
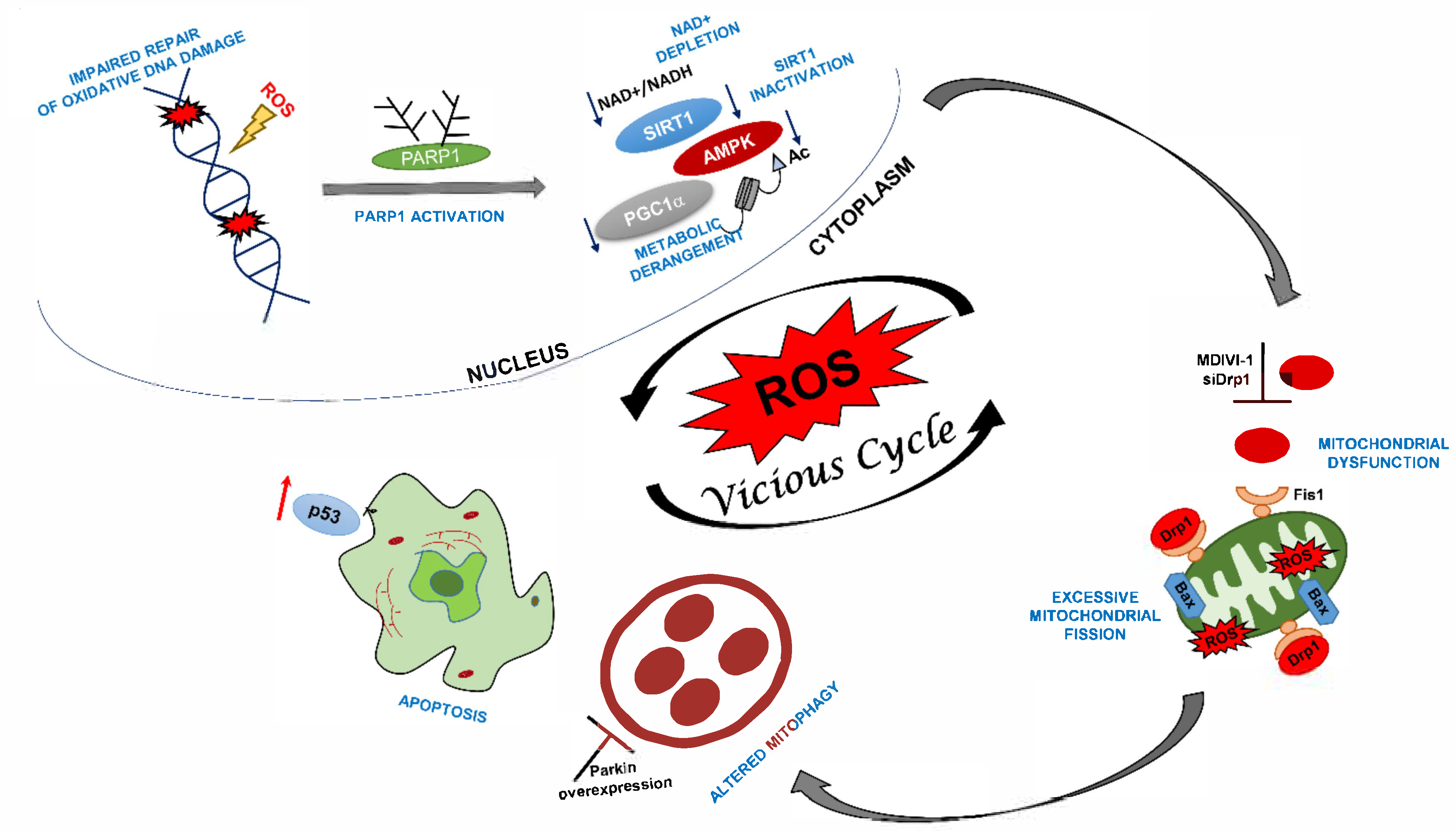
DRP1 Inhibition Rescues Mitochondrial Integrity and Excessive Apoptosis in CS-A Disease Cell Models

 ,
,  , ,
, ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
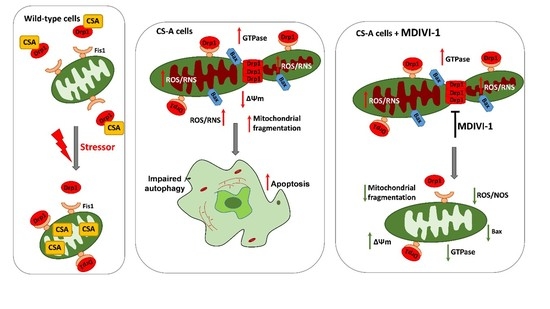
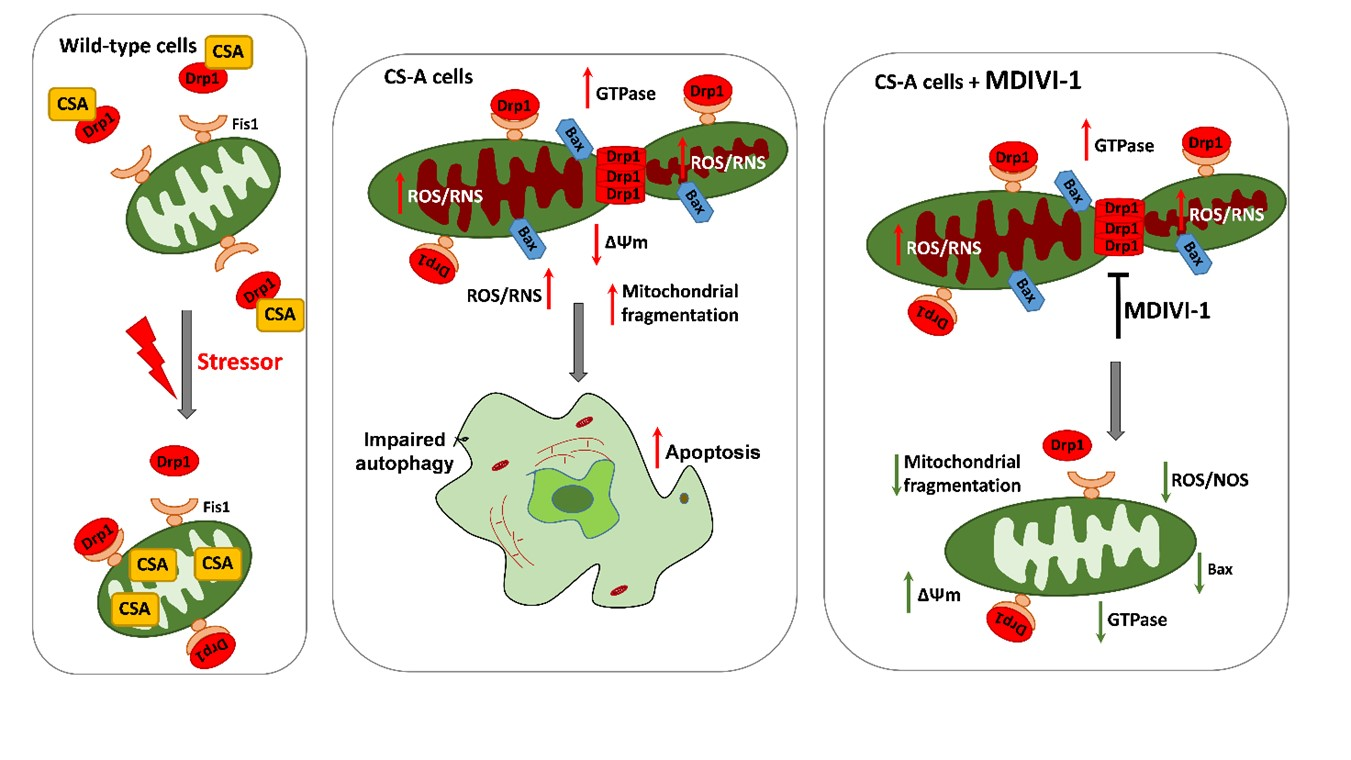
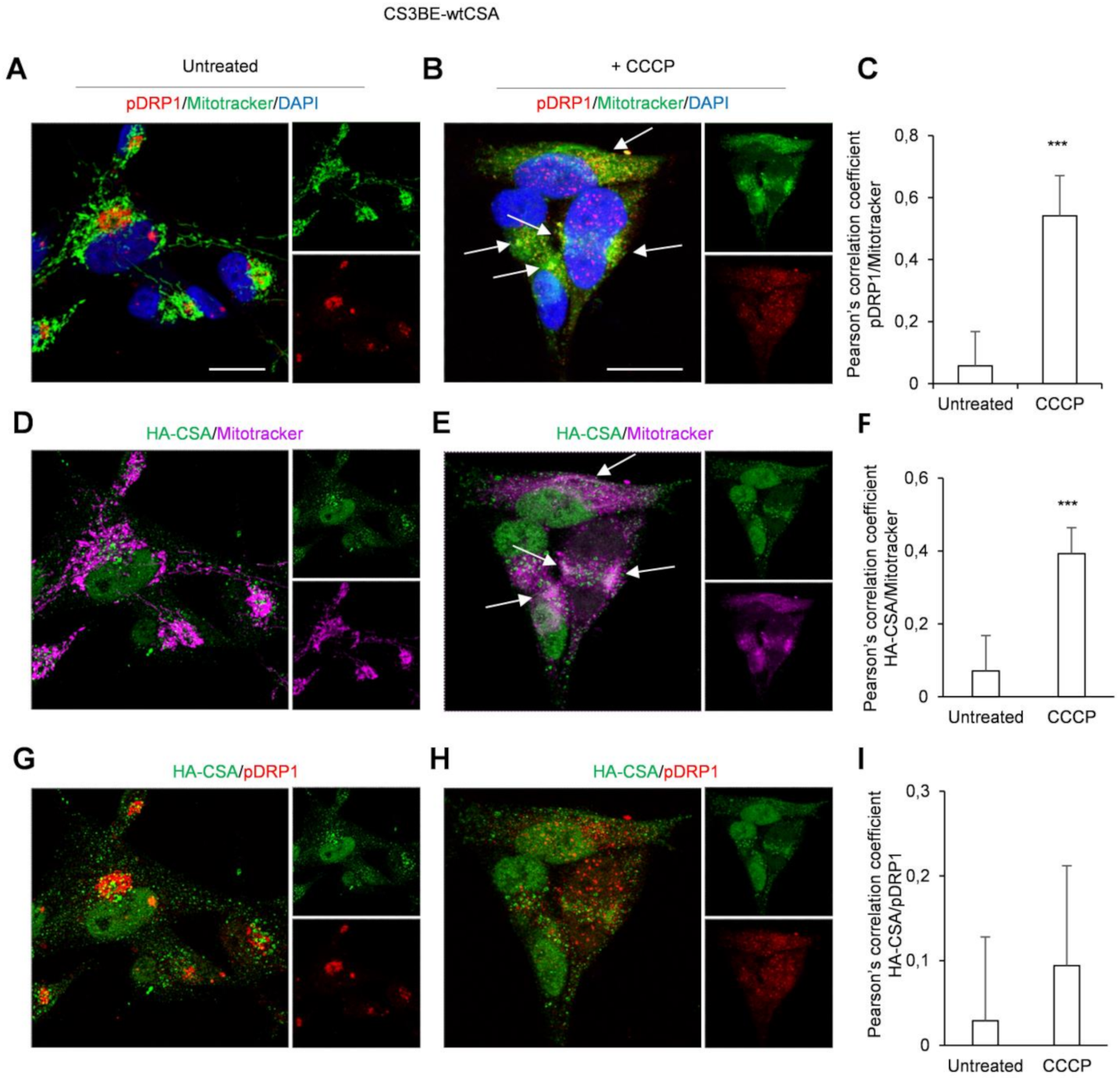
2.1. DRP1 and CSA Translocate to Mitochondria after CCCP Treatment
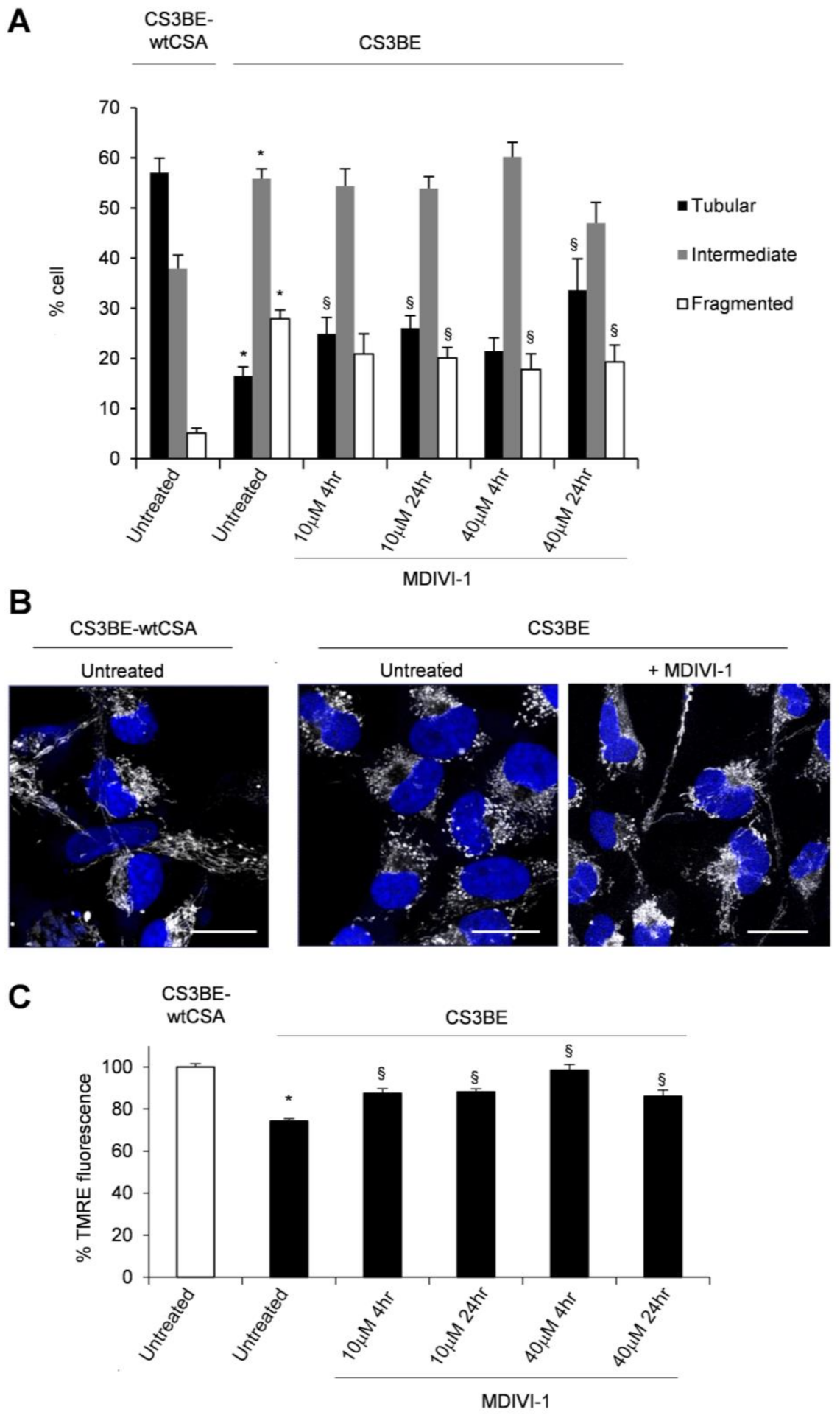
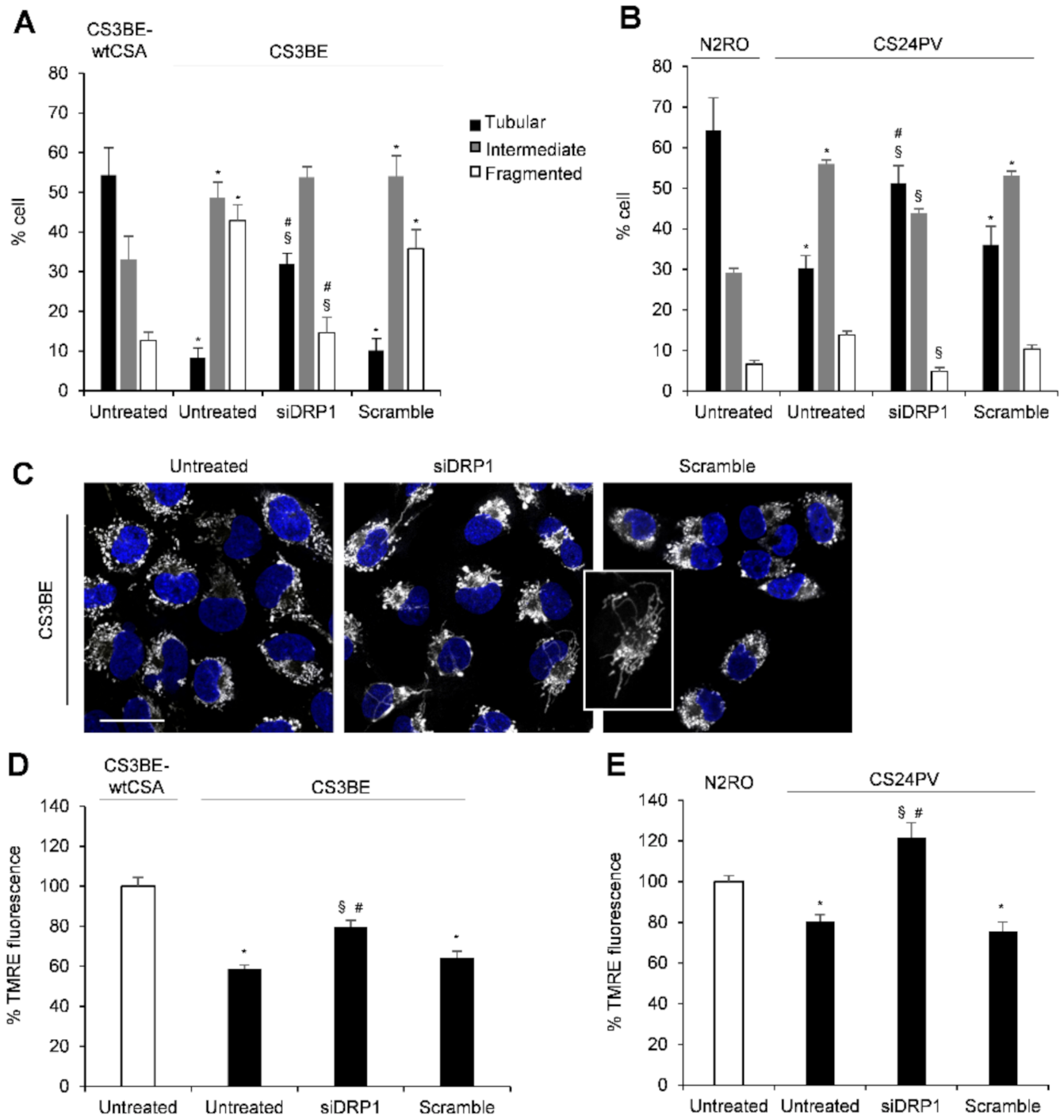
2.2. Inhibition of DRP1 Ameliorates the Dysfunctional Mitochondrial Phenotype of CS-A Cells
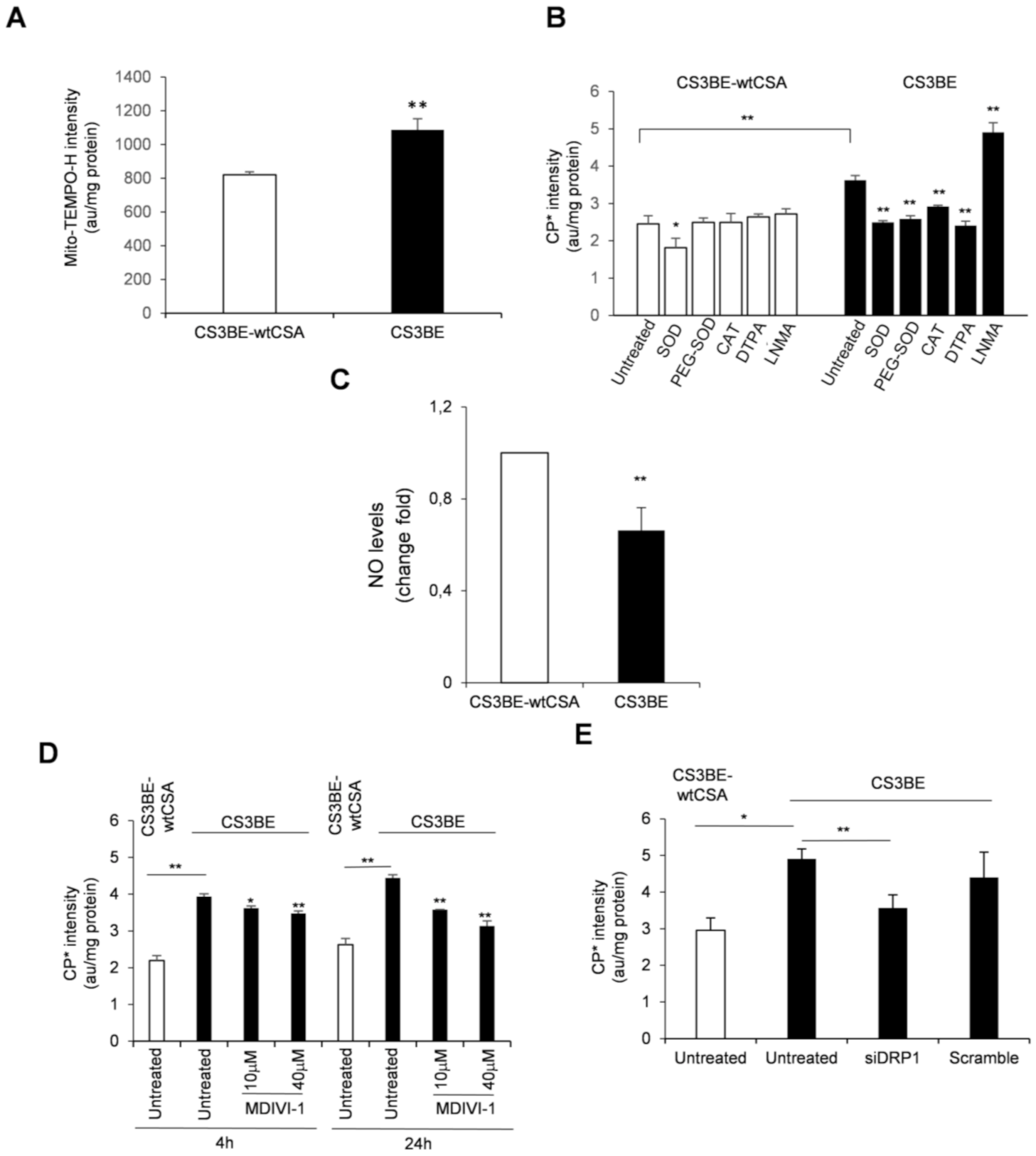
2.3. Reactive Oxygen and Nitrogen Species Are in Excess in CS-A cells and Decrease Ipon DRP1 Inhibition
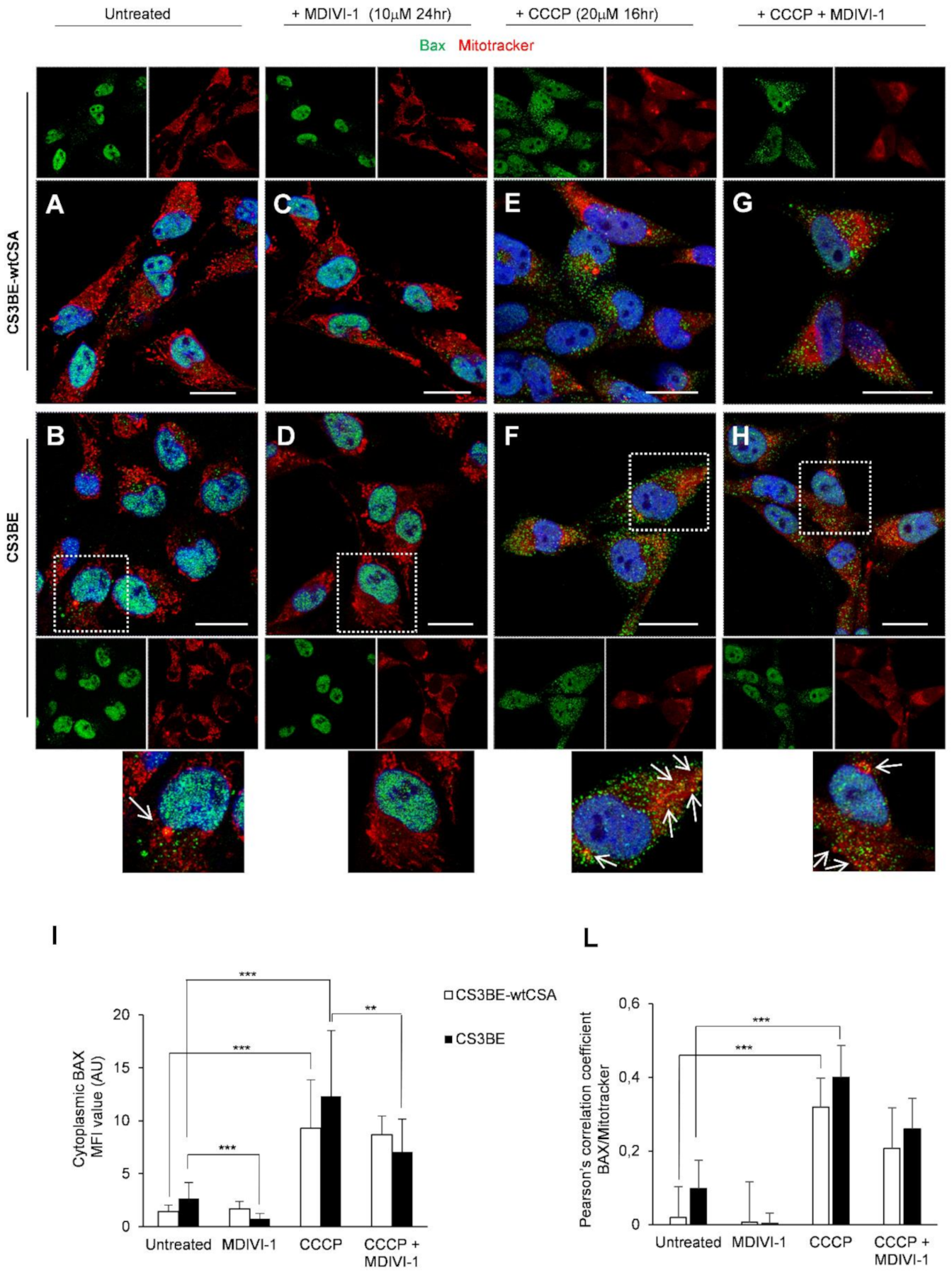
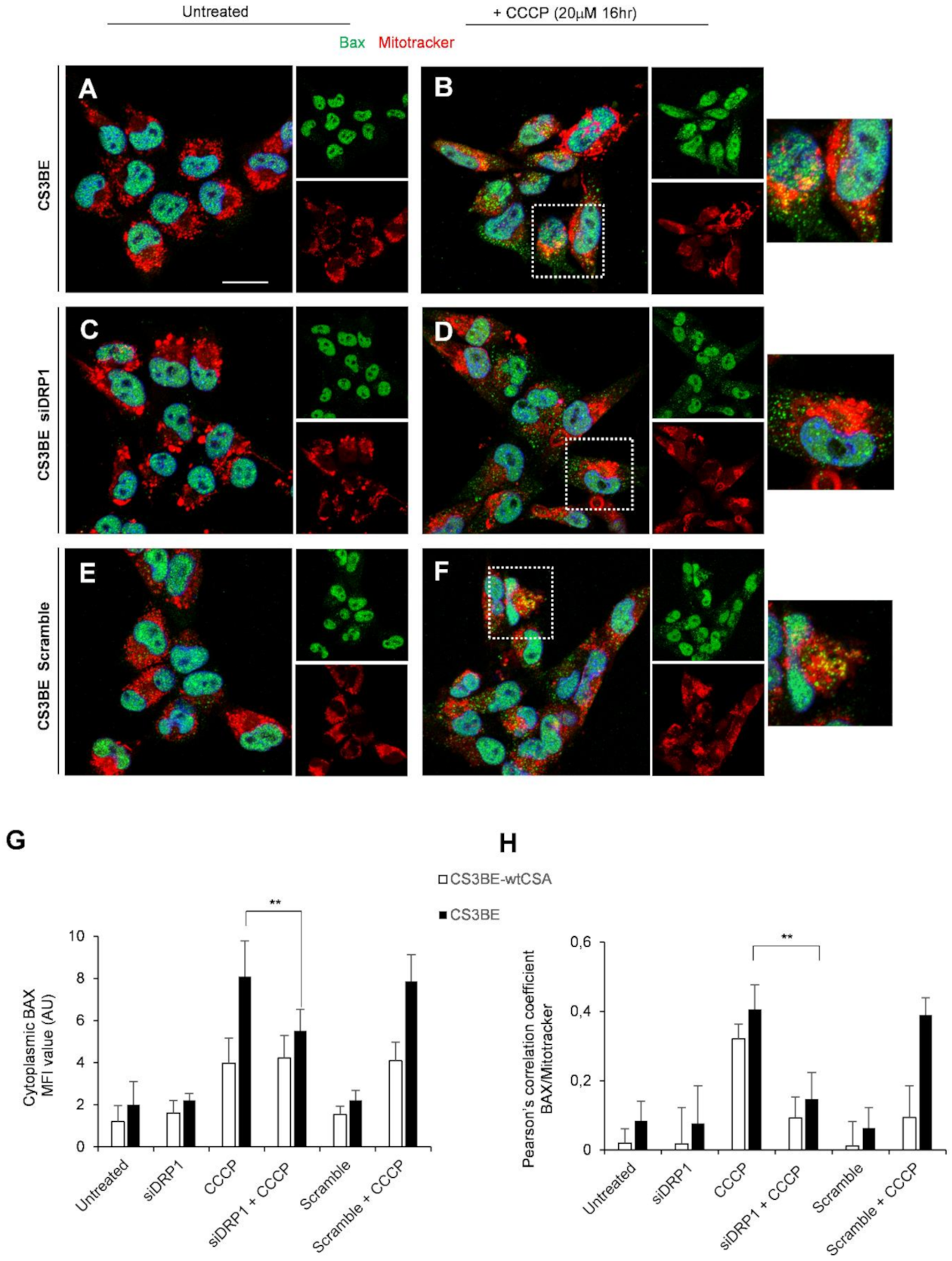
2.4. DRP1 Inhibition Is Able to Reduce the Apoptotic Rate of CS-A Cells
3. Discussion
4. Materials and Methods
4.1. Cell Cultures and Treatment Conditions
4.2. Small Interfering RNA (siRNA) Treatment of CS Cells
4.3. Confocal Laser Scanning Microscopy (CLSM) Analyses
4.4. Fluorescence Video-Imaging Analysis of Mitochondria Parameters
4.5. Electron Paramagnetic Resonance (EPR) Measurement of ROS Levels
4.6. NO Detection by DAF-FM Staining
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tuo, J.; Müftüoglu, M.; Chen, C.; Jaruga, P.; Selzer, R.R.; Brosh, R.M.; Rodriguez, H.; Dizdaroglu, M.; Bohr, V.A. The Cockayne Syndrome Group B Gene Product Is Involved in General Genome Base Excision Repair of 8-Hydroxyguanine in DNA. J. Biol. Chem. 2001, 276, 45772–45779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Errico, M.; Parlanti, E.; Teson, M.; Degan, P.; Lemma, T.; Calcagnile, A.; Iavarone, I.; Jaruga, P.; Ropolo, M.; Pedrini, A.M.; et al. The role of CSA in the response to oxidative DNA damage in human cells. Oncogene 2007, 26, 4336–4343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foresta, M.; Ropolo, M.; Degan, P.; Pettinati, I.; Know, Y.W.; Damonte, G.; Poggi, A.; Frosina, G. Defective repair of 5-hydroxy-2′-deoxycytidine in Cockayne syndrome cells and its complementation by Escherichia coli formamidopyrimidine DNA glycosylase and endonuclease III. Free Radic. Biol. Med. 2010, 48, 681–690. [Google Scholar] [CrossRef] [PubMed]
- Pascucci, B.; Lemma, T.; Iorio, E.; Giovannini, S.; Vaz, B.; Iavarone, I.; Calcagnile, A.; Narciso, L.; Degan, P.; Podo, F.; et al. An altered redox balance mediates the hypersensitivity of Cockayne syndrome primary fibroblasts to oxidative stress. Aging Cell 2012, 11, 520–529. [Google Scholar] [CrossRef]
- Krokidis, M.G.; D’Errico, M.; Pascucci, B.; Parlanti, E.; Masi, A.; Ferreri, C.; Chatgilialoglu, C. Correction: Krokidis, M.G.; et al. Oxygen-Dependent Accumulation of Purine DNA Lesions in Cockayne Syndrome Cells. Cells 2020, 9, 1671. [Google Scholar] [CrossRef]
- Osenbroch, P.O.; Auk-Emblem, P.; Halsne, R.; Strand, J.; Forstrøm, R.J.; Van der Pluijm, I.; Eide, L. Accumulation of mitochondrial DNA damage and bioenergetic dysfunction in CSB defective cells. FEBS J. 2009, 276, 2811–2821. [Google Scholar] [CrossRef] [Green Version]
- Scheibye-Knudsen, M.; Ramamoorthy, M.; Sykora, P.; Maynard, S.; Lin, P.C.; Minor, R.K.; Wilson, D.M.; Cooper, M.; Spencer, R.; de Cabo, R.; et al. Cockayne syndrome group B protein prevents the accumulation of damaged mitochondria by promoting mitochondrial autophagy. J. Exp. Med. 2012, 209, 855–869. [Google Scholar] [CrossRef] [Green Version]
- Cleaver, J.E.; Brennan-Minnella, A.M.; Swanson, R.A.; Fong, K.; Chen, J.; Chou, K.; Chen, Y.; Revet, I.; Bezrookove, V. Mitochondrial reactive oxygen species are scavenged by Cockayne syndrome B protein in human fibroblasts without nuclear DNA damage. Proc. Natl. Acad. Sci. USA 2014, 111, 13487–13492. [Google Scholar] [CrossRef] [Green Version]
- Chatre, L.; Biard, D.S.F.; Sarasin, A.; Ricchetti, M. Reversal of mitochondrial defects with CSB-dependent serine protease inhibitors in patient cells of the progeroid Cockayne syndrome. Proc. Natl. Acad. Sci. USA 2015, 112, E2910–E2919. [Google Scholar] [CrossRef] [Green Version]
- Kamenisch, Y.; Fousteri, M.; Knoch, J.; Von Thaler, A.K.; Fehrenbacher, B.; Kato, H.; Becker, T.; Dollé, M.E.T.; Kuiper, R.; Majora, M.; et al. Proteins of nucleotide and base excision repair pathways interact in mitochondria to protect from loss of subcutaneous fat, a hallmark of aging. J. Exp. Med. 2010, 207, 379–390. [Google Scholar] [CrossRef]
- Aamann, M.D.; Sorensen, M.M.; Hvitby, C.; Berquist, B.R.; Muftuoglu, M.; Tian, J.; Souza-Pinto, N.C.; Scheibye-Knudsen, M.; Wilson, D.M.; Stevnsner, T.; et al. Cockayne syndrome group B protein promotes mitochondrial DNA stability by supporting the DNA repair association with the mitochondrial membrane. FASEB J. 2010, 24, 2334–2346. [Google Scholar] [CrossRef] [Green Version]
- Berquist, B.; Canugovi, C.; Sykora, P.; Wilson, D.M.; Bohr, V.A. Human Cockayne syndrome B protein reciprocally communicates with mitochondrial proteins and promotes transcriptional elongation. Nucleic Acids Res. 2012, 40, 8392–8405. [Google Scholar] [CrossRef] [Green Version]
- Pascucci, B.; D’Errico, M.; Romagnoli, A.; De Nuccio, C.; Savino, M.; Pietraforte, D.; Lanzafame, M.; Calcagnile, A.S.; Fortini, P.; Baccarini, S.; et al. Overexpression of parkin rescues the defective mitochondrial phenotype and the increased apoptosis of Cockayne Syndrome A cells. Oncotarget 2017, 8, 102852–102867. [Google Scholar] [CrossRef] [Green Version]
- Frank, S.; Gaume, B.; Bergmann-Leitner, E.S.; Leitner, W.W.; Robert, E.G.; Catez, F.; Smith, C.L.; Youle, R.J. The Role of Dynamin-Related Protein 1, a Mediator of Mitochondrial Fission, in Apoptosis. Dev. Cell 2001, 1, 515–525. [Google Scholar] [CrossRef] [Green Version]
- Cho, B.; Choi, S.Y.; Cho, H.M.; Kim, H.J.; Sun, W. Physiological and Pathological Significance of Dynamin-Related Protein 1 (Drp1)-Dependent Mitochondrial Fission in the Nervous System. Exp. Neurobiol. 2013, 2, 149–157. [Google Scholar] [CrossRef]
- Bossy-Wetzel, E.; Barsoum, M.J.; Godzik, A.; Schwarzenbacher, R.; Lipton, S.A. Mitochondrial fission in apoptosis, neurodegeneration and aging. Curr. Opin. Cell Biol. 2003, 15, 706–716. [Google Scholar] [CrossRef]
- Shirendeb, U.P.; Calkins, M.J.; Manczak, M.; Anekonda, V.; Dufour, B.; McBride, J.L.; Mao, P.; Reddy, P.H. Mutant Huntingtin’s interaction with mitochondrial protein Drp1 impairs mitochondrial biogenesis and causes defective axonal transport and synaptic degeneration in Huntington’s disease. Hum. Mol. Genet. 2012, 21, 406–420. [Google Scholar] [CrossRef]
- Winklhofer, K.F.; Haass, C. Mitochondrial dysfunction in Parkinson’s disease. Biochim. Biophys. Acta 2010, 1802, 29–44. [Google Scholar] [CrossRef]
- Manczak, M.; Calkins, M.J.; Reddy, P.H. Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: Implications for neuronal damage. Hum. Mol. Genet. 2011, 20, 2495–2509. [Google Scholar] [CrossRef]
- Cassidy-Stone, A.; Chipuk, J.E.; Ingerman, E.; Song, C.; Yoo, C.; Kuwana, T.; Kurth, M.J.; Shaw, J.T.; Hinshaw, J.E.; Green, D.R.; et al. Chemical Inhibition of the Mitochondrial Division Dynamin Reveals Its Role in Bax/Bak-Dependent Mitochondrial Outer Membrane Permeabilization. Dev. Cell 2008, 14, 193–204. [Google Scholar] [CrossRef] [Green Version]
- Dikalov, S.I.; Kirilyuk, I.A.; Voinov, M.; Grigor’ev, I.A. EPR detection of cellular and mitochondrial superoxide using cyclic hydroxylamines. Free Radic. Res. 2011, 45, 417–430. [Google Scholar] [CrossRef] [PubMed]
- Kojima, H.; Nakatsubo, N.; Kikuchi, K.; Kawahara, S.; Kirino, Y.; Nagoshi, H.; Hirata, Y.; Nagano, T. Detection and Imaging of Nitric Oxide with Novel Fluorescent Indicators: Diaminofluoresceins. Anal. Chem. 1998, 70, 2446–2453. [Google Scholar] [CrossRef] [PubMed]
- D’Errico, M.; Teson, M.; Calcagnile, A.; Nardo, T.; De Luca, N.; Lazzari, C.; Soddu, S.; Zambruno, G.; Stefanini, M.; Dogliotti, E. Differential role of transcription-coupled repair in UVB-induced response of human fibroblasts and keratinocytes. Cancer Res. 2005, 65, 432–438. [Google Scholar] [PubMed]
- Frontini, M.; Proietti-De-Santis, L. Interaction between the Cockayne syndrome B and p53 proteins: Implications for aging. Aging 2012, 4, 89–97. [Google Scholar] [CrossRef] [Green Version]
- Bradsher, J.; Auriol, J.; De Santis, L.P.; Iben, S.; Vonesch, J.L.; Grummt, I.; Egly, J.M. CSB is a component of RNA pol I transcription. Mol. Cell 2002, 10, 819–829. [Google Scholar] [CrossRef]
- Koch, S.; Gonzalez, O.G.; Assfalg, R.; Schelling, A.; Schäfer, P.; Scharffetter-Kochanek, K.; Iben, S. Cockayne syndrome protein A is a transcription factor of RNA polymerase I and stimulates ribosomal biogenesis and growth. Cell Cycle 2014, 13, 2029–2037. [Google Scholar] [CrossRef] [Green Version]
- Shah, S.I.; Paine, J.; Perez, C.; Ullah, G. Mitochondrial fragmentation and network architecture in degenerative diseases. PLoS ONE 2019, 14, e0223014. [Google Scholar] [CrossRef]
- Bordt, E.A.; Clerc, P.; Roelofs, B.A.; Saladino, A.J.; Tretter, L.; Adam-Vizi, V.; Cherok, E.; Khalil, A.; Yadava, N.; Ge, S.X.; et al. The Putative Drp1 Inhibitor mdivi-1 Is a Reversible Mitochondrial Complex I Inhibitor that Modulates Reactive Oxygen Species. Dev. Cell 2017, 40, 583–594. [Google Scholar] [CrossRef] [Green Version]
- Parlanti, E.; Pietraforte, D.; Iorio, E.; Visentin, S.; De Nuccio, C.; Zijno, A.; D’Errico, M.; Simonelli, V.; Sanchez, M.; Fattibene, P.; et al. An altered redox balance and increased genetic instability characterize primary fibroblasts derived from xeroderma pigmentosum group A patients. Mutat. Res. Mol. Mech. Mutagen. 2015, 782, 34–43. [Google Scholar] [CrossRef]
- Krokidis, M.G.; Parlanti, E.; D’Errico, M.; Pascucci, B.; Pino, A.; Alimonti, A.; Pietraforte, D.; Masi, A.; Ferreri, C.; Chatgilialoglu, C. Purine DNA Lesions at Different Oxygen Concentration in DNA Repair-Impaired Human Cells (EUE-siXPA). Cells 2019, 8, 1377. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Zhang, Y.; Jiang, X.; Zhang, H.; Gao, Z.; Li, Y.; Fu, R.; Li, L.; Li, J.; Cui, H.; et al. ROS-mediated activation and mitochondrial translocation of CaMKII contributes to Drp1-dependent mitochondrial fission and apoptosis in triple-negative breast cancer cells by isorhamnetin and chloroquine. J. Exp. Clin. Cancer Res. 2019, 38, 225. [Google Scholar] [CrossRef]
- Zorova, L.D.; Popkov, V.A.; Plotnikov, E.Y.; Silachev, D.N.; Pevzner, I.B.; Jankauskas, S.S.; Babenko, V.A.; Zorov, S.D.; Balakireva, A.V.; Juhaszova, M.; et al. Mitochondrial membrane potential. Anal. Biochem. 2018, 552, 50–59. [Google Scholar] [CrossRef]
- Cordisco, S.; Tinaburri, L.; Teson, M.; Orioli, D.; Cardin, R.; Degan, P.; Stefanini, M.; Zambruno, G.; Guerra, L.; Dellambra, E. Cockayne Syndrome Type A Protein Protects Primary Human Keratinocytes from Senescence. J. Invest. Dermatol. 2019, 139, 38–50. [Google Scholar] [CrossRef] [Green Version]
- Crochemore, C.; Fernández-Molina, C.; Montagne, B.; Salles, A.; Ricchetti, M. CSB promoter downregulation via histone H3 hypoacetylation is an early determinant of replicative senescence. Nat. Commun. 2019, 10, 5. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Sesaki, H.; Qi, X. Drp1 stabilizes p53 on the mitochondria to trigger necrosis under oxidative stress conditions in vitro and in vivo. Biochem. J. 2010, 461, 137–146. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Youle, R. Cell biology: Form follows function for mitochondria. Nature 2016, 530, 288–289. [Google Scholar] [CrossRef]
- Okur, M.N.; Fang, E.F.; Fivenson, E.M.; Tiwari, V.; Croteau, D.L.; Bohr, V.A. Cockayne syndrome proteins CSA and CSB maintain mitochondrial homeostasis through NAD + signaling. Aging Cell 2020, 19, e13268. [Google Scholar] [CrossRef]
- Scheibye-Knudsen, M.; Mitchell, S.J.; Fang, E.F.; Iyama, T.; Ward, T.; Wang, J.; Dunn, C.A.; Singh, N.; Veith, S.; Hasan-Olive, M.M.; et al. A high-fat diet and NAD+ activate sirt1 to rescue premature aging in cockayne syndrome. Cell Metab. 2014, 20, 840–855. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Lee, J.Y.; Park, K.J.; Kim, W.H.; Roh, G.S. A mitochondrial division inhibitor, Mdivi-1, inhibits mitochondrial fragmentation and attenuates kainic acid-induced hippocampal cell death. BMC Neurosci. 2016, 17, 33. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Kim, C.; Park, S. Mdivi-1 protects adult rat hippocampal neural stem cells against palmitate-induced oxidative stress and apoptosis. Int. J. Mol. Sci. 2017, 18, 1947. [Google Scholar] [CrossRef]
- Reddy, P.H.; Manczak, M.; Yin, X. Mitochondria-Division Inhibitor 1 Protects Against Amyloid-β induced Mitochondrial Fragmentation and Synaptic Damage in Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 58, 147–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Yin, J.; Ma, X.; Zhao, F.; Siedlak, S.L.; Wang, Z.; Torres, S.; Fujioka, H.; Xu, Y.; Perry, G.; et al. Inhibition of mitochondrial fragmentation protects against Alzheimer’s disease in rodent model. Hum. Mol. Genet. 2017, 26, 4118–4131. [Google Scholar] [CrossRef] [Green Version]
- Manczak, M.; Reddy, P.H. Mitochondrial division inhibitor 1 protects against mutant huntingtin-induced abnormal mitochondrial dynamics and neuronal damage in Huntington’s disease. Hum. Mol. Genet. 2015, 24, 7308–7325. [Google Scholar] [CrossRef] [Green Version]
- Zhang, N.; Wang, S.; Li, Y.; Che, L.; Zhao, Q. A selective inhibitor of Drp1, mdivi-1, acts against cerebral ischemia/reperfusion injury via an anti-apoptotic pathway in rats. Neurosci. Lett. 2013, 535, 104–109. [Google Scholar] [CrossRef] [PubMed]
- Alupei, M.C.; Mait, P.; Esser, P.R.; Krikki, I.; Tuorto, F.; Parlato, R.; Penzo, M.; Schelling, A.; Laugel, V.; Montanaro, L.; et al. Loss of Proteostasis is a Pathomechanism in Cockayne Syndrome. Cell Rep. 2018, 23, 1612–1619. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.W.; Jeong, Y.E.; Wong, M.; Martin, L.J. DNA damage accumulates and responses are engaged in human ALS brain and spinal motor neurons and DNA repair is activatable in iPSC-derived motor neurons with SOD1 mutations. Acta Neuropathol. Commun. 2020, 8, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips, T.; Leeuwenburgh, C. Muscle fiber-specific apoptosis and TNF-α signaling in sarcopenia are attenuated by life-long calorie restriction. FASEB J. 2005, 19, 668–670. [Google Scholar] [CrossRef] [PubMed]
- Vermeij, W.P.; Hoeijmakers, J.H.J.; Pothof, J. Genome Integrity in Aging: Human Syndromes, Mouse Models, and Therapeutic Options. Annu. Rev. Pharmacol. Toxicol. 2016, 56, 427–445. [Google Scholar] [CrossRef]
- Majora, M.; Sondenheimer, K.; Knechten, M.; Uthe, I.; Esser, C.; Schiavi, A.; Ventura, N.; Krutmann, J. HDAC inhibition improves autophagic and lysosomal function to prevent loss of subcutaneous fat in a mouse model of Cockayne syndrome. Sci. Transl. Med. 2018, 10, eaam7510. [Google Scholar] [CrossRef] [Green Version]
- Pascucci, B.; Fragale, A.; Marabitti, V.; Leuzzi, G.; Calcagnile, A.S.; Parlanti, E.; Franchitto, A.; Dogliotti, E.; D’Errico, M. CSA and CSB play a role in the response to DNA breaks. Oncotarget 2018, 9, 11581–11591. [Google Scholar] [CrossRef] [Green Version]
- Rizza, S.; Montagna, C.; Cardaci, S.; Maiani, E.; Di Giacomo, G.; Sanchez-Quiles, V.; Blagoev, B.; Rasola, A.; De Zio, D.; Stamler, J.S.; et al. S-nitrosylation of the mitochondrial chaperone TRAP1 sensitizes hepatocellular carcinoma cells to inhibitors of succinate dehydrogenase. Cancer Res. 2016, 76, 4170–4182. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pascucci, B.; Spadaro, F.; Pietraforte, D.; Nuccio, C.D.; Visentin, S.; Giglio, P.; Dogliotti, E.; D’Errico, M. DRP1 Inhibition Rescues Mitochondrial Integrity and Excessive Apoptosis in CS-A Disease Cell Models. Int. J. Mol. Sci. 2021, 22, 7123. https://doi.org/10.3390/ijms22137123
Pascucci B, Spadaro F, Pietraforte D, Nuccio CD, Visentin S, Giglio P, Dogliotti E, D’Errico M. DRP1 Inhibition Rescues Mitochondrial Integrity and Excessive Apoptosis in CS-A Disease Cell Models. International Journal of Molecular Sciences. 2021; 22(13):7123. https://doi.org/10.3390/ijms22137123
Chicago/Turabian StylePascucci, Barbara, Francesca Spadaro, Donatella Pietraforte, Chiara De Nuccio, Sergio Visentin, Paola Giglio, Eugenia Dogliotti, and Mariarosaria D’Errico. 2021. "DRP1 Inhibition Rescues Mitochondrial Integrity and Excessive Apoptosis in CS-A Disease Cell Models" International Journal of Molecular Sciences 22, no. 13: 7123. https://doi.org/10.3390/ijms22137123