Deletion of p53 and Hyper-Activation of PIK3CA in Keratin-15+ Stem Cells Lead to the Development of Spontaneous Squamous Cell Carcinoma



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. HNSCC Patients with Double Genetic Alterations in PIK3CA and TP53 Exhibited Worse Prognosis and More Immunosuppressive TME
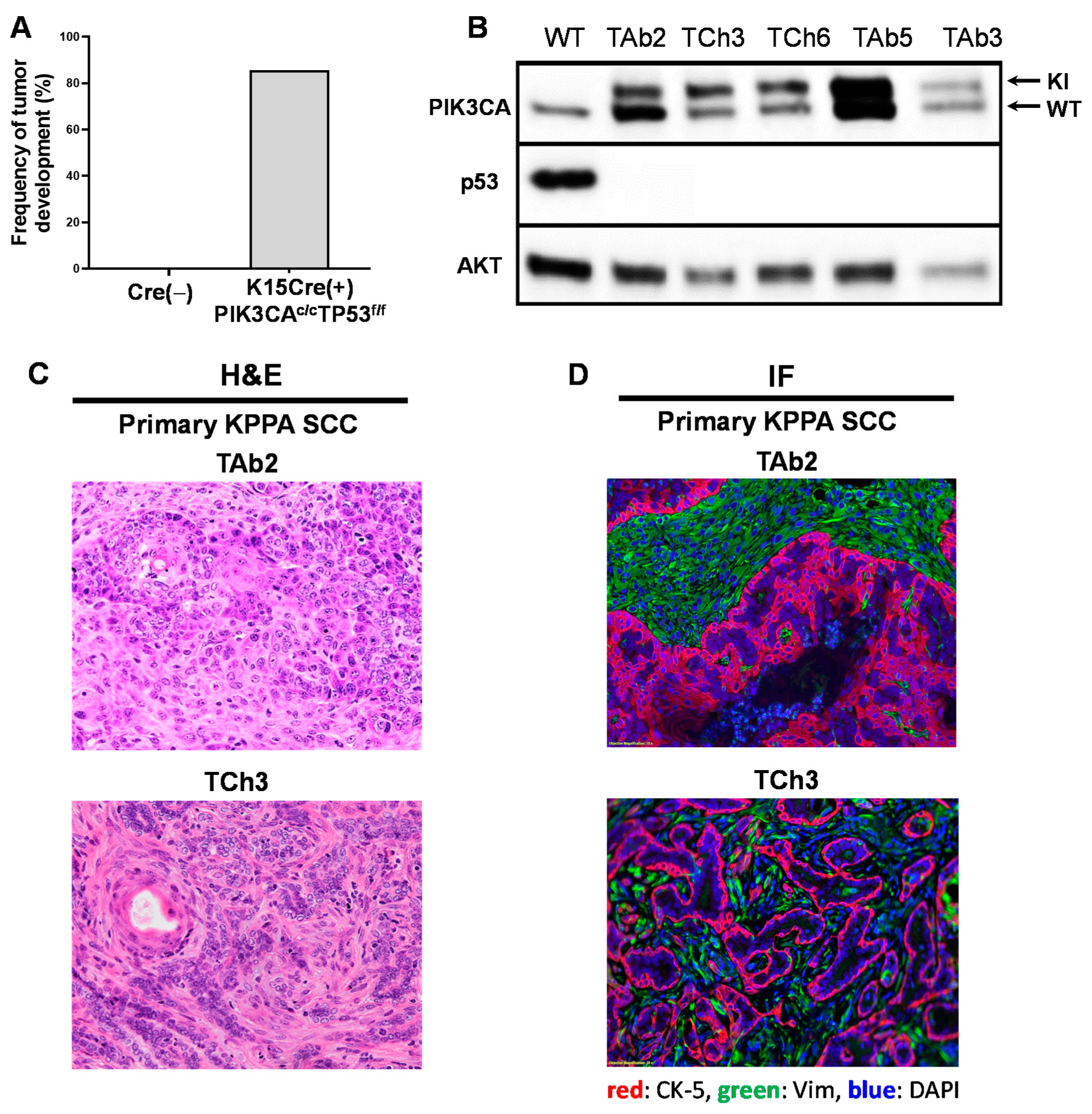
2.2. A Genetic Mouse Model of PIK3CA Hyperactivation and p53 Deletion in K15+ Cells
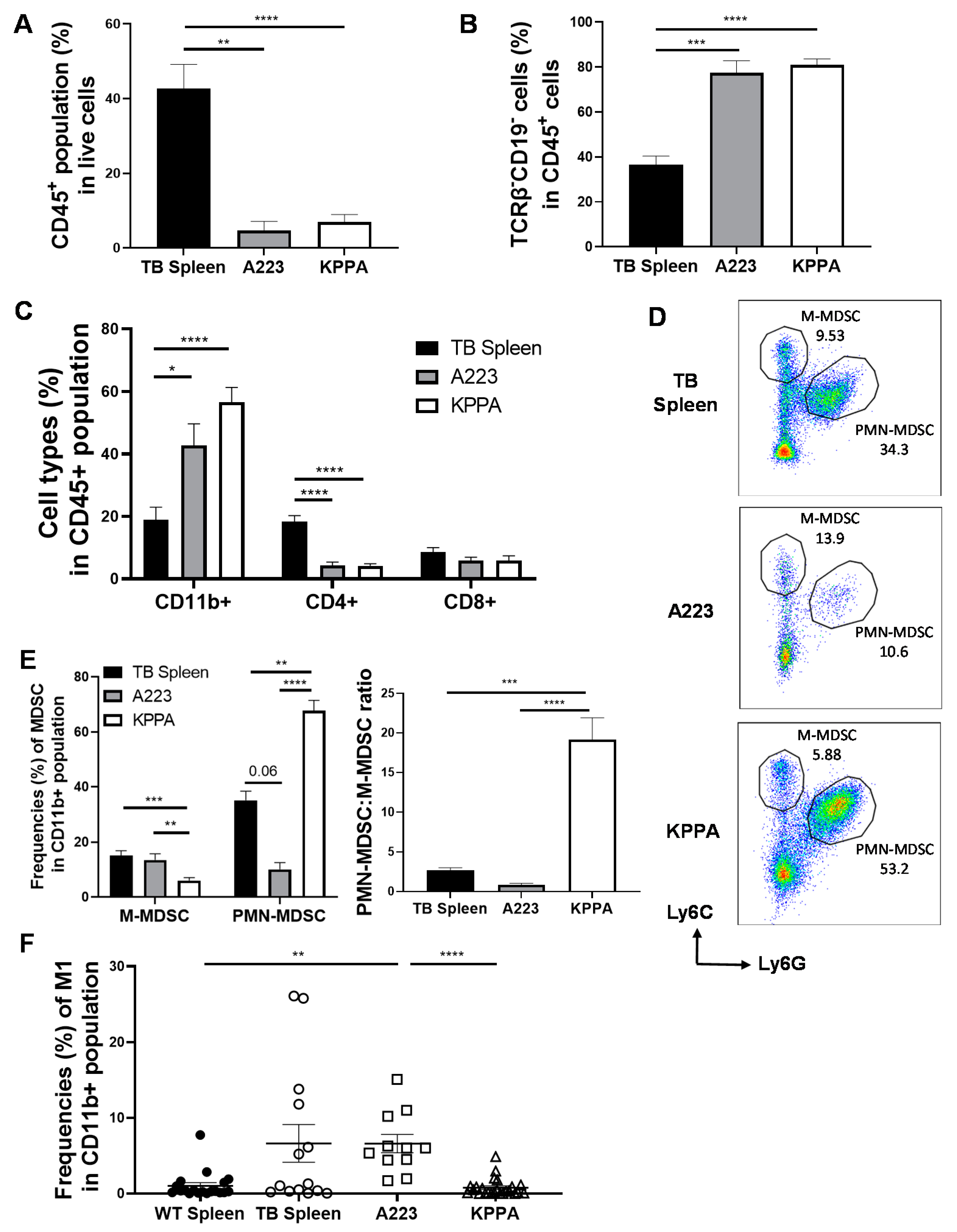
2.3. Characterization of the Immune TME in KPPA Tumors
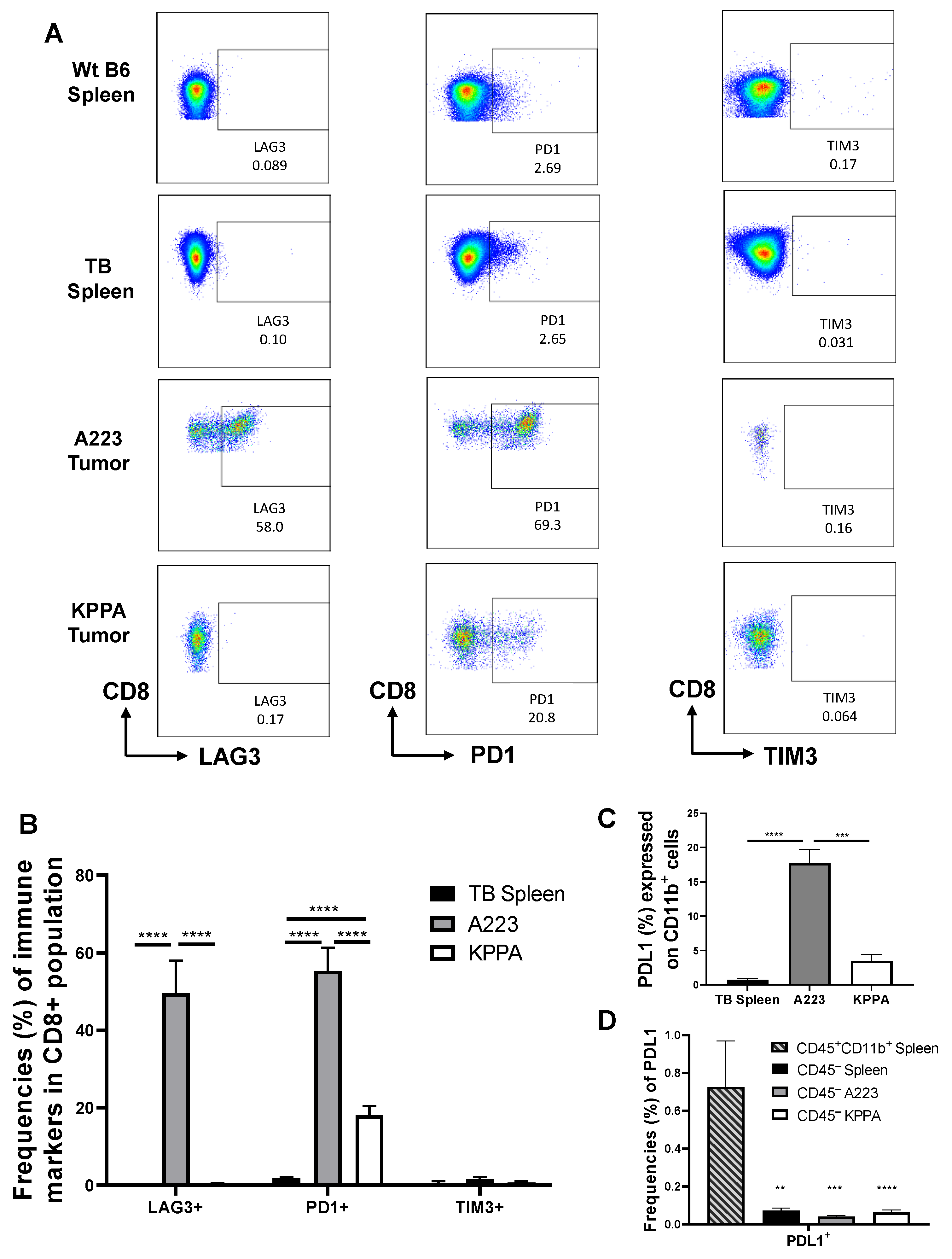
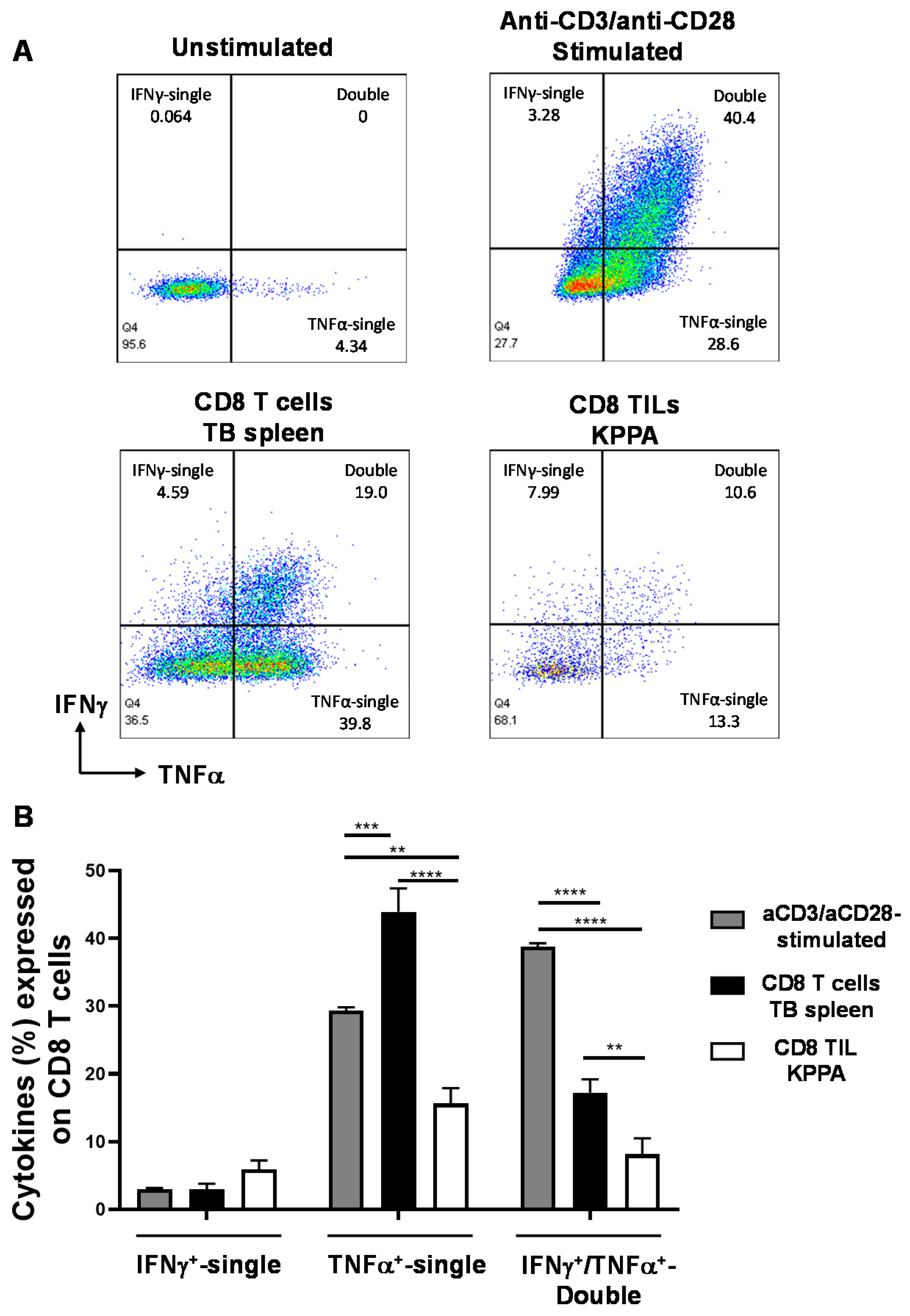
2.4. Dysfunctional TILs in KPPA Tumors Suggest an Immunosuppressive TME
3. Discussion
4. Materials and Methods
4.1. Analysis of Patient Samples Obtained by TCGA
4.2. Mouse Models
4.3. Western Blot and Flow Cytometry
4.4. Histology Analysis
4.5. Statistical Analysis of Murine Samples
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, C.; Marsit, C.J.; McClean, M.D.; Nelson, H.H.; Christensen, B.C.; Haddad, R.I.; Clark, J.R.; Wein, R.O.; Grillone, G.A.; Houseman, E.A.; et al. Biomarkers of HPV in head and neck squamous cell carcinoma. Cancer Res. 2012, 72, 5004–5013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Xia, R.H.; Ye, D.X.; Li, J. Human Papillomavirus 16 Infection and TP53 Mutation: Two Distinct Pathogeneses for Oropharyngeal Squamous Cell Carcinoma in an Eastern Chinese Population. PLoS ONE 2016, 11, e0164491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas, N. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [Google Scholar] [CrossRef] [Green Version]
- Perez Sayans, M.; Chamorro Petronacci, C.M.; Lorenzo Pouso, A.I.; Padin Iruegas, E.; Blanco Carrion, A.; Suarez Penaranda, J.M.; Garcia Garcia, A. Comprehensive Genomic Review of TCGA Head and Neck Squamous Cell Carcinomas (HNSCC). J. Clin. Med. 2019, 8, 1896. [Google Scholar] [CrossRef] [Green Version]
- Du, L.; Chen, X.; Cao, Y.; Lu, L.; Zhang, F.; Bornstein, S.; Li, Y.; Owens, P.; Malkoski, S.; Said, S.; et al. Overexpression of PIK3CA in murine head and neck epithelium drives tumor invasion and metastasis through PDK1 and enhanced TGFbeta signaling. Oncogene 2016, 35, 4641–4652. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.; Kang, H.; Mehra, R. Targeting phosphoinositide 3-kinase (PI3K) in head and neck squamous cell carcinoma (HNSCC). Cancers Head Neck 2018, 3, 3. [Google Scholar] [CrossRef] [Green Version]
- Du, L.; Shen, J.; Weems, A.; Lu, S.L. Role of phosphatidylinositol-3-kinase pathway in head and neck squamous cell carcinoma. J. Oncol. 2012, 2012, 450179. [Google Scholar] [CrossRef] [Green Version]
- Tan, B.S.; Tiong, K.H.; Choo, H.L.; Chung, F.F.; Hii, L.W.; Tan, S.H.; Yap, I.K.; Pani, S.; Khor, N.T.; Wong, S.F.; et al. Mutant p53-R273H mediates cancer cell survival and anoikis resistance through AKT-dependent suppression of BCL2-modifying factor (BMF). Cell Death Dis. 2015, 6, e1826. [Google Scholar] [CrossRef]
- De Pauw, I.; Lardon, F.; Van den Bossche, J.; Baysal, H.; Fransen, E.; Deschoolmeester, V.; Pauwels, P.; Peeters, M.; Vermorken, J.B.; Wouters, A. Simultaneous targeting of EGFR, HER2, and HER4 by afatinib overcomes intrinsic and acquired cetuximab resistance in head and neck squamous cell carcinoma cell lines. Mol. Oncol. 2018, 12, 830–854. [Google Scholar] [CrossRef] [Green Version]
- Bieging, K.T.; Mello, S.S.; Attardi, L.D. Unravelling mechanisms of p53-mediated tumour suppression. Nat. Rev. Cancer 2014, 14, 359–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castellanos, M.R.; Pan, Q. Novel p53 therapies for head and neck cancer. World J. Otorhinolaryngol. Head Neck Surg. 2016, 2, 68–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savar, A.; Acin, S.; Gonzalez, C.L.; El-Sawy, T.; Mejia, O.; Li, Z.; Esmaeli, B.; Lacy-Hulbert, A.; El-Naggar, A.K.; McCarty, J.H.; et al. Loss of epithelial p53 and αv integrin cooperate through Akt to induce squamous cell carcinoma yet prevent remodeling of the tumor microenvironment. Oncogene 2015, 34, 516–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Ruiter, E.J.; Ooft, M.L.; Devriese, L.A.; Willems, S.M. The prognostic role of tumor infiltrating T-lymphocytes in squamous cell carcinoma of the head and neck: A systematic review and meta-analysis. OncoImmunology 2017, 6, e1356148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Wu, V.H.; Allevato, M.M.; Gilardi, M.; He, Y.; Luis Callejas-Valera, J.; Vitale-Cross, L.; Martin, D.; Amornphimoltham, P.; McDermott, J.; et al. Syngeneic animal models of tobacco-associated oral cancer reveal the activity of in situ anti-CTLA-4. Nat. Commun. 2019, 10, 5546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.M.Y.; Krinsky, A.L.; Woolaver, R.A.; Wang, X.; Chen, Z.; Wang, J.H. Tumor immune microenvironment in head and neck cancers. Mol. Carcinog. 2020, 59, 766–774. [Google Scholar] [CrossRef] [PubMed]
- Allavena, P.; Mantovani, A. Immunology in the clinic review series; focus on cancer: Tumour-associated macrophages: Undisputed stars of the inflammatory tumour microenvironment. Clin. Exp. Immunol. 2012, 167, 195–205. [Google Scholar] [CrossRef]
- Chen, Y.; Song, Y.; Du, W.; Gong, L.; Chang, H.; Zou, Z. Tumor-associated macrophages: An accomplice in solid tumor progression. J. Biomed. Sci. 2019, 26, 78. [Google Scholar] [CrossRef]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.; Khodadoust, M.S.; Liu, C.L.; Newman, A.M.; Alizadeh, A.A. Profiling Tumor Infiltrating Immune Cells with CIBERSORT. Methods Mol. Biol. 2018, 1711, 243–259. [Google Scholar]
- Morris, R.J.; Liu, Y.; Marles, L.; Yang, Z.; Trempus, C.; Li, S.; Lin, J.S.; Sawicki, J.A.; Cotsarelis, G. Capturing and profiling adult hair follicle stem cells. Nat. Biotechnol. 2004, 22, 411–417. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, L.; Sasaki, Y.; Calado, D.P.; Zhang, B.; Paik, J.H.; DePinho, R.A.; Kutok, J.L.; Kearney, J.F.; Otipoby, K.L.; Rajewsky, K. PI3 kinase signals BCR-dependent mature B cell survival. Cell 2009, 139, 573–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jonkers, J.; Meuwissen, R.; van der Gulden, H.; Peterse, H.; van der Valk, M.; Berns, A. Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nat. Genet 2001, 29, 418–425. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.K.; Kadoishi, T.; Wang, X.; Driver, E.; Chen, Z.; Wang, X.J.; Wang, J.H. Squamous cell carcinomas escape immune surveillance via inducing chronic activation and exhaustion of CD8+ T Cells co-expressing PD-1 and LAG-3 inhibitory receptors. Oncotarget 2016, 7, 81341–81356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, R.A.; Neiman, J.M.; Reddi, A.; Han, G.; Birlea, S.; Mitra, D.; Dionne, L.; Fernandez, P.; Murao, K.; Bian, L.; et al. Epithelial stem cell mutations that promote squamous cell carcinoma metastasis. J. Clin. Investig. 2013, 123, 4390–4404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bronte, V.; Brandau, S.; Chen, S.H.; Colombo, M.P.; Frey, A.B.; Greten, T.F.; Mandruzzato, S.; Murray, P.J.; Ochoa, A.; Ostrand-Rosenberg, S.; et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat. Commun. 2016, 7, 12150. [Google Scholar] [CrossRef] [Green Version]
- Damuzzo, V.; Pinton, L.; Desantis, G.; Solito, S.; Marigo, I.; Bronte, V.; Mandruzzato, S. Complexity and challenges in defining myeloid-derived suppressor cells. Cytom. B Clin. Cytom. 2015, 88, 77–91. [Google Scholar] [CrossRef]
- Rose, S.; Misharin, A.; Perlman, H. A novel Ly6C/Ly6G-based strategy to analyze the mouse splenic myeloid compartment. Cytom. A 2012, 81, 343–350. [Google Scholar] [CrossRef] [Green Version]
- Jablonski, K.A.; Amici, S.A.; Webb, L.M.; Ruiz-Rosado Jde, D.; Popovich, P.G.; Partida-Sanchez, S.; Guerau-de-Arellano, M. Novel Markers to Delineate Murine M1 and M2 Macrophages. PLoS ONE 2015, 10, e0145342. [Google Scholar] [CrossRef] [Green Version]
- Dilek, N.; Vuillefroy de Silly, R.; Blancho, G.; Vanhove, B. Myeloid-derived suppressor cells: Mechanisms of action and recent advances in their role in transplant tolerance. Front. Immunol. 2012, 3, 208. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Li, Q.; Qin, L.; Zhao, S.; Wang, J.; Chen, X. Transition of tumor-associated macrophages from MHC class II(hi) to MHC class II(low) mediates tumor progression in mice. BMC Immunol. 2011, 12, 43. [Google Scholar] [CrossRef] [PubMed]
- De Groot, R.; Van Loenen, M.M.; Guislain, A.; Nicolet, B.P.; Freen-Van Heeren, J.J.; Verhagen, O.; Van Den Heuvel, M.M.; De Jong, J.; Burger, P.; Van Der Schoot, C.E.; et al. Polyfunctional tumor-reactive T cells are effectively expanded from non-small cell lung cancers, and correlate with an immune-engaged T cell profile. Oncoimmunology 2019, 8, e1648170. [Google Scholar] [CrossRef] [PubMed]
- Egelston, C.A.; Avalos, C.; Tu, T.Y.; Simons, D.L.; Jimenez, G.; Jung, J.Y.; Melstrom, L.; Margolin, K.; Yim, J.H.; Kruper, L.; et al. Human breast tumor-infiltrating CD8(+) T cells retain polyfunctionality despite PD-1 expression. Nat. Commun. 2018, 9, 4297. [Google Scholar] [CrossRef] [Green Version]
- Philip, M.; Fairchild, L.; Sun, L.; Horste, E.L.; Camara, S.; Shakiba, M.; Scott, A.C.; Viale, A.; Lauer, P.; Merghoub, T.; et al. Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature 2017, 545, 452–456. [Google Scholar] [CrossRef]
- Priyadharshini, B.; Welsh, R.M.; Greiner, D.L.; Gerstein, R.M.; Brehm, M.A. Maturation-dependent licensing of naive T cells for rapid TNF production. PLoS ONE 2010, 5, e15038. [Google Scholar] [CrossRef] [PubMed] [Green Version]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, S.M.Y.; Li, B.; Nicklawsky, A.G.; Krinsky, A.L.; Brunetti, T.; Woolaver, R.A.; Wang, X.; Chen, Z.; Young, C.D.; Gao, D.; et al. Deletion of p53 and Hyper-Activation of PIK3CA in Keratin-15+ Stem Cells Lead to the Development of Spontaneous Squamous Cell Carcinoma. Int. J. Mol. Sci. 2020, 21, 6585. https://doi.org/10.3390/ijms21186585
Chen SMY, Li B, Nicklawsky AG, Krinsky AL, Brunetti T, Woolaver RA, Wang X, Chen Z, Young CD, Gao D, et al. Deletion of p53 and Hyper-Activation of PIK3CA in Keratin-15+ Stem Cells Lead to the Development of Spontaneous Squamous Cell Carcinoma. International Journal of Molecular Sciences. 2020; 21(18):6585. https://doi.org/10.3390/ijms21186585
Chicago/Turabian StyleChen, Samantha M. Y., Bian Li, Andrew G. Nicklawsky, Alexandra L. Krinsky, Tonya Brunetti, Rachel A. Woolaver, Xiaoguang Wang, Zhangguo Chen, Christian D. Young, Dexiang Gao, and et al. 2020. "Deletion of p53 and Hyper-Activation of PIK3CA in Keratin-15+ Stem Cells Lead to the Development of Spontaneous Squamous Cell Carcinoma" International Journal of Molecular Sciences 21, no. 18: 6585. https://doi.org/10.3390/ijms21186585