
p-Coumaric Acid Enhances Hypothalamic Leptin Signaling and Glucose Homeostasis in Mice via Differential Effects on AMPK Activation

 ,
,  , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
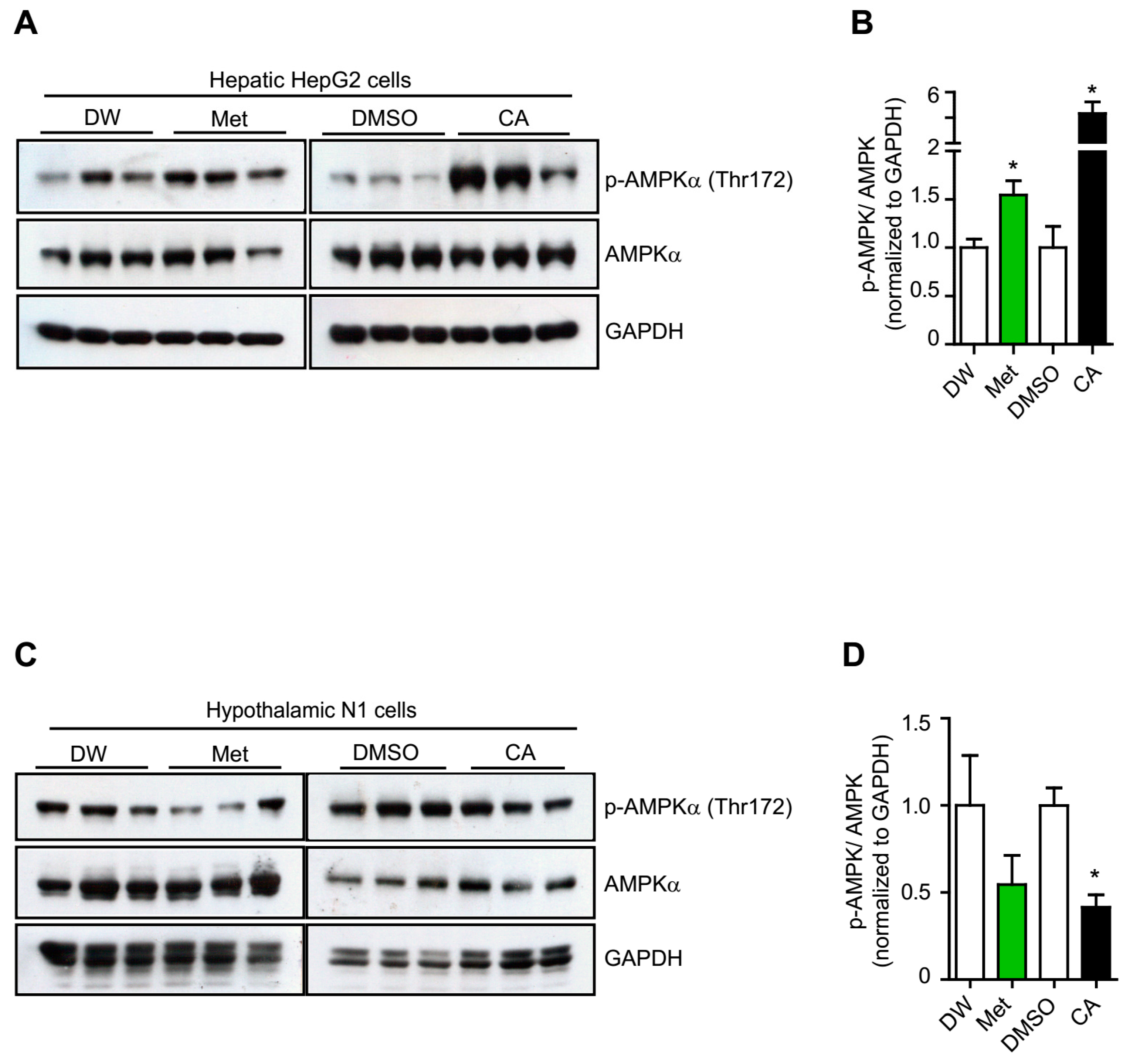
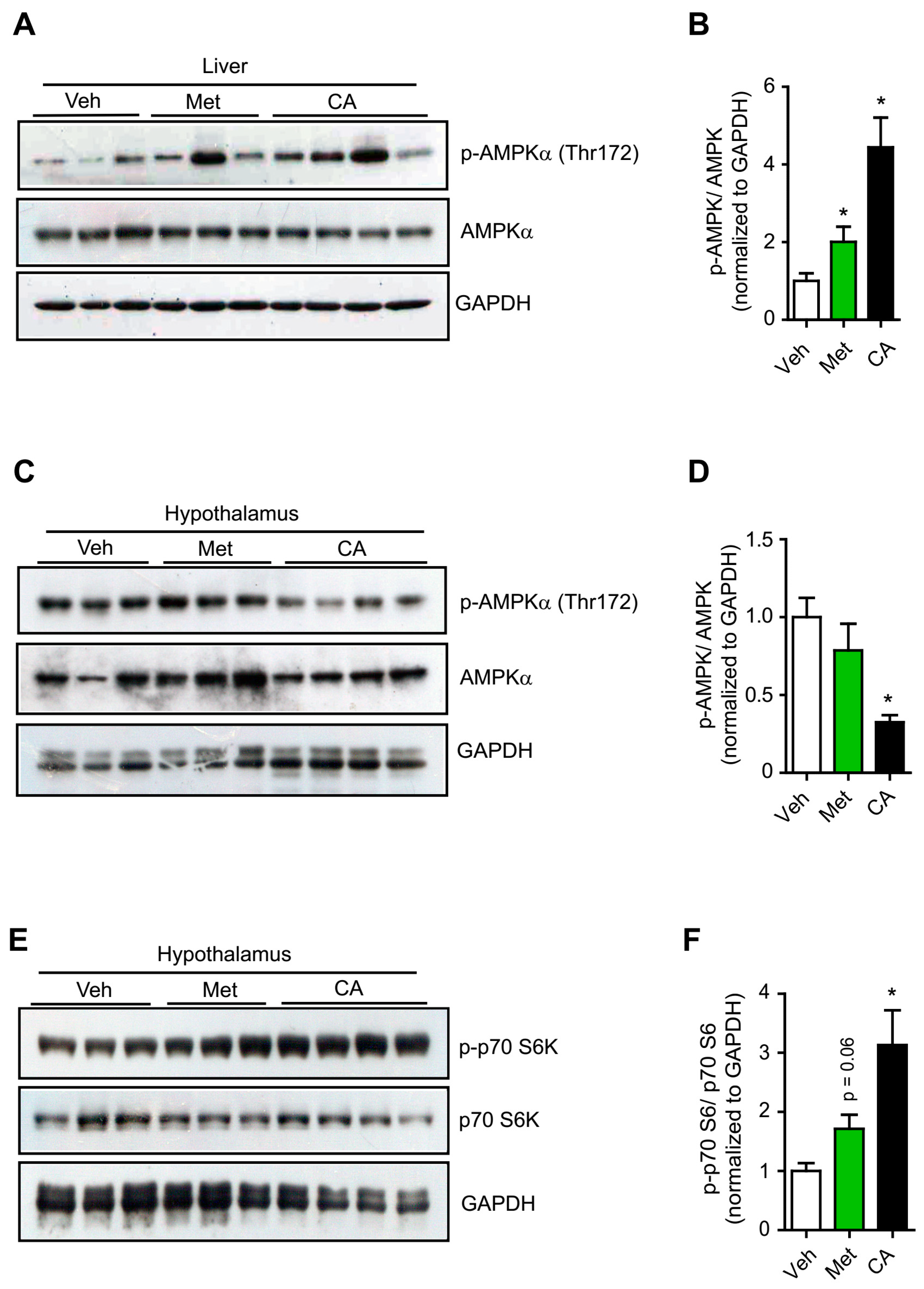
2.1. p-Coumaric Acid Exhibited Differential Effects on Peripheral and Central AMPK Activation
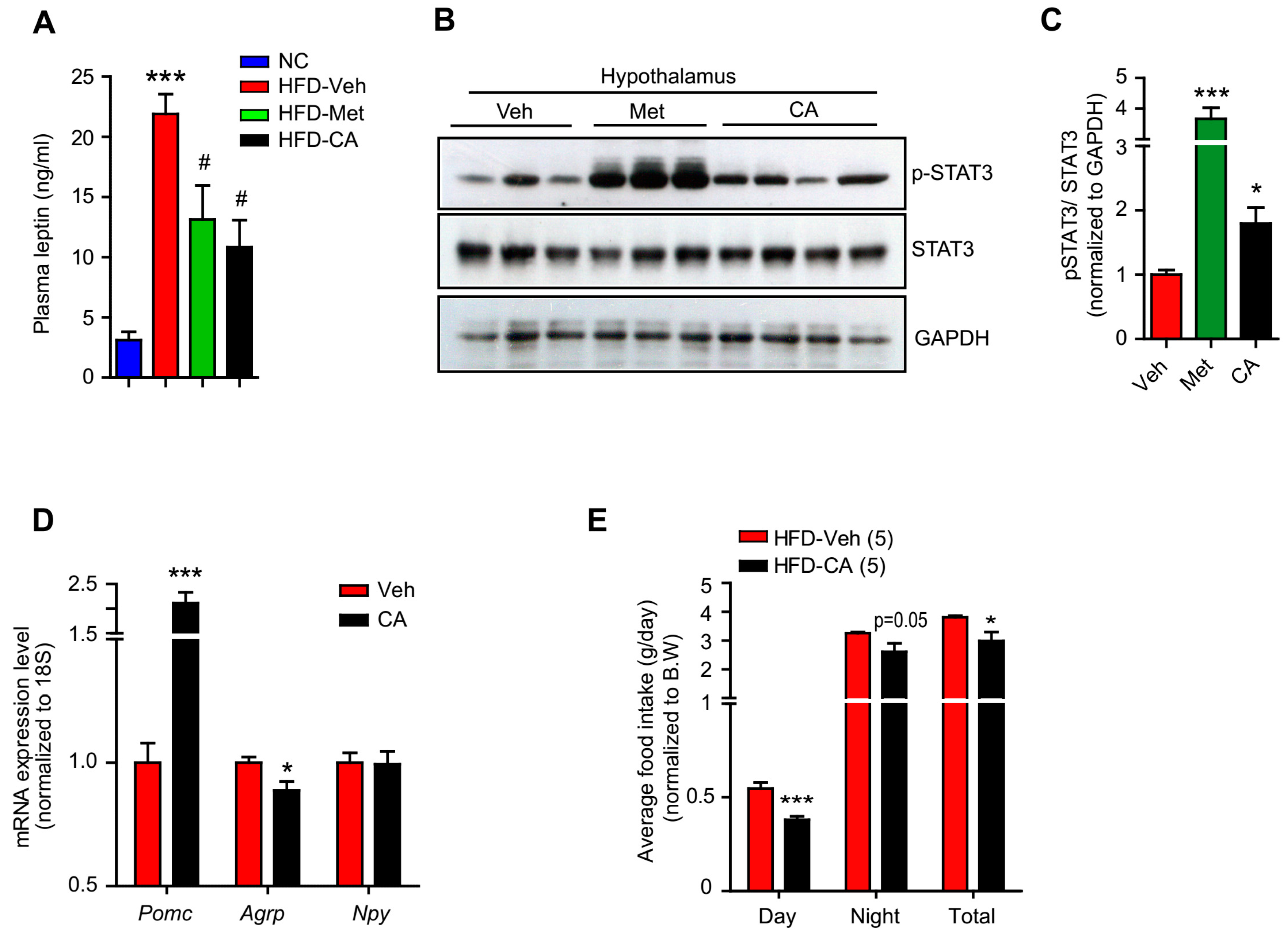
2.2. p-Coumaric Acid Treatment Enhanced Hypothalamic Leptin Signaling
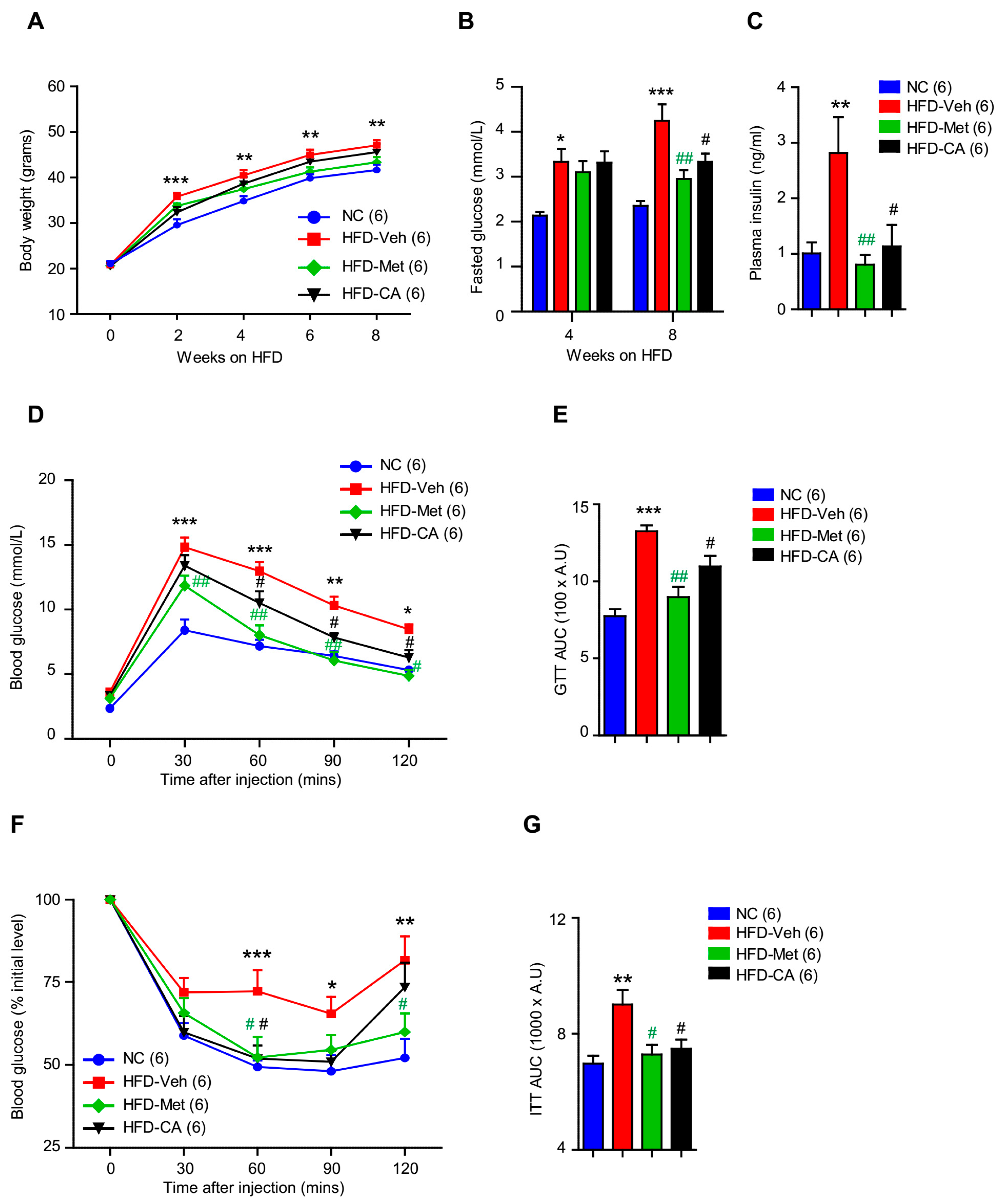
2.3. p-Coumaric Acid Treatment Improved Whole-Body Glucose Homeostasis
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture and Chemical Treatment
4.3. Animals
4.4. Dissecting and Harvesting Hypothalamus Tissues
4.5. Food Intake Measurement
4.6. Insulin and Leptin Measurement
4.7. GTT and ITT Experiments
4.8. SDS-PAGE and Western Blotting
4.9. RNA Isolation and Real-Time qPCR
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Garcia, D.; Shaw, R.J. AMPK: Mechanisms of Cellular Energy Sensing and Restoration of Metabolic Balance. Mol. Cell 2017, 66, 789–800. [Google Scholar] [CrossRef] [Green Version]
- O’Neill, H.M.; Holloway, G.P.; Steinberg, G.R. AMPK regulation of fatty acid metabolism and mitochondrial biogenesis: Implications for obesity. Mol. Cell. Endocrinol. 2013, 366, 135–151. [Google Scholar] [CrossRef]
- Minokoshi, Y.; Alquier, T.; Furukawa, N.; Kim, Y.B.; Lee, A.; Xue, B.; Mu, J.; Foufelle, F.; Ferre, P.; Birnbaum, M.J.; et al. AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature 2004, 428, 569–574. [Google Scholar] [CrossRef]
- Namkoong, C.; Kim, M.S.; Jang, P.G.; Han, S.M.; Park, H.S.; Koh, E.H.; Lee, W.J.; Kim, J.Y.; Park, I.S.; Park, J.Y.; et al. Enhanced hypothalamic AMP-activated protein kinase activity contributes to hyperphagia in diabetic rats. Diabetes 2005, 54, 63–68. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.S.; Lam, C.K.; Chari, M.; Cheung, G.W.; Kokorovic, A.; Gao, S.; Leclerc, I.; Rutter, G.A.; Lam, T.K. Hypothalamic AMP-activated protein kinase regulates glucose production. Diabetes 2010, 59, 2435–2443. [Google Scholar] [CrossRef] [Green Version]
- Dagon, Y.; Hur, E.; Zheng, B.; Wellenstein, K.; Cantley, L.C.; Kahn, B.B. p70S6 kinase phosphorylates AMPK on serine 491 to mediate leptin’s effect on food intake. Cell Metab. 2012, 16, 104–112. [Google Scholar] [CrossRef] [Green Version]
- Ruderman, N.B.; Carling, D.; Prentki, M.; Cacicedo, J.M. AMPK, insulin resistance, and the metabolic syndrome. J. Clin. Investig. 2013, 123, 2764–2772. [Google Scholar] [CrossRef]
- Lopez, M.; Nogueiras, R.; Tena-Sempere, M.; Dieguez, C. Hypothalamic AMPK: A canonical regulator of whole-body energy balance. Nat. Rev. Endocrinol. 2016, 12, 421–432. [Google Scholar] [CrossRef]
- Steinberg, G.R.; Carling, D. AMP-activated protein kinase: The current landscape for drug development. Nat. Rev. Drug Discov. 2019, 18, 527–551. [Google Scholar] [CrossRef]
- Grahame Hardie, D. Regulation of AMP-activated protein kinase by natural and synthetic activators. Acta Pharm. Sin. B 2016, 6, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Tungmunnithum, D.; Thongboonyou, A.; Pholboon, A.; Yangsabai, A. Flavonoids and Other Phenolic Compounds from Medicinal Plants for Pharmaceutical and Medical Aspects: An Overview. Medicines 2018, 5, 93. [Google Scholar] [CrossRef]
- Wang, S.; Moustaid-Moussa, N.; Chen, L.; Mo, H.; Shastri, A.; Su, R.; Bapat, P.; Kwun, I.; Shen, C.L. Novel insights of dietary polyphenols and obesity. J. Nutr. Biochem. 2014, 25, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Pei, K.; Ou, J.; Huang, J.; Ou, S. p-Coumaric acid and its conjugates: Dietary sources, pharmacokinetic properties and biological activities. J. Sci. Food Agric. 2016, 96, 2952–2962. [Google Scholar] [CrossRef]
- Abdel-Moneim, A.; El-Twab, S.M.A.; Yousef, A.I.; Reheim, E.S.A.; Ashour, M.B. Modulation of hyperglycemia and dyslipidemia in experimental type 2 diabetes by gallic acid and p-coumaric acid: The role of adipocytokines and PPARgamma. Biomed. Pharmacother. 2018, 105, 1091–1097. [Google Scholar] [CrossRef]
- Shen, Y.; Song, X.; Li, L.; Sun, J.; Jaiswal, Y.; Huang, J.; Liu, C.; Yang, W.; Williams, L.; Zhang, H.; et al. Protective effects of p-coumaric acid against oxidant and hyperlipidemia-an in vitro and in vivo evaluation. Biomed. Pharmacother. 2019, 111, 579–587. [Google Scholar] [CrossRef]
- Yoon, S.A.; Kang, S.I.; Shin, H.S.; Kang, S.W.; Kim, J.H.; Ko, H.C.; Kim, S.J. p-Coumaric acid modulates glucose and lipid metabolism via AMP-activated protein kinase in L6 skeletal muscle cells. Biochem. Biophys. Res. Commun. 2013, 432, 553–557. [Google Scholar] [CrossRef]
- Kang, S.W.; Kang, S.I.; Shin, H.S.; Yoon, S.A.; Kim, J.H.; Ko, H.C.; Kim, S.J. Sasa quelpaertensis Nakai extract and its constituent p-coumaric acid inhibit adipogenesis in 3T3-L1 cells through activation of the AMPK pathway. Food Chem. Toxicol. 2013, 59, 380–385. [Google Scholar] [CrossRef]
- Kim, J.H.; Kang, S.I.; Shin, H.S.; Yoon, S.A.; Kang, S.W.; Ko, H.C.; Kim, S.J. Sasa quelpaertensis and p-coumaric acid attenuate oleic acid-induced lipid accumulation in HepG2 cells. Biosci. Biotechnol. Biochem. 2013, 77, 1595–1598. [Google Scholar] [CrossRef] [Green Version]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Investig. 2001, 108, 1167–1174. [Google Scholar] [CrossRef]
- Shaw, R.J.; Lamia, K.A.; Vasquez, D.; Koo, S.H.; Bardeesy, N.; Depinho, R.A.; Montminy, M.; Cantley, L.C. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science 2005, 310, 1642–1646. [Google Scholar] [CrossRef] [Green Version]
- Sefried, S.; Haring, H.U.; Weigert, C.; Eckstein, S.S. Suitability of hepatocyte cell lines HepG2, AML12 and THLE-2 for investigation of insulin signalling and hepatokine gene expression. Open Biol. 2018, 8, 180147. [Google Scholar] [CrossRef] [Green Version]
- Chau-Van, C.; Gamba, M.; Salvi, R.; Gaillard, R.C.; Pralong, F.P. Metformin inhibits adenosine 5’-monophosphate-activated kinase activation and prevents increases in neuropeptide Y expression in cultured hypothalamic neurons. Endocrinology 2007, 148, 507–511. [Google Scholar] [CrossRef]
- Stevanovic, D.; Janjetovic, K.; Misirkic, M.; Vucicevic, L.; Sumarac-Dumanovic, M.; Micic, D.; Starcevic, V.; Trajkovic, V. Intracerebroventricular administration of metformin inhibits ghrelin-induced Hypothalamic AMP-kinase signalling and food intake. Neuroendocrinology 2012, 96, 24–31. [Google Scholar] [CrossRef]
- Dalvi, P.S.; Nazarians-Armavil, A.; Tung, S.; Belsham, D.D. Immortalized neurons for the study of hypothalamic function. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 300, R1030–R1052. [Google Scholar] [CrossRef] [Green Version]
- Cai, F.; Gyulkhandanyan, A.V.; Wheeler, M.B.; Belsham, D.D. Glucose regulates AMP-activated protein kinase activity and gene expression in clonal, hypothalamic neurons expressing proopiomelanocortin: Additive effects of leptin or insulin. J. Endocrinol. 2007, 192, 605–614. [Google Scholar] [CrossRef] [Green Version]
- Konishi, Y.; Hitomi, Y.; Yoshioka, E. Intestinal absorption of p-coumaric and gallic acids in rats after oral administration. J. Agric. Food Chem. 2004, 52, 2527–2532. [Google Scholar] [CrossRef]
- Meng, Z.; Wang, W.; Xing, D.M.; Lei, F.; Lan, J.Q.; Du, L.J. Pharmacokinetic study of p-coumaric acid in mouse after oral administration of extract of Ananas comosus L. leaves. Biomed. Chromatogr. BMC 2006, 20, 951–955. [Google Scholar] [CrossRef]
- Gasperotti, M.; Passamonti, S.; Tramer, F.; Masuero, D.; Guella, G.; Mattivi, F.; Vrhovsek, U. Fate of microbial metabolites of dietary polyphenols in rats: Is the brain their target destination? ACS Chem. Neurosci. 2015, 6, 1341–1352. [Google Scholar] [CrossRef] [Green Version]
- Howell, J.J.; Hellberg, K.; Turner, M.; Talbott, G.; Kolar, M.J.; Ross, D.S.; Hoxhaj, G.; Saghatelian, A.; Shaw, R.J.; Manning, B.D. Metformin Inhibits Hepatic mTORC1 Signaling via Dose-Dependent Mechanisms Involving AMPK and the TSC Complex. Cell Metab. 2017, 25, 463–471. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Park, E.Y.; Oh, M.J.; Park, S.S.; Shin, K.H.; Choi, S.H.; Chun, B.G.; Kim, D.H. Central administration of metformin into the third ventricle of C57BL/6 mice decreases meal size and number and activates hypothalamic S6 kinase. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 305, R499–R505. [Google Scholar] [CrossRef]
- Horman, S.; Vertommen, D.; Heath, R.; Neumann, D.; Mouton, V.; Woods, A.; Schlattner, U.; Wallimann, T.; Carling, D.; Hue, L.; et al. Insulin antagonizes ischemia-induced Thr172 phosphorylation of AMP-activated protein kinase alpha-subunits in heart via hierarchical phosphorylation of Ser485/491. J. Biol. Chem. 2006, 281, 5335–5340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.W.; Kim, J.Y.; Park, Y.H.; Park, S.Y.; Won, K.C.; Choi, K.H.; Huh, J.Y.; Moon, K.H. Metformin restores leptin sensitivity in high-fat-fed obese rats with leptin resistance. Diabetes 2006, 55, 716–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aubert, G.; Mansuy, V.; Voirol, M.J.; Pellerin, L.; Pralong, F.P. The anorexigenic effects of metformin involve increases in hypothalamic leptin receptor expression. Metab.Clin. Exp. 2011, 60, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Buettner, C.; Pocai, A.; Muse, E.D.; Etgen, A.M.; Myers, M.G., Jr.; Rossetti, L. Critical role of STAT3 in leptin’s metabolic actions. Cell Metab. 2006, 4, 49–60. [Google Scholar] [CrossRef] [Green Version]
- Varela, L.; Horvath, T.L. Leptin and insulin pathways in POMC and AgRP neurons that modulate energy balance and glucose homeostasis. EMBO Rep. 2012, 13, 1079–1086. [Google Scholar] [CrossRef]
- Munzberg, H.; Huo, L.; Nillni, E.A.; Hollenberg, A.N.; Bjorbaek, C. Role of signal transducer and activator of transcription 3 in regulation of hypothalamic proopiomelanocortin gene expression by leptin. Endocrinology 2003, 144, 2121–2131. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Chen, B.; Shen, J.; Wan, L.; Zhu, Y.; Yi, T.; Xiao, Z. The Beneficial Effects of Quercetin, Curcumin, and Resveratrol in Obesity. Oxidative Med. Cell. Longev. 2017, 2017, 1459497. [Google Scholar] [CrossRef]
- Carrasco-Pozo, C.; Cires, M.J.; Gotteland, M. Quercetin and Epigallocatechin Gallate in the Prevention and Treatment of Obesity: From Molecular to Clinical Studies. J. Med. Food 2019, 22, 753–770. [Google Scholar] [CrossRef]
- Ford, R.J.; Fullerton, M.D.; Pinkosky, S.L.; Day, E.A.; Scott, J.W.; Oakhill, J.S.; Bujak, A.L.; Smith, B.K.; Crane, J.D.; Blumer, R.M.; et al. Metformin and salicylate synergistically activate liver AMPK, inhibit lipogenesis and improve insulin sensitivity. Biochem. J. 2015, 468, 125–132. [Google Scholar] [CrossRef] [Green Version]
- Sakamula, R.; Thong-Asa, W. Neuroprotective effect of p-coumaric acid in mice with cerebral ischemia reperfusion injuries. Metab. Brain Dis. 2018, 33, 765–773. [Google Scholar] [CrossRef]
- Da Silva, A.A.; do Carmo, J.M.; Hall, J.E. CNS Regulation of Glucose Homeostasis: Role of the Leptin-Melanocortin System. Curr. Diabetes Rep. 2020, 20, 29. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Guo, J.; You, Y.; Zhan, J.; Huang, W. p-Coumaric acid prevents obesity via activating thermogenesis in brown adipose tissue mediated by mTORC1-RPS6. FASEB J. 2020, 34, 7810–7824. [Google Scholar] [CrossRef] [PubMed]
- Santoro, A.; Mattace Raso, G.; Meli, R. Drug targeting of leptin resistance. Life Sci. 2015, 140, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Roman, E.A.; Reis, D.; Romanatto, T.; Maimoni, D.; Ferreira, E.A.; Santos, G.A.; Torsoni, A.S.; Velloso, L.A.; Torsoni, M.A. Central leptin action improves skeletal muscle AKT, AMPK, and PGC1 alpha activation by hypothalamic PI3K-dependent mechanism. Mol. Cell. Endocrinol. 2010, 314, 62–69. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, L.V.; Nguyen, K.D.A.; Ma, C.-T.; Nguyen, Q.-T.; Nguyen, H.T.H.; Yang, D.-J.; Tran, T.L.; Kim, K.W.; Doan, K.V. p-Coumaric Acid Enhances Hypothalamic Leptin Signaling and Glucose Homeostasis in Mice via Differential Effects on AMPK Activation. Int. J. Mol. Sci. 2021, 22, 1431. https://doi.org/10.3390/ijms22031431
Nguyen LV, Nguyen KDA, Ma C-T, Nguyen Q-T, Nguyen HTH, Yang D-J, Tran TL, Kim KW, Doan KV. p-Coumaric Acid Enhances Hypothalamic Leptin Signaling and Glucose Homeostasis in Mice via Differential Effects on AMPK Activation. International Journal of Molecular Sciences. 2021; 22(3):1431. https://doi.org/10.3390/ijms22031431
Chicago/Turabian StyleNguyen, Linh V., Khoa D. A. Nguyen, Chi-Thanh Ma, Quoc-Thai Nguyen, Huong T. H. Nguyen, Dong-Joo Yang, Trung Le Tran, Ki Woo Kim, and Khanh V. Doan. 2021. "p-Coumaric Acid Enhances Hypothalamic Leptin Signaling and Glucose Homeostasis in Mice via Differential Effects on AMPK Activation" International Journal of Molecular Sciences 22, no. 3: 1431. https://doi.org/10.3390/ijms22031431