
Genomic Uracil and Aberrant Profile of Demethylation Intermediates in Epigenetics and Hematologic Malignancies


Abstract
:1. Introduction
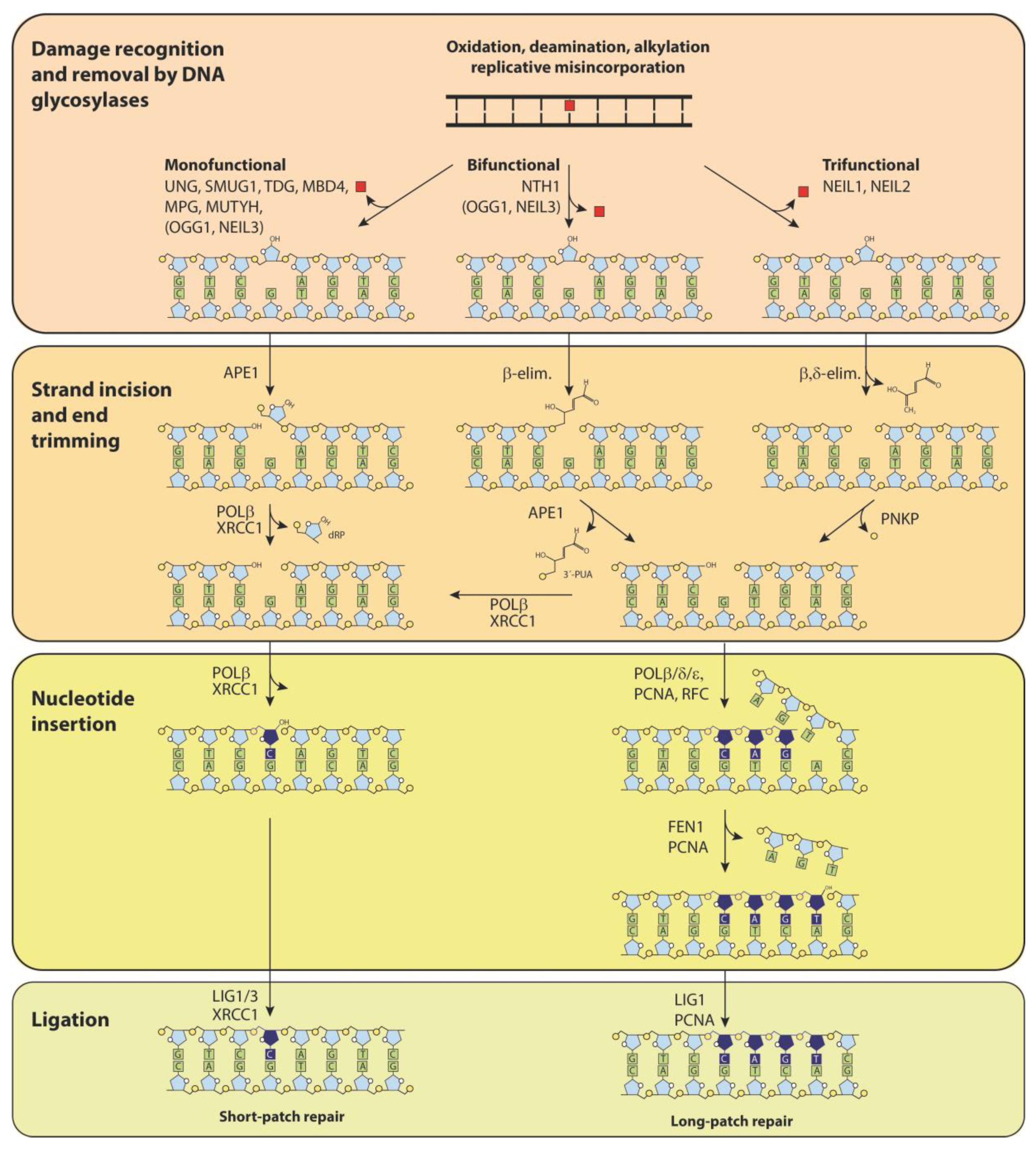
2. DNA Glycosylases—More Than Initiators of Base Excision Repair (BER)
3. The Mutagenicity of U:G Mismatches and U:A Pairs in DNA
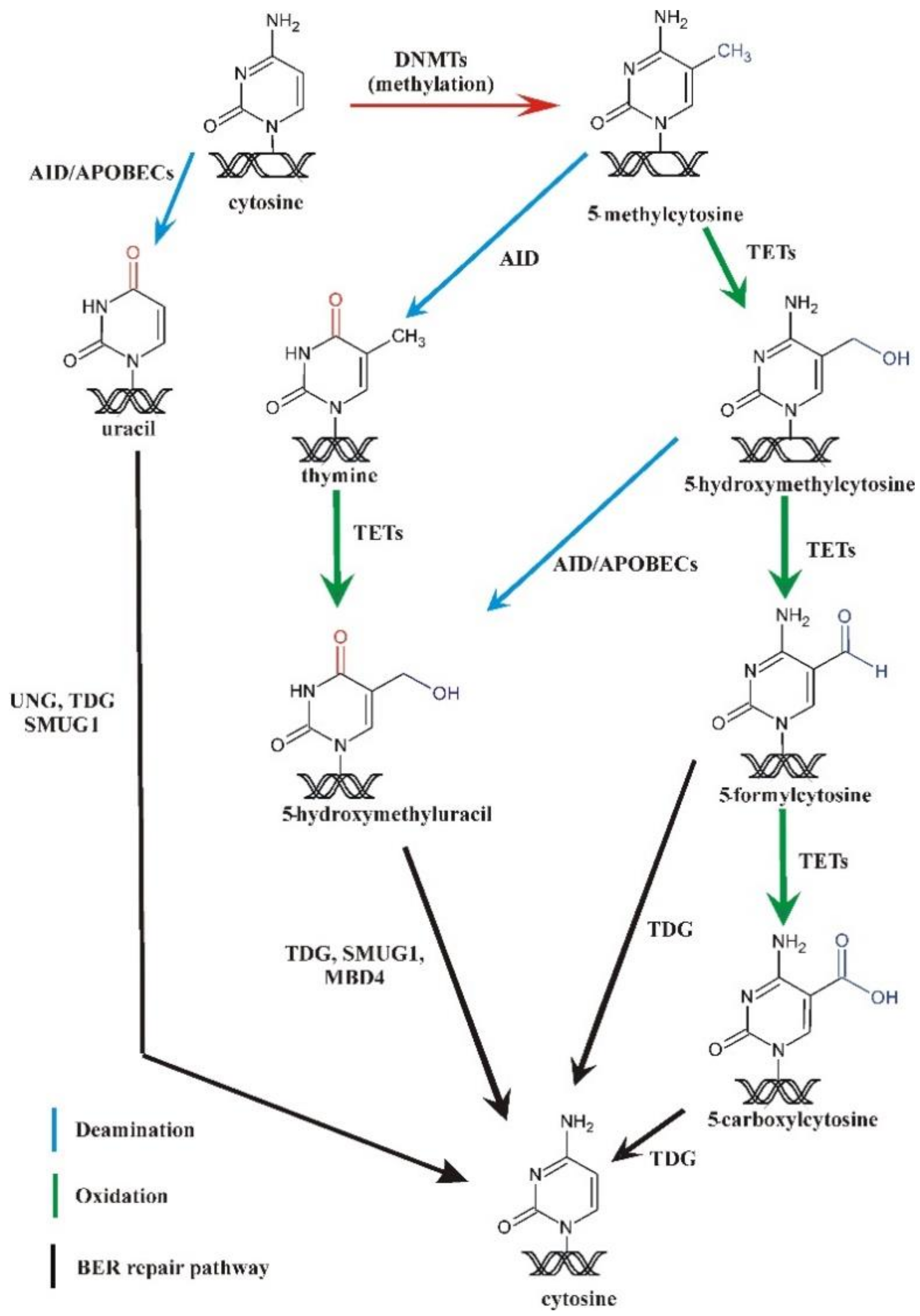
4. Methylation and Demethylation of DNA
5. Genomic Uracil, Uracil Repair Deficiency and B-Cell Malignancy
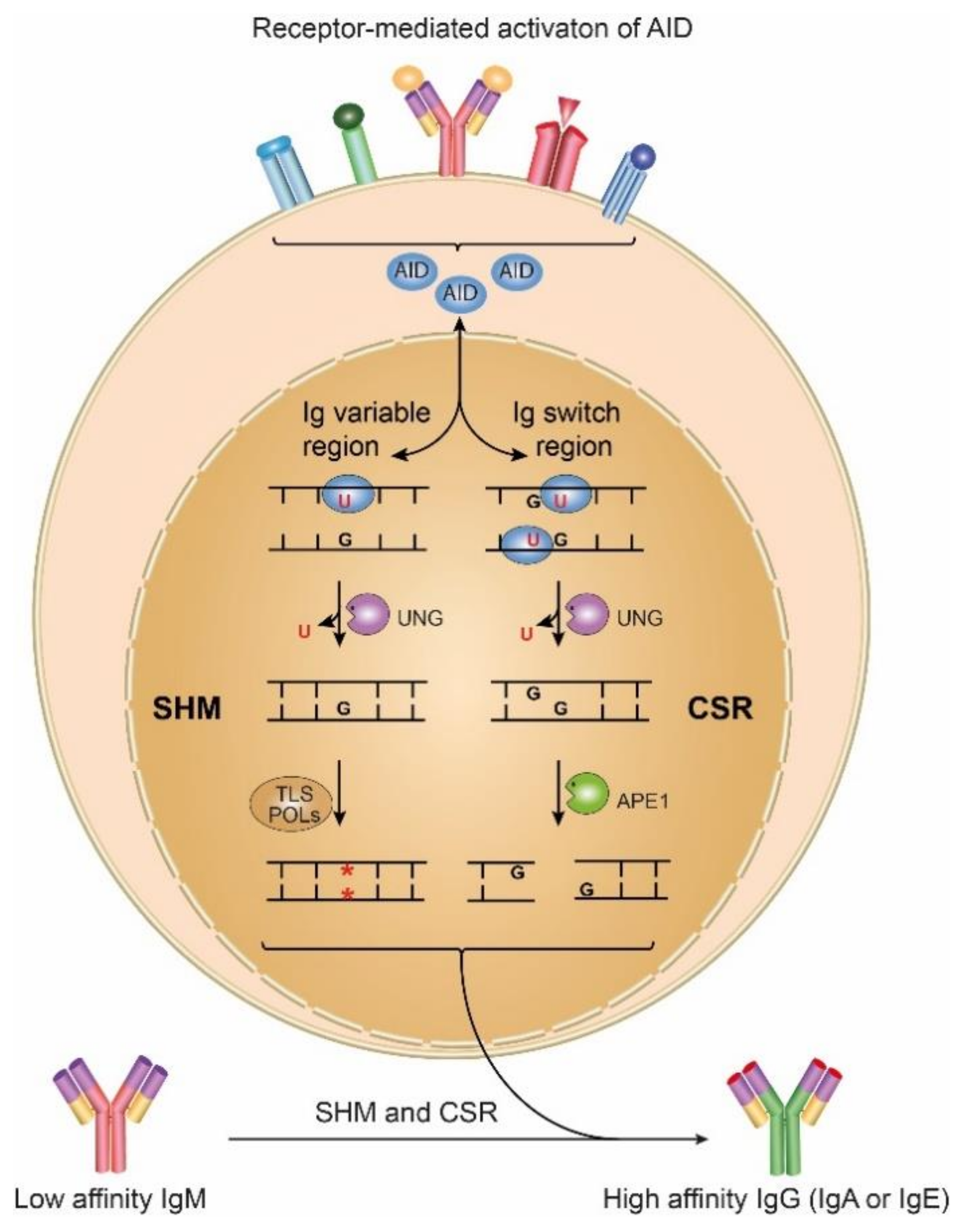
6. Activation-Induced Cytosine Deaminase as a Source of Uracil in DNA: A Link to Genome Instability
7. Aberrant AID Expression and Malignant Development
8. Involvement of AID and APOBEC Enzymes in MM Development
9. Is There a Link between Active Demethylation and Malignant Transformation?
10. What Has Been Learned about 5hmC in Tumor Development?
11. Pattern of DNA Epigenetic Modifications in Myeloid Malignances and MM
12. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Olinski, R.; Gackowski, D.; Cooke, M.S. Endogenously generated DNA nucleobase modifications source, and significance as possible biomarkers of malignant transformation risk, and role in anticancer therapy. Biochim. Biophys. Acta Rev. Cancer 2018, 1869, 29–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krokan, H.E.; Saetrom, P.; Aas, P.A.; Pettersen, H.S.; Kavli, B.; Slupphaug, G. Error-free versus mutagenic processing of genomic uracil--relevance to cancer. DNA Repair 2014, 19, 38–47. [Google Scholar] [CrossRef] [Green Version]
- Moore, S.P.; Toomire, K.J.; Strauss, P.R. DNA modifications repaired by base excision repair are epigenetic. DNA Repair 2013, 12, 1152–1158. [Google Scholar] [CrossRef] [PubMed]
- Hailer-Morrison, M.K.; Kotler, J.M.; Martin, B.D.; Sugden, K.D. Oxidized guanine lesions as modulators of gene transcription. Altered p50 binding affinity and repair shielding by 7,8-dihydro-8-oxo-2′-deoxyguanosine lesions in the NF-kappaB promoter element. Biochemistry 2003, 42, 9761–9770. [Google Scholar] [CrossRef] [PubMed]
- Muramatsu, M.; Sankaranand, V.S.; Anant, S.; Sugai, M.; Kinoshita, K.; Davidson, N.O.; Honjo, T. Specific expression of activation-induced cytidine deaminase (AID), a novel member of the RNA-editing deaminase family in germinal center B cells. J. Biol. Chem. 1999, 274, 18470–18476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.; Schatz, D.G. Balancing AID and DNA repair during somatic hypermutation. Trends Immunol. 2009, 30, 173–181. [Google Scholar] [CrossRef]
- Krokan, H.E.; Bjoras, M. Base excision repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012583. [Google Scholar] [CrossRef]
- Sarno, A.; Lundbaek, M.; Liabakk, N.B.; Aas, P.A.; Mjelle, R.; Hagen, L.; Sousa, M.M.L.; Krokan, H.E.; Kavli, B. Uracil-DNA glycosylase UNG1 isoform variant supports class switch recombination and repairs nuclear genomic uracil. Nucleic Acids Res. 2019, 47, 4569–4585. [Google Scholar] [CrossRef]
- Kyle, R.A.; Durie, B.G.M.; Rajkumar, S.V.; Landgren, O.; Blade, J.; Merlini, G.; Kroger, N.; Einsele, H.; Vesole, D.H.; Dimopoulos, M.; et al. Monoclonal gammopathy of undetermined significance (MGUS) and smoldering (asymptomatic) multiple myeloma: IMWG consensus perspectives risk factors for progression and guidelines for monitoring and management. Leukemia 2010, 24, 1121–1127. [Google Scholar] [CrossRef] [Green Version]
- Alsoe, L.; Sarno, A.; Carracedo, S.; Domanska, D.; Dingler, F.; Lirussi, L.; SenGupta, T.; Tekin, N.B.; Jobert, L.; Alexandrov, L.B.; et al. Uracil Accumulation and Mutagenesis Dominated by Cytosine Deamination in CpG Dinucleotides in Mice Lacking UNG and SMUG1. Sci. Rep. 2017, 7, 7199. [Google Scholar] [CrossRef]
- Lirussi, L.; Demir, O.; You, P.; Sarno, A.; Amaro, R.E.; Nilsen, H. RNA Metabolism Guided by RNA Modifications: The Role of SMUG1 in rRNA Quality Control. Biomolecules 2021, 11, 76. [Google Scholar] [CrossRef]
- Pan, L.; Hao, W.; Zheng, X.; Zeng, X.; Ahmed Abbasi, A.; Boldogh, I.; Ba, X. OGG1-DNA interactions facilitate NF-kappaB binding to DNA targets. Sci. Rep. 2017, 7, 43297. [Google Scholar] [CrossRef] [Green Version]
- Fleming, A.M.; Ding, Y.; Burrows, C.J. Oxidative DNA damage is epigenetic by regulating gene transcription via base excision repair. Proc. Natl. Acad. Sci. USA 2017, 114, 2604–2609. [Google Scholar] [CrossRef]
- Wang, W.; Ma, Y.; Huang, M.; Liang, W.; Zhao, X.; Li, Q.; Wang, S.; Hu, Z.; He, L.; Gao, T.; et al. Asymmetrical arginine dimethylation of histone H4 by 8-oxog/OGG1/PRMT1 is essential for oxidative stress-induced transcription activation. Free Radic Biol. Med. 2021, 164, 175–186. [Google Scholar] [CrossRef]
- Rolseth, V.; Luna, L.; Olsen, A.K.; Suganthan, R.; Scheffler, K.; Neurauter, C.G.; Esbensen, Y.; Kusnierczyk, A.; Hildrestrand, G.A.; Graupner, A.; et al. No cancer predisposition or increased spontaneous mutation frequencies in NEIL DNA glycosylases-deficient mice. Sci. Rep. 2017, 7, 4384. [Google Scholar] [CrossRef]
- Canugovi, C.; Yoon, J.S.; Feldman, N.H.; Croteau, D.L.; Mattson, M.P.; Bohr, V.A. Endonuclease VIII-like 1 (NEIL1) promotes short-term spatial memory retention and protects from ischemic stroke-induced brain dysfunction and death in mice. Proc. Natl. Acad. Sci. USA 2012, 109, 14948–14953. [Google Scholar] [CrossRef] [Green Version]
- Scheffler, K.; Bjoras, K.O.; Bjoras, M. Diverse functions of DNA glycosylases processing oxidative base lesions in brain. DNA Repair 2019, 81, 102665. [Google Scholar] [CrossRef]
- Liu, D.; Croteau, D.L.; Souza-Pinto, N.; Pitta, M.; Tian, J.; Wu, C.; Jiang, H.; Mustafa, K.; Keijzers, G.; Bohr, V.A.; et al. Evidence that OGG1 glycosylase protects neurons against oxidative DNA damage and cell death under ischemic conditions. J. Cereb. Blood Flow Metab. 2011, 31, 680–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Endres, M.; Biniszkiewicz, D.; Sobol, R.W.; Harms, C.; Ahmadi, M.; Lipski, A.; Katchanov, J.; Mergenthaler, P.; Dirnagl, U.; Wilson, S.H.; et al. Increased postischemic brain injury in mice deficient in uracil-DNA glycosylase. J. Clin. Investig. 2004, 113, 1711–1721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortazar, D.; Kunz, C.; Selfridge, J.; Lettieri, T.; Saito, Y.; MacDougall, E.; Wirz, A.; Schuermann, D.; Jacobs, A.L.; Siegrist, F.; et al. Embryonic lethal phenotype reveals a function of TDG in maintaining epigenetic stability. Nature 2011, 470, 419–423. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.H.; Xu, G.F.; Gu, T.P.; Chen, G.D.; Han, B.B.; Xu, Z.M.; Bjoras, M.; Krokan, H.E.; Xu, G.L.; Du, Y.R. Uracil-DNA Glycosylase UNG Promotes Tet-mediated DNA Demethylation. J. Biol. Chem. 2016, 291, 731–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wist, E.; Unhjem, O.; Krokan, H. Accumulation of small fragments of DNA in isolated HeLa cell nuclei due to transient incorporation of dUMP. Biochim. Biophys. Acta 1978, 520, 253–270. [Google Scholar] [CrossRef]
- Lindahl, T.; Nyberg, B. Heat-induced deamination of cytosine residues in deoxyribonucleic acid. Biochemistry 1974, 13, 3405–3410. [Google Scholar] [CrossRef] [PubMed]
- Kavli, B.; Otterlei, M.; Slupphaug, G.; Krokan, H.E. Uracil in DNA--general mutagen, but normal intermediate in acquired immunity. DNA Repair 2007, 6, 505–516. [Google Scholar] [CrossRef]
- Slupphaug, G.; Eftedal, I.; Kavli, B.; Bharati, S.; Helle, N.M.; Haug, T.; Levine, D.W.; Krokan, H.E. Properties of a recombinant human uracil-DNA glycosylase from the UNG gene and evidence that UNG encodes the major uracil-DNA glycosylase. Biochemistry 1995, 34, 128–138. [Google Scholar] [CrossRef]
- Tarantino, M.E.; Dow, B.J.; Drohat, A.C.; Delaney, S. Nucleosomes and the three glycosylases: High, medium, and low levels of excision by the uracil DNA glycosylase superfamily. DNA Repair 2018, 72, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Guillet, M.; Van Der Kemp, P.A.; Boiteux, S. dUTPase activity is critical to maintain genetic stability in Saccharomyces cerevisiae. Nucleic Acids Res. 2006, 34, 2056–2066. [Google Scholar] [CrossRef]
- Collura, A.; Kemp, P.A.; Boiteux, S. Abasic sites linked to dUTP incorporation in DNA are a major cause of spontaneous mutations in absence of base excision repair and Rad17-Mec3-Ddc1 (9-1-1) DNA damage checkpoint clamp in Saccharomyces cerevisiae. DNA Repair 2012, 11, 294–303. [Google Scholar] [CrossRef]
- Auerbach, P.; Bennett, R.A.; Bailey, E.A.; Krokan, H.E.; Demple, B. Mutagenic specificity of endogenously generated abasic sites in Saccharomyces cerevisiae chromosomal DNA. Proc. Natl. Acad. Sci. USA 2005, 102, 17711–17716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennett, S.E.; Sung, J.S.; Mosbaugh, D.W. Fidelity of uracil-initiated base excision DNA repair in DNA polymerase beta-proficient and -deficient mouse embryonic fibroblast cell extracts. J. Biol. Chem. 2001, 276, 42588–42600. [Google Scholar] [CrossRef] [Green Version]
- Akbari, M.; Pena-Diaz, J.; Andersen, S.; Liabakk, N.B.; Otterlei, M.; Krokan, H.E. Extracts of proliferating and non-proliferating human cells display different base excision pathways and repair fidelity. DNA Repair 2009, 8, 834–843. [Google Scholar] [CrossRef]
- Slupphaug, G.; Kavli, B.; Krokan, H.E. Routes to uracil in DNA. In Genomic Uracil-Evolution, Biology, Immunology and Disease; Slupphaug, G., Krokan, H.E., Eds.; World Scientific Publishing Co.: London, UK, 2018; pp. 47–88. [Google Scholar]
- Kennedy, E.M.; Daddacha, W.; Slater, R.; Gavegnano, C.; Fromentin, E.; Schinazi, R.F.; Kim, B. Abundant non-canonical dUTP found in primary human macrophages drives its frequent incorporation by HIV-1 reverse transcriptase. J. Biol. Chem. 2011, 286, 25047–25055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nilsen, H.; Rosewell, I.; Robins, P.; Skjelbred, C.F.; Andersen, S.; Slupphaug, G.; Daly, G.; Krokan, H.E.; Lindahl, T.; Barnes, D.E. Uracil-DNA glycosylase (UNG)-deficient mice reveal a primary role of the enzyme during DNA replication. Mol. Cell 2000, 5, 1059–1065. [Google Scholar] [CrossRef]
- Otterlei, M.; Warbrick, E.; Nagelhus, T.A.; Haug, T.; Slupphaug, G.; Akbari, M.; Aas, P.A.; Steinsbekk, K.; Bakke, O.; Krokan, H.E. Post-replicative base excision repair in replication foci. EMBO J. 1999, 18, 3834–3844. [Google Scholar] [CrossRef] [PubMed]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef] [Green Version]
- Bhutani, N.; Burns, D.M.; Blau, H.M. DNA Demethylation Dynamics. Cell 2011, 146, 866–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, S.H.; Jacobsen, S.E.; Reik, W. Epigenetic Reprogramming in Plant and Animal Development. Science 2010, 330, 622–627. [Google Scholar] [CrossRef] [Green Version]
- Kriaucionis, S.; Heintz, N. The Nuclear DNA Base 5-Hydroxymethylcytosine Is Present in Purkinje Neurons and the Brain. Science 2009, 324, 929–930. [Google Scholar] [CrossRef] [Green Version]
- Tahiliani, M.; Koh, K.P.; Shen, Y.H.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-Methylcytosine to 5-Hydroxymethylcytosine in Mammalian DNA by MLL Partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef] [Green Version]
- Globisch, D.; Munzel, M.; Muller, M.; Michalakis, S.; Wagner, M.; Koch, S.; Bruckl, T.; Biel, M.; Carell, T. Tissue Distribution of 5-Hydroxymethylcytosine and Search for Active Demethylation Intermediates. PLoS ONE 2010, 5. [Google Scholar] [CrossRef] [Green Version]
- Cadet, J.; Wagner, J.R. TET enzymatic oxidation of 5-methylcytosine, 5-hydroxymethylcytosine and 5-formylcytosine. Mutat. Res. Genet. Toxicol. Environ. Mutagen 2014, 764–765, 18–35. [Google Scholar] [CrossRef]
- Pfaffeneder, T.; Spada, F.; Wagner, M.; Brandmayr, C.; Laube, S.K.; Eisen, D.; Truss, M.; Steinbacher, J.; Hackner, B.; Kotljarova, O.; et al. Tet oxidizes thymine to 5-hydroxymethyluracil in mouse embryonic stem cell DNA. Nat. Chem. Biol. 2014, 10, 574–581. [Google Scholar] [CrossRef]
- Rai, K.; Huggins, I.J.; James, S.R.; Karpf, A.R.; Jones, D.A.; Cairns, B.R. DNA demethylation in zebrafish involves the coupling of a deaminase, a glycosylase, and gadd45. Cell 2008, 135, 1201–1212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, F.; Peat, J.; Burgess, H.; Rada, C.; Reik, W.; Dean, W. Active demethylation in mouse zygotes involves cytosine deamination and base excision repair. Epigenet. Chromatin 2013, 6, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grin, I.; Ishchenko, A.A. An interplay of the base excision repair and mismatch repair pathways in active DNA demethylation. Nucleic Acids Res. 2016, 44, 3713–3727. [Google Scholar] [CrossRef]
- Franchini, D.M.; Chan, C.F.; Morgan, H.; Incorvaia, E.; Rangam, G.; Dean, W.; Santos, F.; Reik, W.; Petersen-Mahrt, S.K. Processive DNA demethylation via DNA deaminase-induced lesion resolution. PLoS ONE 2014, 9, e97754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maiti, A.; Drohat, A.C. Thymine DNA glycosylase can rapidly excise 5-formylcytosine and 5-carboxylcytosine: Potential implications for active demethylation of CpG sites. J. Biol. Chem. 2011, 286, 35334–35338. [Google Scholar] [CrossRef] [Green Version]
- He, Y.F.; Li, B.Z.; Li, Z.; Liu, P.; Wang, Y.; Tang, Q.; Ding, J.; Jia, Y.; Chen, Z.; Li, L.; et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 2011, 333, 1303–1307. [Google Scholar] [CrossRef] [Green Version]
- Slyvka, A.; Mierzejewska, K.; Bochtler, M. Nei-like 1 (NEIL1) excises 5-carboxylcytosine directly and stimulates TDG-mediated 5-formyl and 5-carboxylcytosine excision. Sci. Rep. 2017, 7, 9001. [Google Scholar] [CrossRef] [Green Version]
- Schomacher, L.; Han, D.; Musheev, M.U.; Arab, K.; Kienhofer, S.; von Seggern, A.; Niehrs, C. Neil DNA glycosylases promote substrate turnover by Tdg during DNA demethylation. Nat. Struct. Mol. Biol. 2016, 23, 116–124. [Google Scholar] [CrossRef] [Green Version]
- Schuermann, D.; Weber, A.R.; Schar, P. Active DNA demethylation by DNA repair: Facts and uncertainties. DNA Repair 2016, 44, 92–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spada, F.; Schiffers, S.; Kirchner, A.; Zhang, Y.; Arista, G.; Kosmatchev, O.; Korytiakova, E.; Rahimoff, R.; Ebert, C.; Carell, T. Active turnover of genomic methylcytosine in pluripotent cells. Nat. Chem. Biol. 2020, 16, 1411–1419. [Google Scholar] [CrossRef]
- Yildirim, O.; Li, R.W.; Hung, J.H.; Chen, P.B.; Dong, X.J.; Ee, L.S.; Weng, Z.P.; Rando, O.J.; Fazzio, T.G. Mbd3/NURD Complex Regulates Expression of 5-Hydroxymethylcytosine Marked Genes in Embryonic Stem Cells. Cell 2011, 147, 1498–1510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kudo, Y.; Tateishi, K.; Yamamoto, K.; Yamamoto, S.; Asaoka, Y.; Ijichi, H.; Nagae, G.; Yoshida, H.; Aburatani, H.; Koike, K. Loss of 5-hydroxymethylcytosine is accompanied with malignant cellular transformation. Cancer Sci. 2012, 103, 670–676. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Gao, H. Hydroxymethylation and tumors: Can 5-hydroxymethylation be used as a marker for tumor diagnosis and treatment? Hum. Genom. 2020, 14, 15. [Google Scholar] [CrossRef] [PubMed]
- Galashevskaya, A.; Sarno, A.; Vagbo, C.B.; Aas, P.A.; Hagen, L.; Slupphaug, G.; Krokan, H.E. A robust, sensitive assay for genomic uracil determination by LC/MS/MS reveals lower levels than previously reported. DNA Repair 2013, 12, 699–706. [Google Scholar] [CrossRef] [Green Version]
- Hagen, L.; Pena-Diaz, J.; Kavli, B.; Otterlei, M.; Slupphaug, G.; Krokan, H.E. Genomic uracil and human disease. Exp. Cell Res. 2006, 312, 2666–2672. [Google Scholar] [CrossRef] [PubMed]
- Rada, C.; Williams, G.T.; Nilsen, H.; Barnes, D.E.; Lindahl, T.; Neuberger, M.S. Immunoglobulin isotype switching is inhibited and somatic hypermutation perturbed in UNG-deficient mice. Curr. Biol. 2002, 12, 1748–1755. [Google Scholar] [CrossRef] [Green Version]
- Imai, K.; Slupphaug, G.; Lee, W.I.; Revy, P.; Nonoyama, S.; Catalan, N.; Yel, L.; Forveille, M.; Kavli, B.; Krokan, H.E.; et al. Human uracil-DNA glycosylase deficiency associated with profoundly impaired immunoglobulin class-switch recombination. Nat. Immunol. 2003, 4, 1023–1028. [Google Scholar] [CrossRef]
- Chaim, I.A.; Nagel, Z.D.; Jordan, J.J.; Mazzucato, P.; Ngo, L.P.; Samson, L.D. In vivo measurements of interindividual differences in DNA glycosylases and APE1 activities. Proc. Natl. Acad. Sci. USA 2017, 114, E10379–E10388. [Google Scholar] [CrossRef] [Green Version]
- Ramiro, A.R.; Jankovic, M.; Callen, E.; Difilippantonio, S.; Chen, H.T.; McBride, K.M.; Eisenreich, T.R.; Chen, J.; Dickins, R.A.; Lowe, S.W.; et al. Role of genomic instability and p53 in AID-induced c-myc-Igh translocations. Nature 2006, 440, 105–109. [Google Scholar] [CrossRef] [Green Version]
- Gu, X.; Booth, C.J.; Liu, Z.; Strout, M.P. AID-associated DNA repair pathways regulate malignant transformation in a murine model of BCL6-driven diffuse large B-cell lymphoma. Blood 2016, 127, 102–112. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, H.S.; Galashevskaya, A.; Doseth, B.; Sousa, M.M.; Sarno, A.; Visnes, T.; Aas, P.A.; Liabakk, N.B.; Slupphaug, G.; Saetrom, P.; et al. AID expression in B-cell lymphomas causes accumulation of genomic uracil and a distinct AID mutational signature. DNA Repair 2015, 25, 60–71. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, I.M.; Hiai, H.; Kakazu, N.; Yamada, S.; Muramatsu, M.; Kinoshita, K.; Honjo, T. Constitutive expression of AID leads to tumorigenesis. J. Exp. Med. 2003, 197, 1173–1181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Endo, Y.; Marusawa, H.; Kinoshita, K.; Morisawa, T.; Sakurai, T.; Okazaki, I.M.; Watashi, K.; Shimotohno, K.; Honjo, T.; Chiba, T. Expression of activation-induced cytidine deaminase in human hepatocytes via NF-kappa B signaling. Oncogene 2007, 26, 5587–5595. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, Y.; Marusawa, H.; Kinoshita, K.; Endo, Y.; Kou, T.; Morisawa, T.; Azuma, T.; Okazaki, I.M.; Honjo, T.; Chiba, T. Helicobacter pylori infection triggers aberrant expression of activation-induced cytidine deaminase in gastric epithelium. Nat. Med. 2007, 13, 470–476. [Google Scholar] [CrossRef] [Green Version]
- Marusawa, H. Aberrant AID expression and human cancer development. Int. J. Biochem. Cell B 2008, 40, 1399–1402. [Google Scholar] [CrossRef] [PubMed]
- Greeve, J.; Philipsen, A.; Krause, K.; Klapper, W.; Heidom, K.; Castle, B.E.; Janda, J.; Marcu, K.B.; Parwaresch, R. Expression of activation-induced cytidine deaminase in human B-cell non-Hodgkin lymphomas. Blood 2003, 101, 3574–3580. [Google Scholar] [CrossRef]
- Chesi, M.; Robbiani, D.F.; Sebag, M.; Chng, W.J.; Affer, M.; Tiedemann, R.; Valdez, R.; Palmer, S.E.; Haas, S.S.; Stewart, A.K.; et al. AID-dependent activation of a MYC transgene induces multiple myeloma in a conditional mouse model of post-germinal center malignancies. Cancer Cell 2008, 13, 167–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burns, M.B.; Lackey, L.; Carpenter, M.A.; Rathore, A.; Land, A.M.; Leonard, B.; Refsland, E.W.; Kotandeniya, D.; Tretyakova, N.; Nikas, J.B.; et al. APOBEC3B is an enzymatic source of mutation in breast cancer. Nature 2013, 494, 366–370. [Google Scholar] [CrossRef]
- Refsland, E.W.; Harris, R.S. The APOBEC3 Family of Retroelement Restriction Factors. Curr. Top. Microbiol. 2013, 371, 1–27. [Google Scholar] [CrossRef] [Green Version]
- Hoang, P.H.; Cornish, A.J.; Dobbins, S.E.; Kaiser, M.; Houlston, R.S. Mutational processes contributing to the development of multiple myeloma. Blood Cancer J. 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Maura, F.; Petljak, M.; Lionetti, M.; Cifola, I.; Liang, W.; Pinatel, E.; Alexandrov, L.B.; Fullam, A.; Martincorena, I.; Dawson, K.J.; et al. Biological and prognostic impact of APOBEC-induced mutations in the spectrum of plasma cell dyscrasias and multiple myeloma cell lines. Leukemia 2018, 32, 1043–1047. [Google Scholar] [CrossRef] [Green Version]
- Bolli, N.; Maura, F.; Minvielle, S.; Gloznik, D.; Szalat, R.; Fullam, A.; Martincorena, I.; Dawson, K.J.; Samur, M.K.; Zamora, J.; et al. Genomic patterns of progression in smoldering multiple myeloma. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef]
- Feinberg, A.P.; Vogelstein, B. Hypomethylation Distinguishes Genes of Some Human Cancers from Their Normal Counterparts. Nature 1983, 301, 89–92. [Google Scholar] [CrossRef] [PubMed]
- Casalino, L.; Verde, P. Multifaceted Roles of DNA Methylation in Neoplastic Transformation, from Tumor Suppressors to EMT and Metastasis. Genes 2020, 11, 922. [Google Scholar] [CrossRef]
- Aran, D.; Sabato, S.; Hellman, A. DNA methylation of distal regulatory sites characterizes dysregulation of cancer genes. Genome Biol. 2013, 14, R21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groh, S.; Schotta, G. Silencing of endogenous retroviruses by heterochromatin. Cell Mol. Life Sci. 2017, 74, 2055–2065. [Google Scholar] [CrossRef] [PubMed]
- Kiel, M.J.; Sahasrabuddhe, A.A.; Rolland, D.C.M.; Velusamy, T.; Chung, F.; Schaller, M.; Bailey, N.G.; Betz, B.L.; Miranda, R.N.; Porcu, P.; et al. Genomic analyses reveal recurrent mutations in epigenetic modifiers and the JAK-STAT pathway in Sezary syndrome. Nat. Commun. 2015, 6, 8470. [Google Scholar] [CrossRef] [PubMed]
- Lopusna, K.; Nowialis, P.; Opavska, J.; Abraham, A.; Riva, A.; Haney, S.L.; Opavsky, R. Decreases in different Dnmt3b activities drive distinct development of hematologic malignancies in mice. J. Biol. Chem. 2021, 100285. [Google Scholar] [CrossRef]
- Lorsbach, R.B.; Moore, J.; Mathew, S.; Raimondi, S.C.; Mukatira, S.T.; Downing, J.R. TET1, a member of a novel protein family, is fused to MLL in acute myeloid leukemia containing the t(10;11)(q22;q23). Leukemia 2003, 17, 637–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, M.; Huang, Y.; Jankowska, A.M.; Pape, U.J.; Tahiliani, M.; Bandukwala, H.S.; An, J.; Lamperti, E.D.; Koh, K.P.; Ganetzky, R.; et al. Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature 2010, 468, 839–843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Liu, M. Distribution of 5-hydroxymethylcytosine in different human tissues. J. Nucleic Acids 2011, 2011, 870726. [Google Scholar] [CrossRef] [Green Version]
- Jin, S.G.; Jiang, Y.; Qiu, R.X.; Rauch, T.A.; Wang, Y.S.; Schackert, G.; Krex, D.; Lu, Q.; Pfeifer, G.P. 5-Hydroxymethylcytosine Is Strongly Depleted in Human Cancers but Its Levels Do Not Correlate with IDH1 Mutations. Cancer Res. 2011, 71, 7360–7365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lian, C.G.; Xu, Y.F.; Ceol, C.; Wu, F.Z.; Larson, A.; Dresser, K.; Xu, W.Q.; Tan, L.; Hu, Y.G.; Zhan, Q.; et al. Loss of 5-Hydroxymethylcytosine Is an Epigenetic Hallmark of Melanoma. Cell 2012, 150, 1135–1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Liu, Y.; Bai, F.; Zhang, J.Y.; Ma, S.H.; Liu, J.; Xu, Z.D.; Zhu, H.G.; Ling, Z.Q.; Ye, D.; et al. Tumor development is associated with decrease of TET gene expression and 5-methylcytosine hydroxylation. Oncogene 2013, 32, 663–669. [Google Scholar] [CrossRef] [Green Version]
- Cimmino, L.; Abdel-Wahab, O.; Levine, R.L.; Aifantis, I. TET Family Proteins and Their Role in Stem Cell Differentiation and Transformation. Cell Stem Cell 2011, 9, 193–204. [Google Scholar] [CrossRef] [Green Version]
- Bennett, M.T.; Rodgers, M.T.; Hebert, A.S.; Ruslander, L.E.; Eisele, L.; Drohat, A.C. Specificity of human thymine DNA glycosylase depends on N-glycosidic bond stability. J. Am. Chem. Soc. 2006, 128, 12510–12519. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.U.; Su, Y.J.; Zhong, C.; Ming, G.L.; Song, H.J. Hydroxylation of 5-Methylcytosine by TET1 Promotes Active DNA Demethylation in the Adult Brain. Cell 2011, 145, 423–434. [Google Scholar] [CrossRef] [Green Version]
- Celik, H.; Kramer, A.; Challen, G.A. DNA methylation in normal and malignant hematopoiesis. Int. J. Hematol. 2016, 103, 617–626. [Google Scholar] [CrossRef] [Green Version]
- Hoang, N.M.; Rui, L. DNA methyltransferases in hematological malignancies. J. Genet. Genom. 2020, 47, 361–372. [Google Scholar] [CrossRef]
- Jankowska, A.M.; Szpurka, H.; Tiu, R.V.; Makishima, H.; Afable, M.; Huh, J.; O’Keefe, C.L.; Ganetzky, R.; McDevitt, M.A.; Maciejewski, J.P. Loss of heterozygosity 4q24 and TET2 mutations associated with myelodysplastic/myeloproliferative neoplasms. Blood 2009, 113, 6403–6410. [Google Scholar] [CrossRef] [Green Version]
- Tefferi, A.; Pardanani, A.; Lim, K.H.; Abdel-Wahab, O.; Lasho, T.L.; Patel, J.; Gangat, N.; Finke, C.M.; Schwager, S.; Mullally, A.; et al. TET2 mutations and their clinical correlates in polycythemia vera, essential thrombocythemia and myelofibrosis. Leukemia 2009, 23, 905–911. [Google Scholar] [CrossRef] [Green Version]
- Delhommeau, F.; Dupont, S.; Della Valle, V.; James, C.; Trannoy, S.; Masse, A.; Kosmider, O.; Le Couedic, J.P.; Robert, F.; Alberdi, A.; et al. Mutation in TET2 in Myeloid Cancers. N. Engl. J. Med. 2009, 360, 2289–2301. [Google Scholar] [CrossRef]
- Walker, B.A.; Mavrommatis, K.; Wardell, C.P.; Ashby, T.C.; Bauer, M.; Davies, F.E.; Rosenthal, A.; Wang, H.W.; Qu, P.P.; Hoering, A.; et al. Identification of novel mutational drivers reveals oncogene dependencies in multiple myeloma. Blood 2018, 132, 587–597. [Google Scholar] [CrossRef]
- Li, Z.; Cai, X.; Cai, C.L.; Wang, J.; Zhang, W.; Petersen, B.E.; Yang, F.C.; Xu, M. Deletion of Tet2 in mice leads to dysregulated hematopoietic stem cells and subsequent development of myeloid malignancies. Blood 2011, 118, 4509–4518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Y.; Li, X.; Cassady, K.; Zou, Z.; Zhang, X. TET2 Function in Hematopoietic Malignancies, Immune Regulation, and DNA Repair. Front. Oncol. 2019, 9, 210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gackowski, D.; Gawronski, M.; Kerr, C.; Radivoyevitch, T.; Zarakowska, E.; Starczak, M.; Abakir, A.; Ruzov, A.; Maciejewski, J.P.; Olinski, R. 5-formylcytosine and 5-hydroxymethyluracil as surrogate markers of TET2 and SF3B1 mutations in myelodysplastic syndrome, respectively. Haematologica 2020, 105, E213–E215. [Google Scholar] [CrossRef] [PubMed]
- Gackowski, D.; Starczak, M.; Zarakowska, E.; Modrzejewska, M.; Szpila, A.; Banaszkiewicz, Z.; Olinski, R. Accurate, Direct, and High-Throughput Analyses of a Broad Spectrum of Endogenously Generated DNA Base Modifications with Isotope-Dilution Two-Dimensional Ultraperformance Liquid Chromatography with Tandem Mass Spectrometry: Possible Clinical Implication. Anal. Chem. 2016, 88, 12128–12136. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.L.; Lu, J.Y.; Cheng, J.D.; Rao, Q.H.; Li, Z.; Hou, H.F.; Lou, Z.Y.; Zhang, L.; Li, W.; Gong, W.; et al. Structural insight into substrate preference for TET- mediated oxidation. Nature 2015, 527, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.Y.; Torabifard, H.; Crawford, D.J.; DeNizio, J.E.; Cao, X.J.; Garcia, B.A.; Cisneros, G.A.; Kohli, R.M. Mutations along a TET2 active site scaffold stall oxidation at 5-hydroxymethylcytosine. Nat. Chem. Biol. 2017, 13, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Iurlaro, M.; McInroy, G.R.; Burgess, H.E.; Dean, W.; Raiber, E.A.; Bachman, M.; Beraldi, D.; Balasubramanian, S.; Reik, W. In vivo genome-wide profiling reveals a tissue-specific role for 5-formylcytosine. Genome Biol. 2016, 17, 141. [Google Scholar] [CrossRef] [Green Version]
- Raiber, E.A.; Portella, G.; Martinez Cuesta, S.; Hardisty, R.; Murat, P.; Li, Z.; Iurlaro, M.; Dean, W.; Spindel, J.; Beraldi, D.; et al. 5-Formylcytosine organizes nucleosomes and forms Schiff base interactions with histones in mouse embryonic stem cells. Nat. Chem. 2018, 10, 1258–1266. [Google Scholar] [CrossRef]
- Mailankody, S.; Pfeiffer, R.M.; Kristinsson, S.Y.; Korde, N.; Bjorkholm, M.; Goldin, L.R.; Turesson, I.; Landgren, O. Risk of acute myeloid leukemia and myelodysplastic syndromes after multiple myeloma and its precursor disease (MGUS). Blood 2011, 118, 4086–4092. [Google Scholar] [CrossRef] [Green Version]
- Alberge, J.B.; Magrangeas, F.; Wagner, M.; Denie, S.; Guerin-Charbonnel, C.; Campion, L.; Attal, M.; Avet-Loiseau, H.; Carell, T.; Moreau, P.; et al. DNA hydroxymethylation is associated with disease severity and persists at enhancers of oncogenic regions in multiple myeloma. Clin. Epigenet. 2020, 12, 163. [Google Scholar] [CrossRef]
- Chiu, B.C.-H.; Zhang, Z.; Karpus, J.; Derman, B.; Zeng, C.; Stepniak, E.; Chiu, R.; Spinelli, J.; Jakubowiak, A.; He, C.; et al. Abstract PR03: Genome-wide 5-hydroxymethylcytosine profiles in circulating cell-free DNA and survival in patients with multiple myeloma. Clin. Cancer Res. 2020, 26, PR03. [Google Scholar] [CrossRef]
- Zhang, W.Q.; Qu, J.; Liu, G.H.; Belmonte, J.C.I. The ageing epigenome and its rejuvenation. Nat. Rev. Mol. Cell Biol. 2020, 21, 137–150. [Google Scholar] [CrossRef]
- Busque, L.; Patel, J.P.; Figueroa, M.E.; Vasanthakumar, A.; Provost, S.; Hamilou, Z.; Mollica, L.; Li, J.; Viale, A.; Heguy, A.; et al. Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat. Genet. 2012, 44, 1179–1181. [Google Scholar] [CrossRef] [PubMed]
- De Smedt, E.; Lui, H.; Maes, K.; De Veirman, K.; Menu, E.; Vanderkerken, K.; De Bruyne, E. The Epigenome in Multiple Myeloma: Impact on Tumor Cell Plasticity and Drug Response. Front. Oncol. 2018, 8. [Google Scholar] [CrossRef]
- Paiva, B.; Puig, N.; Cedena, M.T.; de Jong, B.G.; Ruiz, Y.; Rapado, I.; Martinez-Lopez, J.; Cordon, L.; Alignani, D.; Delgado, J.A.; et al. Differentiation stage of myeloma plasma cells: Biological and clinical significance. Leukemia 2017, 31, 382–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsagaratou, A. Unveiling the regulation of NKT17 cell differentiation and function. Mol. Immunol. 2019, 105, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Lio, C.W.J.; Shukla, V.; Samaniego-Castruita, D.; Gonzalez-Avalos, E.; Chakraborty, A.; Yue, X.J.; Schatz, D.G.; Ay, F.; Rao, A. TET enzymes augment activation-induced deaminase (AID) expression via 5-hydroxymethylcytosine modifications at the Aicda superenhancer. Sci. Immunol. 2019, 4. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Enzyme | Subcellular Localization | Substrates and (Minor Substrates) | Mouse Knockout | Human Disease |
|---|---|---|---|---|
| UNG * | Nuclei and mitochondria | U, 5-FU in ss and dsDNA, U:A and U:G context (alloxan, 5-hydroxyuracil, isodialuric acid) | Partial defect in CSR, skewed SHM, B-cell lymphomas | Complete defect in CSR, HIGM syndrome, infections, lymphoid hyperplasia |
| SMUG1 | Nuclei | 5-hmU, U:G > U:A > ssU, 5-FU, εC in ss and dsDNA | Viable and fertile. SMUG1/UNG/MSH triple k.o. reduced longevity | Unknown |
| TDG | Nuclei | U:G > T:G, (5-hmU in dsDNA, 5-FU) | Embryonic lethal, epigenetic role in development | Unknown |
| MBD4 | Nuclei | U:G and T:G, 5-hmU in CpG context (εC, 5-FU in dsDNA) | Viable and fertile, C to T transitions, intestinal neoplasia | Mutated in carcinomas with microsatellite instability |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olinski, R.; Slupphaug, G.; Foksinski, M.; Krokan, H.E. Genomic Uracil and Aberrant Profile of Demethylation Intermediates in Epigenetics and Hematologic Malignancies. Int. J. Mol. Sci. 2021, 22, 4212. https://doi.org/10.3390/ijms22084212
Olinski R, Slupphaug G, Foksinski M, Krokan HE. Genomic Uracil and Aberrant Profile of Demethylation Intermediates in Epigenetics and Hematologic Malignancies. International Journal of Molecular Sciences. 2021; 22(8):4212. https://doi.org/10.3390/ijms22084212
Chicago/Turabian StyleOlinski, Ryszard, Geir Slupphaug, Marek Foksinski, and Hans Einar Krokan. 2021. "Genomic Uracil and Aberrant Profile of Demethylation Intermediates in Epigenetics and Hematologic Malignancies" International Journal of Molecular Sciences 22, no. 8: 4212. https://doi.org/10.3390/ijms22084212