
CHOP Pro-Apoptotic Transcriptional Program in Response to ER Stress Is Hacked by Zika Virus

 , , , , and
, , , , and 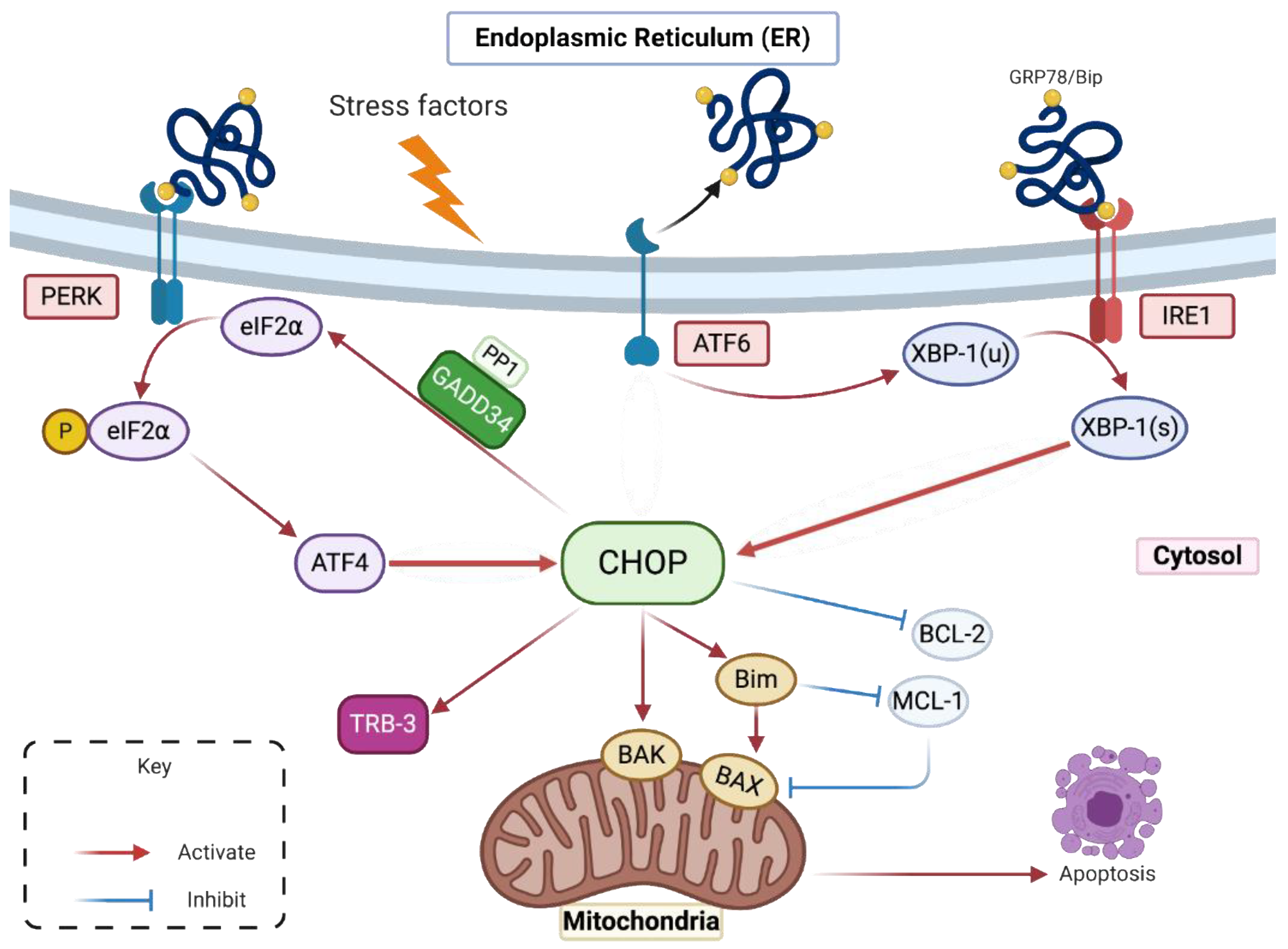{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
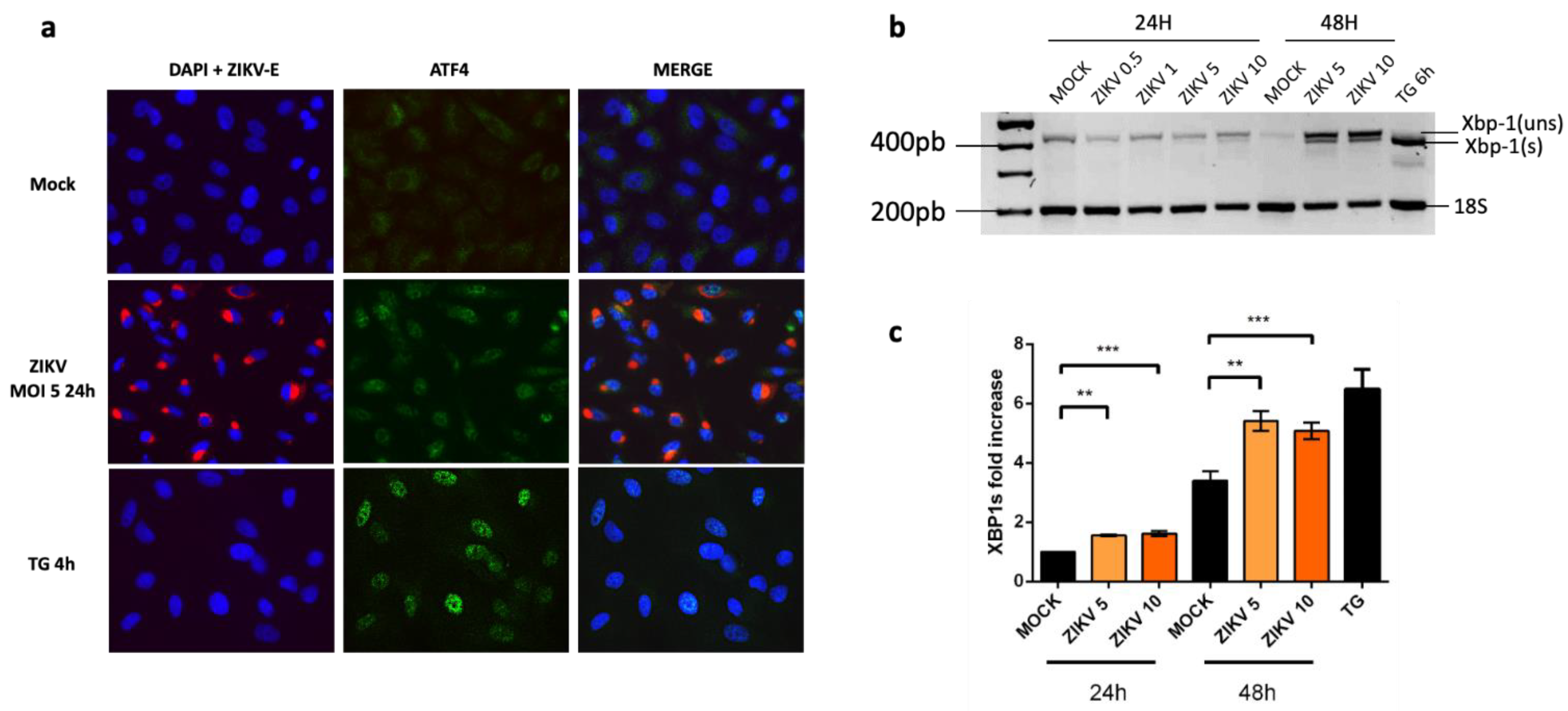
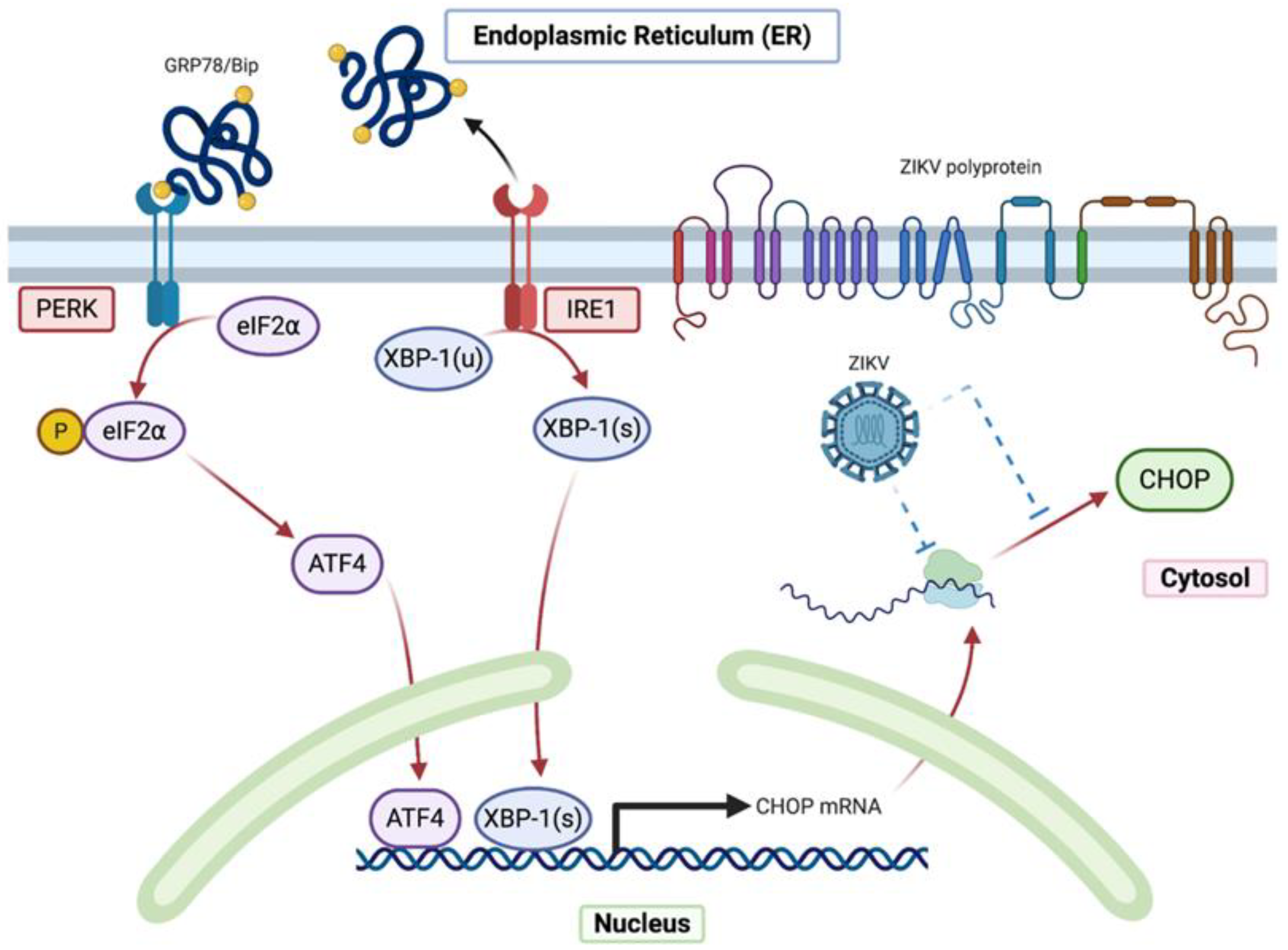
2.1. Despite an Incomplete UPR, the Factors That Govern the Transcriptional Activation of CHOP Are Present in ZIKV-Infected Cells
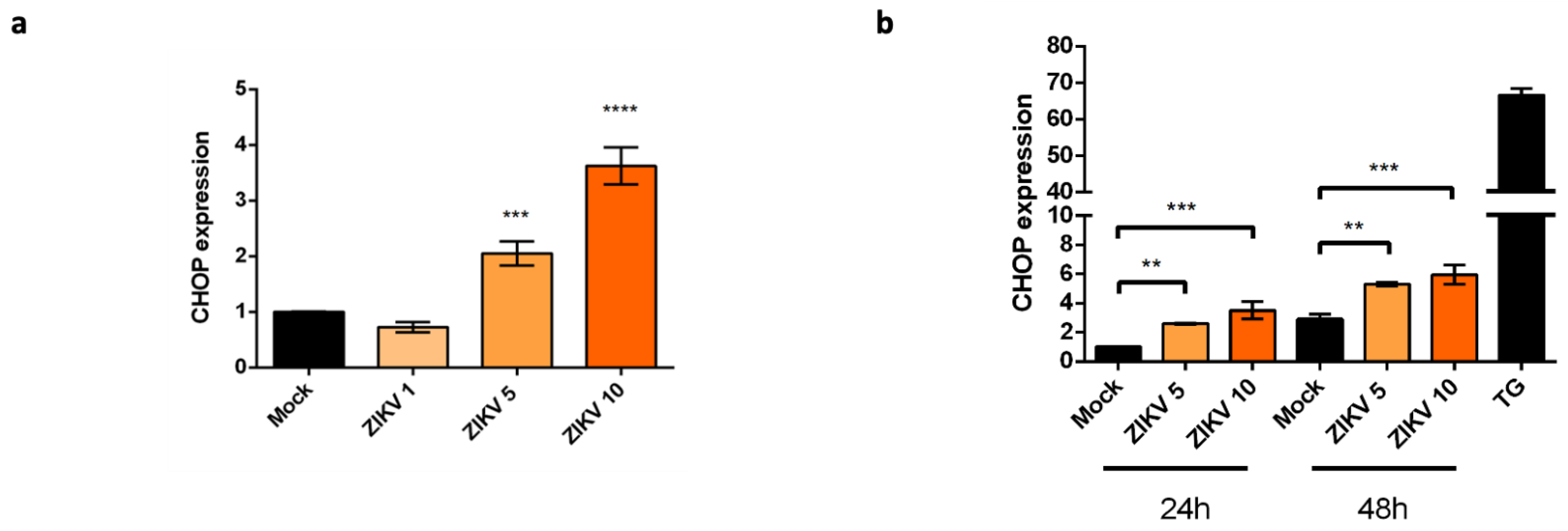
2.2. Upregulation of CHOP Transcriptional Activity in A549 Cells Infected by ZIKV
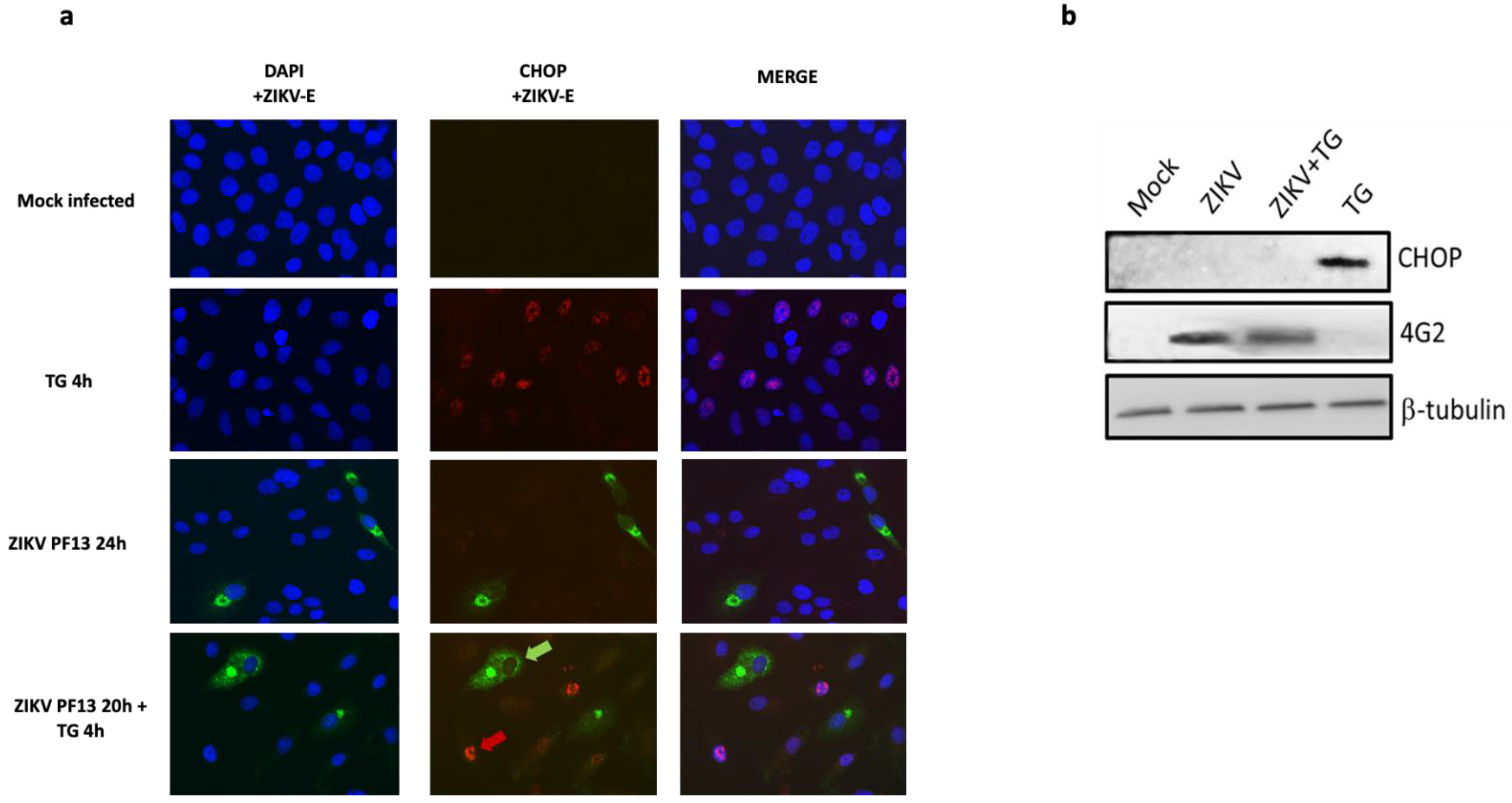
2.3. ZIKV Inhibits CHOP Protein Expression
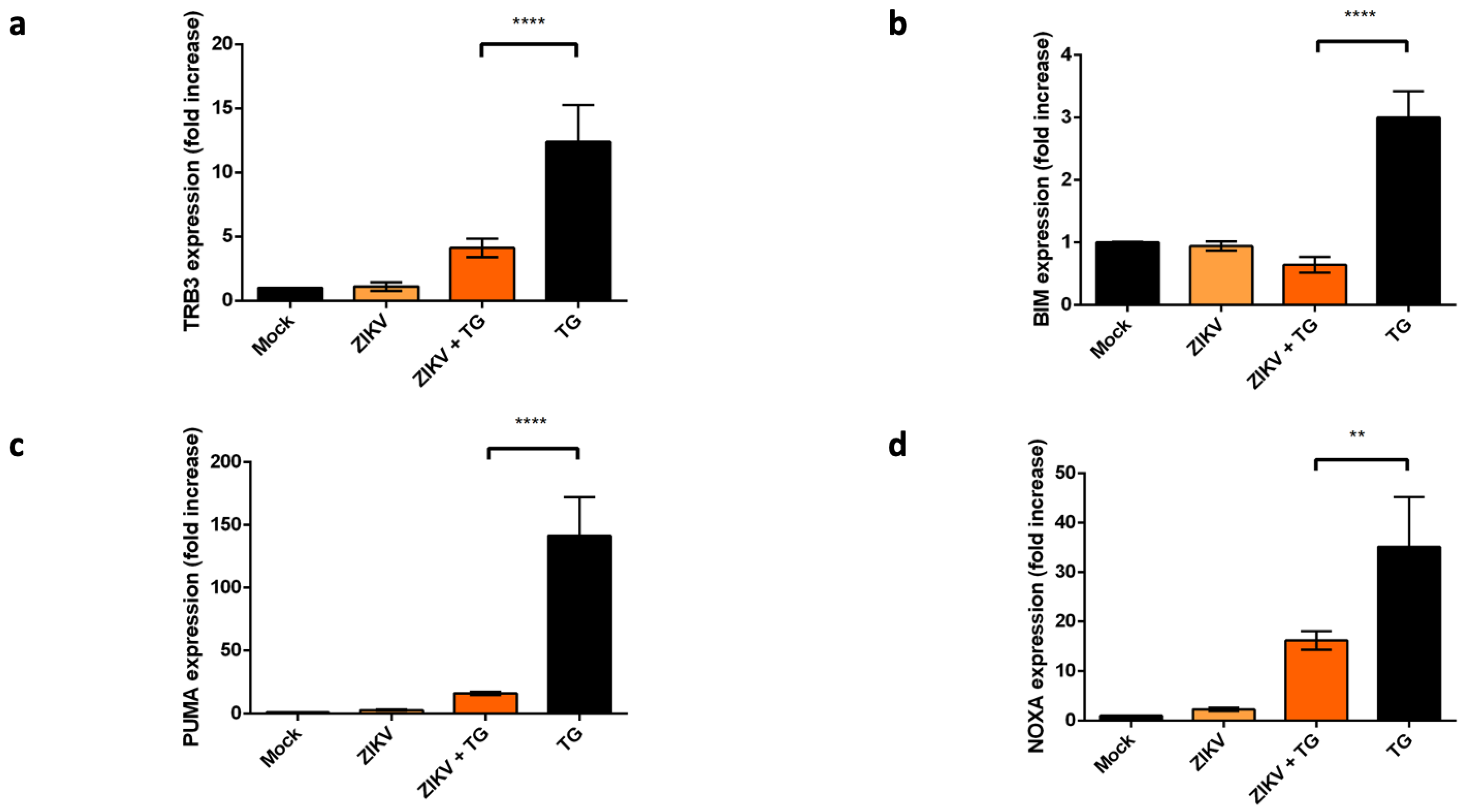
2.4. ZIKV Controls the Transcriptional Activity of CHOP-Dependent Genes
3. Discussion
4. Materials and Methods
4.1. Virus, Cell Culture, Antibodies and Reagents
4.2. RNA Extraction and qRT-PCR
4.3. Immunofluorescence Assay
4.4. Cell Extracts Preparation and Western Blotting Optimization
4.5. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Faria, N.R.; da Silva Azevedo, R.D.; Kraemer, M.U.G.; Souza, R.; Cunha, M.S.; Hill, S.C.; Thézé, J.; Bonsall, M.B.; Bowden, T.A.; Rissanen, I.; et al. Zika Virus in the Americas: Early Epidemiological and Genetic Findings. Science 2016, 352, 345–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakkas, H.; Bozidis, P.; Giannakopoulos, X.; Sofikitis, N.; Papadopoulou, C. An Update on Sexual Transmission of Zika Virus. Pathogens 2018, 7, 66. [Google Scholar] [CrossRef] [Green Version]
- Sager, G.; Gabaglio, S.; Sztul, E.; Belov, G.A. Role of Host Cell Secretory Machinery in Zika Virus Life Cycle. Viruses 2018, 10, 559. [Google Scholar] [CrossRef] [Green Version]
- Ropidi, M.I.M.; Khazali, A.S.; Rashid, N.N.; Yusof, R. Endoplasmic reticulum: A focal point of Zika virus infection. J. Biomed. Sci. 2020, 27, 27. [Google Scholar] [CrossRef] [Green Version]
- Lin, D.L.; Inoue, T.; Chen, Y.-J.; Chang, A.; Tsai, B.; Tai, A.W. The ER Membrane Protein Complex Promotes Biogenesis of Dengue and Zika Virus Non-Structural Multi-Pass Transmembrane Proteins to Support Infection. Cell Rep. 2019, 27, 1666–1674. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, D.S.; Blower, M.D. The Endoplasmic Reticulum: Structure, Function and Response to Cellular Signaling. Cell Mol. Life Sci. 2016, 73, 79–94. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Bailly-Maitre, B.; Reed, J.C. Endoplasmic Reticulum Stress: Cell Life and Death Decisions. J. Clin. Investig. 2005, 115, 2656–2664. [Google Scholar] [CrossRef] [Green Version]
- Schröder, M.; Kaufman, R.J. ER Stress and the Unfolded Protein Response. Mutat. Res. 2005, 569, 29–63. [Google Scholar] [CrossRef]
- Corazzari, M.; Gagliardi, M.; Fimia, G.M.; Piacentini, M. Endoplasmic Reticulum Stress, Unfolded Protein Response, and Cancer Cell Fate. Front. Oncol. 2017, 7. [Google Scholar] [CrossRef] [Green Version]
- Zhu, G.; Lee, A.S. Role of the Unfolded Protein Response, GRP78 and GRP94 in Organ Homeostasis: UPR and GRPs regulate organ homeostasis. J. Cell. Physiol. 2015, 230, 1413–1420. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.H.; Ploegh, H.L.; Weissman, J.S. Road to Ruin: Targeting Proteins for Degradation in the Endoplasmic Reticulum. Science 2011, 334, 1086–1090. [Google Scholar] [CrossRef] [Green Version]
- Bergmann, T.J.; Molinari, M. Three Branches to Rule Them All? UPR Signalling in Response to Chemically versus Misfolded Proteins-Induced ER Stress. Biol. Cell 2018, 110, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Iurlaro, R.; Muñoz-Pinedo, C. Cell Death Induced by Endoplasmic Reticulum Stress. FEBS J. 2016, 283, 2640–2652. [Google Scholar] [CrossRef] [Green Version]
- Marciniak, S.J.; Yun, C.Y.; Oyadomari, S.; Novoa, I.; Zhang, Y.; Jungreis, R.; Nagata, K.; Harding, H.P.; Ron, D. CHOP Induces Death by Promoting Protein Synthesis and Oxidation in the Stressed Endoplasmic Reticulum. Genes Dev. 2004, 18, 3066–3077. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Tu, H.-C.; Ren, D.; Takeuchi, O.; Jeffers, J.R.; Zambetti, G.P.; Hsieh, J.J.-D.; Cheng, E.H.-Y. Stepwise Activation of BAX and BAK by TBID, BIM, and PUMA Initiates Mitochondrial Apoptosis. Mol. Cell 2009, 36, 487–499. [Google Scholar] [CrossRef] [Green Version]
- Ohoka, N.; Yoshii, S.; Hattori, T.; Onozaki, K.; Hayashi, H. TRB3, a Novel ER Stress-Inducible Gene, Is Induced via ATF4–CHOP Pathway and Is Involved in Cell Death. EMBO J. 2005, 24, 1243–1255. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Lee, B.; Lee, A.S. Endoplasmic Reticulum Stress-Induced Apoptosis: Multiple Pathways and Activation of P53-up-Regulated Modulator of Apoptosis (PUMA) and NOXA by P53. J. Biol. Chem. 2006, 281, 7260–7270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, H.; Tian, M.; Ding, C.; Yu, S. The C/EBP Homologous Protein (CHOP) Transcription Factor Functions in Endoplasmic Reticulum Stress-Induced Apoptosis and Microbial Infection. Front. Immunol. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, J.; Kaufman, R.J. Physiological/Pathological Ramifications of Transcription Factors in the Unfolded Protein Response. Genes Dev. 2017, 31, 1417–1438. [Google Scholar] [CrossRef] [Green Version]
- DuRose, J.B.; Scheuner, D.; Kaufman, R.J.; Rothblum, L.I.; Niwa, M. Phosphorylation of Eukaryotic Translation Initia-Tion Factor 2α Coordinates RRNA Transcription and Translation Inhibition during Endoplasmic Reticulum Stress. Mol. Cell. Biol. 2009, 29, 4295–4307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blázquez, A.-B.; Escribano-Romero, E.; Merino-Ramos, T.; Saiz, J.-C.; Martín-Acebes, M.A. Stress Responses in Fla-Vivirus-Infected Cells: Activation of Unfolded Protein Response and Autophagy. Front. Microbiol. 2014, 5. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, A. Virus-induced ER stress and the unfolded protein response. Front. Plant Sci. 2012, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gladwyn-Ng, I.; Cordón-Barris, L.; Alfano, C.; Creppe, C.; Couderc, T.; Morelli, G.; Thelen, N.; America, M.; Bessières, B.; Encha-Razavi, F.; et al. Stress-Induced Unfolded Protein Response Contributes to Zika Virus–Associated Microcephaly. Nat. Neurosci. 2018, 21, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Tan, Z.; Zhang, W.; Sun, J.; Fu, Z.; Ke, X.; Zheng, C.; Zhang, Y.; Li, P.; Liu, Y.; Hu, Q.; et al. ZIKV Infection Activates the IRE1-XBP1 and ATF6 Pathways of Unfolded Protein Response in Neural Cells. J. Neuroinflamm. 2018, 15, 275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turpin, J.; Frumence, E.; Harrabi, W.; Haddad, J.G.; El Kalamouni, C.; Desprès, P.; Krejbich-Trotot, P.; Viranaïcken, W. Zika Virus Subversion of Chaperone GRP78/BiP Expression in A549 Cells during UPR Activation. Biochimie 2020, 175, 99–105. [Google Scholar] [CrossRef]
- Chan, S.-W.; Egan, P.A. Hepatitis C Virus Envelope Proteins Regulate CHOP via Induction of the Unfolded Protein Response. FASEB J. 2005, 19, 1510–1512. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.-Y.; Hsu, Y.-W.; Liao, C.-L.; Lin, Y.-L. Flavivirus Infection Activates the XBP1 Pathway of the Unfolded Protein Response To Cope with Endoplasmic Reticulum Stress. J. Virol. 2006, 80, 11868–11880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambrose, R.L.; Mackenzie, J.M. Flaviviral Regulation of the Unfolded Protein Response: Can Stress Be Beneficial? Future Virol. 2013, 8, 1095–1109. [Google Scholar] [CrossRef]
- Benbrook, D.M.; Long, A. Integration of Autophagy, Proteasomal Degradation, Unfolded Protein Response and Apoptosis. Exp. Oncol. 2012, 34, 286–297. [Google Scholar]
- Turpin, J.; Frumence, E.; Desprès, P.; Viranaicken, W.; Krejbich-Trotot, P. The ZIKA Virus Delays Cell Death through the Anti-Apoptotic Bcl-2 Family Proteins. Cells 2019, 8, 1338. [Google Scholar] [CrossRef] [Green Version]
- Gardner, B.M.; Pincus, D.; Gotthardt, K.; Gallagher, C.M.; Walter, P. Endoplasmic Reticulum Stress Sensing in the Un-Folded Protein Response. Cold Spring Harb. Perspect. Biol. 2013, 5, 013169. [Google Scholar] [CrossRef] [Green Version]
- Rozpędek, W.; Pytel, D.; Mucha, B.; Leszczyńska, H.; Diehl, J.A.; Majsterek, I. The Role of the PERK/EIF2α/ATF4/CHOP Signaling Pathway in Tumor Progression during Endoplasmic Reticulum Stress. Curr. Mol. Med. 2016, 16, 533–544. [Google Scholar] [CrossRef]
- Han, J.; Back, S.H.; Hur, J.; Lin, Y.-H.; Gildersleeve, R.; Shan, J.; Yuan, C.L.; Krokowski, D.; Wang, S.; Hatzoglou, M.; et al. ER-Stress-Induced Transcriptional Regulation Increases Protein Synthesis Leading to Cell Death. Nat. Cell Biol. 2013, 15, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Tirasophon, W.; Shen, X.; Michalak, M.; Prywes, R.; Okada, T.; Yoshida, H.; Mori, K.; Kaufman, R.J. IRE1-Mediated Unconventional MRNA Splicing and S2P-Mediated ATF6 Cleavage Merge to Regulate XBP1 in Signaling the Unfolded Protein Response. Genes Dev. 2002, 16, 452–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Xing, P.; Cui, W.; Wang, W.; Cui, Y.; Ying, G.; Wang, X.; Li, B. Acute Endoplasmic Reticulum Stress-Independent Unconventional Splicing of XBP1 MRNA in the Nucleus of Mammalian Cells. Int. J. Mol. Sci. 2015, 16, 13302–13321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urano, F.; Bertolotti, A.; Ron, D. IRE1 and efferent signaling from the endoplasmic reticulum. J. Cell Sci. 2000, 113 Pt 21, 3697–3702. [Google Scholar]
- Turpin, J.; Frumence, E.; Safadi, D.E.; Meilhac, O.; Krejbich-Trotot, P.; Viranaïcken, W. Improvement of Immunodetection of the Transcription Factor C/EBP Homologous Protein by Western Blot. Anal. Biochem. 2020, 601, 113775. [Google Scholar] [CrossRef] [PubMed]
- Anelli, T.; Sitia, R. Protein Quality Control in the Early Secretory Pathway. EMBO J. 2008, 27, 315–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wärri, A.; Cook, K.L.; Hu, R.; Jin, L.; Zwart, A.; Soto-Pantoja, D.R.; Liu, J.; Finkel, T.; Clarke, R. Autophagy and Unfolded Protein Response (UPR) Regulate Mammary Gland Involution by Restraining Apoptosis-Driven Irreversible Changes. Cell Death Discov. 2018, 4, 40. [Google Scholar] [CrossRef]
- Chen, Y.; Gui, D.; Chen, J.; He, D.; Luo, Y.; Wang, N. Down-Regulation of PERK-ATF4-CHOP Pathway by Astragaloside IV Is Associated with the Inhibition of Endoplasmic Reticulum Stress-Induced Podocyte Apoptosis in Diabetic Rats. Cell Physiol. Biochem. 2014, 33, 1975–1987. [Google Scholar] [CrossRef]
- Mackenzie, J.M.; Westaway, E.G. Assembly and Maturation of the Flavivirus Kunjin Virus Appear to Occur in the Rough Endoplasmic Reticulum and along the Secretory Pathway, Respectively. J. Virol. 2001, 75, 10787–10799. [Google Scholar] [CrossRef] [Green Version]
- Zhao, D.; Yang, J.; Han, K.; Liu, Q.; Wang, H.; Liu, Y.; Huang, X.; Zhang, L.; Li, Y. The Unfolded Protein Response Induced by Tembusu Virus Infection. BMC Vet. Res. 2019, 15, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medigeshi, G.R.; Lancaster, A.M.; Hirsch, A.J.; Briese, T.; Lipkin, W.I.; DeFilippis, V.; Früh, K.; Mason, P.W.; Nikolich-Zugich, J.; Nelson, J.A. West Nile Virus Infection Activates the Unfolded Protein Response, Leading to CHOP Induction and Apoptosis. J. Virol. 2007, 81, 10849–10860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambrose, R.L.; Mackenzie, J.M. West Nile Virus Differentially Modulates the Unfolded Protein Response To Facilitate Replication and Immune Evasion. J. Virol. 2011, 85, 2723–2732. [Google Scholar] [CrossRef] [Green Version]
- Peña, J.; Harris, E. Dengue Virus Modulates the Unfolded Protein Response in a Time-dependent Manner. J. Biol. Chem. 2011, 286, 14226–14236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciccaglione, A.R.; Costantino, A.; Tritarelli, E.; Marcantonio, C.; Equestre, M.; Marziliano, N.; Rapicetta, M. Activation of Endoplasmic Reticulum Stress Response by Hepatitis C Virus Proteins. Arch. Virol. 2005, 150, 1339–1356. [Google Scholar] [CrossRef]
- Su, H.-L.; Liao, C.-L.; Lin, Y.-L. Japanese Encephalitis Virus Infection Initiates Endoplasmic Reticulum Stress and an Un-Folded Protein Response. J. Virol. 2002, 76, 4162–4171. [Google Scholar] [CrossRef] [Green Version]
- Bhuvanakantham, R.; Chong, M.-K.; Ng, M.-L. Specific Interaction of Capsid Protein and Importin-α/β Influences West Nile Virus Production. Biochem. Biophys. Res. Commun. 2009, 389, 63–69. [Google Scholar] [CrossRef]
- Goh, G.; Dunker, A.; Foster, J.; Uversky, V. Zika and Flavivirus Shell Disorder: Virulence and Fetal Morbidity. Biomolecules 2019, 9, 710. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Gu, Y.; Qi, B.; Zhang, Y.; Li, X.; Fang, W. Porcine Circovirus Type 2 Capsid Protein Induces Unfolded Protein Response with Subsequent Activation of Apoptosis. J. Zhejiang Univ. Sci. B 2017, 18, 316–323. [Google Scholar] [CrossRef] [Green Version]
- Airo, A.M.; Urbanowski, M.D.; Lopez-Orozco, J.; You, J.H.; Skene-Arnold, T.D.; Holmes, C.; Yamshchikov, V.; Malik-Soni, N.; Frappier, L.; Hobman, T.C. Expression of Flavivirus Capsids Enhance the Cellular Environment for Viral Replication by Activating Akt-Signalling Pathways. Virology 2018, 516, 147–157. [Google Scholar] [CrossRef]
- Fros, J.J.; Major, L.D.; Scholte, F.E.M.; Gardner, J.; van Hemert, M.J.; Suhrbier, A.; Pijlman, G.P. Chikungunya Virus Non-Structural Protein 2-Mediated Host Shut-off Disables the Unfolded Protein Response. J. Gen. Virol. 2015, 96, 580–589. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Moon, A.; Childs, K.; Goodbourn, S.; Dixon, L.K. The African Swine Fever Virus DP71L Protein Recruits the Protein Phosphatase 1 Catalytic Subunit to Dephosphorylate EIF2alpha and Inhibits CHOP Induction but Is Dispensable for These Activities during Virus Infection. J. Virol. 2010, 84, 10681–10689. [Google Scholar] [CrossRef] [Green Version]
- Hinte, F.; Müller, J.; Brune, W. Viral Mediated Tethering to SEL1L Facilitates ER-Associated Degradation of IRE1. J. Virol. 2021. [Google Scholar] [CrossRef]
- Khongwichit, S.; Sornjai, W.; Jitobaom, K.; Greenwood, M.; Greenwood, M.P.; Hitakarun, A.; Wikan, N.; Murphy, D.; Smith, D.R. A Functional Interaction between GRP78 and Zika Virus E Protein. Sci. Rep. 2021, 11, 393. [Google Scholar] [CrossRef]
- Liao, Y.; Fung, T.S.; Huang, M.; Fang, S.G.; Zhong, Y.; Liu, D.X. Upregulation of CHOP/GADD153 during Coronavirus Infectious Bronchitis Virus Infection Modulates Apoptosis by Restricting Activation of the Extracellular Signal-Regulated Kinase Pathway. J. Virol. 2013, 87, 8124–8134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao-Lormeau, V.-M.; Roche, C.; Teissier, A.; Robin, E.; Berry, A.-L.; Mallet, H.-P.; Sall, A.A.; Musso, D. Zika Virus, French Polynesia, South Pacific, 2013. Emerg. Infect. Dis. 2014, 20, 1084–1086. [Google Scholar] [CrossRef] [PubMed]
- Gadea, G.; Bos, S.; Krejbich-Trotot, P.; Clain, E.; Viranaicken, W.; El-Kalamouni, C.; Mavingui, P.; Desprès, P. A Robust Method for the Rapid Generation of Recombinant Zika Virus Expressing the GFP Reporter Gene. Virology 2016, 497, 157–162. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Turpin, J.; El-Safadi, D.; Lebeau, G.; Frumence, E.; Desprès, P.; Viranaïcken, W.; Krejbich-Trotot, P. CHOP Pro-Apoptotic Transcriptional Program in Response to ER Stress Is Hacked by Zika Virus. Int. J. Mol. Sci. 2021, 22, 3750. https://doi.org/10.3390/ijms22073750
Turpin J, El-Safadi D, Lebeau G, Frumence E, Desprès P, Viranaïcken W, Krejbich-Trotot P. CHOP Pro-Apoptotic Transcriptional Program in Response to ER Stress Is Hacked by Zika Virus. International Journal of Molecular Sciences. 2021; 22(7):3750. https://doi.org/10.3390/ijms22073750
Chicago/Turabian StyleTurpin, Jonathan, Daed El-Safadi, Grégorie Lebeau, Etienne Frumence, Philippe Desprès, Wildriss Viranaïcken, and Pascale Krejbich-Trotot. 2021. "CHOP Pro-Apoptotic Transcriptional Program in Response to ER Stress Is Hacked by Zika Virus" International Journal of Molecular Sciences 22, no. 7: 3750. https://doi.org/10.3390/ijms22073750