lett. 2015, 584, 208–213. [Google Scholar] [CrossRef]
- Eren, İ.; Nazıroğlu, M.; Demirdaş, A. Protective Effects of Lamotrigine, Aripiprazole and Escitalopram on Depression-Induced Oxidative Stress in Rat Brain. Neurochem. Res. 2007, 32, 1188–1195. [Google Scholar] [CrossRef]
- Shalaby, A.; Kamal, S. Effect of Escitalopram on GABA Level and Anti-Oxidant Markers in Prefrontal Cortex and Nucleus Accumbens of Chronic Mild Stress-Exposed Albino Rats. Int. J. Physiol. Pathophysiol. Pharmacol. 2009, 1, 154–161. [Google Scholar]
- Matchkov, V.; Kravtsova, V.; Wiborg, O.; Aalkjaer, C.; Bouzinova, E. Chronic Selective Serotonin Reuptake Inhibition Modulates Endothelial Dysfunction and Oxidative State in Rat Chronic Mild Stress Model of Depression. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2015, 309, R814–R823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucca, G.; Comim, C.; Valvassori, S.; Réus, G.; Vuolo, F.; Petronilho, F.; Dal-Pizzol, F.; Gavioli, E.; Quevedo, J. Effects of Chronic Mild Stress on the Oxidative Parameters in the Rat Brain. Neurochem. Int. 2009, 54, 358–362. [Google Scholar] [CrossRef]
- Wang, C.; Wu, H.; Jing, X.; Meng, Q.; Liu, B.; Zhang, H.; Gao, G. Oxidative Parameters in the Rat Brain of Chronic Mild Stress Model For Depression: Relation to Anhedonia-Like Responses. J. Membr. Biol. 2012, 245, 675–681. [Google Scholar] [CrossRef] [PubMed]
- Fontella, F.; Siqueira, I.; Vasconcellos, A.; Tabajara, A.; Netto, C.; Dalmaz, C. Repeated Restraint Stress Induces Oxidative Damage in Rat Hippocampus. Neurochem. Res. 2005, 30, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, X.; Shigenaga, M.; Yeo, H.; Mori, A.; Ames, B. Immobilization Stress Causes Oxidative Damage to Lipid, Protein, and DNA in the Brain of Rats. FASEB J. 1996, 10, 1532–1538. [Google Scholar] [CrossRef] [PubMed]
- Arent, C.; Réus, G.; Abelaira, H.; Ribeiro, K.; Steckert, A.; Mina, F.; Dal-Pizzol, F.; Quevedo, J. Synergist Effects of N-Acetylcysteine and Deferoxamine Treatment on Behavioral and Oxidative Parameters Induced by Chronic Mild Stress in Rats. Neurochem. Int. 2012, 61, 1072–1080. [Google Scholar] [CrossRef]
- Zhong, H.; Haddjeri, N.; Sánchez, C. Escitalopram, an Antidepressant with an Allosteric Effect at the Serotonin Transporter—A Review of Current Understanding of Its Mechanism of Action. Psychopharmacology 2011, 219, 1–13. [Google Scholar] [CrossRef]
- Mitozo, P.; de Souza, L.; Loch-Neckel, G.; Flesch, S.; Maris, A.; Figueiredo, C.; dos Santos, A.; Farina, M.; Dafre, A. A Study of the Relative Importance of the Peroxiredoxin-, Catalase-, and Glutathione-Dependent Systems in Neural Peroxide Metabolism. Free Radic. Biol. Med. 2011, 51, 69–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szymonik-Lesiuk, S.; Czechowska, G.; Stryjecka-Zimmer, M.; SLomka, M.; MAldro, A.; CeliNski, K.; Wielosz, M. Catalase, Superoxide Dismutase, and Glutathione Peroxidase Activities in Various Rat Tissues After Carbon Tetrachloride Intoxication. J. Hepatobiliary Pancreat. Surg. 2003, 10, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Castañeda, J.R.; Montilla, P.; Padillo, F.J.; Bujalance, I.; Muñoz, M.C.; Muntané, J.; Túnez, I. Role of serotonin in cerebral oxidative stress in rats. Acta Neurobiol. Exp. 2006, 66, 1–6. [Google Scholar]
- Liu, J.; Mori, A. Monoamine Metabolism Provides an Antioxidant Defense in the Brain against Oxidant- and Free Radical-Induced Damage. Arch. Biochem. Biophys. 1993, 302, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Hadi, N.; Malik, A.; Azam, S.; Khan, N.; Iqbal, J. Serotonin–Cu(II)-Mediated DNA Cleavage: Mechanism of Copper Binding by Serotonin. Toxicol. In Vitro 2002, 16, 669–674. [Google Scholar] [CrossRef]
- Azouzi, S.; Santuz, H.; Morandat, S.; Pereira, C.; Côté, F.; Hermine, O.; El Kirat, K.; Colin, Y.; Le Van Kim, C.; Etchebest, C.; et al. Antioxidant and Membrane Binding Properties of Serotonin Protect Lipids from Oxidation. Biophys. J. 2017, 112, 1863–1873. [Google Scholar] [CrossRef] [Green Version]
- Sarikaya, S.B.O.; Gulcin, I. Radical Scavenging and Antioxidant Capacity of Serotonin. Curr. Bioact. Compd. 2013, 9, 143–152. [Google Scholar] [CrossRef]
- McEwen, B.; Gianaros, P. Stress- and Allostasis-Induced Brain Plasticity. Annu. Rev. Med. 2011, 62, 431–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnsten, A. Stress Signalling Pathways That Impair Prefrontal Cortex Structure and Function. Nat. Rev. Neurosci. 2009, 10, 410–422. [Google Scholar] [CrossRef]
- Gobinath, A.; Mahmoud, R.; Galea, L. Influence of Sex and Stress Exposure Across the Lifespan on Endophenotypes of Depression: Focus on Behavior, Glucocorticoids, and Hippocampus. Front. Neurosci. 2015, 8, 420. [Google Scholar] [CrossRef]
- George, M.; Ketter, T.; Post, R. Prefrontal Cortex Dysfunction in Clinical Depression. Depression 1994, 2, 59–72. [Google Scholar] [CrossRef]
- McKinnon, M.C.; Yucel, K.; Nazarov, A.; MacQueen, G.M. A Meta-Analysis Examining Clinical Predictors of Hippocampal Volume in Patients with Major Depressive Disorder. J. Psychiatry Neurosci. 2009, 34, 41–54. [Google Scholar]
- Bessa, J.; Ferreira, D.; Melo, I.; Marques, F.; Cerqueira, J.; Palha, J.; Almeida, O.; Sousa, N. The Mood-Improving Actions of Antidepressants Do Not Depend on Neurogenesis But Are Associated with Neuronal Remodeling. Mol. Psychiatry 2008, 14, 764–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerqueira, J.; Mailliet, F.; Almeida, O.; Jay, T.; Sousa, N. The Prefrontal Cortex as a Key Target of the Maladaptive Response to Stress. J. Neurosci. 2007, 27, 2781–2787. [Google Scholar] [CrossRef] [PubMed]
- Dusi, N.; Barlati, S.; Vita, A.; Brambilla, P. Brain Structural Effects of Antidepressant Treatment in Major Depression. Curr. Neuropharmacol. 2015, 13, 458–465. [Google Scholar] [CrossRef] [PubMed]
- Orlovsky, M.; Dosenko, V.; Spiga, F.; Skibo, G.; Lightman, S. Hippocampus Remodeling by Chronic Stress Accompanied by GR, Proteasome and Caspase-3 Overexpression. Brain Res. 2014, 1593, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Bachis, A.; Cruz, M.; Nosheny, R.; Mocchetti, I. Chronic Unpredictable Stress Promotes Neuronal Apoptosis in the Cerebral Cortex. Neurosci. Lett. 2008, 442, 104–108. [Google Scholar] [CrossRef] [Green Version]
- Filipović, D.; Zlatković, J.; Inta, D.; Bjelobaba, I.; Stojiljkovic, M.; Gass, P. Chronic Isolation Stress Predisposes the Frontal Cortex But Not the Hippocampus to the Potentially Detrimental Release of Cytochrome C from Mitochondria and the Activation of Caspase-3. J. Neurosci. Res. 2011, 89, 1461–1470. [Google Scholar] [CrossRef]
- Wang, J.; Xu, S.; Chen, X.; Wang, L.; Li, J.; Li, G.; Zhang, B. Antidepressant Effect of EGCG Through the Inhibition of Hippocampal Neuroinflammation in Chronic Unpredictable Mild Stress-Induced Depression Rat Model. J. Funct. Foods 2020, 73, 104106. [Google Scholar] [CrossRef]
- Yang, Y.; Hu, Z.; Du, X.; Davies, H.; Huo, X.; Fang, M. Mir-16 and Fluoxetine Both Reverse Autophagic and Apoptotic Change in Chronic Unpredictable Mild Stress Model Rats. Front. Neurosci. 2017, 11, 428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Zheng, L.; Wan, Y.; Chen, Z.; Li, P.; Wang, Y. Metoprolol, N-Acetylcysteine, and Escitalopram Prevents Chronic Unpredictable Mild Stress-Induced Depression by Inhibition of Endoplasmic Reticulum Stress. Front. Psychiatry 2018, 9, 696. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Xu, J.; Lazarovici, P.; Quirion, R.; Zheng, W. cAMP Response Element-Binding Protein (CREB): A Possible Signaling Molecule Link in the Pathophysiology of Schizophrenia. Front. Mol. Neurosci. 2018, 11, 255. [Google Scholar] [CrossRef]
- Nowacka, M.; Obuchowicz, E. BDNF and VEGF in the Pathogenesis of Stress-Induced Affective Diseases: An Insight from Experimental Studies. Pharmacol. Rep. 2013, 65, 535–546. [Google Scholar] [CrossRef]
- Larsen, M.; Mikkelsen, J.; Hay-Schmidt, A.; Sandi, C. Regulation of Brain-Derived Neurotrophic Factor (BDNF) in the Chronic Unpredictable Stress Rat Model and the Effects of Chronic Antidepressant Treatment. J. Psychiatry Res. 2010, 44, 808–816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, M.; Lee, C.; Cho, H.; You, Y.; Lee, B.; Lee, J.; Park, S.; Kim, Y. Effects of Antipsychotic Drugs on the Expression of Synapse-Associated Proteins in the Frontal Cortex of Rats Subjected to Immobilization Stress. Psychiatry Res. 2015, 229, 968–974. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Lee, S.; Kim, J.; Yoon, J.; Kim, Y. Effects of Quetiapine on the Brain-Derived Neurotrophic Factor Expression in the Hippocampus and Neocortex of Rats. Neurosci. Lett. 2006, 402, 25–29. [Google Scholar] [CrossRef]
- Cieślik, K.; Sowa-Kućma, M.; Ossowska, G.; Legutko, B.; Wolak, M.; Opoka, W.; Nowak, G. Chronic Unpredictable Stress-Induced Reduction in the Hippocampal Brain-Derived Neurotrophic Factor (BDNF) Gene Expression Is Antagonized by Zinc Treatment. Pharmacol. Rep. 2011, 63, 537–543. [Google Scholar] [CrossRef]
- Allaman, I.; Papp, M.; Kraftsik, R.; Fiumelli, H.; Magistretti, P.J.; Martin, J.L. Expression of Brain-Derived Neurotrophic Factor Is Not Modulated by Chronic Mild Stress in the Rat Hippocampus and Amygdala. Pharmacol. Rep. 2008, 60, 1001–1007. [Google Scholar]
- Bergström, A.; Jayatissa, M.; Mørk, A.; Wiborg, O. Stress Sensitivity and Resilience in the Chronic Mild Stress Rat Model of Depression; an in Situ Hybridization Study. Brain Res. 2008, 1196, 41–52. [Google Scholar] [CrossRef]
- Schulte-Herbrüggen, O.; Fuchs, E.; Abumaria, N.; Ziegler, A.; Danker-Hopfe, H.; Hiemke, C.; Hellweg, R. Effects of Escitalopram on the Regulation of Brain-Derived Neurotrophic Factor and Nerve Growth Factor Protein Levels in a Rat Model of Chronic Stress. J. Neurosci. Res. 2009, 87, 2551–2560. [Google Scholar] [CrossRef] [PubMed]
- Angelucci, F.; Brenè, S.; Mathé, A. BDNF in Schizophrenia, Depression and Corresponding Animal Models. Mol. Psychiatry 2005, 10, 345–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Zhao, J.; Wang, Z.; Xu, L.; Liu, A.; Du, G. DL0410 Attenuates Oxidative Stress and Neuroinflammation via BDNF/Trkb/ERK/CREB and Nrf2/HO-1 Activation. Int. Immunopharmacol. 2020, 86, 106729. [Google Scholar] [CrossRef]
- Nair, A.; Vaidya, V. Cyclic AMP Response Element Binding Protein and Brain-Derived Neurotrophic Factor: Molecules That Modulate Our Mood? J. Biosci. 2006, 31, 423–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martocchia, A.; Curto, M.; Scaccianoce, S.; Comite, F.; Xenos, D.; Nasca, C.; Falaschi, G.; Ferracuti, S.; Girardi, P.; Nicoletti, F.; et al. Effects of Escitalopram on Serum BDNF Levels in Elderly Patients with Depression: A Preliminary Report. Aging Clin. Exp. Res. 2014, 26, 461–464. [Google Scholar] [CrossRef]
- Wolkowitz, O.; Wolf, J.; Shelly, W.; Rosser, R.; Burke, H.; Lerner, G.; Reus, V.; Nelson, J.; Epel, E.; Mellon, S. Serum BDNF Levels Before Treatment Predict SSRI Response in Depression. Prog. Neuropsychopharmacol. Biol. Psychiatry 2011, 35, 1623–1630. [Google Scholar] [CrossRef] [Green Version]
- Aydemir, C.; Yalcin, E.; Aksaray, S.; Kisa, C.; Yildirim, S.; Uzbay, T.; Goka, E. Brain-Derived Neurotrophic Factor (BDNF) Changes in the Serum of Depressed Women. Prog. Neuropsychopharmacol. Biol. Psychiatry 2006, 30, 1256–1260. [Google Scholar] [CrossRef]
- Park, Y.; Lee, B.; Um, T.; Kim, S. Lower Serum Brain-Derived Neurotrophic Factor Levels Are Associated with Failure to Achieve Remission in Patients with Major Depression After Escitalopram Treatment. Neuropsychiatr. Dis. Treat. 2014, 10, 1393–1398. [Google Scholar] [CrossRef] [Green Version]
- Cowansage, K.; LeDoux, J.; Monfils, M. Brain-Derived Neurotrophic Factor: A Dynamic Gatekeeper of Neural Plasticity. Curr. Mol. Pharmacol. 2010, 3, 12–29. [Google Scholar] [CrossRef]
- Blaze, J.; Roth, T. Exposure to Caregiver Maltreatment Alters Expression Levels of Epigenetic Regulators in the Medial Prefrontal Cortex. Int. J. Dev. Neurosci. 2013, 31, 804–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGill, B.; Bundle, S.; Yaylaoglu, M.; Carson, J.; Thaller, C.; Zoghbi, H. Enhanced Anxiety and Stress-Induced Corticosterone Release Are Associated with Increased Crh Expression in a Mouse Model of Rett Syndrome. Proc. Natl. Acad. Sci. USA 2006, 103, 18267–18272. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Stewart, R.; Kang, H.; Bae, K.; Kim, S.; Shin, I.; Hong, Y.; Ahn, Y.; Jeong, M.; Yoon, J. BDNF Methylation and Depressive Disorder in Acute Coronary Syndrome: The K-DEPACS and Esdepacs Studies. Psychoneuroendocrinology 2015, 62, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Pei, L.; Li, Y.; Zheng, H.; Yang, S.; Wan, Y.; Mao, L.; Xia, Y.; He, Q.; Li, M.; et al. Alleviative Effects of Fluoxetine on Depressive-Like Behaviors by Epigenetic Regulation of BDNF Gene Transcription in Mouse Model of Post-Stroke Depression. Sci. Rep. 2017, 7, 14926. [Google Scholar] [CrossRef] [PubMed]
- Webb, L.; Phillips, K.; Ho, M.; Veldic, M.; Blacker, C. The Relationship between DNA Methylation and Antidepressant Medications: A Systematic Review. Int. J. Mol. Sci. 2020, 21, 826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, N.; Sibille, E. A Putative Functional Role for Oligodendrocytes in Mood Regulation. Transl. Psychiatry 2012, 2, e109. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, S.; Gritti, L.; Crooks, D.; Dombrowski, Y. Oligodendrocytes in Development, Myelin Generation and Beyond. Cells 2019, 8, 1424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, J.; Liang, X.; Zhang, Y.; Chen, L.; Wang, F.; Tan, C.; Luo, Y.; Xiao, Q.; Chao, F.; Zhang, L.; et al. The Effects of Running Exercise on Oligodendrocytes in the Hippocampus of Rats with Depression Induced by Chronic Unpredictable Stress. Brain Res. Bull. 2019, 149, 1–10. [Google Scholar] [CrossRef]
- Wang, J.; Luo, Y.; Tang, J.; Liang, X.; Huang, C.; Gao, Y.; Qi, Y.; Yang, C.; Chao, F.; Zhang, Y.; et al. The Effects of Fluoxetine on Oligodendrocytes in the Hippocampus of Chronic Unpredictable Stress-Induced Depressed Model Rats. J. Comp. Neurol. 2020, 528, 2583–2594. [Google Scholar] [CrossRef]
- Luo, Y.; Xiao, Q.; Wang, J.; Jiang, L.; Hu, M.; Jiang, Y.; Tang, J.; Liang, X.; Qi, Y.; Dou, X.; et al. Running Exercise Protects Oligodendrocytes in the Medial Prefrontal Cortex in Chronic Unpredictable Stress Rat Model. Transl. Psychiatry 2019, 9, 322. [Google Scholar] [CrossRef]
- Wang, J.; Qiao, J.; Zhang, Y.; Wang, H.; Zhu, S.; Zhang, H.; Hartle, K.; Guo, H.; Guo, W.; He, J.; et al. Desvenlafaxine Prevents White Matter Injury and Improves the Decreased Phosphorylation of the Rate-Limiting Enzyme of Cholesterol Synthesis in a Chronic Mouse Model of Depression. J. Neurochem. 2014, 131, 229–238. [Google Scholar] [CrossRef]
- Chetty, S.; Friedman, A.; Taravosh-Lahn, K.; Kirby, E.; Mirescu, C.; Guo, F.; Krupik, D.; Nicholas, A.; Geraghty, A.; Krishnamurthy, A.; et al. Stress and Glucocorticoids Promote Oligodendrogenesis in the Adult Hippocampus. Mol. Psychiatry 2014, 19, 1275–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaul, D.; Schwab, S.; Mechawar, N.; Matosin, N. How Stress Physically Re-Shapes the Brain: Impact on Brain Cell Shapes, Numbers and Connections in Psychiatric Disorders. Neurosci. Biobehav. Rev. 2021, 124, 193–215. [Google Scholar] [CrossRef] [PubMed]
- Salim, S. Oxidative Stress and the Central Nervous System. J. Pharmacol. Exp. Ther. 2016, 360, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Patki, G.; Solanki, N.; Atrooz, F.; Allam, F.; Salim, S. Depression, Anxiety-Like Behavior and Memory Impairment Are Associated with Increased Oxidative Stress and Inflammation in a Rat Model of Social Stress. Brain Res. 2013, 1539, 73–86. [Google Scholar] [CrossRef] [Green Version]
- Felger, J.; Lotrich, F. Inflammatory Cytokines in Depression: Neurobiological Mechanisms and Therapeutic Implications. Neuroscience 2013, 246, 199–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, B.; Cao, R.; Choi, Y.; Cho, H.; Rhee, A.; Hah, C.; Hoyt, K.; Obrietan, K. The CREB/CRE Transcriptional Pathway: Protection against Oxidative Stress-Mediated Neuronal Cell Death. J. Neurochem. 2009, 108, 1251–1265. [Google Scholar] [CrossRef] [Green Version]
- Martinowich, K.; Lu, B. Interaction between BDNF and Serotonin: Role in Mood Disorders. Neuropsychopharmacology 2007, 33, 73–83. [Google Scholar] [CrossRef]
- Couto, F.; Batalha, V.; Valadas, J.; Data-Franca, J.; Ribeiro, J.; Lopes, L. Escitalopram Improves Memory Deficits Induced by Maternal Separation in the Rat. Eur. J. Pharmacol. 2012, 695, 71–75. [Google Scholar] [CrossRef]
- Pandey, D.; Yadav, S.; Mahesh, R.; Rajkumar, R. Depression-Like and Anxiety-Like Behavioural Aftermaths of Impact Accelerated Traumatic Brain Injury in Rats: A Model of Comorbid Depression and Anxiety? Behav. Brain Res. 2009, 205, 436–442. [Google Scholar] [CrossRef]
- Willner, P.; Muscat, R.; Papp, M. Chronic Mild Stress-Induced Anhedonia: A Realistic Animal Model of Depression. Neurosci. Biobehav. Rev. 1992, 16, 525–534. [Google Scholar] [CrossRef]
- Hoffman, K.L. Modeling Neuropsychiatric Disorders in Laboratory Animals, 1st ed.; Woodhead Publishing: Sawston, UK, 2015; pp. 56–57. [Google Scholar]
- Dallé, E.; Daniels, W.; Mabandla, M. Fluvoxamine Maleate Normalizes Striatal Neuronal Inflammatory Cytokine Activity in a Parkinsonian Rat Model Associated with Depression. Behav. Brain Res. 2017, 316, 189–196. [Google Scholar] [CrossRef]
- Hurley, L.; Akinfiresoye, L.; Kalejaiye, O.; Tizabi, Y. Antidepressant Effects of Resveratrol in an Animal Model of Depression. Behav. Brain Res. 2014, 268, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Tucker, L.; McCabe, J. Behavior of Male and Female C57BL/6J Mice Is More Consistent with Repeated Trials in the Elevated Zero Maze than in the Elevated Plus Maze. Front. Behav. Neurosci. 2017, 11, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walf, A.; Frye, C. The Use of the Elevated Plus Maze as an Assay of Anxiety-Related Behavior in Rodents. Nat. Protoc. 2007, 2, 322–328. [Google Scholar] [CrossRef] [Green Version]
- Gamberini, M.; Rodrigues, D.; Rodrigues, D.; Pontes, V. Effects of the Aqueous Extract of Pimpinella Anisum L. Seeds on Exploratory Activity and Emotional Behavior in Rats Using the Open Field and Elevated Plus Maze Tests. J. Ethnopharmacol. 2015, 168, 45–49. [Google Scholar] [CrossRef]
- Sevastre-Berghian, A.; Făgărăsan, V.; Toma, V.; Bâldea, I.; Olteanu, D.; Moldovan, R.; Decea, N.; Filip, G.; Clichici, S. Curcumin Reverses the Diazepam-Induced Cognitive Impairment by Modulation of Oxidative Stress and ERK 1/2/NF-Κb Pathway in Brain. Oxid. Med. Cell. Longev. 2017, 2017, 3037876. [Google Scholar] [CrossRef] [Green Version]
- Conti, M.; Morand, P.C.; Levillain, P.; Lemonnier, A. Improved Fluorometric Determination of Malonaldehyde. Clin. Chem. 1991, 37, 1273–1275. [Google Scholar] [CrossRef] [PubMed]
- Post-White, J.; Ladas, E.; Kelly, K. Advances in the Use of Milk Thistle (Silybum marianum). Integr. Cancer Ther. 2007, 6, 104–109. [Google Scholar] [CrossRef]
- Pippenger, C.; Browne, R.; Armstrong, D. Regulatory Antioxidant Enzymes. Methods Mol. Biol. 1998, 108, 299–313. [Google Scholar] [PubMed]
- GraphPad Software. GraphPad Prism, Version 8.0; Software for Windows; GraphPad Software: San Diego, CA, USA.











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dionisie, V.; Ciobanu, A.M.; Toma, V.A.; Manea, M.C.; Baldea, I.; Olteanu, D.; Sevastre-Berghian, A.; Clichici, S.; Manea, M.; Riga, S.; et al. Escitalopram Targets Oxidative Stress, Caspase-3, BDNF and MeCP2 in the Hippocampus and Frontal Cortex of a Rat Model of Depression Induced by Chronic Unpredictable Mild Stress. Int. J. Mol. Sci. 2021, 22, 7483. https://doi.org/10.3390/ijms22147483
Dionisie V, Ciobanu AM, Toma VA, Manea MC, Baldea I, Olteanu D, Sevastre-Berghian A, Clichici S, Manea M, Riga S, et al. Escitalopram Targets Oxidative Stress, Caspase-3, BDNF and MeCP2 in the Hippocampus and Frontal Cortex of a Rat Model of Depression Induced by Chronic Unpredictable Mild Stress. International Journal of Molecular Sciences. 2021; 22(14):7483. https://doi.org/10.3390/ijms22147483
Chicago/Turabian StyleDionisie, Vlad, Adela Magdalena Ciobanu, Vlad Alexandru Toma, Mihnea Costin Manea, Ioana Baldea, Diana Olteanu, Alexandra Sevastre-Berghian, Simona Clichici, Mirela Manea, Sorin Riga, and et al. 2021. "Escitalopram Targets Oxidative Stress, Caspase-3, BDNF and MeCP2 in the Hippocampus and Frontal Cortex of a Rat Model of Depression Induced by Chronic Unpredictable Mild Stress" International Journal of Molecular Sciences 22, no. 14: 7483. https://doi.org/10.3390/ijms22147483