
Regulatory Role of Sex Hormones in Cardiovascular Calcification


1
The Roslin Institute & R(D)SVS, University of Edinburgh, Easter Bush, Midlothian EH25 9RG, UK
2
Guangzhou Institute of Cardiovascular Disease, Guangdong Key Laboratory of Vascular Diseases, State Key Laboratory of Respiratory Disease, the Second Affiliated Hospital, Guangzhou Medical University, Guangzhou 510260, China
3
University/BHF Centre for Cardiovascular Science, University of Edinburgh, Edinburgh, 47 Little France Crescent, Edinburgh EH16 4TJ, UK
*
Authors to whom correspondence should be addressed.
Int. J. Mol. Sci. 2021, 22(9), 4620; https://doi.org/10.3390/ijms22094620
Submission received: 3 February 2021
/
Revised: 20 April 2021
/
Accepted: 26 April 2021
/
Published: 28 April 2021
(This article belongs to the Special Issue Calcification in Human Pathology 2.0)
Abstract
:Sex differences in cardiovascular disease (CVD), including aortic stenosis, atherosclerosis and cardiovascular calcification, are well documented. High levels of testosterone, the primary male sex hormone, are associated with increased risk of cardiovascular calcification, whilst estrogen, the primary female sex hormone, is considered cardioprotective. Current understanding of sexual dimorphism in cardiovascular calcification is still very limited. This review assesses the evidence that the actions of sex hormones influence the development of cardiovascular calcification. We address the current question of whether sex hormones could play a role in the sexual dimorphism seen in cardiovascular calcification, by discussing potential mechanisms of actions of sex hormones and evidence in pre-clinical research. More advanced investigations and understanding of sex hormones in calcification could provide a better translational outcome for those suffering with cardiovascular calcification.
1. Clinical Consequences of Cardiovascular Calcification
Cardiovascular calcification describes the regulated deposition of mineral in blood vessels (vascular calcification) and heart valves (valvular calcification). Calcification is considered a predictor of risk associated with vascular disease [1], with more than 60% of people over 65 years of age displaying calcification in their cardiovascular system [2]. If left untreated, calcification can lead to a number of significant clinical consequences, including coronary insufficiency, aortic stenosis, and, in severe cases, heart failure. Vascular calcification was previously considered the consequence of passive precipitation of calcium and phosphate in the vascular system due to ageing. However, over the past decades, studies have revealed that cardiovascular calcification is indeed an actively regulated process that shares many similarities with physiological bone formation [3]. Despite extensive characterization of cardiovascular calcification in patients, the precise mechanisms that initiate and regulate calcification are still unclear. There is also a distinct sex difference in patients, with males having a tendency to acquire calcification earlier in life, and females developing calcification post-menopause [4]. Although the mechanism(s) behind this sex difference remain(s) to be fully elucidated, current evidence suggests a strong link between the specific actions of individual sex hormones and cardiovascular calcification. In this review, we seek to complement the recent excellent publication by Zhang et al. [5] by discussing the current understanding of the underlying mechanisms through which estrogens and androgens regulate calcification in blood vessels and valves, including a focus on the role for pre-clinical models in this research.
2. Types of Cardiovascular Calcification
According to its location, cardiovascular calcification can be divided into three major types: atherosclerotic intimal vascular calcification, medial vascular calcification, and aortic valve calcification. Within the scientific literature, cardiovascular and vascular calcification terms are frequently used interchangeably, however it is important to recognize that these are separate processes. The following section addresses these different types of calcification in more detail (Figure 1).
2.1. Atherosclerotic Intimal Calcification
There are two types of calcification—intimal or medial—that occur in large arteries. Intimal calcification occurs as a result of atherosclerotic plaque development [6]. During blood vessel calcification, vascular smooth muscle cells (VSMCs) lose their contractile phenotype and become osteogenic, depositing matrix vesicles that enhance calcification [7]. Intimal calcification in atherosclerotic plaques can either stabilise or destabilise the plaque depending on the pattern of deposition and plaque composition. There is evidence that macrocalcification stabilises the fibrous cap covering the atherosclerotic plaque, whereas microcalcification destabilises the plaque, provoking a pro-inflammatory response causing increased susceptibility to plaque rupture [8]. Plaque rupture is considered a major cause of blood vessel stenosis and ischemia in patients, directly leading to thrombus formation and myocardial infarction [9]; although it has been suggested that superficial erosion of plaques may be more significant than plaque rupture as a cause of thrombosis [10]. Within blood vessels, VSMCs in the media, myofibroblasts in the adventitia and pericytes in the micro vessels can also become calcified [11].
2.2. Medial Calcification
Medial calcification is linked to altered bone and mineral metabolism and is a clinical predictor of coronary artery disease [2]. Additionally, it is highly prevalent in patients with end stage renal disease (ESRD) and diabetes [12]. Medial calcification occurs in vessels without inflammatory or lipid cell infiltration into the vessel wall and occurs along the elastic fibres of blood vessels causing vessel stiffening and decreased compliance [13]. Medial calcification can also occur in rare diseases such as Mönckeberg’s Arteriosclerosis and Kawasaki disease [14]. The blood vessel stiffening (sclerosis) associated with medial calcification can result in increased blood pressure, local ischaemia and a high risk of vascular mortality [15,16]. Furthermore, medial calcification can lead to heart failure due to loss of aortic elasticity [17].
2.3. Valvular Calcification
Calcific aortic valve disease (CAVD) is the most common valve disease in the western world, affects up to 25% of the older population, and currently has no pharmaceutical interventions [18]. The pathogenesis of valve disease is not completely understood and there is a debate as to whether it is a feature of atherosclerosis or is independent of traditional CV risk factors. Endothelial cell disruption and basement membrane damage are evident in early-stage valve disease; one theory proposes that this could be caused by mechanical injury as a result of valve movement [19].
Clinically, progression of valve disease is well-characterised. Initial stages involve endothelial cell damage, infiltration of lipids and macrophages, as well as lipid oxidisation [20]. In later stages fibrosis and calcification cause obstruction of the aortic valve opening [20]. Valve interstitial cells (VICs) have been proposed to play a role in valve calcification. Quiescent VICs (qVICs) are the main cell type in a normal valve and function to maintain physiological valve structure and inhibit angiogenesis. If VICs are injured these cells can become activated. These activated VICs (aVICs) have the capacity to adapt to a dynamic environment through their activation and secretion of proteolytic enzymes mediating extracellular matrix remodelling followed by a normalization of phenotype [19]. When VICs are cultured in osteoblastic culture medium, they differentiate to osteoblastic cells (obVICs). Proteins associated with osteogenesis (including osteopontin, bone sialoprotein, alkaline phosphatase and bone morphogenetic protein (BMP)-2 and -4) have been identified by in vitro studies [21]. ObVICs also produce matrix vesicles which deposit calcium in the valves in the form of hydroxyapatite (HA) crystals. Development of macrocalcification and large calcium nodules is extremely damaging in valves. These nodules appear on the aortic side of the valve and restrict valve movement, causing stenosis and aortic regurgitation [22].
2.4. Pharmaceutical Strategies
Numerous pharmaceutical interventions against medial and intimal vascular calcification have been interrogated, ranging from statins to phosphate binders, with little therapeutic effect [15]. Vascular calcification is difficult to treat due to calcification having multiple causes, some of which are idiopathic. Furthermore, common diseases such as hypertension, chronic kidney disease, osteoporosis and hyperlipidaemia accelerate the progression of vascular calcification (reviewed in Lu et al. [23]). Therefore, future treatment approaches may need to be personalized, and reflect the other co-morbidities of the patient [24].
There are currently no pharmaceutical interventions for valve calcification. Clinically, severe stenosis can only be treated with either a mechanical or bioprosthetic valve replacement. Without surgery patient prognosis is between 2–3 years [25]. Synthetic valves characteristically have a 20-year lifespan, bringing increased mortality risks for patients requiring subsequent valve replacements [26,27]. Following surgery, patients are typically prescribed anticoagulants and/or immunosuppressants although their effectiveness on clinical outcomes and patient haemodynamic is debated [28].
3. Current Understanding of Cardiovascular Calcification
3.1. Calcification Is Similar to Physiological Bone Formation
Accumulating evidence shows that cardiovascular calcification is an active, cell-mediated process that shares many similarities with physiological bone formation [29] (Figure 2). Indeed, mature, lamellar bone, with hematopoietic elements and active bone remodelling, has been identified in up to 15% of calcified arteries [16]. Consistent with these findings, bone specific genes (including alkaline phosphatase (ALP), osteocalcin (OCN), runt-related transcription factor 2 (RUNX2), msh homeobox 2 (MSX2), and SRY-box transcription factor 9 (SOX9)) are significantly induced in calcified human arteries [30]. VSMCs, the predominant cell type in blood vessels, can undergo osteochondrogenic/osteocytic differentiation in the presence of calcifying conditions [31,32]. In vitro studies have demonstrated that high phosphate conditions mimicking the hyperphosphataemia seen in ESRD patients induces VSMC calcification and osteochondrogenic transition [31,33], in which the sodium-dependent phosphate cotransporter PiT1 plays a role. Knockdown of PiT1 expression in VSMCs in vitro significantly reduces phosphate uptake, expression of the osteogenic marker RUNX2, and calcium deposition [33].
Bone morphogenic proteins (BMPs) are regulators of physiological bone formation and have also been proposed to regulate vascular calcification. Previous reports, including studies from our laboratory, have shown that serum levels of BMP2 and BMP9 are elevated in patients with ESRD [34,35]. BMP2 enhances high phosphate-induced VSMC calcification in vitro and osteochondrogenic differentiation through up-regulation of RUNX2 [36]. Suppression of phosphate uptake using a sodium-dependent phosphate cotransporter inhibitor, phosphonoformic acid (PFA), attenuates BMP2-induced VSMC calcification in vitro [36]. To note, there is also evidence that PFA can inhibit calcium phosphate deposition independently of phosphate transport, potentially through a physicochemical mechanism [37].
BMP9 induces VSMC osteogenic differentiation and calcification through the orphan activin receptor-like kinase 1 (ALK1)-mediated pSmad1/5/8 signalling pathway [35]. The Wnt/β-catenin pathway is also involved in BMP9-induced VSMC calcification [38]. Consistent with these observations, pharmacological inhibition of BMP signalling reduces vascular calcification in a murine atherosclerotic model [39]. In contrast, BMP7 treatment efficiently reduces arterial calcification in murine models of atherosclerosis and ESRD [40].
3.2. Loss of Endogenous Inhibitors Induces Vascular Calcification
VSMCs within normal arteries have been shown to express a number of inhibitors, including matrix Gla protein (MGP) and ectonucleotide pyrophosphatase/phosphodiesterase 1 (ENPP1), which protect against ectopic calcification [19]. MGP is a secreted carboxyglutamic acid modified protein [41]. Mice that lack Mgp show arterial calcification [42]. Homozygous loss-of-function of MGP in humans is associated with Keutel’s syndrome, in which patients exhibit excessive calcification of cartilaginous tissues and diffuse arterial calcification [43]. A possible mechanism through which MGP abrogates vascular calcification is by antagonism of BMP signalling. MGP can directly bind to BMP2 and BMP4, thus inhibiting their downstream signalling [44,45]. In accordance with these data, inhibition of BMP signalling using LDN-193189 or ALK3-Fc reduces vascular calcification in Mgp null mice [46]. MGP undergoes γ-glutamate carboxylation, in a vitamin-k dependent mechanism, to fully exert its protective effect on arterial calcification [47]. The vitamin K antagonist warfarin rapidly induces arterial calcification through inactivation of MGP in rodents, which can be rescued by a vitamin K-enriched diet [48].
ENPP1 is an ecto-enzyme that hydrolyzes ATP to generate pyrophosphate (PPi), which acts as a calcium phosphate crystallization inhibitor when high levels of PPi are produced. ENPP1 is widely expressed in various cells including chondrocytes, osteoblasts and VSMCs [49,50]. Tiptoe-walking (ttw/ttw) mice with a natural occurring nonsense truncation mutation in Enpp1, or Enpp1 null mice, develop extensive peri-articular and arterial calcifications, and progressive ectopic ossification of the spinal ligaments [51,52]. Loss-of-function ENPP1 mutations in humans have been associated with Generalized Arterial Calcification of Infancy (GACI) [53,54]. Functional characterization of these ENPP1 mutations demonstrates a complete loss or partial reduction of its enzymatic activity, thereby reducing the generation of PPi [55]. Beyond the regulation of vascular calcification, ENPP1 also plays a role in the pathogenesis of osteoarthritis, atherosclerosis, insulin resistance and diabetes through mechanisms including the production of high levels of PPi [56,57,58].
3.3. Matrix Vesicles and Apoptotic Bodies Promote Cardiovascular Calcification
Matrix vesicles (MVs) are extracellular membrane-derived microparticles (approximately 100 nm in diameter). MVs have been implicated in bone mineralization, where they serve as the initial nucleation sites for hydroxyapatite formation [59]. Over recent years, the role of MVs in the regulation of cardiovascular calcification has attracted extensive attention. Previous work, including data from our laboratory, has reported the accumulation of MVs in calcified human aortic valves [60], aortic medial tissue, and atherosclerotic plaques [61]. These MVs are released by VSMCs, macrophages and valve interstitial cells, which play a role in initiating vascular calcification [60,62,63]. Proteomic analysis demonstrates that MVs released by cardiovascular cells during calcification show up-regulation of the calcium-binding Annexins but reduced expression of calcification inhibitors, including MGP and fetuin A [60,64]. Of note, vascular cell-secreted MVs contain a large amount of microRNAs, which also play a role in regulating vascular calcification [65]. A number of microRNAs identified in MVs released by vascular cells are involved in osteogenic differentiation. For example, miR-30 identified in MVs regulates the expression of osteogenic genes including RUNX2 and SMAD1, and VSMC calcification [66]. In addition, MVs can be taken up by recipient VSMCs, leading to altered MAPK signalling and calcium metabolism that further drives vascular calcification [67].
Apoptosis, also termed programmed cell death, is a tightly regulated, energy dependent process which is mainly regulated by caspases [68]. Apoptotic cells exhibit blebbing, cell shrinkage, and nuclear and DNA fragmentation. Previous studies have shown apoptosis to increase during VSMC [69] and VIC calcification [70] in vitro. Induction of apoptosis using anti-Fas IgM and cycloheximide enhances VSMC calcification, while inhibition of apoptosis using the caspase inhibitor z-VAD-FMK attenuates VSMC calcification in vitro [69]. Furthermore, activation of the Gas6/Axl/Akt survival pathway reduces VSMC calcification [71]. PiT1 is required for high phosphate-induced apoptosis in both VICs [72] and VSMCs [73]. Mechanistically, apoptotic bodies may expose phosphatidylserine on the outer membranes and generate a potential calcium-binding site suitable for HA deposition [69]. In this review we have focused on activation of calcification but it is important to note that calcification is a balance of both continuous activation and lack of resolution of calcification (reviewed in Carracedo et al. [74]).
4. The Role of Sex and Sex Hormones in Cardiovascular Calcification
4.1. Sex difference Exists in Cardiovascular Calcification
Sex differences and effects of sex hormones have been associated with cardiovascular calcification. Men have a 2-fold increased risk, compared with women, of developing CAVD [75]. Clinical studies of CAVD have demonstrated that for the same degree of stenosis, male patients have more calcification than females, whereas female patients have more fibrosis [76,77]. Furthermore, studies have shown that valve disease does not develop until after menopause in women [4].
Males also develop vascular disease, including atherosclerosis, earlier than females [78]. Circulating levels of testosterone, the predominant sex hormone in men, are positively associated with vascular calcification [79]. In elderly male patients with stable coronary artery disease, serum testosterone levels are inversely associated with vascular calcification [80]. Additionally, females with polycystic ovary disease (PCD) develop elevated levels of testosterone and have an increased risk of cardiovascular disease [81]. Over the past decades, it has become accepted that women are protected against cardiovascular disease; however, these protective effects are lost post-menopause. The most established hormonal change in postmenopausal women is the decline of estrogen levels [82], which is considered a causal factor for the increased incidence of vascular disease [83]. Serum levels of estrogen are negatively associated with vascular calcification [84]. Whilst established dogma highlights testosterone as a risk factor for calcification and estrogen as cardioprotective, the specific pathways underpinning how these sex hormones induce calcification have yet to be fully elucidated. Our current knowledge on the mechanisms through which sex hormones and their receptors interact with calcification, is summarised below.
4.2. Estrogen and Activation of the Estrogen Receptor Prevents Calcification
Clinical studies have demonstrated that estrogen replacement therapy (ERT) reduces cardiovascular disease in postmenopausal women [85] and that the effectiveness of ERT may be dependent on the “timing” of estrogen delivery relative to the age of menopause onset (within 6 years of menopause onset) [86]. Recently, the controlled Women’s Health Initiative Study reported that long-term estrogen therapy reduced vascular calcification in postmenopausal women aged 50 to 59 years [87], consistent with a protective role. Studies employing estrogen replacement in ovariectomy-induced vascular calcification and osteoporosis in Apoe−/− mice further corroborate these data [88].
In VSMCs, the expression of estrogen receptor α (ERα) is greater than that of estrogen receptor β (ERβ). Estrogen mainly acts through ERα (Figure 3) to inhibit RANKL signalling, thereby reducing the osteogenic differentiation and calcification of VSMCs by upregulating BMP and downregulating MGP [88]. In addition, estrogen can inhibit vascular calcification through regulation of a range of molecular and cellular events, including hypoxia-induced factor-1α signalling [89], autophagy [90] and estrogen receptor α-dependent growth arrest-specific gene 6 transactivation [91]. Conversely, exogenous estrogen application has been reported to enhance vascular calcification in bovine aortic medial cells (in vitro) [92] and in aged male and female ApoE−/− mice (in vivo) [93]. These studies suggest a complex role of estrogen in vascular calcification. Estrogen can be generated in blood vessels by aromatase-mediated conversion of testosterone [94] and therefore, the activity of aromatase in blood vessels may also have a role in mediating the effects of estrogen on vascular calcification and warrants future investigation.
4.3. Testosterone Is a Risk Factor for Cardiovascular Calcification
Low levels of circulating testosterone have been associated with increased coronary artery calcification in non-obese Korean men [95]. However, the recent Framingham Heart Study reports no significant association between testosterone and vascular calcification after model adjustment for other vascular risk factors in community-dwelling men [96]. In post-menopausal women, higher free serum testosterone is associated with coronary artery calcium progression [97]. Experimental studies show conflicting results on the effect of testosterone in calcification. In both male and female Apoe−/− mice, testosterone administration increases atherosclerotic calcification, which involves both androgen receptor- and estrogen receptor-mediated pathways [98]. Consistent with these results, our laboratory has previously shown that deletion of androgen receptor in VSMCs prevents testosterone-induced VSMC calcification in vitro [99]. Contrasting studies report an inhibitory effect of testosterone on VSMC calcification, which is mediated through androgen receptor-dependent transactivation of growth arrest-specific gene 6 signalling [100]. This discrepancy may reflect the complex actions of testosterone within different in vitro models. Clinical studies have shown that, in men, low endogenous testosterone levels are associated with cardiovascular disease [101] but also that high testosterone (for example with anabolic steroid use) has adverse cardiovascular effects [102]. This type of U-shaped relationship has been recognized with other hormones: for example, with corticosteroids where insufficiency causes Addison’s disease but excess causes Cushing’s syndrome [103].
5. Sex Hormones Mediate Cellular Signalling Pathways in the Cardiovascular System
5.1. Estrogen Signalling and Cardiovascular Function
Estrogen regulates cardiovascular function through several ERs, including ERα, ERβ and the orphan G-protein-coupled receptor (GPR30) (Figure 3) [104,105]. Upon binding to estrogen, ERs undergo conformational change, leading to ER dimerization and binding to consensus (5′GGTCAnnnTGACC 3′) estrogen response element (ERE) sites on nuclear DNA. Co-regulators are then recruited to the estrogen-ER complex to activate or inhibit gene expression [106]. Estrogen also has the capacity to regulate gene expression indirectly by altering the activity of other transcription factors (such as AP1 and Sp1) [107,108]. In the absence of estrogen, ER at specific serine sites can be phosphorylated by Epidermal Growth Factor (EGF), thereby mediating the stimulatory effects of EGF [109]. In addition to its genomic action, estrogen also induces acute non-genomic signalling pathways. Estrogen can bind to cell membranes to rapidly induce PI3K signalling. Furthermore, estrogen activates PI3K and MAPK through binding to GPR30 [110]. Functional classical ERs and non-classical GPR30 are widely expressed by vascular cells, including endothelial cells, VSMCs and cardiomyocytes [111]. Through estrogen’s genomic and non-genomic mechanisms, regulation of a wide range of cardiovascular processes has been reported, such as: cardiac hypertrophy and failure, ischemic heart diseases, vascular injury and atherosclerosis [112,113].
5.2. Testosterone Signalling and Cardiovascular Function
Testosterone can regulate a range of cardiovascular functions and phenotypes, including vasodilation and vasoconstriction and intima-media thickness [114]. Similar to estrogen, testosterone induces both genomic and non-genomic actions. Testosterone binds to the androgen receptor (AR) to regulate gene expression through binding with the androgen response element (ARE) [115]. It also induces rapid non-genomic effects through binding to membrane androgen receptors or sensors, leading to the activation of a range of intracellular signalling molecules including calcium (Ca2+), nitric oxide (NO), PKA, PKC, and MAPK [116]. The AR is expressed in several vascular cell types, including VSMCs and endothelial cells [117], and has been shown to be by expressed within the aortic valve (although the identity of valve cells expressing AR remains unclear). Testosterone can also be converted to 5α-dihydrotestosterone (DHT; which has a higher binding activity to the AR) by the cytochrome P-450 enzyme, 5α-reductase [118]. As testosterone can be converted into estrogen through aromatase, AR activation is also regulated by the cell-specific profile of metabolic products of testosterone [94]. Aromatase has been observed in vascular endothelial cells and 5α-reductase has been shown to be expressed in VSMCs [119,120]. Aromatase expression has been observed in the aortic sinus of mice, but the cell-type expressing the enzyme is unknown [98].
6. Animal Models Offer Insights into Sex Differences in Cardiovascular Calcification
Females are typically under-represented in pre-clinical studies. Indeed, there is concerning evidence that within biological and medical research, 80% of studies that specified sex used only male subjects [121]. This lack of inclusion of female models likely contributes to the poorer treatment outcomes for women and a reduced understanding of sex differences in the calcification process [122]. In this section we discuss the use of both male and female in vivo models in calcification research.
6.1. Rodent Models
The human cardiovascular system comprises a complex arrangement of specialized structures with distinct functions. The current use of small rodents as the main model of human diseases is widespread. They are relatively cost effective and easy to maintain. Furthermore, rodent models offer opportunities for genetic manipulation and pre-clinical imaging techniques, making them indispensable for elucidating the mechanisms underpinning cardiovascular disease [123]. Nonetheless, limitations do exist, and significant vascular differences are apparent between humans and rodents.
Rodent models are highly resistant to vascular calcification. In order to investigate cardiovascular calcification in rodents, the pathological process has to be artificially induced, typically through diet, genetic manipulation or mechanical injury [124]. The most commonly used model of vascular and valvular calcification is the Apoe−/− mouse model fed a “western” diet [125]. However, calcification does not present uniformly across all animals within a cohort. Additional genetic models of calcification include mice lacking low density lipoprotein receptor (Ldlr) (intimal calcification) [126], Enpp1(medial) [127], Mgp (medial) [42] and ATP binding cassette subfamily C Member 6 (Abcc6) (medial) [128,129]. These models primarily display aortic calcification but valvular calcification has also been observed in the Ldlr−/− and Apoe−/− mice [130]. Vascular calcification is a clinical consequence of ESRD, therefore it is reassuring that many models of this condition (including dietary addition of adenine, vitamin D administration and surgical induction of kidney insufficiency through 5/6 nephrectomy) have also been developed to interrogate the process of vascular calcification [131].
Exposure of rats to warfarin also produces calcification in the aorta and aortic valves [132]. However, inconsistent levels of calcification are typically observed in these rodent models, highlighting the requirement for more refined options [133].
A number of approaches can be employed to investigate the sex hormone effect on cardiovascular calcification in vivo. Sex hormone receptors can be ablated; the Ar−/− mouse has been utilized to investigate the effect of testosterone in vascular calcification [99]. Furthermore, studies involving mice lacking ERα and ERβ, have elucidated that these receptors are necessary for estrogen-mediated inhibition of the vascular injury response [134]. A physiological approach to investigating the effects of sex hormone involves the surgical removal (ovariectomy or castration) of sexual organs. Indeed, ovariectomised mice have been used to investigate bone calcification and show impaired bone formation [135].
Interestingly, sex differences in the atherosclerotic phenotype observed in Apoe−/− mice have been well-defined, whereby females tend to have larger plaques and increased calcification in those plaques [136]. Conversely, in human patients, females typically have smaller plaques and less calcification within the plaques [137]. Whilst investigations are frequently limited by the focus on a single sex, those rodent studies that have employed males and females to interrogate cardiovascular calcification pathways (Table 1), have produced divergent findings; highlighting the constraints of these animal models, and the importance of investigating both sexes in pre-clinical studies.
6.2. Large Animal Models
An inherent resistance to the development of cardiovascular calcification offers a crucial limitation to the employment of small animal models to investigate pathways underpinning this pathological process [146,147]. Subsequently novel therapeutic interventions for vascular or valvular calcification require pre-clinical testing in both small and large animal models to assess their suitability for clinical application.
Rabbits are a frequently employed, and highly appropriate, animal model of valvular calcification, due to the tri-layered composition of their valve leaflets (similar composition to humans) [148]. Methods of inducing calcification in rabbits include diet (e.g., vitamin D and long-term cholesterol treatment), genetic manipulation (e.g., LDLR mutated Watanabe rabbits), and surgical intervention (e.g., aortic balloon injury) [149,150,151,152]. However, studies have typically focused on male rabbits, with an extremely limited number of calcification studies including female animals. A recent study assessing both sexes in a matrix metallopeptidase 9 (MMP-9) overexpression model of atherosclerosis, revealed that both male and female rabbits developed calcified lesions in the aortic arch, with qualitative assessment suggesting greater calcification in males [153].
Porcine and ovine models are commonly used in investigations of aortic stenosis, with the majority of these studies examining synthetic or bioprosthetic valve replacements. An advantage of these larger animal models is the relative size of the vascular structures compared to humans, which permits the implementation of surgical procedures routinely employed in the clinic. Indeed, porcine valves have been an established option for human valve replacement for over 20 years [154]. Porcine models are highly appropriate due to the closely comparable valve anatomy, haemodynamic profiles and lipid composition to human patients [155]. They also show age-dependent insulin resistance [155] and are susceptible to the development of calcified plaques through exposure to a high fat diet [156]. The fibrosa layer in porcine aortic valves has similar features to the human fibrosa, including regions rich in collagen, elastin and proteoglycans [157]. As in humans, porcine aortic valves are more vulnerable to calcification on the aortic side, with molecular investigations confirming that valve endothelial cells (VECs) derived from the aortic side express osteogenic regulators including BMP4 [158]. Indeed, porcine VICs have been used to elucidate genetic sexual dimorphism, with recent microarray analysis of bovine cells highlighting the over-representation of cell pathways including cell death, proliferation, cell-to-cell signalling and movement in male-derived VICs compared to female-derived VICs [159,160]. Future aortic calcification studies are, however, required to confirm these findings in vivo. Due to their size and husbandry needs, when compared to smaller models, employing large animals will involve higher costs. Despite this, their importance in the field of human diseases is evident and will undoubtedly illuminate new biological pathways and mechanisms to facilitate the refinement of therapeutic strategies against cardiovascular calcification.
7. Future Perspectives
Despite testosterone being an established risk factor for cardiovascular calcification (and many other vascular diseases) its clinical impact is unclear and its mechanism of action in cardiovascular calcification remains to be fully understood. Whilst females are believed to be ‘cardio-protected’, pre-menopausal patients are severely under-represented in pre-clinical studies, which may contribute to our lack of understanding of the mechanisms underpinning the cardioprotective role of estrogens. Whether these sex hormones directly contribute to the increased vascular calcification observed in postmenopausal women remains to be investigated. Furthermore, elucidating these mechanisms is hampered not only by the limitations of pre-clinical models, but also by the severe under-representation of the female sex in pre-clinical research. Further research is essential to bridge the knowledge gap between the cellular mechanisms of calcification and the clinical sex risk factors, in order to ensure equitable treatment approaches for patients of both sexes.
Funding
H.J.W is supported by funding from the Biotechnology and Biological Science Research Council (BBSRC, no. BB/M010996/1) in the form of an EASTBIO Studentship Grant. V.E.M is supported by funding from the BBSRC in the form of an Institute Strategic Programme Grant ((BB/J004316/1). D.Z is supported by funding from the National Natural Science Foundation for Young Scientists of China (No. 81800428), The ‘Yangcheng Scholar’ Grant of Guangzhou (No. 202032768), The Innovation Project of Department of Education of Guangdong Province (No. 2017KTSCX158) and Guangdong Natural Science Foundation (No. 2018A030310178) and Science and Technology Projects of Guangzhou (No. 201904010289). P.W.F.H.’s research is supported by finding from the MRC (Confidence in Concept; MRC/CIC7/68), British Heart Foundation (RG/20/5/34796; PG/16/90/32518; REA3200409) and NC3Rs (NC/V001302/1).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Liu, W.; Zhang, Y.; Yu, C.-M.; Ji, Q.-W.; Cai, M.; Zhao, Y.-X.; Zhou, Y.-J. Current understanding of coronary artery calcification. J. Geriatr. Cardiol. 2015, 12, 668–675. [Google Scholar]
- Chen, Q.; Wang, Z.-Y.; Chen, L.-Y.; Hu, H.-Y. Roles of High Mobility Group Box 1 in Cardiovascular Calcification. Cell. Physiol. Biochem. 2017, 42, 427–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shanahan, C.M.; Cary, N.R.B.; Salisbury, J.R.; Proudfoot, D.; Weissberg, P.L.; Edmonds, M.E. Medial Localization of Mineralization-Regulating Proteins in Association with Mönckeberg’s Sclerosis. Circulation 1999, 100, 2168–2176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christian, R.C.; Harrington, S.; Edwards, W.D.; Oberg, A.L.; Fitzpatrick, L.A. Estrogen Status Correlates with the Calcium Content of Coronary Atherosclerotic Plaques in Women. J. Clin. Endocrinol. Metab. 2002, 87, 1062–1067. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Miller, V.M.; Miller, J.D. Influences of Sex and Estrogen in Arterial and Valvular Calcification. Front. Endocrinol. 2019, 10, 622. [Google Scholar] [CrossRef]
- Amann, K. Media Calcification and Intima Calcification Are Distinct Entities in Chronic Kidney Disease: Figure 1. Clin. J. Am. Soc. Nephrol. 2008, 3, 1599–1605. [Google Scholar] [CrossRef] [Green Version]
- Steitz, S.A.; Speer, M.Y.; Curinga, G.; Yang, H.-Y.; Haynes, P.A.; Aebersold, R.; Schinke, T.; Karsenty, G.; Giachelli, C.M. Smooth Muscle Cell Phenotypic Transition Associated with Calcification. Circ. Res. 2001, 89, 1147–1154. [Google Scholar] [CrossRef]
- Nadra, I.; Mason, J.C.; Philippidis, P.; Florey, O.; Smythe, C.D.; McCarthy, G.M.; Landis, R.C.; Haskard, D.O. Proinflammatory Activation of Macrophages by Basic Calcium Phosphate Crystals via Protein Kinase C and MAP Kinase Pathways. Circ. Res. 2005, 96, 1248–1256. [Google Scholar] [CrossRef] [Green Version]
- Bentzon, J.F.; Otsuka, F.; Virmani, R.; Falk, E. Mechanisms of Plaque Formation and Rupture. Circ. Res. 2014, 114, 1852–1866. [Google Scholar] [CrossRef]
- Libby, P.; Pasterkamp, G. Requiem for the ‘vulnerable plaque’. Eur. Heart J. 2015, 36, 2984–2987. [Google Scholar] [CrossRef] [Green Version]
- Bardeesi, A.S.A.; Gao, J.; Zhang, K.; Yu, S.; Wei, M.; Liu, P.; Huang, H. A novel role of cellular interactions in vascular calcification. J. Transl. Med. 2017, 15, 1–8. [Google Scholar] [CrossRef]
- Manzoor, S.; Ahmed, S.; Ali, A.; Han, K.H.; Sechopoulos, I.; O’Neill, A.; Fei, B.; O’Neill, W.C. Progression of Medial Arterial Calcification in CKD. Kidney Int. Rep. 2018, 3, 1328–1335. [Google Scholar] [CrossRef] [Green Version]
- Ho, C.Y.; Shanahan, C.M. Medial Arterial Calcification. Arter. Thromb. Vasc. Biol. 2016, 36, 1475–1482. [Google Scholar] [CrossRef] [Green Version]
- Lanzer, P.; Boehm, M.; Sorribas, V.; Thiriet, M.; Janzen, J.; Zeller, T.; Hilaire, C.S.; Shanahan, C. Medial vascular calcification revisited: Review and perspectives. Eur. Heart J. 2014, 35, 1515–1525. [Google Scholar] [CrossRef]
- Wu, M.; Rementer, C.; Giachelli, C.M. Vascular Calcification: An Update on Mechanisms and Challenges in Treatment. Calcif. Tissue Int. 2013, 93, 365–373. [Google Scholar] [CrossRef]
- Mohler, E.R.; Gannon, F.; Reynolds, C.; Zimmerman, R.; Keane, M.G.; Kaplan, F.S. Bone Formation and Inflammation in Cardiac Valves. Circulation 2001, 103, 1522–1528. [Google Scholar] [CrossRef] [PubMed]
- Ooyama, T.; Sakamato, H. Elastase in the Prevention of Arterial Ageing and the Treatment of Atherosclerosis. Novartis Found. Symposia 2007, 192, 307–320. [Google Scholar] [CrossRef]
- Schlotter, F.; Halu, A.; Pham, T.; Rogers, M.A.; Sharma, A.; Seidman, C.E.; Loscalzo, J.; Seidman, J.G.; Aikawa, M.; Singh, S.A.; et al. Spatiotemporal Multi-Omics Mapping Generates a Molecular Atlas of the Aortic Valve and Reveals Networks Driving Disease. Circulation 2018, 138, 377–393. [Google Scholar] [CrossRef] [PubMed]
- Bäck, M.; Aranyi, T.; Cancela, M.L.; Carracedo, M.; Conceição, N.; Leftheriotis, G.; Macrae, V.; Martin, L.; Nitschke, Y.; Pasch, A.; et al. Endogenous Calcification Inhibitors in the Prevention of Vascular Calcification: A Consensus Statement From the COST Action EuroSoftCalcNet. Front. Cardiovasc. Med. 2019, 5, 196. [Google Scholar] [CrossRef]
- Small, A.; Kiss, D.; Giri, J.; Anwaruddin, S.; Siddiqi, H.; Guerraty, M.; Chirinos, J.A.; Ferrari, G.; Rader, D.J. Biomarkers of Calcific Aortic Valve Disease. Arter. Thromb. Vasc. Biol. 2017, 37, 623–632. [Google Scholar] [CrossRef] [Green Version]
- Liu, A.C.; Joag, V.R.; Gotlieb, A.I. The Emerging Role of Valve Interstitial Cell Phenotypes in Regulating Heart Valve Pathobiology. Am. J. Pathol. 2007, 171, 1407–1418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Normand, J.; Loire, R.; Zambartas, C. The anatomical aspects of adult aortic stenosis. Eur. Heart J. 1988, 9, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.-C.; Wu, C.-C.; Yen, J.-F.; Liu, W.-C. Vascular Calcification and Renal Bone Disorders. Sci. World J. 2014, 2014, 1–20. [Google Scholar] [CrossRef]
- Tinica, G.; Chistol, R.O.; Enache, M.; Constantin, M.M.L.; Ciocoiu, M.; Furnica, C. Long-term graft patency after coronary artery bypass grafting: Effects of morphological and pathophysiological factors. Anatol. J. Cardiol. 2018, 20, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Badran, A.A.; Vohra, H.A.; Livesey, S.A. Unoperated severe aortic stenosis: Decision making in an adult UK-based population. Ann. R. Coll. Surg. Engl. 2012, 94, 416–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toker, M.E.; Eren, E.; Guler, M.; Kirali, K.; Yanartas, M.; Balkanay, M.; Yakut, C. Second and third cardiac valve reoperations: Factors influencing death and long-term survival. Tex. Heart Inst. J. 2009, 36, 557–562. [Google Scholar]
- Ranganath, N.K.; Koeckert, M.S.; Smith, D.E.; Hisamoto, K.; Loulmet, D.F.; Galloway, A.C.; Grossi, E.A. Aggressive tissue aortic valve replacement in younger patients and the risk of re-replacement: Implications from microsimulation analysis. J. Thorac. Cardiovasc. Surg. 2019, 158, 39–45.e1. [Google Scholar] [CrossRef]
- Chakravarty, T.; Patel, A.; Kapadia, S.; Raschpichler, M.; Smalling, R.W.; Szeto, W.Y.; Abramowitz, Y.; Cheng, W.; Douglas, P.S.; Hahn, R.T.; et al. Anticoagulation After Surgical or Transcatheter Bioprosthetic Aortic Valve Replacement. J. Am. Coll. Cardiol. 2019, 74, 1190–1200. [Google Scholar] [CrossRef]
- Chen, Y.; Zhao, X.; Wu, H. Arterial Stiffness. Arter. Thromb. Vasc. Biol. 2020, 40, 1078–1093. [Google Scholar] [CrossRef]
- Tyson, K.L.; Reynolds, J.L.; McNair, R.; Zhang, Q.; Weissberg, P.L.; Shanahan, C.M. Osteo/Chondrocytic Transcription Factors and Their Target Genes Exhibit Distinct Patterns of Expression in Human Arterial Calcification. Arter. Thromb. Vasc. Biol. 2003, 23, 489–494. [Google Scholar] [CrossRef] [Green Version]
- Zhu, D.; MacKenzie, N.C.W.; Millán, J.L.; Farquharson, C.; Macrae, V.E. The Appearance and Modulation of Osteocyte Marker Expression during Calcification of Vascular Smooth Muscle Cells. PLoS ONE 2011, 6, e19595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Speer, M.Y.; Yang, H.-Y.; Brabb, T.; Leaf, E.; Look, A.; Lin, W.-L.; Frutkin, A.; Dichek, D.; Giachelli, C.M. Smooth Muscle Cells Give Rise to Osteochondrogenic Precursors and Chondrocytes in Calcifying Arteries. Circ. Res. 2009, 104, 733–741. [Google Scholar] [CrossRef]
- Li, X.; Yang, H.-Y.; Giachelli, C.M. Role of the Sodium-Dependent Phosphate Cotransporter, Pit-1, in Vascular Smooth Muscle Cell Calcification. Circ. Res. 2006, 98, 905–912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalfino, G.; Simone, S.; Porreca, S.; Cosola, C.; Balestra, C.; Manno, C.; Schena, F.P.; Grandaliano, G.; Pertosa, G. Bone morphogenetic protein-2 may represent the molecular link between oxidative stress and vascular stiffness in chronic kidney disease. Atherosclerosis 2010, 211, 418–423. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; MacKenzie, N.C.W.; Shanahan, C.M.; Shroff, R.C.; Farquharson, C.; Macrae, V.E. BMP-9 regulates the osteoblastic differentiation and calcification of vascular smooth muscle cells through an ALK1 mediated pathway. J. Cell. Mol. Med. 2015, 19, 165–174. [Google Scholar] [CrossRef]
- Li, X.; Yang, H.-Y.; Giachelli, C.M. BMP-2 promotes phosphate uptake, phenotypic modulation, and calcification of human vascular smooth muscle cells. Atherosclerosis 2008, 199, 271–277. [Google Scholar] [CrossRef] [Green Version]
- Villa-Bellosta, R.; Sorribas, V. Phosphonoformic Acid Prevents Vascular Smooth Muscle Cell Calcification by Inhibiting Calcium-Phosphate Deposition. Arter. Thromb. Vasc. Biol. 2009, 29, 761–766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, F.; Wang, H.; Ren, W.-Y.; Ma, Y.; Liao, Y.-P.; Zhu, J.-H.; Cui, J.; Deng, Z.-L.; Su, Y.-X.; Gan, H.; et al. BMP9/COX-2 axial mediates high phosphate-induced calcification in vascular smooth muscle cells via Wnt/β-catenin pathway. J. Cell. Biochem. 2018, 119, 2851–2863. [Google Scholar] [CrossRef]
- Derwall, M.S.; Malhotra, R.; Lai, C.S.; Beppu, Y.; Aikawa, E.; Seehra, J.S.; Zapol, W.M.; Bloch, K.D.; Yu, P.B. Inhibition of Bone Morphogenetic Protein Signaling Reduces Vascular Calcification and Atherosclerosis. Arter. Thromb. Vasc. Biol. 2012, 32, 613–622. [Google Scholar] [CrossRef] [Green Version]
- Davies, M.R.; Lund, R.J.; Hruska, K.A. BMP-7 Is an Efficacious Treatment of Vascular Calcification in a Murine Model of Atherosclerosis and Chronic Renal Failure. J. Am. Soc. Nephrol. 2003, 14, 1559–1567. [Google Scholar] [CrossRef] [Green Version]
- Mori, K.; Shioi, A.; Jono, S.; Nishizawa, Y.; Morii, H. Expression of matrix Gla protein (MGP) in an in vitro model of vascular calcification. FEBS Lett. 1998, 433, 19–22. [Google Scholar] [CrossRef] [Green Version]
- Luo, G.; Ducy, P.; McKee, M.D.; Pinero, G.J.; Loyer, E.; Behringer, R.R.; Karsenty, G. Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nat. Cell Biol. 1997, 386, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Munroe, P.P.; Olgunturk, R.R.; Fryns, J.-P.J.-P.; Van Maldergem, L.; Ziereisen, F.; Yuksel, B.B.; Gardiner, M.R.; Chung, E.E. Mutations in the gene encoding the human matrix Gla protein cause Keutel syndrome. Nat. Genet. 1999, 21, 142–144. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Zebboudj, A.F.; Shao, E.; Perez, M.; Boström, K. Regulation of Bone Morphogenetic Protein-4 by Matrix GLA Protein in Vascular Endothelial Cells Involves Activin-like Kinase Receptor 1. J. Biol. Chem. 2006, 281, 33921–33930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zebboudj, A.F.; Imura, M.; Boström, K. Matrix GLA Protein, a Regulatory Protein for Bone Morphogenetic Protein-2. J. Biol. Chem. 2002, 277, 4388–4394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malhotra, R.; Burke, M.F.; Hoeft, K.; Mayeur, C.; Jiramongkolchai, P.; Kumar, R.; Buys, E.S.; Yu, P.B.; Bloch, K.D.; Bloch, D.B.; et al. Inhibition of Bone Morphogenetic Protein Signal Transduction Prevents the Medial Vascular Calcification Associated with Matrix Gla Protein Deficiency. PLoS ONE 2015, 10, e0117098. [Google Scholar] [CrossRef] [PubMed]
- Cranenburg, E.C.M.; Van Spaendonck-Zwarts, K.Y.; Bonafé, L.; Crettol, L.M.; Rödiger, L.A.; Dikkers, F.G.; Van Essen, A.J.; Superti-Furga, A.; Alexandrakis, E.; Vermeer, C.; et al. Circulating matrix γ-carboxyglutamate protein (MGP) species are refractory to vitamin K treatment in a new case of Keutel syndrome. J. Thromb. Haemost. 2011, 9, 1225–1235. [Google Scholar] [CrossRef] [PubMed]
- Schurgers, L.J.; Spronk, H.M.H.; Soute, B.A.M.; Schiffers, P.M.; Demey, J.G.R.; Vermeer, C. Regression of warfarin-induced medial elastocalcinosis by high intake of vitamin K in rats. Blood 2006, 109, 2823–2831. [Google Scholar] [CrossRef]
- Goding, J.W.; Terkeltaub, R.; Maurice, M.; Deterre, P.; Sali, A.; Belli, S.I. Ecto-phosphodiesterase/pyrophosphatase of lymphocytes and non-lymphoid cells: Structure and function of the PC-1 family. Immunol. Rev. 1998, 161, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Nitschke, Y.; Yan, Y.; Buers, I.; Kintziger, K.; Askew, K.; Rutsch, F. ENPP1-Fc prevents neointima formation in generalized arterial calcification of infancy through the generation of AMP. Exp. Mol. Med. 2018, 50, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Okawa, A.; Nakamura, I.; Goto, S.; Moriya, H.; Nakamura, Y.; Ikegawa, S. Mutation in Npps in a mouse model of ossification of the posterior longitudinal ligament of the spine. Nat. Genet. 1998, 19, 271–273. [Google Scholar] [CrossRef] [PubMed]
- MacKenzie, N.C.W.; Zhu, D.; Milne, E.M.; Hof, R.V.T.; Martin, A.; Quarles, D.L.; Millán, J.L.; Farquharson, C.; Macrae, V.E. Altered Bone Development and an Increase in FGF-23 Expression in Enpp1−/− Mice. PLoS ONE 2012, 7, e32177. [Google Scholar] [CrossRef]
- Rutsch, F.; Böyer, P.; Nitschke, Y.; Ruf, N.; Lorenz-Depierieux, B.; Wittkampf, T.; Weissen-Plenz, G.; Fischer, R.-J.; Mughal, Z.; Gregory, J.W.; et al. Hypophosphatemia, Hyperphosphaturia, and Bisphosphonate Treatment Are Associated With Survival Beyond Infancy in Generalized Arterial Calcification of Infancy. Circ. Cardiovasc. Genet. 2008, 1, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Ruf, N.; Uhlenberg, B.; Terkeltaub, R.; Nürnberg, P.; Rutsch, F. The mutational spectrum ofENPP1as arising after the analysis of 23 unrelated patients with generalized arterial calcification of infancy (GACI). Hum. Mutat. 2004, 25, 98. [Google Scholar] [CrossRef]
- Stella, J.; Buers, I.; Van De Wetering, K.; Höhne, W.; Rutsch, F.; Nitschke, Y. Effects of Different Variants in theENPP1Gene on the Functional Properties of Ectonucleotide Pyrophosphatase/Phosphodiesterase Family Member 1. Hum. Mutat. 2016, 37, 1190–1201. [Google Scholar] [CrossRef] [PubMed]
- Roberts, F.; Zhu, D.; Farquharson, C.; Macrae, V.E. ENPP1 in the Regulation of Mineralization and Beyond. Trends Biochem. Sci. 2019, 44, 616–628. [Google Scholar] [CrossRef]
- Huesa, C.; Zhu, D.; Glover, J.D.; Ferron, M.; Karsenty, G.; Milne, E.M.; Millan, J.L.; Ahmed, S.F.; Farquharson, C.; Morton, N.M.; et al. Deficiency of the bone mineralization inhibitor NPP1 protects mice against obesity and diabetes. Dis. Model. Mech. 2014, 7, 1341–1350. [Google Scholar] [CrossRef] [Green Version]
- Roberts, F.L.; Rashdan, N.A.; Phadwal, K.; Markby, G.R.; Dillon, S.; Zoll, J.; Berger, J.; Milne, E.; Orriss, I.R.; Karsenty, G.; et al. Osteoblast-specific deficiency of ectonucleotide pyrophosphatase or phosphodiesterase-1 engenders insulin resistance in high-fat diet fed mice. J. Cell. Physiol. 2021, 236, 4614–4624. [Google Scholar] [CrossRef]
- Golub, E.E. Role of matrix vesicles in biomineralization. Biochim. Biophys. Acta Gen. Subj. 2009, 1790, 1592–1598. [Google Scholar] [CrossRef] [Green Version]
- Cui, L.; Rashdan, N.A.; Zhu, N.; Milne, E.M.; Ajuh, P.; Milne, G.; Helfrich, M.H.; Lim, K.; Prasad, S.; Lerman, D.A.; et al. End stage renal disease-induced hypercalcemia may promote aortic valve calcification via Annexin VI enrichment of valve interstitial cell derived-matrix vesicles. J. Cell. Physiol. 2017, 232, 2985–2995. [Google Scholar] [CrossRef] [Green Version]
- Hutcheson, J.D.; Goettsch, C.; Bertazzo, S.S.; Maldonado, N.; Ruiz, J.L.; Goh, W.; Yabusaki, K.; Faits, T.; Bouten, C.C.; Franck, G.; et al. Genesis and growth of extracellular-vesicle-derived microcalcification in atherosclerotic plaques. Nat. Mater. 2016, 15, 335–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, N.X.; O’Neill, K.D.; Chen, X.; Moe, S.M. Annexin-Mediated Matrix Vesicle Calcification in Vascular Smooth Muscle Cells. J. Bone Miner. Res. 2008, 23, 1798–1805. [Google Scholar] [CrossRef] [Green Version]
- New, S.E.P.; Goettsch, C.; Aikawa, M.; Marchini, J.F.; Shibasaki, M.; Yabusaki, K.; Libby, P.; Shanahan, C.M.; Croce, K.; Aikawa, E. Macrophage-Derived Matrix Vesicles. Circ. Res. 2013, 113, 72–77. [Google Scholar] [CrossRef]
- Kapustin, A.N.; Chatrou, M.L.L.; Drozdov, I.; Zheng, Y.; Davidson, S.M.; Soong, D.; Furmanik, M.; Sanchis, P.; De Rosales, R.T.M.; Alvarez-Hernandez, D.; et al. Vascular Smooth Muscle Cell Calcification Is Mediated by Regulated Exosome Secretion. Circ. Res. 2015, 116, 1312–1323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goettsch, C.; Hutcheson, J.D.; Aikawa, E. MicroRNA in Cardiovascular Calcification. Circ. Res. 2013, 112, 1073–1084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaturvedi, P.; Chen, N.X.; O’Neill, K.; McClintick, J.N.; Moe, S.M.; Janga, S.C. Differential miRNA Expression in Cells and Matrix Vesicles in Vascular Smooth Muscle Cells from Rats with Kidney Disease. PLoS ONE 2015, 10, e0131589. [Google Scholar] [CrossRef] [Green Version]
- Chen, N.X.; O’Neill, K.D.; Moe, S.M. Matrix vesicles induce calcification of recipient vascular smooth muscle cells through multiple signaling pathways. Kidney Int. 2018, 93, 343–354. [Google Scholar] [CrossRef] [Green Version]
- Tsuchiya, K.; Nakajima, S.; Hosojima, S.; Nguyen, D.T.; Hattori, T.; Le, T.M.; Hori, O.; Mahib, M.R.; Yamaguchi, Y.; Miura, M.; et al. Caspase-1 initiates apoptosis in the absence of gasdermin D. Nat. Commun. 2019, 10, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Proudfoot, D.; Skepper, J.N.; Hegyi, L.; Bennett, M.R.; Shanahan, C.M.; Weissberg, P.L. Apoptosis Regulates Human Vascular Calcification In Vitro. Circ. Res. 2000, 87, 1055–1062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Gordillo-Martinez, F.; Jiang, L.; He, P.; Hong, W.; Wei, X.; Staines, K.A.; Macrae, V.E.; Zhang, C.; Yu, D.; et al. Zinc ameliorates human aortic valve calcification through GPR39 mediated ERK1/2 signalling pathway. Cardiovasc. Res. 2021, 117, 820–835. [Google Scholar] [CrossRef]
- Kim, H.; Kim, H.-J.; Lee, K.; Kim, J.-M.; Kim, H.S.; Kim, J.-R.; Ha, C.-M.; Choi, Y.-K.; Lee, S.J.; Kim, J.-Y.; et al. α-Lipoic acid attenuates vascular calcification via reversal of mitochondrial function and restoration of Gas6/Axl/Akt survival pathway. J. Cell. Mol. Med. 2011, 16, 273–286. [Google Scholar] [CrossRef] [PubMed]
- El Husseini, D.; Boulanger, M.-C.; Fournier, D.; Mahmut, A.; Bossé, Y.; Pibarot, P.; Mathieu, P. High Expression of the Pi-Transporter SLC20A1/Pit1 in Calcific Aortic Valve Disease Promotes Mineralization through Regulation of Akt-1. PLoS ONE 2013, 8, e53393. [Google Scholar] [CrossRef]
- Chavkin, N.W.; Chia, J.J.; Crouthamel, M.H.; Giachelli, C.M. Phosphate uptake-independent signaling functions of the type III sodium-dependent phosphate transporter, PiT-1, in vascular smooth muscle cells. Exp. Cell Res. 2015, 333, 39–48. [Google Scholar] [CrossRef] [Green Version]
- Carracedo, M.; Artiach, G.; Arnardottir, H.; Bäck, M. The resolution of inflammation through omega-3 fatty acids in atherosclerosis, intimal hyperplasia, and vascular calcification. Semin. Immunopathol. 2019, 41, 757–766. [Google Scholar] [CrossRef] [Green Version]
- Gomel, M.A.; Lee, R.; Grande-Allen, K.J. Comparing the Role of Mechanical Forces in Vascular and Valvular Calcification Progression. Front. Cardiovasc. Med. 2019, 5, 197. [Google Scholar] [CrossRef]
- Simard, L.; Côté, N.; Dagenais, F.; Mathieu, P.; Couture, C.; Trahan, S.; Bossé, Y.; Mohammadi, S.; Pagé, S.; Joubert, P.; et al. Sex-Related Discordance Between Aortic Valve Calcification and Hemodynamic Severity of Aortic Stenosis. Circ. Res. 2017, 120, 681–691. [Google Scholar] [CrossRef]
- Nitsche, C.; Koschutnik, M.; Kammerlander, A.; Hengstenberg, C.; Mascherbauer, J. Gender-specific differences in valvular heart disease. Wien. Klin. Wochenschr. 2020, 132, 61–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stout, R.W. Atherosclerosis in males and females. Horm. Atheroscler. 1982, 97–111. [Google Scholar] [CrossRef]
- Pérez-López, F.R.; Larrad-Mur, L.; Kallen, A.; Chedraui, P.; Taylor, H.S. Review: Gender Differences in Cardiovascular Disease: Hormonal and Biochemical Influences. Reprod. Sci. 2010, 17, 511–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, J.; Ge, Y.; Shao, Y.; Xuan, T.; Xia, S.; Li, M. Low serum testosterone level was associated with extensive coronary artery calcification in elderly male patients with stable coronary artery disease. Coron. Artery Dis. 2015, 26, 437–441. [Google Scholar] [CrossRef]
- Calderon-Margalit, R.; Siscovick, D.; Merkin, S.S.; Wang, E.; Daviglus, M.L.; Schreiner, P.J.; Sternfeld, B.; Williams, O.D.; Lewis, C.E.; Azziz, R.; et al. Prospective Association of Polycystic Ovary Syndrome With Coronary Artery Calcification and Carotid-Intima-Media Thickness. Arter. Thromb. Vasc. Biol. 2014, 34, 2688–2694. [Google Scholar] [CrossRef] [Green Version]
- Zhu, D.; Li, X.; Macrae, V.E.; Simoncini, T.; Fu, X. Extragonadal Effects of Follicle-Stimulating Hormone on Osteoporosis and Cardiovascular Disease in Women during Menopausal Transition. Trends Endocrinol. Metab. 2018, 29, 571–580. [Google Scholar] [CrossRef] [Green Version]
- Dalal, P.K.; Agarwal, M. Postmenopausal syndrome. Indian J. Psychiatry 2015, 57, 222–232. [Google Scholar] [CrossRef] [PubMed]
- Nakao, J.; Orimo, H.; Ooyama, T.; Shiraki, M. Low serum estradiol levels in subjects with arterial calcification. Atherosclerosis 1979, 34, 469–474. [Google Scholar] [CrossRef]
- Lopez-Pier, M.A.; Lipovka, Y.; Koppinger, M.P.; Harris, P.R.; Konhilas, J.P. The clinical impact of estrogen loss on cardio-vascular disease in menopausal females. Med. Res. Arch. 2018, 6, 1663. [Google Scholar] [PubMed]
- Hodis, H.N.; Mack, W.J.; Shoupe, D.; Azen, S.P.; Stanczyk, F.Z.; Hwang-Levine, J.; Budoff, M.J.; Henderson, V.W. Methods and baseline cardiovascular data from the Early versus Late Intervention Trial with Estradiol testing the menopausal hormone timing hypothesis. Menopause 2015, 22, 391–401. [Google Scholar] [CrossRef] [Green Version]
- Manson, J.E.; Allison, M.A.; Rossouw, J.E.; Carr, J.J.; Langer, R.D.; Hsia, J.; Kuller, L.H.; Cochrane, B.B.; Hunt, J.R.; Ludlam, S.E.; et al. Estrogen Therapy and Coronary-Artery Calcification. N. Engl. J. Med. 2007, 356, 2591–2602. [Google Scholar] [CrossRef]
- Osako, M.K.; Nakagami, H.; Koibuchi, N.; Shimizu, H.; Nakagami, F.; Koriyama, H.; Shimamura, M.; Miyake, T.; Rakugi, H.; Morishita, R. Estrogen Inhibits Vascular Calcification via Vascular RANKL System. Circ. Res. 2010, 107, 466–475. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Zhao, Q.; Chen, Z.; Geng, Y.-J.; Zhang, W.; Zhou, Q.; Yang, W.; Liu, Q.; Liu, H. Estrogen inhibits vascular calcification in rats via hypoxia-induced factor-1α signaling. Vascular 2020, 28, 465–474. [Google Scholar] [CrossRef]
- Peng, Y.-Q.; Xiong, D.; Lin, X.; Cui, R.-R.; Xu, F.; Zhong, J.-Y.; Zhu, T.; Wu, F.; Mao, M.-Z.; Liao, X.-B.; et al. Oestrogen Inhibits Arterial Calcification by Promoting Autophagy. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nanao-Hamai, M.; Son, B.-K.; Hashizume, T.; Ogawa, S.; Akishita, M. Protective effects of estrogen against vascular calcification via estrogen receptor α-dependent growth arrest-specific gene 6 transactivation. Biochem. Biophys. Res. Commun. 2016, 480, 429–435. [Google Scholar] [CrossRef]
- Balica, M.; Bostrom, K.I.; Shin, V.; Tillisch, K.; Demer, L.L. Calcifying Subpopulation of Bovine Aortic Smooth Muscle Cells Is Responsive to 17 & beta-Estradiol. Circulation 1997, 95, 1954–1960. [Google Scholar] [CrossRef]
- McRobb, L.S.; McGrath, K.C.; Tsatralis, T.; Liong, E.C.; Tan, J.T.; Hughes, G.; Handelsman, D.J.; Heather, A.K. Estrogen Receptor Control of Atherosclerotic Calcification and Smooth Muscle Cell Osteogenic Differentiation. Arter. Thromb. Vasc. Biol. 2017, 37, 1127–1137. [Google Scholar] [CrossRef] [Green Version]
- Harada, N.; Sasano, H.; Murakami, H.; Ohkuma, T.; Nagura, H.; Takagi, Y. Localized expression of aromatase in human vascular tissues. Circ. Res. 1999, 84, 1285–1291. [Google Scholar] [CrossRef]
- Park, B.-J.; Shim, J.-Y.; Lee, Y.-J.; Lee, J.-H.; Lee, H.-R. Inverse relationship between bioavailable testosterone and subclinical coronary artery calcification in non-obese Korean men. Asian J. Androl. 2012, 14, 612–615. [Google Scholar] [CrossRef] [Green Version]
- Travison, T.G.; O’Donnell, C.J.; Bhasin, S.; Massaro, J.M.; Hoffmann, U.; Vasan, R.S.; D’Agostino, R.B.; Basaria, S. Circulating Sex Steroids and Vascular Calcification in Community-Dwelling Men: The Framingham Heart Study. J. Clin. Endocrinol. Metab. 2016, 101, 2160–2167. [Google Scholar] [CrossRef] [Green Version]
- Subramanya, V.; Zhao, D.; Ouyang, P.; Ying, W.; Vaidya, D.; Ndumele, C.E.; Heckbert, S.R.; Budoff, M.J.; Post, W.S.; Michos, E.D. Association of endogenous sex hormone levels with coronary artery calcium progression among post-menopausal women in the Multi-Ethnic Study of Atherosclerosis (MESA). J. Cardiovasc. Comput. Tomogr. 2019, 13, 41–47. [Google Scholar] [CrossRef] [PubMed]
- McRobb, L.; Handelsman, D.J.; Heather, A.K. Androgen-Induced Progression of Arterial Calcification in Apolipoprotein E-Null Mice Is Uncoupled from Plaque Growth and Lipid Levels. Endocrinology 2009, 150, 841–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, D.; Hadoke, P.W.F.; Wu, J.; Vesey, A.T.; Lerman, D.A.; Dweck, M.R.; Newby, D.E.; Smith, L.B.; Macrae, V.E. Ablation of the androgen receptor from vascular smooth muscle cells demonstrates a role for testosterone in vascular calcification. Sci. Rep. 2016, 6, 24807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Son, B.-K.; Akishita, M.; Iijima, K.; Ogawa, S.; Maemura, K.; Yu, J.; Takeyama, K.; Kato, S.; Eto, M.; Ouchi, Y. Androgen Receptor-dependent Transactivation of Growth Arrest-specific Gene 6 Mediates Inhibitory Effects of Testosterone on Vascular Calcification. J. Biol. Chem. 2010, 285, 7537–7544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akishita, M.; Hashimoto, M.; Ohike, Y.; Ogawa, S.; Iijima, K.; Eto, M.; Ouchi, Y. Low testosterone level as a predictor of cardiovascular events in Japanese men with coronary risk factors. Atherosclerosis 2010, 210, 232–236. [Google Scholar] [CrossRef]
- Achar, S.; Rostamian, A.; Narayan, S.M. Cardiac and Metabolic Effects of Anabolic-Androgenic Steroid Abuse on Lipids, Blood Pressure, Left Ventricular Dimensions, and Rhythm. Am. J. Cardiol. 2010, 106, 893–901. [Google Scholar] [CrossRef] [Green Version]
- Moisan, M.-P.; Castanon, N. Emerging Role of Corticosteroid-Binding Globulin in Glucocorticoid-Driven Metabolic Disorders. Front. Endocrinol. 2016, 7, 160. [Google Scholar] [CrossRef] [Green Version]
- Kuiper, G.G.; Enmark, E.; Pelto-Huikko, M.; Nilsson, S.; Gustafsson, J.A. Cloning of a novel receptor expressed in rat prostate and ovary. Proc. Natl. Acad. Sci. USA 1996, 93, 5925–5930. [Google Scholar] [CrossRef] [Green Version]
- Filardo, E.J.; Quinn, J.A.; Frackelton, A.R.; Bland, K.I. Estrogen Action Via the G Protein-Coupled Receptor, GPR30: Stimulation of Adenylyl Cyclase and cAMP-Mediated Attenuation of the Epidermal Growth Factor Receptor-to-MAPK Signaling Axis. Mol. Endocrinol. 2002, 16, 70–84. [Google Scholar] [CrossRef]
- Murphy, E. Estrogen Signaling and Cardiovascular Disease. Circ. Res. 2011, 109, 687–696. [Google Scholar] [CrossRef] [Green Version]
- Jakacka, M.; Ito, M.; Weiss, J.; Chien, P.-Y.; Gehm, B.D.; Jameson, J.L. Estrogen Receptor Binding to DNA Is Not Required for Its Activity through the Nonclassical AP1 Pathway. J. Biol. Chem. 2001, 276, 13615–13621. [Google Scholar] [CrossRef] [Green Version]
- Burns, K.A.; Li, Y.; Arao, Y.; Petrovich, R.M.; Korach, K.S. Selective Mutations in Estrogen Receptor α D-domain Alters Nuclear Translocation and Non-estrogen Response Element Gene Regulatory Mechanisms. J. Biol. Chem. 2011, 286, 12640–12649. [Google Scholar] [CrossRef] [Green Version]
- Curtis, S.W.; Washburn, T.F.; Sewall, C.; DiAugustine, R.P.; Lindzey, J.; Couse, J.F.; Korach, K.S. Physiological coupling of growth factor and steroid receptor signaling pathways: Estrogen receptor knockout mice lack estrogen-like response to epidermal growth factor. Proc. Natl. Acad. Sci. USA 1996, 93, 12626–12630. [Google Scholar] [CrossRef] [Green Version]
- Simoncini, T.; Hafezi-Moghadam, A.; Brazil, D.P.; Ley, K.; Chin, W.W.; Liao, J.K. Interaction of oestrogen receptor with the regulatory subunit of phosphatidylinositol-3-OH kinase. Nat. Cell Biol. 2000, 407, 538–541. [Google Scholar] [CrossRef]
- Mendelsohn, M.E.; Karas, R.H. The Protective Effects of Estrogen on the Cardiovascular System. N. Engl. J. Med. 1999, 340, 1801–1811. [Google Scholar] [CrossRef]
- Ueda, K.; Adachi, Y.; Liu, P.; Fukuma, N.; Takimoto, E. Regulatory Actions of Estrogen Receptor Signaling in the Cardiovascular System. Front. Endocrinol. 2020, 10, 909. [Google Scholar] [CrossRef]
- Teoh, J.-P.; Li, X.; Simoncini, T.; Zhu, D.; Fu, X. Estrogen-Mediated Gaseous Signaling Molecules in Cardiovascular Disease. Trends Endocrinol. Metab. 2020, 31, 773–784. [Google Scholar] [CrossRef]
- Kloner, R.A.; Carson, C.; Dobs, A.; Kopecky, S.; Mohler, E.R. Testosterone and Cardiovascular Disease. J. Am. Coll. Cardiol. 2016, 67, 545–557. [Google Scholar] [CrossRef]
- Tostes, R.C.; Carneiro, F.S.; Carvalho, M.H.C.; Reckelhoff, J.F. Reactive oxygen species: Players in the cardiovascular effects of testosterone. Am. J. Physiol. Integr. Comp. Physiol. 2016, 310, R1–R14. [Google Scholar] [CrossRef] [Green Version]
- Thomas, P. Membrane Androgen Receptors Unrelated to Nuclear Steroid Receptors. Endocrinology 2019, 160, 772–781. [Google Scholar] [CrossRef] [PubMed]
- Gonzales, R.J.; Ansar, S.; Duckles, S.P.; Krause, D.N. Androgenic/Estrogenic Balance in the Male Rat Cerebral Circulation: Metabolic Enzymes and Sex Steroid Receptors. Br. J. Pharmacol. 2007, 27, 1841–1852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brinkmann, A.O. Molecular Mechanisms of Androgen Action—A Historical Perspective. Adv. Struct. Saf. Stud. 2011, 776, 3–24. [Google Scholar] [CrossRef]
- Fujimoto, R.; Morimoto, I.; Morita, E.; Sugimoto, H.; Ito, Y.; Eto, S. Androgen receptors, 5 alpha-reductase activity and androgen-dependent proliferation of vascular smooth muscle cells. J. Steroid Biochem. Mol. Biol. 1994, 50, 169–174. [Google Scholar] [CrossRef]
- Mukherjee, T.K.; Dinh, H.; Chaudhuri, G.; Nathan, L. Testosterone attenuates expression of vascular cell adhesion molecule-1 by conversion to estradiol by aromatase in endothelial cells: Implications in atherosclerosis. Proc. Natl. Acad. Sci. USA 2002, 99, 4055–4060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beery, A.K.; Zucker, I. Sex bias in neuroscience and biomedical research. Neurosci. Biobehav. Rev. 2011, 35, 565–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beery, A.K. Inclusion of females does not increase variability in rodent research studies. Curr. Opin. Behav. Sci. 2018, 23, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Becher, O.J.; Holland, E.C. Genetically Engineered Models Have Advantages over Xenografts for Preclinical Studies. Cancer Res. 2006, 66, 3355–3359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sider, K.L.; Blaser, M.C.; Simmons, C.A. Animal Models of Calcific Aortic Valve Disease. Int. J. Inflamm. 2011, 2011, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rattazzi, M.; Bennett, B.J.; Bea, F.; Kirk, E.A.; Ricks, J.L.; Speer, M.; Schwartz, S.M.; Giachelli, C.M.; Rosenfeld, M.E. Calcification of Advanced Atherosclerotic Lesions in the Innominate Arteries of ApoE-Deficient Mice. Arter. Thromb. Vasc. Biol. 2005, 25, 1420–1425. [Google Scholar] [CrossRef] [Green Version]
- Awan, Z.; Denis, M.; Bailey, D.; Giaid, A.; Prat, A.; Goltzman, D.; Seidah, N.G.; Genest, J. The LDLR deficient mouse as a model for aortic calcification and quantification by micro-computed tomography. Atherosclerosis 2011, 219, 455–462. [Google Scholar] [CrossRef]
- Watanabe, R.; Fujita, N.; Sato, Y.; Kobayashi, T.; Morita, M.; Oike, T.; Miyamoto, K.; Kuro-O, M.; Michigami, T.; Fukumoto, S.; et al. Enpp1 is an anti-aging factor that regulates Klotho under phosphate overload conditions. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Kauffenstein, G.; Pizard, A.; Le Corre, Y.; Vessières, E.; Grimaud, L.; Toutain, B.; Labat, C.; Mauras, Y.; Gorgels, T.G.; Bergen, A.A.; et al. Disseminated arterial calcification and enhanced myogenic response are associated with abcc6 deficiency in a mouse model of pseudoxanthoma elasticum. Arter. Thromb. Vasc. Biol. 2014, 34, 1045–1056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrmann, J.; Babic, M.; Tölle, M.; Van Der Giet, M.; Schuchardt, M. Research Models for Studying Vascular Calcification. Int. J. Mol. Sci. 2020, 21, 2204. [Google Scholar] [CrossRef] [Green Version]
- Scatena, M.; Jackson, M.F.; Speer, M.Y.; Leaf, E.M.; Wallingford, M.C.; Giachelli, C.M. Increased Calcific Aortic Valve Disease in response to a diabetogenic, procalcific diet in the LDLr −/− ApoB 100/100 mouse model. Cardiovasc. Pathol. 2018, 34, 28–37. [Google Scholar] [CrossRef]
- Liao, R.; Wang, L.; Li, J.; Sun, S.; Xiong, Y.; Li, Y.; Han, M.; Jiang, H.; Anil, M.; Su, B. Vascular calcification is associated with Wnt-signaling pathway and blood pressure variability in chronic kidney disease rats. Nephrology 2019, 25, 264–272. [Google Scholar] [CrossRef]
- Price, P.A.; Faus, S.A.; Williamson, M.K. Warfarin Causes Rapid Calcification of the Elastic Lamellae in Rat Arteries and Heart Valves. Arter. Thromb. Vasc. Biol. 1998, 18, 1400–1407. [Google Scholar] [CrossRef] [Green Version]
- Shobeiri, N.; Adams, M.; Holden, R. Vascular Calcification in Animal Models of CKD: A Review. Am. J. Nephrol. 2010, 31, 471–481. [Google Scholar] [CrossRef] [PubMed]
- Karas, R.H.; Schulten, H.; Pare, G.; Aronovitz, M.J.; Ohlsson, C.; Gustafsson, J.-A.; Mendelsohn, M.E. Effects of Estrogen on the Vascular Injury Response in Estrogen Receptor α,β (Double) Knockout Mice. Circ. Res. 2001, 89, 534–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Kuhn, G.; Schirmer, M.; Müller, R.; Ruffoni, D. Impaired bone formation in ovariectomized mice reduces implant integration as indicated by longitudinal in vivo micro-computed tomography. PLoS ONE 2017, 12, e0184835. [Google Scholar] [CrossRef] [PubMed]
- Marek, I.; Canu, M.; Cordasic, N.; Rauh, M.; Volkert, G.; Fahlbusch, F.B.; Rascher, W.; Hilgers, K.F.; Hartner, A.; Menendez-Castro, C. Sex differences in the development of vascular and renal lesions in mice with a simultaneous deficiency of Apoe and the integrin chain Itga8. Biol. Sex Differ. 2017, 8, 1–13. [Google Scholar] [CrossRef]
- Ferencik, M. Insights Into Coronary Plaque Microstructure Differences Between Women and Men. Circ. Cardiovasc. Imaging 2016, 9, 005343. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Shah, T.A.; Yurkow, E.J.; Rogers, M.B. Post-transcriptional regulation of aortic calcification in KLOTHO deficient mice: Impact of miR-145 and miR-378. Biorivex. 2020. pre-print. [Google Scholar] [CrossRef]
- Shah, T.A.; Tang, Y.; Yurkow, E.J.; Rogers, M. B Post-Transcriptional Bone Morphogenetic Protein 2 (BMP2) Gene Regulation in Aorta. Biorivex. 2019. pre-print. [Google Scholar] [CrossRef]
- Pereira, T.M.; Nogueira, B.V.; Lima, L.C.; Porto, M.L.; Arruda, J.A.; Vasquez, E.C.; Meyrelles, S.S. Cardiac and vascular changes in elderly atherosclerotic mice: The influence of gender. Lipids Health Dis. 2010, 9, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiao, J.H.; Xie, P.Z.; Fishbein, M.C.; Kreuzer, J.; Drake, T.A.; Demer, L.L.; Lusis, A.J. Pathology of atheromatous lesions in inbred and genetically engineered mice. Genetic determination of arterial calcification. Arterioscler Thromb. 1994, 14, 1480–1497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.S.; Martin, L.J.; Schadt, E.E.; Meng, H.; Wang, X.; Zhao, W.; Ingram-Drake, L.; Nebohacova, M.; Mehrabian, M.; Drake, T.A.; et al. Disruption of the aortic elastic lamina and medial calcification share genetic determinants in mice. Circ Cardiovasc Genet. 2009, 2, 573–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, T.; Akishita, M.; Kozaki, K.; Toba, K.; Orimo, H.; Ouchi, Y. Influence of sex and estrogen on vitamin D-induced arterial calcification in rats. Geriatr Gerontol Int. 2002, 3, 145–149. [Google Scholar] [CrossRef]
- Kohn, M.H.; Price, R.E.; Pelz, H.J. A cardiovascular phenotype in warfarin-resistant Vkorc1 mutant rats. Artery Res. 2008, 2, 138–147. [Google Scholar] [CrossRef] [Green Version]
- Ng, K.; Hildreth, C.M.; Avolio, A.P.; Phillips, J.K. Angiotensin-converting enzyme inhibitor limits pulse-wave velocity and aortic calcification in a rat model of cystic renal disease. Am J Physiol Renal Physiol. 2011, 301, F959–F966. [Google Scholar] [CrossRef] [Green Version]
- Dixon, J.A.; Spinale, F.G. Large Animal Models of Heart Failure. Circ. Heart Fail. 2009, 2, 262–271. [Google Scholar] [CrossRef] [Green Version]
- Warren, H.S.; Fitting, C.; Hoff, E.; Adib-Conquy, M.; Beasley-Topliffe, L.; Tesini, B.; Liang, X.; Valentine, C.; Hellman, J.; Hayden, D.; et al. Resilience to Bacterial Infection: Difference between Species Could Be Due to Proteins in Serum. J. Infect. Dis. 2010, 201, 223–232. [Google Scholar] [CrossRef] [Green Version]
- Cimini, M.; Boughner, D.R.; Ronald, J.A.; Aldington, L.; Rogers, K.A. Development of aortic valve sclerosis in a rabbit model of atherosclerosis: An immunohistochemical and histological study. J. Heart Valve Dis. 2005, 14, 365–375. [Google Scholar]
- Rajamannan, N.M.; Subramaniam, M.; Caira, F.; Stock, S.R.; Spelsberg, T.C. Atorvastatin inhibits hypercholesterolemia-induced calcification in the aortic valves via the Lrp5 receptor pathway. Circulation 2005, 112, I229–I234. [Google Scholar]
- Marechaux, S.; Corseaux, D.; Vincentelli, A.; Richardson, M.; Ung, A.; Susen, S.; Zawadzki, C.; Beregi, J.-P.; Ezekowitz, M.D.; Jude, B.; et al. Identification of tissue factor in experimental aortic valve sclerosis. Cardiovasc. Pathol. 2009, 18, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.; Lee, S.; Song, J.-K.; Kim, S.-M.; Lee, E.-J.; Lee, S.R.; Kim, D.-H.; Jang, J.Y.; Kang, S.-W.; Lee, K.-U.; et al. Dipeptidyl Peptidase-4 Induces Aortic Valve Calcification by Inhibiting Insulin-Like Growth Factor-1 Signaling in Valvular Interstitial Cells. Circulation 2017, 135, 1935–1950. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Nie, M.; Li, J.; Xu, Z.; Zhang, M.; Yan, Y.; Feng, T.; Zhao, X.; Zhao, Q. Effect of pioglitazone on inflammation and calcification in atherosclerotic rabbits. Herz 2017, 43, 733–740. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Waqar, A.B.; Nishijima, K.; Ning, B.; Kitajima, S.; Matsuhisa, F.; Chen, L.; Liu, E.; Koike, T.; Yu, Y.; et al. Macrophage-derived MMP-9 enhances the progression of atherosclerotic lesions and vascular calcification in transgenic rabbits. J. Cell. Mol. Med. 2020, 24, 4261–4274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beute, T.J.; Goehler, M.; Parker, J.; Boeve, T.; Heiser, J.; Murphy, E.; Timek, T.; Willekes, C.L. Long-Term Outcomes of Mosaic Versus Perimount Mitral Replacements: 17-Year Follow-Up of 940 Implants. Ann. Thorac. Surg. 2020, 110, 508–515. [Google Scholar] [CrossRef] [PubMed]
- Badin, J.K.; Progar, V.; Pareddy, A.; Cagle, J.; Alloosh, M.; Sturek, M. Effect of Age on Diabetogenicity of Alloxan in Ossabaw Miniature Swine. Comp. Med. 2019, 69, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Van Ditzhuijzen, N.S.; Heuvel, M.V.D.; Nieman, K.; Mulder, M.T.; Zijlstra, F.; Duncker, D.J.; Van Beusekom, H.M.; Regar, E.; Sorop, O.; Rossi, A.; et al. Serial Coronary Imaging of Early Atherosclerosis Development in Fast-Food-Fed Diabetic and Nondiabetic Swine. JACC Basic Transl. Sci. 2016, 1, 449–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sider, K.L.; Zhu, C.; Kwong, A.V.; Mirzaei, Z.; De Langé, C.F.; Simmons, C.A. Evaluation of a porcine model of early aortic valve sclerosis. Cardiovasc. Pathol. 2014, 23, 289–297. [Google Scholar] [CrossRef]
- Simmons, C.A.; Grant, G.R.; Manduchi, E.; Davies, P.F. Spatial Heterogeneity of Endothelial Phenotypes Correlates With Side-Specific Vulnerability to Calcification in Normal Porcine Aortic Valves. Circ. Res. 2005, 96, 792–799. [Google Scholar] [CrossRef] [Green Version]
- McCoy, C.M.; Nicholas, D.Q.; Masters, K.S. Sex-Related Differences in Gene Expression by Porcine Aortic Valvular Interstitial Cells. PLoS ONE 2012, 7, e39980. [Google Scholar] [CrossRef]
- Masjedi, S.; Lei, Y.; Patel, J.; Ferdous, Z. Sex-related differences in matrix remodeling and early osteogenic markers in aortic valvular interstitial cells. Heart Vessel. 2016, 32, 217–228. [Google Scholar] [CrossRef]
Figure 1.
Comparison of types of cardiovascular calcification. The most common types of cardiovascular calcification are medial, intimal and valve. This figure displays both the normal and disease state for each of these diseases. Medial calcification occurs in the tunica media of the aorta, around the smooth muscle cells. Intimal calcification occurs within an atherosclerotic plaque in the intima of the artery. Valve interstitial cells, which could have a role in valve calcification, mostly accumulate in the aortic side of the valve.
Figure 1.
Comparison of types of cardiovascular calcification. The most common types of cardiovascular calcification are medial, intimal and valve. This figure displays both the normal and disease state for each of these diseases. Medial calcification occurs in the tunica media of the aorta, around the smooth muscle cells. Intimal calcification occurs within an atherosclerotic plaque in the intima of the artery. Valve interstitial cells, which could have a role in valve calcification, mostly accumulate in the aortic side of the valve.

Figure 2.
Vascular calcification mechanisms. In individuals with chronic kidney disease (CKD) high serum phosphate induces osteogenic transition and calcification of vascular smooth muscle cells (VSMCs) through the sodium-dependent phosphate cotransporter, Pit-1. In addition, normal blood vessels express a number of calcification inhibitors including ectonucleotide pyrophosphatase/phosphodiesterase 1 (ENPP1) and matrix Gla protein (MGP). Loss of these inhibitors promotes osteogenic transition and calcification of VSMCs. RUNX2, ALP and BMP2 become elevated which promotes osteoblast-like redifferentiation in SMCs. Matrix vesicles and apoptotic bodies also play a role in vascular calcification. Pi-phosphate RUNX2- runt-related transcription factor 2, ALP-alkaline phosphatase Bmp2-bone morphogenic protein 2.
Figure 2.
Vascular calcification mechanisms. In individuals with chronic kidney disease (CKD) high serum phosphate induces osteogenic transition and calcification of vascular smooth muscle cells (VSMCs) through the sodium-dependent phosphate cotransporter, Pit-1. In addition, normal blood vessels express a number of calcification inhibitors including ectonucleotide pyrophosphatase/phosphodiesterase 1 (ENPP1) and matrix Gla protein (MGP). Loss of these inhibitors promotes osteogenic transition and calcification of VSMCs. RUNX2, ALP and BMP2 become elevated which promotes osteoblast-like redifferentiation in SMCs. Matrix vesicles and apoptotic bodies also play a role in vascular calcification. Pi-phosphate RUNX2- runt-related transcription factor 2, ALP-alkaline phosphatase Bmp2-bone morphogenic protein 2.

Figure 3.
Estrogen binds to estrogen receptors (ERs), which act as transcription factors to regulate the expression of genes containing estrogen response elements (ERE). In addition, estrogen also rapidly induces phosphoinositide 3-kinase (PI3K) and mitogen-activated protein kinase (MAPK) signaling through the orphan G-protein coupled receptor (GPR30). Through genomic and non-genomic pathways, estrogen regulates a wide range of vascular cell functions, including proliferation, migration and senescence. E2- Estradiol. Testosterone acts in a similar mechanism, binding to the androgen receptor (AR), which act as transcription factors to regulate the expression of genes containing the androgen response elements (ARE). Testosterone is also believed to have non-genomic effects, although these pathways are less clear than estrogens.
Figure 3.
Estrogen binds to estrogen receptors (ERs), which act as transcription factors to regulate the expression of genes containing estrogen response elements (ERE). In addition, estrogen also rapidly induces phosphoinositide 3-kinase (PI3K) and mitogen-activated protein kinase (MAPK) signaling through the orphan G-protein coupled receptor (GPR30). Through genomic and non-genomic pathways, estrogen regulates a wide range of vascular cell functions, including proliferation, migration and senescence. E2- Estradiol. Testosterone acts in a similar mechanism, binding to the androgen receptor (AR), which act as transcription factors to regulate the expression of genes containing the androgen response elements (ARE). Testosterone is also believed to have non-genomic effects, although these pathways are less clear than estrogens.


{kind=link}
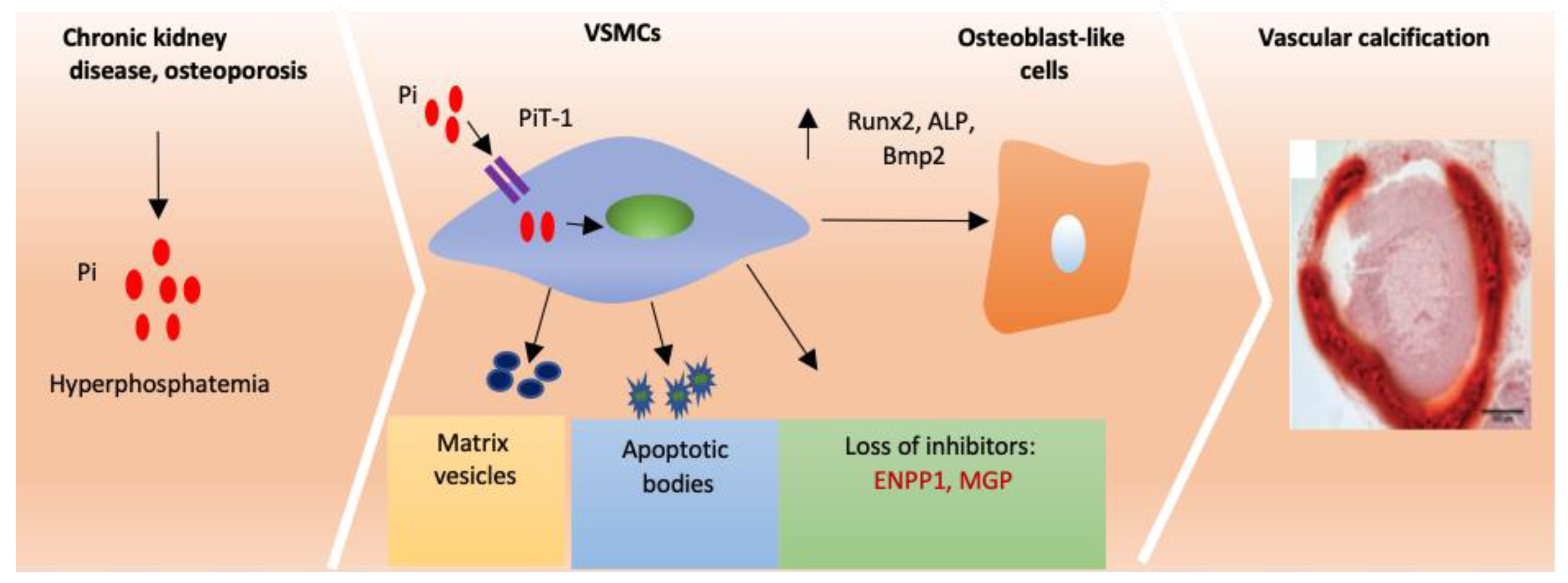{kind=link}
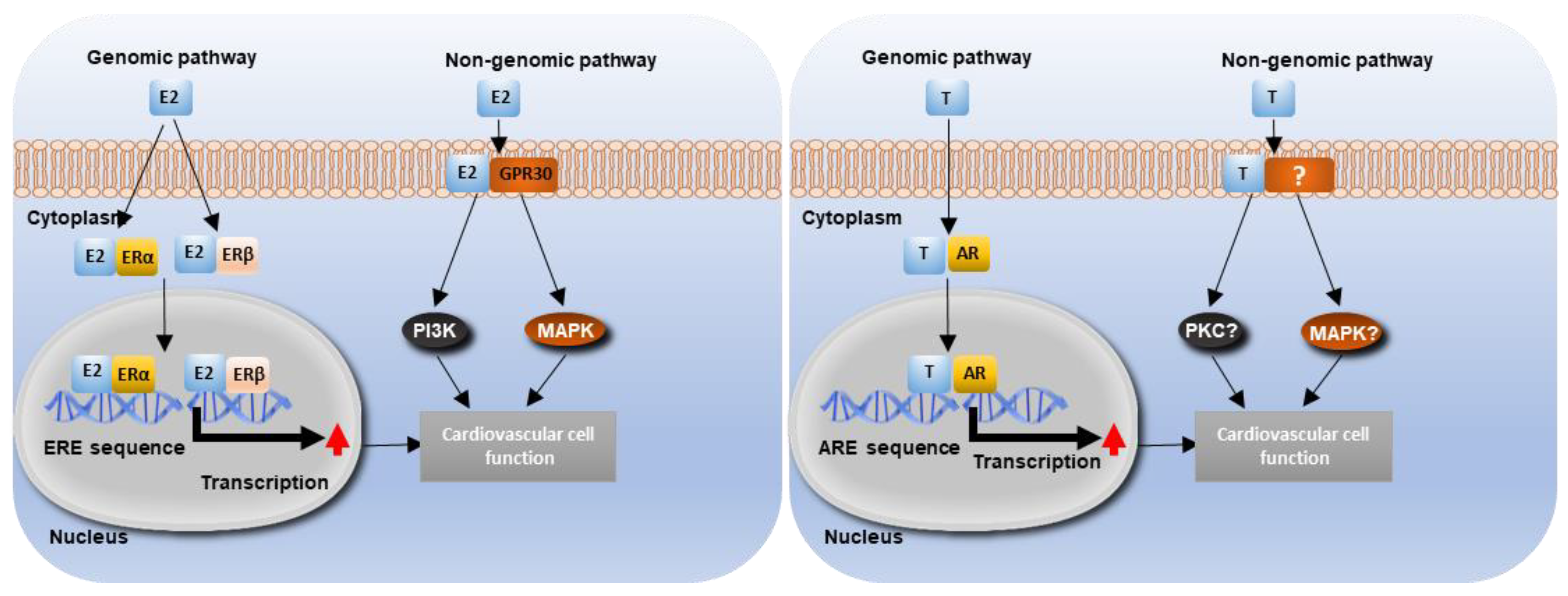{kind=link}
Table 1.
Sex differences in rodent cardiovascular calcification studies. This table summarises key findings of cardiovascular calcification studies that have investigated both sexes, including the model and sex difference. In mice, increased calcification has typically been reported in females compared to males whereas in rats lower calcification has been observed in females. ApoE—Apolipoprotein E, DHT—Dihydrotestosterone, ↑—increased.
Table 1.
Sex differences in rodent cardiovascular calcification studies. This table summarises key findings of cardiovascular calcification studies that have investigated both sexes, including the model and sex difference. In mice, increased calcification has typically been reported in females compared to males whereas in rats lower calcification has been observed in females. ApoE—Apolipoprotein E, DHT—Dihydrotestosterone, ↑—increased.
| Model | Method of Calcification Induction | Type of Calcification | Sex Differences in Calcification | Treatment | Ref |
|---|---|---|---|---|---|
| Mouse | |||||
| ApoE−/− | Aged to 36 weeks | Vascular and valvular | ↑ calcification in females (aortic sinus) | 17β-estradiol | [93] |
| ApoE−/− | Crossed with Itga8−/− | Vascular | ↑ calcification in females | [93] | |
| Klotho−/− | Vascular | None | MicroRNA-145 and microRNA-378a | [138] | |
| Klotho−/− | Vascular | None | None | [139] | |
| ApoE−/− | Vascular, aortic sinus | ↑ calcification in females | Testosterone and DHT | [98] | |
| ApoE−/− | 18 months | Vascular | More vascular calcification in males | [140] | |
| ApoE−/− | Uraemia | Vascular | ↑ calcification in females | [141] | |
| C57BL/6J | Vascular | None | [141] | ||
| ApoA-II | Vascular | None | [141] | ||
| ApoE−/− | Vascular | ↑ calcification in females | [141] | ||
| ApoE−/− | Hyperlipidaemic diet | Vascular | ↑ calcification in females (medial arteries) | [142] | |
| Rat | |||||
| Fisher | 1α-Hydroxyvitamin D3 | Vascular | ↑ calcification in males | [143] | |
| Wistar | Vascular | ↑ calcification in males | [144] | ||
| Lewis Polycystic Kidney | Vascular | None | Perindopril | [145] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Woodward, H.J.; Zhu, D.; Hadoke, P.W.F.; MacRae, V.E. Regulatory Role of Sex Hormones in Cardiovascular Calcification. Int. J. Mol. Sci. 2021, 22, 4620. https://doi.org/10.3390/ijms22094620
AMA Style
Woodward HJ, Zhu D, Hadoke PWF, MacRae VE. Regulatory Role of Sex Hormones in Cardiovascular Calcification. International Journal of Molecular Sciences. 2021; 22(9):4620. https://doi.org/10.3390/ijms22094620
Chicago/Turabian StyleWoodward, Holly J., Dongxing Zhu, Patrick W. F. Hadoke, and Victoria E. MacRae. 2021. "Regulatory Role of Sex Hormones in Cardiovascular Calcification" International Journal of Molecular Sciences 22, no. 9: 4620. https://doi.org/10.3390/ijms22094620
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.