The Down-Regulation of Clusterin Expression Enhances the αSynuclein Aggregation Process

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Generation and Characterization of SH-Syn Cells
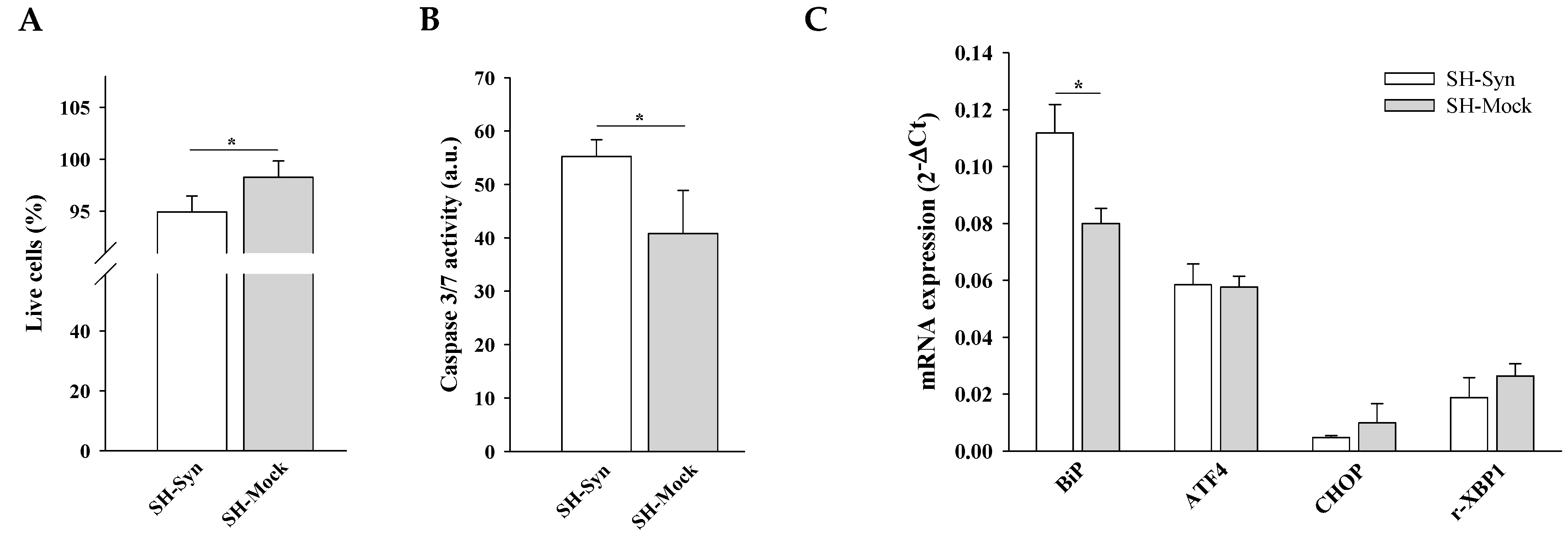
2.2. Effects of αSyn Overexpression on Cell Viability and UPR Induction
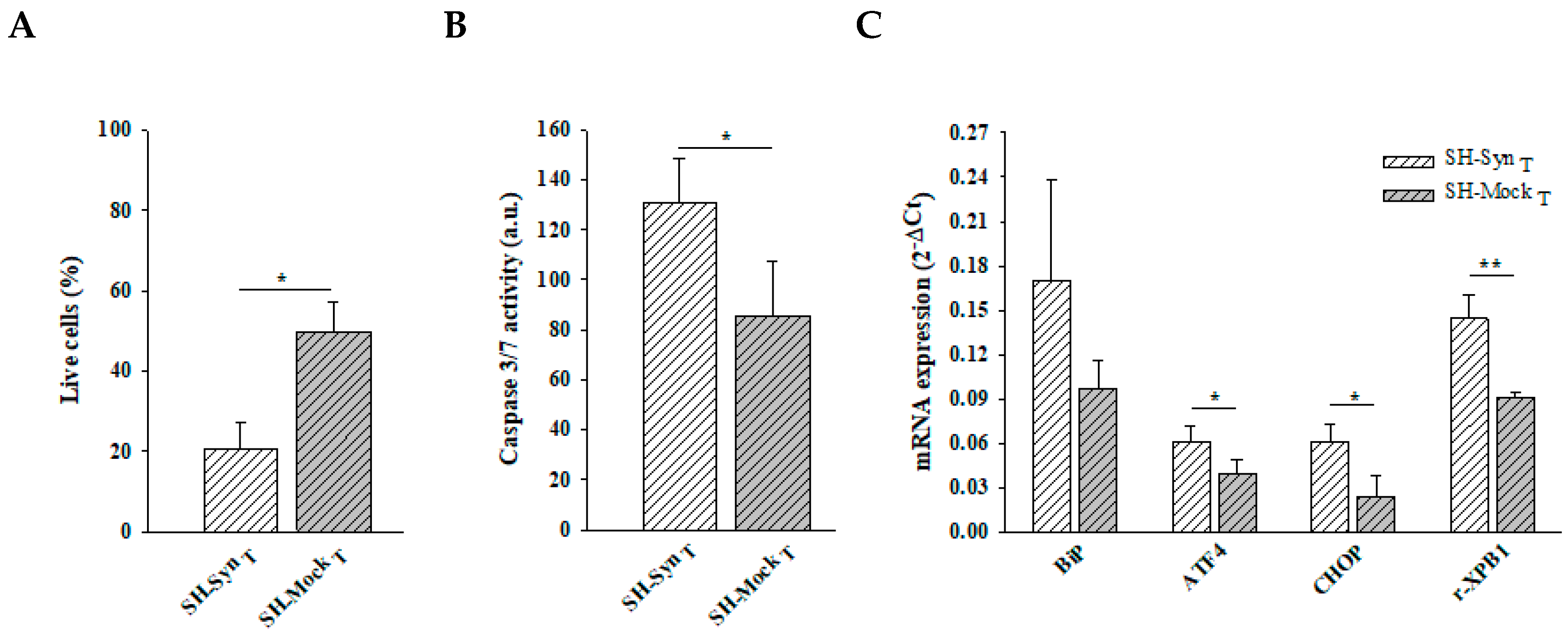
2.3. Effects of Proteasome Inhibition on αSyn Aggregation, Cell Viability, Executioner 3/7 Caspase Activity, and UPR Induction
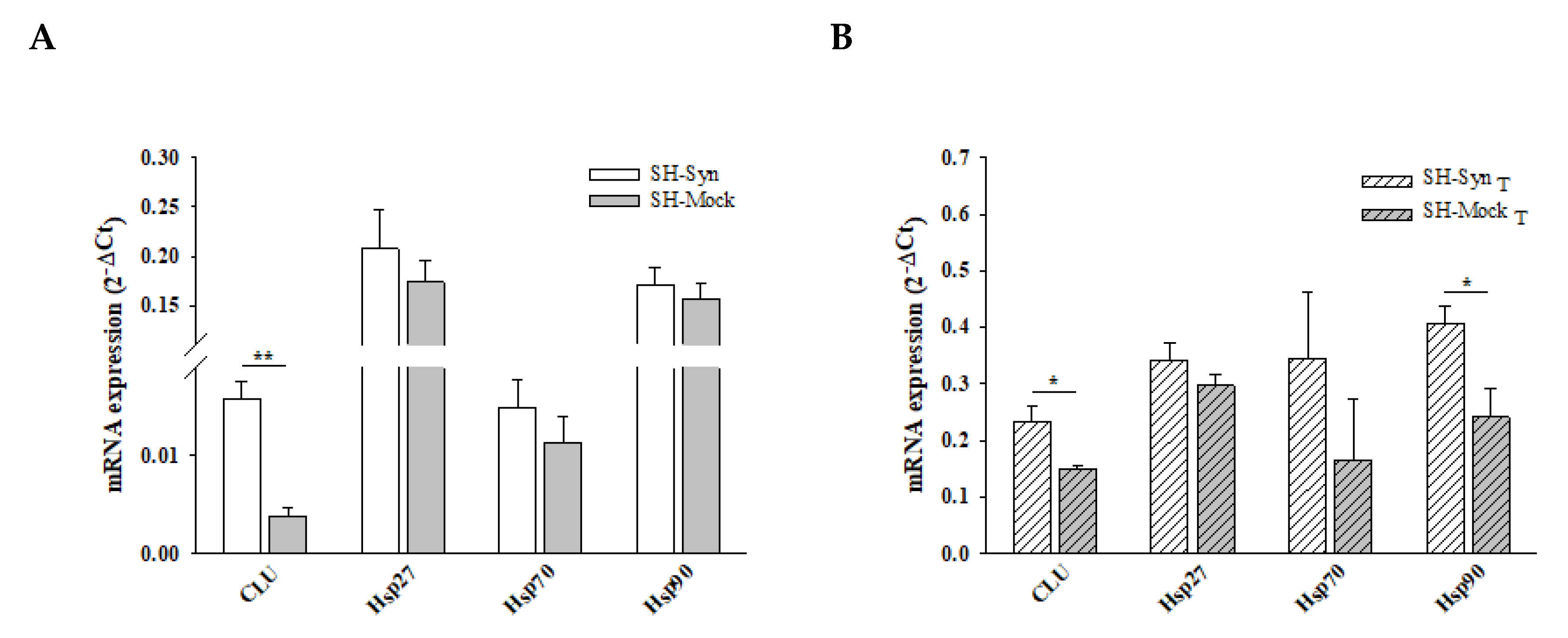
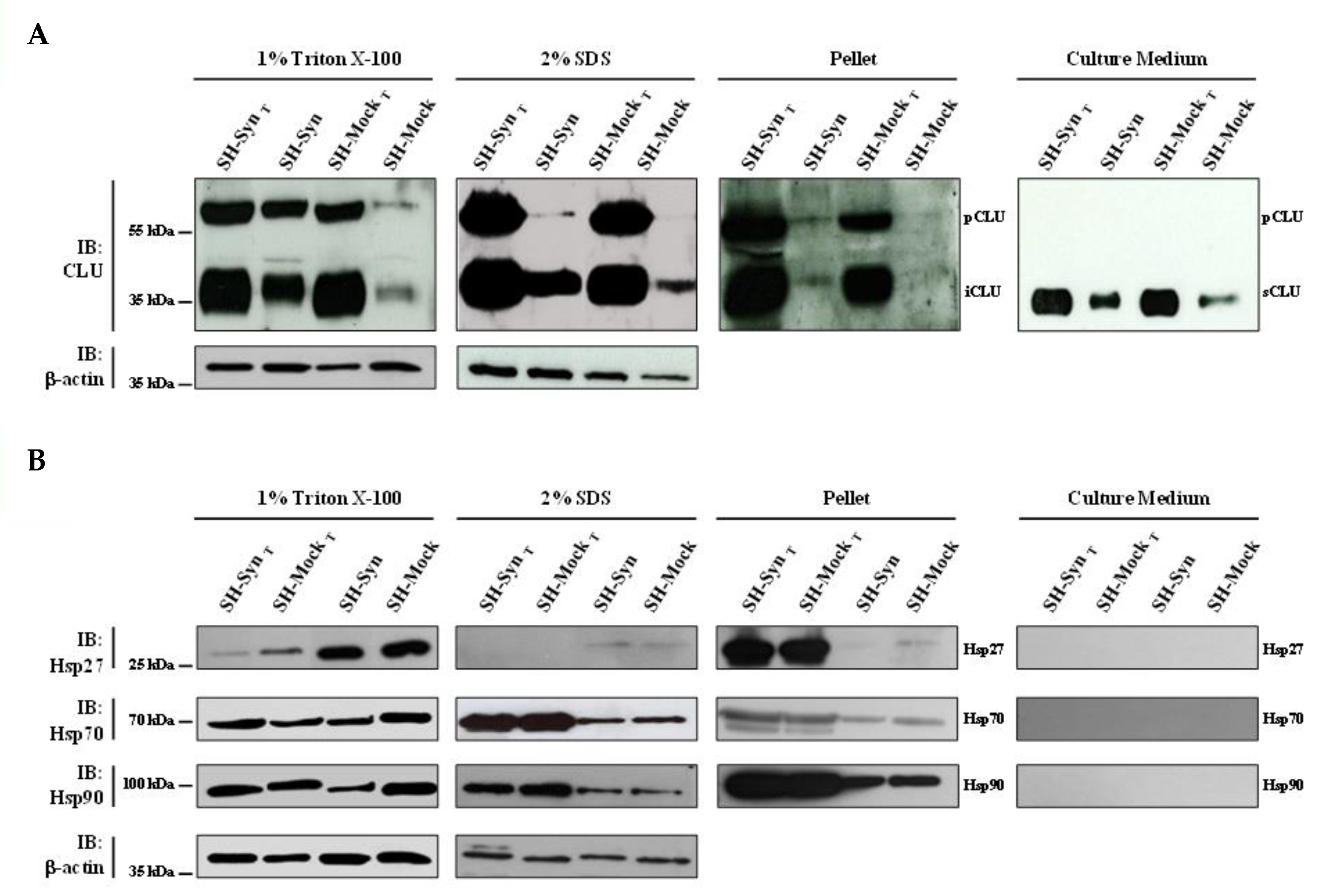
2.4. Expression Analyses of CLU and Other Molecular Chaperones in SH-Syn
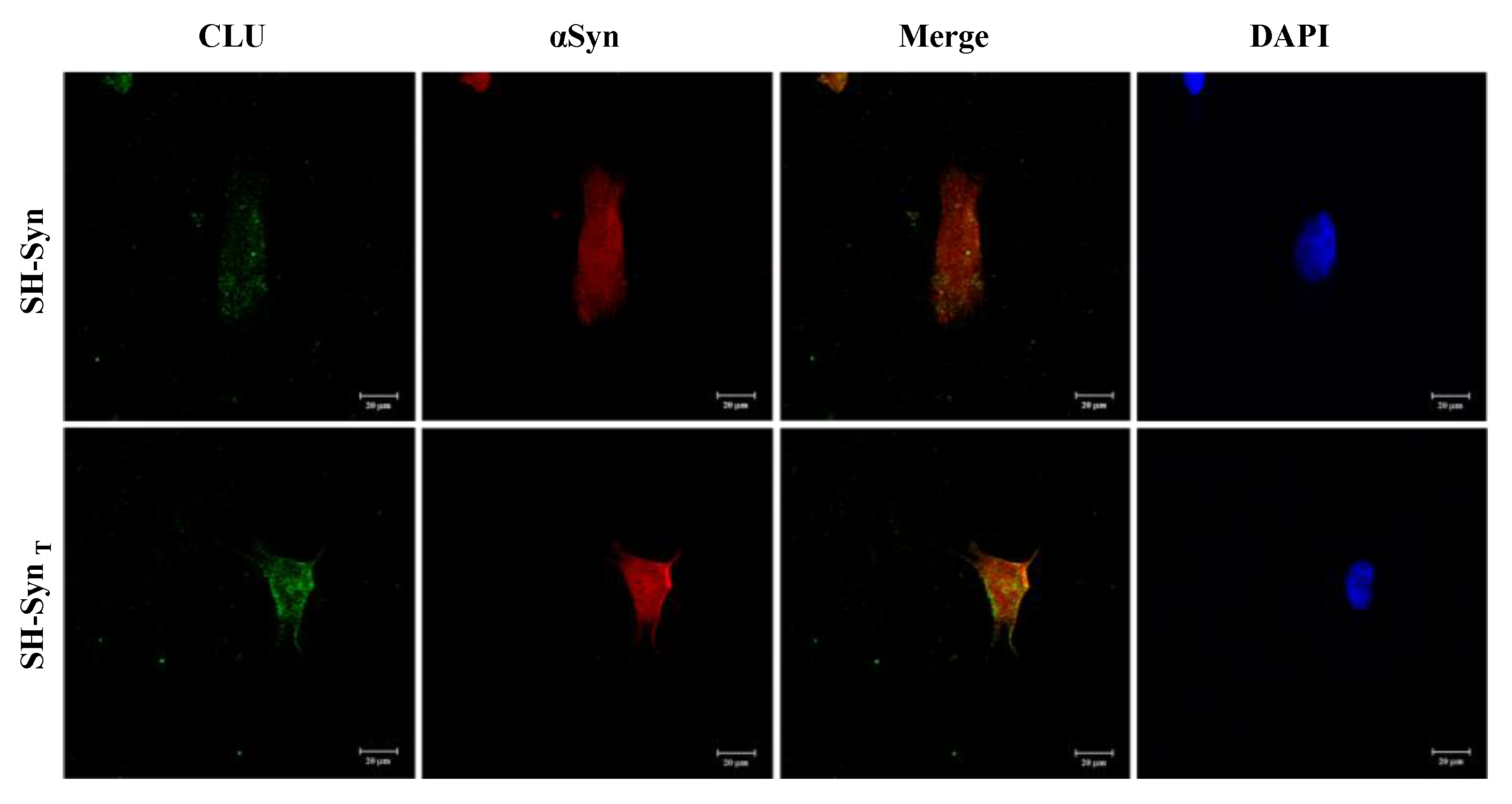
2.5. CLU and αSyn Intracellular Localization and Interaction
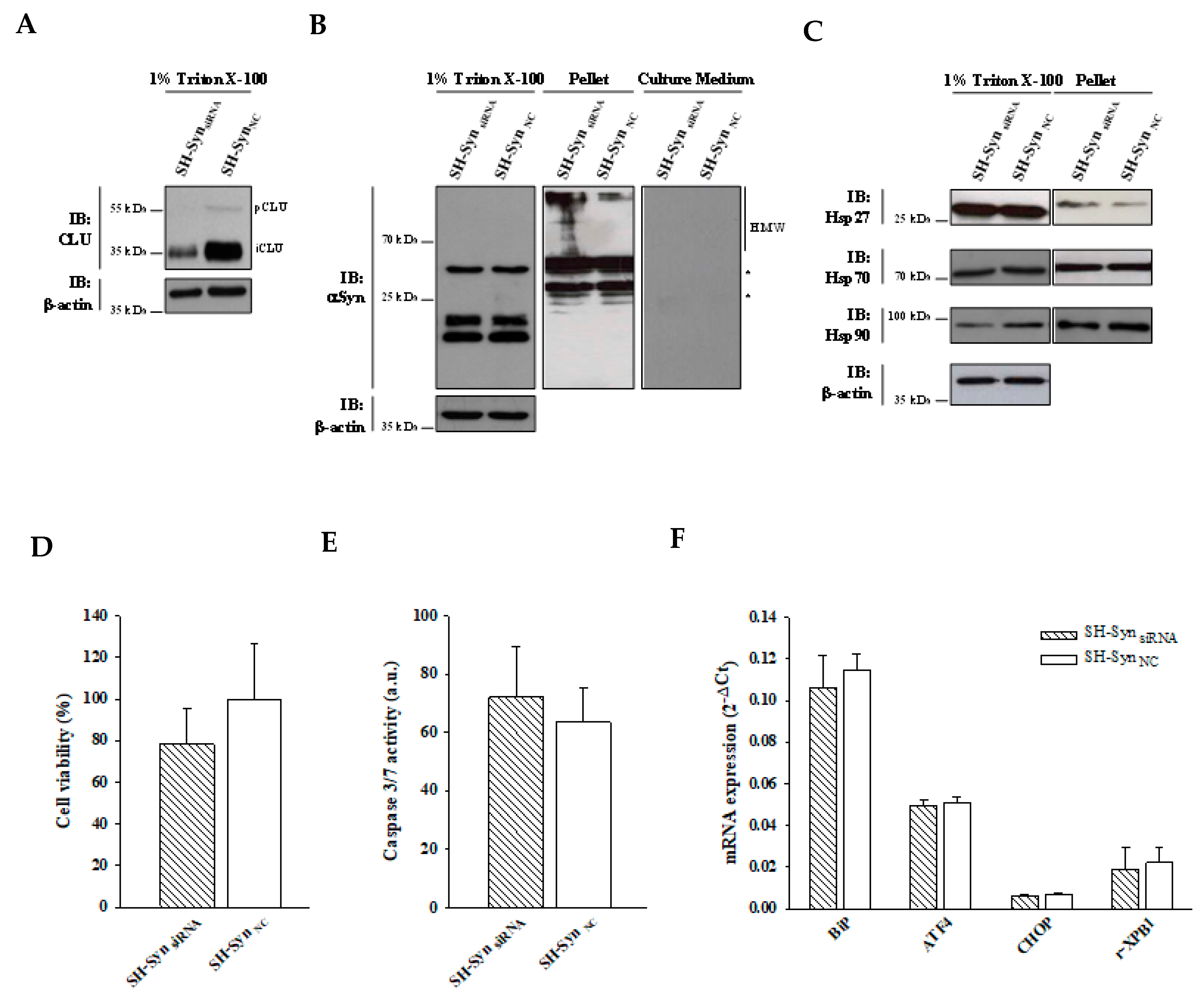
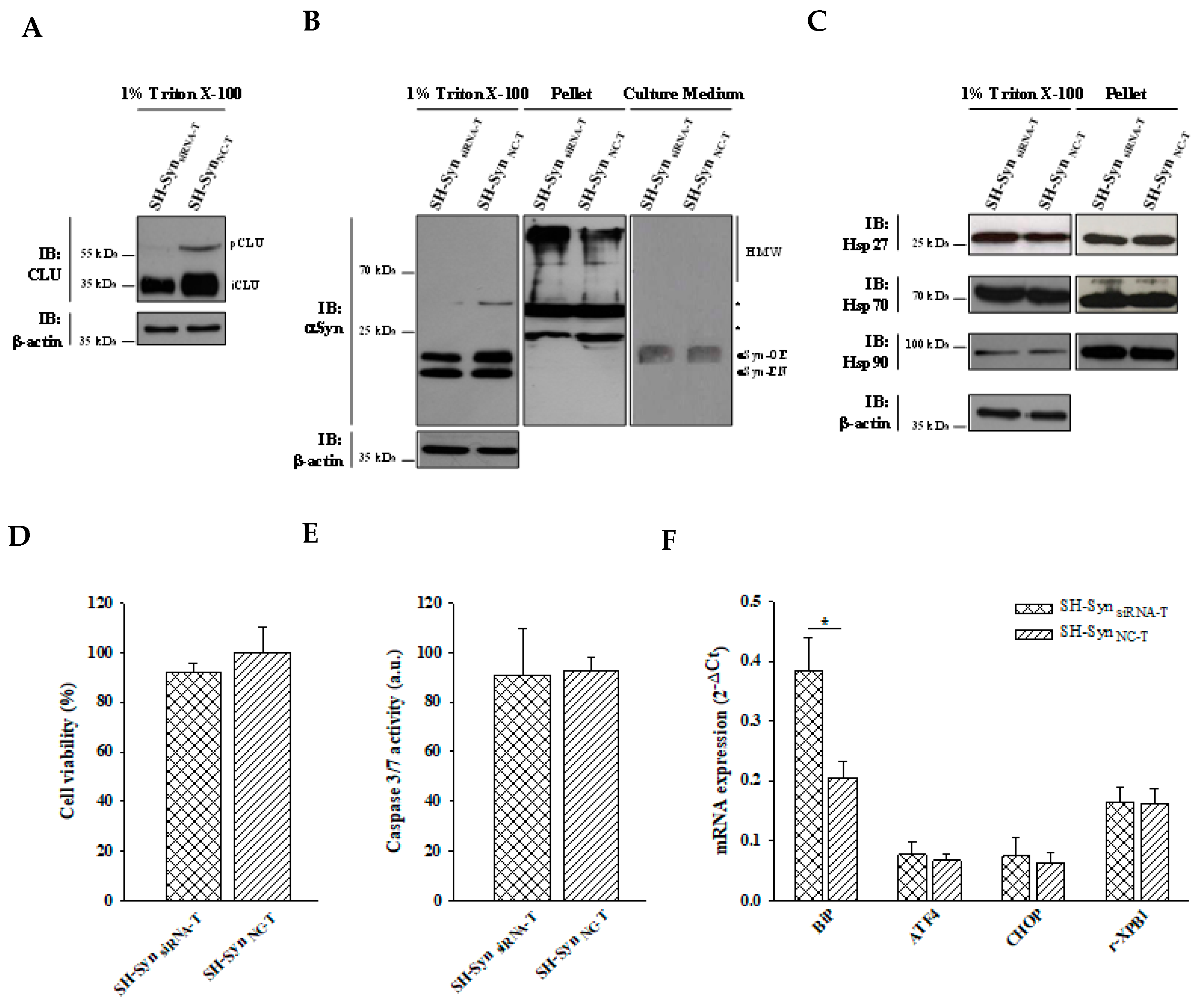
2.6. Effects of CLU Down-Regulation
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Transfection
4.2. Cell Viability Assay
4.3. Trypan Blue Staining
4.4. Cell Proliferation Assay
4.5. siRNA Experiments
4.6. Caspases Assay
4.7. RNA Extraction and qPCR
4.8. Sequential Extraction and Western Blot Analyses
4.9. Double-Immunofluorescence Analysis
4.10. Co-Immunoprecipitation Assay
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| αSyn | αSynuclein |
| αSyn-EN | Endogenous Syn |
| αSyn-OE | Overexpressed Syn |
| Aβ | Amyloid-beta peptide |
| AD | Alzheimer’s Disease |
| ATF4 | Activating transcription factor 4 |
| BiP | Binding Immunoglobulin Protein |
| CHOP | C/EBP homologous protein |
| CLU | Clusterin |
| co-IP | Co-immunoprecipitation |
| DAPI | 4′dye-6˗diamidino˗2˗phenylindole |
| GAPDH | Glyceraldehyde 3˗phosphate dehydrogenase |
| GWAS | Genome-Wide Association Study |
| HMW | High molecular weight |
| HSF-1 | Heat shock factor-1 |
| IB | Immunoblotted |
| iCLU | Intracellular CLU |
| IP | Immunoprecipitated fraction |
| IRE1α | Inositol-requiring enzyme 1 alpha |
| LB | Lewy bodies |
| LN | Lewy neuritis |
| MES | Mesencephalic/neuroblastoma hybrid cell line |
| pCLU | CLU precursor protein |
| PD | Parkinson’s Disease |
| PERK | PKR-like ER kinase |
| qPCR | Quantitative real-time PCR |
| r-XBP1 | XBP1us/XBP1 |
| sCLU | CLU secreted protein |
| SH-SY5Y | Human neuroblastoma cell line |
| SH-Mock | SH-SY5Y cells transfected with pHM6-mock |
| SH-MockT | MG132 treated control cells |
| SH-Syn | SH-SY5Y cells transfected with pHM6-αSyn-wt |
| SH-SynNC | NC-transfected SH-Syn |
| SH-SynNC-T | MG132 treated NC-transfected SH-Syn |
| SH-SynsiRNA | CLU siRNA-transfected SH-Syn |
| SH-SynsiRNA-T | MG132 treated CLU siRNA-transfected SH-Syn |
| SH-SynT | MG132 treated SH-Syn |
| UPR | Unfolded Protein Response |
| XBP1 | X-box binding protein 1 |
| XBP1us | X-box binding protein 1 unconventional spliced form |
References
- Jayaraj, G.G.; Hipp, M.S.; Ulrich Hartl, F. Functional modules of the proteostasis network. Cold Spring Harb. Perspect. Biol. 2020, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newton, T.M.; Duce, J.A.; Bayle, E.D. The proteostasis network provides targets for neurodegeneration. Br. J. Pharm. 2019, 176, 3508–3514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kampinga, H.H.; Bergink, S. Heat shock proteins as potential targets for protective strategies in neurodegeneration. Lancet Neurol. 2016, 15, 748–759. [Google Scholar] [CrossRef]
- Douglas, P.M.; Dillin, A. Protein homeostasis and aging in neurodegeneration. J. Cell Biol. 2010, 190, 719–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lashuel, H.A.; Overk, C.R.; Oueslati, A.; Masliah, E. The many faces of α-synuclein: From structure and toxicity to therapeutic target. Nat. Rev. Neurosci. 2013, 14, 38–48. [Google Scholar] [CrossRef] [Green Version]
- Goedert, M.; Jakes, R.; Spillantini, M.G. The Synucleinopathies: Twenty Years on. J. Parkinsons. Dis. 2017, 7, S53–S71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steiner, J.A.; Quansah, E.; Brundin, P. The concept of alpha-synuclein as a prion-like protein: Ten years after. Cell Tissue Res. 2018, 373, 161–173. [Google Scholar] [CrossRef]
- Goedert, M.; Masuda-Suzukake, M.; Falcon, B. Like prions: The propagation of aggregated tau and α-synuclein in neurodegeneration. Brain 2017, 140, 266–278. [Google Scholar] [CrossRef] [Green Version]
- Brundin, P.; Dave, K.D.; Kordower, J.H. Therapeutic approaches to target alpha-synuclein pathology. Exp. Neurol. 2017, 298, 225–235. [Google Scholar] [CrossRef]
- Cox, D.; Carver, J.A.; Ecroyd, H. Preventing α-synuclein aggregation: The role of the small heat-shock molecular chaperone proteins. Biochim. Biophys. Acta Mol. Basis Dis. 2014, 1842, 1830–1843. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.K.; Priya, S. Expanding role of molecular chaperones in regulating α-synuclein misfolding; implications in Parkinson’s disease. Cell. Mol. Life Sci. 2017, 74, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Tissue-based map of the human proteome. Science. 2015, 347. [Google Scholar] [CrossRef] [PubMed]
- Rizzi, F.; Bettuzzi, S. The clusterin paradigm in prostate and breast carcinogenesis. Endocr. Relat. Cancer 2010, 17, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Rohne, P.; Prochnow, H.; Koch-Brandt, C. The CLU-files: Disentanglement of a mystery. Biomol. Concepts 2016, 7, 1–15. [Google Scholar] [CrossRef]
- Trougakos, I.P. The molecular chaperone apolipoprotein J/Clusterin as a sensor of oxidative stress: Implications in therapeutic approaches—A mini-review. Gerontology 2013, 59, 514–523. [Google Scholar] [CrossRef]
- Rohne, P.; Prochnow, H.; Wolf, S.; Renner, B.; Koch-Brandt, C. The chaperone activity of clusterin is dependent on glycosylation and redox environment. Cell. Physiol. Biochem. 2014, 34, 1626–1639. [Google Scholar] [CrossRef]
- Humphreys, D.T.; Carver, J.A.; Easterbrook-Smith, S.B.; Wilson, M.R. Clusterin has chaperone-like activity similar to that of small heat shock proteins. J. Biol. Chem. 1999, 274, 6875–6881. [Google Scholar] [CrossRef] [Green Version]
- Charnay, Y.; Imhof, A.; Vallet, P.G.; Kovari, E.; Bouras, C.; Giannakopoulos, P. Clusterin in neurological disorders: Molecular perspectives and clinical relevance. Brain Res. Bull. 2012, 88, 434–443. [Google Scholar] [CrossRef]
- Cordero-Llana, O.; Scott, S.A.; Maslen, S.L.; Anderson, J.M.; Boyle, J.; Chowhdury, R.R.; Tyers, P.; Barker, R.A.; Kelly, C.M.; Rosser, A.E.; et al. Clusterin secreted by astrocytes enhances neuronal differentiation from human neural precursor cells. Cell Death Differ. 2011, 18, 907–913. [Google Scholar] [CrossRef] [Green Version]
- Gregory, J.M.; Whiten, D.R.; Brown, R.A.; Barros, T.P.; Kumita, J.R.; Yerbury, J.J.; Satapathy, S.; McDade, K.; Smith, C.; Luheshi, L.M.; et al. Clusterin protects neurons against intracellular proteotoxicity. Acta Neuropathol. Commun. 2017, 5, 81. [Google Scholar] [CrossRef]
- Manavalan, A.; Mishra, M.; Feng, L.; Sze, S.K.; Akatsu, H.; Heese, K. Brain site-specific proteome changes in aging-related dementia. Exp. Mol. Med. 2013, 45, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Roussotte, F.F.; Gutman, B.A.; Madsen, S.K.; Colby, J.B.; Thompson, P.M. Combined effects of Alzheimer risk variants in the CLU and ApoE genes on ventricular expansion patterns in the elderly. J. Neurosci. 2014, 34, 6537–6545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zlokovic, B.V.; Martel, C.L.; Matsubara, E.; Mccomb, J.G.; Zheng, G.; Mccluskey, R.T.; Frangione, B.; Ghiso, J. Glycoprotein 330/megalin: Probable role in receptor-mediated transport of apolipoprotein J alone and in a complex with Alzheimer disease amyloid β at the blood-brain and blood-cerebrospinal fluid barriers. Proc. Natl. Acad. Sci. USA 1996, 93, 4229–4234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foster, E.M.; Dangla-Valls, A.; Lovestone, S.; Ribe, E.M.; Buckley, N.J. Clusterin in Alzheimer’s disease: Mechanisms, genetics, and lessons from other pathologies. Front. Neurosci. 2019, 13, 1–27. [Google Scholar] [CrossRef] [Green Version]
- Harold, D.; Abraham, R.; Hollingworth, P.; Sims, R.; Gerrish, A.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; Dowzell, K.; Williams, A.; et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat. Genet. 2009, 41, 1088–1093. [Google Scholar] [CrossRef] [Green Version]
- Lambert, J.C.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; Jun, G.; DeStefano, A.L.; Bis, J.C.; Beecham, G.W.; et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 2013, 45, 1452–1458. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, K.; Doh-ura, K.; Wakisaka, Y.; Iwaki, T. Clusterin/apolipoprotein J is associated with cortical Lewy bodies: Immunohistochemical study in cases with α-synucleinopathies. Acta Neuropathol. 2002, 104, 225–230. [Google Scholar] [CrossRef]
- Zhou, Y.; Gu, G.; Goodlett, D.R.; Zhang, T.; Pan, C.; Montine, T.J.; Montine, K.S.; Aebersold, R.H.; Zhang, J. Analysis of α-synuclein-associated proteins by quantitative proteomics. J. Biol. Chem. 2004, 279, 39155–39164. [Google Scholar] [CrossRef] [Green Version]
- Vranová, H.P.; Mareš, J.; Nevrlý, M.; Stejskal, D.; Zapletalová, J.; Hluštík, P.; Kaňovský, P. CSF markers of neurodegeneration in Parkinson’s disease. J. Neural Transm. 2010, 117, 1177–1181. [Google Scholar] [CrossRef]
- Zhang, X.; Yin, X.; Yu, H.; Liu, X.; Yang, F.; Yao, J.; Jin, H.; Yang, P. Quantitative proteomic analysis of serum proteins in patients with Parkinson’s disease using an isobaric tag for relative and absolute quantification labeling, two-dimensional liquid chromatography, and tandem mass spectrometry. Analyst 2012, 137, 490–495. [Google Scholar] [CrossRef]
- Maarouf, C.L.; Beach, T.G.; Adler, C.H.; Shill, H.A.; Sabbagh, M.N.; Wu, T.; Walker, D.G.; Kokjohn, T.A.; Roher, A.E. Cerebrospinal fluid biomarkers of neuropathologically diagnosed Parkinson’s disease subjects. Neurol. Res. 2012, 34, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Huang, X.; Park, Y.Y.; Hollenbeck, A.; Chen, H. An exploratory study on CLU, CR1 and PICALM and Parkinson disease. PLoS ONE 2011, 6, e24211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myöhänen, T.T.; Hannula, M.J.; Van Elzen, R.; Gerard, M.; Van Der Veken, P.; García-Horsman, J.A.; Baekelandt, V.; Männistö, P.T.; Lambeir, A.M. A prolyl oligopeptidase inhibitor, KYP-2047, reduces α-synuclein protein levels and aggregates in cellular and animal models of Parkinson’s disease. Br. J. Pharm. 2012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, O.W.; Chung, K.K.K. The role of alpha-synuclein oligomerization and aggregation in cellular and animal models of Parkinson’s disease. PLoS ONE 2012. [Google Scholar] [CrossRef] [PubMed]
- Bandopadhyay, R. Sequential extraction of soluble and insoluble alpha-synuclein from parkinsonian brains. J. Vis. Exp. 2016. [Google Scholar] [CrossRef] [PubMed]
- Karampetsou, M.; Ardah, M.T.; Semitekolou, M.; Polissidis, A.; Samiotaki, M.; Kalomoiri, M.; Majbour, N.; Xanthou, G.; El-Agnaf, O.M.A.; Vekrellis, K. Phosphorylated exogenous alpha-synuclein fibrils exacerbate pathology and induce neuronal dysfunction in mice. Sci. Rep. 2017. [Google Scholar] [CrossRef]
- Hetz, C.; Papa, F.R. The Unfolded Protein Response and Cell Fate Control. Mol. Cell 2018, 69, 169–181. [Google Scholar] [CrossRef] [Green Version]
- Bentea, E.; Verbruggen, L.; Massie, A. The Proteasome Inhibition Model of Parkinson’s Disease. J. Parkinsons. Dis. 2017, 7, 31–63. [Google Scholar] [CrossRef] [Green Version]
- Bellucci, A.; Navarria, L.; Zaltieri, M.; Falarti, E.; Bodei, S.; Sigala, S.; Battistin, L.; Spillantini, M.; Missale, C.; Spano, P. Induction of the unfolded protein response by α-synuclein in experimental models of Parkinson’s disease. J. Neurochem. 2011, 116, 588–605. [Google Scholar] [CrossRef]
- Colla, E.; Coune, P.; Liu, Y.; Pletnikova, O.; Troncoso, J.C.; Iwatsubo, T.; Schneider, B.L.; Lee, M.K. Endoplasmic reticulum stress is important for the manifestations of α-synucleinopathy in vivo. J. Neurosci. 2012, 32, 3306–3320. [Google Scholar] [CrossRef]
- Perfeito, R.; Ribeiro, M.; Rego, A.C. Alpha-synuclein-induced oxidative stress correlates with altered superoxide dismutase and glutathione synthesis in human neuroblastoma SH-SY5Y cells. Arch. Toxicol. 2017, 91, 1245–1259. [Google Scholar] [CrossRef] [PubMed]
- Yamada, M.; Iwatsubo, T.; Mizuno, Y.; Mochizuki, H. Overexpression of α-synuclein in rat substantia nigra results in loss of dopaminergic neurons, phosphorylation of α-synuclein and activation of caspase-9: Resemblance to pathogenetic changes in Parkinson’s disease. J. Neurochem. 2004, 91, 451–461. [Google Scholar] [CrossRef]
- Venderova, K.; Park, D.S. Programmed Cell Death in Parkinson’s Disease. Cold Spring Harb. Perspect. Med. 2020, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Hung, K.C.; Huang, H.J.; Wang, Y.T.; Lin, A.M.Y. Baicalein attenuates α-synuclein aggregation, inflammasome activation and autophagy in the MPP+-treated nigrostriatal dopaminergic system in vivo. J. Ethnopharmacol. 2016, 194, 522–529. [Google Scholar] [CrossRef] [PubMed]
- Colla, E. Linking the endoplasmic reticulum to Parkinson’s disease and alpha-synucleinopathy. Front. Neurosci. 2019, 13, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Hammad, S.M.; Ranganathan, S.; Loukinova, E.; Twal, W.O.; Argraves, W.S. Interaction of apolipoprotein J-amyloid β-peptide complex with low density lipoprotein receptor-related protein-2/megalin. A mechanism to prevent pathological accumulation of amyloid β-peptide. J. Biol. Chem. 1997, 272, 18644–18649. [Google Scholar] [CrossRef] [Green Version]
- Wojtas, A.M.; Kang, S.S.; Olley, B.M.; Gatherer, M.; Shinohara, M.; Lozano, P.A.; Liu, C.C.; Kurti, A.; Baker, K.E.; Dickson, D.W.; et al. Loss of clusterin shifts amyloid deposition to the cerebrovasculature via disruption of perivascular drainage pathways. Proc. Natl. Acad. Sci. USA 2017, 114, E6962–E6971. [Google Scholar] [CrossRef] [Green Version]
- Yerbury, J.J.; Poon, S.; Meehan, S.; Thompson, B.; Kumita, J.R.; Dobson, C.M.; Wilson, M.R. The extracellular chaperone clusterin influences amyloid formation and toxicity by interacting with prefibrillar structures. FASEB J. 2007, 21, 2312–2322. [Google Scholar] [CrossRef]
- Whiten, D.R.; Cox, D.; Horrocks, M.H.; Taylor, C.G.; De, S.; Flagmeier, P.; Tosatto, L.; Kumita, J.R.; Ecroyd, H.; Dobson, C.M.; et al. Single-Molecule Characterization of the Interactions between Extracellular Chaperones and Toxic α-Synuclein Oligomers. Cell Rep. 2018, 23, 3492–3500. [Google Scholar] [CrossRef]
- Zhang, Z.; Cheng, Y. MiR-16-1 promotes the aberrant α -synuclein accumulation in parkinson disease via targeting heat shock protein 70. Sci. World J. 2014, 2014. [Google Scholar] [CrossRef] [Green Version]
- Salganik, M.; Sergeyev, V.G.; Shinde, V.; Meyers, C.A.; Gorbatyuk, M.S.; Lin, J.H.; Zolotukhin, S.; Gorbatyuk, O.S. The loss of glucose-regulated protein 78 (GRP78) during normal aging or from siRNA knockdown augments human alpha-synuclein (α-syn) toxicity to rat nigral neurons. Neurobiol. Aging 2015, 36, 2213–2223. [Google Scholar] [CrossRef] [PubMed] [Green Version]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lenzi, C.; Ramazzina, I.; Russo, I.; Filippini, A.; Bettuzzi, S.; Rizzi, F. The Down-Regulation of Clusterin Expression Enhances the αSynuclein Aggregation Process. Int. J. Mol. Sci. 2020, 21, 7181. https://doi.org/10.3390/ijms21197181
Lenzi C, Ramazzina I, Russo I, Filippini A, Bettuzzi S, Rizzi F. The Down-Regulation of Clusterin Expression Enhances the αSynuclein Aggregation Process. International Journal of Molecular Sciences. 2020; 21(19):7181. https://doi.org/10.3390/ijms21197181
Chicago/Turabian StyleLenzi, Chiara, Ileana Ramazzina, Isabella Russo, Alice Filippini, Saverio Bettuzzi, and Federica Rizzi. 2020. "The Down-Regulation of Clusterin Expression Enhances the αSynuclein Aggregation Process" International Journal of Molecular Sciences 21, no. 19: 7181. https://doi.org/10.3390/ijms21197181