Primary Humoral Immune Deficiencies: Overlooked Mimickers of Chronic Immune-Mediated Gastrointestinal Diseases in Adults

 , , ,
, , ,  , and
, and
Abstract
:1. Introduction
2. Selective Immunoglobulin A Deficiency
2.1. SIgAD: Epidemiology and Diagnostic Criteria
2.2. SIgAD: Etiology
2.3. SIgAD: Symptoms and Clinical Course
3. Common Variable Immunodeficiency
3.1. CVID: Definition and Epidemiology
3.2. CVID: Diagnostics and Criteria
3.3. CVID: Etiology
3.4. CVID: Symptoms and Clinical Course
4. SIgAD Progression to CVID
5. Celiac Disease
5.1. CeD: Definition and Epidemiology
5.2. CeD: Diagnostics
5.3. CeD: Symptoms
6. Celiac Disease in Selective IgA Deficiency
6.1. CeD in SIgAD: Epidemiology
6.2. CeD in SIgAD: Etiology
6.3. CeD in SIgAD: Diagnostic Difficulties
6.4. CeD in SIgAD: Symptoms
6.5. CeD in SIgAD: Treatment
7. Celiac Disease in Common Variable Immunodeficiency
7.1. CeD in CVID: Epidemiology
7.2. CeD in CVID: Etiology
7.3. CeD in CVID: Diagnostic Difficulties
7.4. CeD in CVID: Symptoms
7.5. CeD in CVID: Treatment
8. Inflammatory Bowel Disease
8.1. Ulcerative Colitis
8.1.1. UC: Definition and Epidemiology
8.1.2. UC: Etiology
8.1.3. UC: Symptoms
8.1.4. UC: Diagnostics
8.1.5. UC: Treatment
8.2. Crohn’s Disease
8.2.1. CD: Definition and Epidemiology
8.2.2. CD: Etiology
8.2.3. CD: Symptoms
8.2.4. CD: Diagnostics
8.2.5. CD: Treatment
9. Inflammatory Bowel Disease in Selective IgA Deficiency
9.1. IBD in SIgAD: Epidemiology
9.2. IBD in SIgAD: Etiology
9.3. IBD in SIgAD: Diagnostic Difficulties, Symptoms, and Treatment
10. Inflammatory Bowel Disease in Common Variable Immunodeficiency
10.1. IBD in CVID: Epidemiology
10.2. IBD in CVID: Etiology
10.3. IBD in CVID: Diagnostic Difficulties and Symptoms
10.4. IBD in CVID: Treatment
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CeD | Celiac disease |
| IBD | Inflammatory bowel disease |
| PID | Primary immunodeficiency |
| SIgAD | Selective IgA disease |
| CVID | Common variable immunodeficiency |
| GI | gastrointestinal |
| PAGID | Pan-American Group for Immunodeficiency |
| ESID | European Society for Immunodeficiencies |
| CSR | class switching recombination |
| Treg | regulatory T-cell |
| TGB-β | transforming growth factor-beta |
| BCR | B-cell receptor |
| DC | dendritic cell |
| MHC | major histocompatibility complex |
| Tfh | T follicular helper cell |
| HLA | human leukocyte antigen |
| SHM | somatic hypermutation |
| TLR | tool-like receptor |
| IFN-γ | interferon-gamma |
| Breg | regulatory B-cell |
| TNF | tumor necrosis factor |
| ILC | innate lymphoid cell |
| NK | natural-killer |
| GD | granulomatous disease |
| GFD | gluten-free diet |
| TG2 | transglutaminase-2 |
| EMA | endomysial antibodies |
| DGP | deamidated gliadin peptides |
| ESPGHAN | European Society Paediatric Gastroenterology, Hepatology and Nutrition |
| IEL | intraepithelial lymphocytes |
| ESsCD | European Society for the Study of Coeliac Disease |
| APC | antigen-presenting cell |
| TCRγδ | T-cell receptor γδ |
| pANCA | Perinuclear antineutrophil cytoplasmic |
| BSG | British Society of Gastroenterology |
| ACG | American College of Gastroenterology |
| ASCA | anti-Saccharomyces cerevisiae antibody |
References
- Molodecky, N.A.; Soon, I.S.; Rabi, D.M.; Ghali, W.A.; Ferris, M.; Chernoff, G.; Benchimol, E.I.; Panaccione, R.; Ghosh, S.; Barkema, H.W.; et al. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology 2012, 142, 46–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubio-Tapia, A.; Kyle, R.A.; Kaplan, E.L.; Johnson, D.R.; Page, W.; Erdtmann, F.; Brantner, T.L.; Kim, W.R.; Phelps, T.K.; Lahr, B.D.; et al. Increased prevalence and mortality in undiagnosed celiac disease. Gastroenterology 2009, 137, 88–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebwohl, B.; Sanders, D.S.; Green, P.H.R. Coeliac disease. Lancet Lond. Engl. 2018, 391, 70–81. [Google Scholar] [CrossRef]
- Torres, J.; Mehandru, S.; Colombel, J.-F.; Peyrin-Biroulet, L. Crohn’s disease. Lancet Lond. Engl. 2017, 389, 1741–1755. [Google Scholar] [CrossRef]
- Ungaro, R.; Mehandru, S.; Allen, P.B.; Peyrin-Biroulet, L.; Colombel, J.-F. Ulcerative colitis. Lancet Lond. Engl. 2017, 389, 1756–1770. [Google Scholar] [CrossRef]
- Agarwal, S.; Cunningham-Rundles, C. Gastrointestinal Manifestations and Complications of Primary Immunodeficiency Disorders. Immunol. Allergy Clin. N. Am. 2019, 39, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Azizi, G.; Ziaee, V.; Tavakol, M.; Alinia, T.; Yazdai, R.; Mohammadi, H.; Abolhassani, H.; Aghamohammadi, A. Approach to the Management of Autoimmunity in Primary Immunodeficiency. Scand. J. Immunol. 2017, 85, 13–29. [Google Scholar] [CrossRef] [Green Version]
- Schwimmer, D.; Glover, S. Primary Immunodeficiency and the Gut. Gastroenterol. Clin. N. Am. 2019, 48, 199–220. [Google Scholar] [CrossRef]
- Abolhassani, H.; Rezaei, N.; Mohammadinejad, P.; Mirminachi, B.; Hammarstrom, L.; Aghamohammadi, A. Important differences in the diagnostic spectrum of primary immunodeficiency in adults versus children. Expert Rev. Clin. Immunol. 2015, 11, 289–302. [Google Scholar] [CrossRef]
- Boyle, J.M.; Buckley, R.H. Population prevalence of diagnosed primary immunodeficiency diseases in the United States. J. Clin. Immunol. 2007, 27, 497–502. [Google Scholar] [CrossRef] [Green Version]
- Mansouri, D.; Adimi, P.; Mirsaedi, M.; Mansouri, N.; Tabarsi, P.; Amiri, M.; Jamaati, H.R.; Motavasseli, M.; Baghaii, N.; Cheraghvandi, A.; et al. Primary immune deficiencies presenting in adults: Seven years of experience from Iran. J. Clin. Immunol. 2005, 25, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Srinivasa, B.T.; Alizadehfar, R.; Desrosiers, M.; Shuster, J.; Pai, N.P.; Tsoukas, C.M. Adult primary immune deficiency: What are we missing? Am. J. Med. 2012, 125, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.; Mayer, L. Gastrointestinal manifestations in primary immune disorders. Inflamm. Bowel Dis. 2010, 16, 703–711. [Google Scholar] [CrossRef] [PubMed]
- Uhlig, H.H.; Schwerd, T.; Koletzko, S.; Shah, N.; Kammermeier, J.; Elkadri, A.; Ouahed, J.; Wilson, D.C.; Travis, S.P.; Turner, D.; et al. The diagnostic approach to monogenic very early onset inflammatory bowel disease. Gastroenterology 2014, 147, 990–1007. [Google Scholar] [CrossRef] [Green Version]
- Tegtmeyer, D.; Seidl, M.; Gerner, P.; Baumann, U.; Klemann, C. Inflammatory bowel disease caused by primary immunodeficiencies-Clinical presentations, review of literature, and proposal of a rational diagnostic algorithm. Pediatr. Allergy Immunol. Off. Publ. Eur. Soc. Pediatr. Allergy Immunol. 2017, 28, 412–429. [Google Scholar] [CrossRef]
- Modell, V.; Orange, J.S.; Quinn, J.; Modell, F. Global report on primary immunodeficiencies: 2018 update from the Jeffrey Modell Centers Network on disease classification, regional trends, treatment modalities, and physician reported outcomes. Immunol. Res. 2018, 66, 367–380. [Google Scholar] [CrossRef]
- Al-Attas, R.A.; Rahi, A.H. Primary antibody deficiency in Arabs: First report from eastern Saudi Arabia. J. Clin. Immunol. 1998, 18, 368–371. [Google Scholar] [CrossRef]
- Pan-Hammarström, Q.; Hammarström, L. Antibody deficiency diseases. Eur. J. Immunol. 2008, 38, 327–333. [Google Scholar] [CrossRef]
- Singh, K.; Chang, C.; Gershwin, M.E. IgA deficiency and autoimmunity. Autoimmun. Rev. 2014, 13, 163–177. [Google Scholar] [CrossRef]
- Jorgensen, G.H.; Gardulf, A.; Sigurdsson, M.I.; Sigurdardottir, S.T.; Thorsteinsdottir, I.; Gudmundsson, S.; Hammarström, L.; Ludviksson, B.R. Clinical symptoms in adults with selective IgA deficiency: A case-control study. J. Clin. Immunol. 2013, 33, 742–747. [Google Scholar] [CrossRef]
- Yel, L. Selective IgA deficiency. J. Clin. Immunol. 2010, 30, 10–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conley, M.E.; Notarangelo, L.D.; Etzioni, A. Diagnostic criteria for primary immunodeficiencies. Representing PAGID (Pan-American Group for Immunodeficiency) and ESID (European Society for Immunodeficiencies). Clin. Immunol. 1999, 93, 190–197. [Google Scholar] [CrossRef] [PubMed]
- European Society for Immunodeficiencies. Available online: https://esid.org/Working-Parties/Registry-Working-Party/Diagnosis-criteria (accessed on 24 June 2020).
- Hammarström, L.; Vorechovsky, I.; Webster, D. Selective IgA deficiency (SIgAD) and common variable immunodeficiency (CVID). Clin. Exp. Immunol. 2000, 120, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Abolhassani, H.; Gharib, B.; Shahinpour, S.; Masoom, S.N.; Havaei, A.; Mirminachi, B.; Arandi, N.; Torabi-Sagvand, B.; Khazaei, H.A.; Mohammadi, J.; et al. Autoimmunity in patients with selective IgA deficiency. J. Investig. Allergol. Clin. Immunol. 2015, 25, 112–119. [Google Scholar]
- Bonilla, F.A.; Khan, D.A.; Ballas, Z.K.; Chinen, J.; Frank, M.M.; Hsu, J.T.; Keller, M.; Kobrynski, L.J.; Komarow, H.D.; Mazer, B.; et al. Practice parameter for the diagnosis and management of primary immunodeficiency. J. Allergy Clin. Immunol. 2015, 136, 1186–1205. [Google Scholar] [CrossRef] [Green Version]
- Woof, J.M.; Kerr, M.A. The function of immunoglobulin A in immunity. J. Pathol. 2006, 208, 270–282. [Google Scholar] [CrossRef] [Green Version]
- Soheili, H.; Abolhassani, H.; Arandi, N.; Khazaei, H.A.; Shahinpour, S.; Hirbod-Mobarakeh, A.; Rezaei, N.; Aghamohammadi, A. Evaluation of natural regulatory T cells in subjects with selective IgA deficiency: From senior idea to novel opportunities. Int. Arch. Allergy Immunol. 2013, 160, 208–214. [Google Scholar] [CrossRef]
- Cunningham-Rundles, C. Physiology of IgA and IgA deficiency. J. Clin. Immunol. 2001, 21, 303–309. [Google Scholar] [CrossRef]
- Bagheri, Y.; Sanaei, R.; Yazdani, R.; Shekarabi, M.; Falak, R.; Mohammadi, J.; Abolhassani, H.; Aghamohammadi, A. The Heterogeneous Pathogenesis of Selective Immunoglobulin A Deficiency. Int. Arch. Allergy Immunol. 2019, 179, 231–246. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yunis, D.; Irigoyen, M.; Kitchens, B.; Bottaro, A.; Alt, F.W.; Alper, C.A. Discordance between IgA switching at the DNA level and IgA expression at the mRNA level in IgA-deficient patients. Clin. Immunol. 1999, 91, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Yazdani, R.; Fatholahi, M.; Ganjalikhani-Hakemi, M.; Abolhassani, H.; Azizi, G.; Hamid, K.M.; Rezaei, N.; Aghamohammadi, A. Role of apoptosis in common variable immunodeficiency and selective immunoglobulin A deficiency. Mol. Immunol. 2016, 71, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Conley, M.E.; Cooper, M.D. Immature IgA B cells in IgA-deficient patients. N. Engl. J. Med. 1981, 305, 495–497. [Google Scholar] [CrossRef] [PubMed]
- Nilssen, D.E.; Friman, V.; Theman, K.; Björkander, J.; Kilander, A.; Holmgren, J.; Hanson, L.A.; Brandtzaeg, P. B-cell activation in duodenal mucosa after oral cholera vaccination in IgA deficient subjects with or without IgG subclass deficiency. Scand. J. Immunol. 1993, 38, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Hammarström, L.; Lönnqvist, B.; Ringdén, O.; Smith, C.I.; Wiebe, T. Transfer of IgA deficiency to a bone-marrow-grafted patient with aplastic anaemia. Lancet Lond. Engl. 1985, 1, 778–781. [Google Scholar] [CrossRef]
- Grosserichter-Wagener, C.; Franco-Gallego, A.; Ahmadi, F.; Moncada-Vélez, M.; Dalm, V.A.; Rojas, J.L.; Orrego, J.C.; Correa Vargas, N.; Hammarström, L.; Schreurs, M.W.; et al. Defective formation of IgA memory B cells, Th1 and Th17 cells in symptomatic patients with selective IgA deficiency. Clin. Transl. Immunol. 2020, 9, e1130. [Google Scholar] [CrossRef]
- Azizi, G.; Tavakol, M.; Rafiemanesh, H.; Kiaee, F.; Yazdani, R.; Heydari, A.; Abouhamzeh, K.; Anvari, P.; Mohammadikhajehdehi, S.; Sharifia, L.; et al. Autoimmunity in a cohort of 471 patients with primary antibody deficiencies. Expert Rev. Clin. Immunol. 2017, 13, 1099–1106. [Google Scholar] [CrossRef]
- Lemarquis, A.L.; Einarsdottir, H.K.; Kristjansdottir, R.N.; Jonsdottir, I.; Ludviksson, B.R. Transitional B Cells and TLR9 Responses Are Defective in Selective IgA Deficiency. Front. Immunol. 2018, 9, 909. [Google Scholar] [CrossRef] [Green Version]
- Asano, T.; Kaneko, H.; Terada, T.; Kasahara, Y.; Fukao, T.; Kasahara, K.; Kondo, N. Molecular analysis of B-cell differentiation in selective or partial IgA deficiency. Clin. Exp. Immunol. 2004, 136, 284–290. [Google Scholar] [CrossRef]
- Yazdani, R.; Azizi, G.; Abolhassani, H.; Aghamohammadi, A. Selective IgA Deficiency: Epidemiology, Pathogenesis, Clinical Phenotype, Diagnosis, Prognosis and Management. Scand. J. Immunol. 2017, 85, 3–12. [Google Scholar] [CrossRef]
- Borte, S.; Pan-Hammarström, Q.; Liu, C.; Sack, U.; Borte, M.; Wagner, U.; Graf, D.; Hammarström, L. Interleukin-21 restores immunoglobulin production ex vivo in patients with common variable immunodeficiency and selective IgA deficiency. Blood 2009, 114, 4089–4098. [Google Scholar] [CrossRef]
- Jorgensen, G.H.; Ornolfsson, A.E.; Johannesson, A.; Gudmundsson, S.; Janzi, M.; Wang, N.; Hammarström, L.; Ludviksson, B.R. Association of immunoglobulin A deficiency and elevated thyrotropin-receptor autoantibodies in two Nordic countries. Hum. Immunol. 2011, 72, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Price, P.; Witt, C.; Allcock, R.; Sayer, D.; Garlepp, M.; Kok, C.C.; French, M.; Mallal, S.; Christiansen, F. The genetic basis for the association of the 8.1 ancestral haplotype (A1, B8, DR3) with multiple immunopathological diseases. Immunol. Rev. 1999, 167, 257–274. [Google Scholar] [CrossRef] [PubMed]
- Ramanujam, R.; Piehl, F.; Pirskanen, R.; Gregersen, P.K.; Hammarström, L. Concomitant autoimmunity in myasthenia gravis-lack of association with IgA deficiency. J. Neuroimmunol. 2011, 236, 118–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rioux, J.D.; Goyette, P.; Vyse, T.J.; Hammarström, L.; Fernando, M.M.A.; Green, T.; De Jager, P.L.; Foisy, S.; Wang, J.; Leslie, S.; et al. Mapping of multiple susceptibility variants within the MHC region for 7 immune-mediated diseases. Proc. Natl. Acad. Sci. USA 2009, 106, 18680–18685. [Google Scholar] [CrossRef] [Green Version]
- Mohammadi, J.; Hammarstrom, L. Genetic Association in Familial Common Variable Immunodeficiency (CVID) and IgA Deficiency (IgAD). Immunol. Genet. J. 2018, 1, 24–33. [Google Scholar] [CrossRef]
- Abolhassani, H.; Aghamohammadi, A.; Hammarström, L. Monogenic mutations associated with IgA deficiency. Expert Rev. Clin. Immunol. 2016, 12, 1321–1335. [Google Scholar] [CrossRef]
- Latiff, A.H.A.; Kerr, M.A. The clinical significance of immunoglobulin A deficiency. Ann. Clin. Biochem. 2007, 44, 131–139. [Google Scholar] [CrossRef] [Green Version]
- Saiga, T.; Hashimoto, K.; Kimura, N.; Ono, H.; Hiai, H. Trisomy 10p and translocation of 10q to 4p associated with selective dysgenesis of IgA-producing cells in lymphoid tissue. Pathol. Int. 2007, 57, 37–42. [Google Scholar] [CrossRef]
- Lewkonia, R.M.; Lin, C.C.; Haslam, R.H. Selective IgA deficiency with 18q+ and 18q- karyotypic anomalies. J. Med. Genet. 1980, 17, 453–456. [Google Scholar] [CrossRef] [Green Version]
- Schatorjé, E.; van der Flier, M.; Seppänen, M.; Browning, M.; Morsheimer, M.; Henriet, S.; Neves, J.F.; Vinh, D.C.; Alsina, L.; Grumach, A.; et al. Primary immunodeficiency associated with chromosomal aberration—An ESID survey. Orphanet J. Rare Dis. 2016, 11, 110. [Google Scholar] [CrossRef] [Green Version]
- Dostal, A.; Linnankivi, T.; Somer, M.; Kähkönen, M.; Litzman, J.; Tienari, P. Mapping susceptibility gene locus for IgA deficiency at del(18)(q22.3-q23); report of familial cryptic chromosome t(18q; 10p) translocations. Int. J. Immunogenet. 2007, 34, 143–147. [Google Scholar] [CrossRef] [PubMed]
- Hanley-Lopez, J.; Estabrooks, L.L.; Stiehm, R. Antibody deficiency in Wolf-Hirschhorn syndrome. J. Pediatr. 1998, 133, 141–143. [Google Scholar] [CrossRef]
- Kubinak, J.L.; Round, J.L. Do antibodies select a healthy microbiota? Nat. Rev. Immunol. 2016, 16, 767–774. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, A.D.; Khan, M.A.W.; Chatzistamou, I.; Chamseddine, D.; Williams-Kang, K.; Perry, M.; Enos, R.; Murphy, A.; Gomez, G.; Aladhami, A.; et al. Gut Antibody Deficiency in a Mouse Model of CVID Results in Spontaneous Development of a Gluten-Sensitive Enteropathy. Front. Immunol. 2019, 10, 2484. [Google Scholar] [CrossRef] [Green Version]
- Catanzaro, J.R.; Strauss, J.D.; Bielecka, A.; Porto, A.F.; Lobo, F.M.; Urban, A.; Schofield, W.B.; Palm, N.W. IgA-deficient humans exhibit gut microbiota dysbiosis despite secretion of compensatory IgM. Sci. Rep. 2019, 9, 13574. [Google Scholar] [CrossRef]
- Yazdani, R.; Latif, A.; Tabassomi, F.; Abolhassani, H.; Azizi, G.; Rezaei, N.; Aghamohammadi, A. Clinical phenotype classification for selective immunoglobulin A deficiency. Expert Rev. Clin. Immunol. 2015, 11, 1245–1254. [Google Scholar] [CrossRef]
- Oxelius, V.A.; Laurell, A.B.; Lindquist, B.; Golebiowska, H.; Axelsson, U.; Björkander, J.; Hanson, L.A. IgG subclasses in selective IgA deficiency: Importance of IgG2-IgA deficiency. N. Engl. J. Med. 1981, 304, 1476–1477. [Google Scholar] [CrossRef]
- Björkander, J.; Bake, B.; Oxelius, V.A.; Hanson, L.A. Impaired lung function in patients with IgA deficiency and low levels of IgG2 or IgG3. N. Engl. J. Med. 1985, 313, 720–724. [Google Scholar] [CrossRef]
- Urm, S.-H.; Yun, H.D.; Fenta, Y.A.; Yoo, K.H.; Abraham, R.S.; Hagan, J.; Juhn, Y.J. Asthma and risk of selective IgA deficiency or common variable immunodeficiency: A population-based case-control study. Mayo Clin. Proc. 2013, 88, 813–821. [Google Scholar] [CrossRef] [Green Version]
- Pabst, O. New concepts in the generation and functions of IgA. Nat. Rev. Immunol. 2012, 12, 821–832. [Google Scholar] [CrossRef]
- Agarwal, S.; Mayer, L. Diagnosis and treatment of gastrointestinal disorders in patients with primary immunodeficiency. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2013, 11, 1050–1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarmiento, E.; Mora, R.; Rodríguez-Mahou, M.; Rodríguez-Molina, J.; Fernández-Cruz, E.; Carbone, J. Autoimmune disease in primary antibody deficiencies. Allergol. Immunopathol. 2005, 33, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Erkoçoğlu, M.; Metin, A.; Kaya, A.; Özcan, C.; Akan, A.; Civelek, E.; Çapanoğlu, M.; Giniş, T.; Kocabaş, C.N. Allergic and autoimmune disorders in families with selective IgA deficiency. Turk. J. Med. Sci. 2017, 47, 592–598. [Google Scholar] [CrossRef]
- Aghamohammadi, A.; Abolhassani, H.; Biglari, M.; Abolmaali, S.; Moazzami, K.; Tabatabaeiyan, M.; Asgarian-Omran, H.; Parvaneh, N.; Mirahmadian, M.; Rezaei, N. Analysis of switched memory B cells in patients with IgA deficiency. Int. Arch. Allergy Immunol. 2011, 156, 462–468. [Google Scholar] [CrossRef] [PubMed]
- Shkalim, V.; Monselize, Y.; Segal, N.; Zan-Bar, I.; Hoffer, V.; Garty, B.Z. Selective IgA deficiency in children in Israel. J. Clin. Immunol. 2010, 30, 761–765. [Google Scholar] [CrossRef] [PubMed]
- Jacob, C.M.A.; Pastorino, A.C.; Fahl, K.; Carneiro-Sampaio, M.; Monteiro, R.C. Autoimmunity in IgA deficiency: Revisiting the role of IgA as a silent housekeeper. J. Clin. Immunol. 2008, 28 (Suppl. 1), S56–S61. [Google Scholar] [CrossRef]
- Odineal, D.D.; Gershwin, M.E. The Epidemiology and Clinical Manifestations of Autoimmunity in Selective IgA Deficiency. Clin. Rev. Allergy Immunol. 2020, 58, 107–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, N.; Hammarström, L. IgA deficiency: What is new? Curr. Opin. Allergy Clin. Immunol. 2012, 12, 602–608. [Google Scholar] [CrossRef] [PubMed]
- Vorechovský, I.; Webster, A.D.; Plebani, A.; Hammarström, L. Genetic linkage of IgA deficiency to the major histocompatibility complex: Evidence for allele segregation distortion, parent-of-origin penetrance differences, and the role of anti-IgA antibodies in disease predisposition. Am. J. Hum. Genet. 1999, 64, 1096–1109. [Google Scholar] [CrossRef] [Green Version]
- Nechvatalova, J.; Pikulova, Z.; Stikarovska, D.; Pesak, S.; Vlkova, M.; Litzman, J. B-lymphocyte subpopulations in patients with selective IgA deficiency. J. Clin. Immunol. 2012, 32, 441–448. [Google Scholar] [CrossRef]
- Haimila, K.; Einarsdottir, E.; de Kauwe, A.; Koskinen, L.L.E.; Pan-Hammarström, Q.; Kaartinen, T.; Kurppa, K.; Ziberna, F.; Not, T.; Vatta, S.; et al. The shared CTLA4-ICOS risk locus in celiac disease, IgA deficiency and common variable immunodeficiency. Genes Immun. 2009, 10, 151–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bronson, P.G.; Chang, D.; Bhangale, T.; Seldin, M.F.; Ortmann, W.; Ferreira, R.C.; Urcelay, E.; Pereira, L.F.; Martin, J.; Plebani, A.; et al. Common variants at PVT1, ATG13-AMBRA1, AHI1 and CLEC16A are associated with selective IgA deficiency. Nat. Genet. 2016, 48, 1425–1429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domínguez, O.; Giner, M.T.; Alsina, L.; Martín, M.A.; Lozano, J.; Plaza, A.M. Clinical phenotypes associated with selective IgA deficiency: A review of 330 cases and a proposed follow-up protocol. An. Pediatr. Barc. Spain 2012, 76, 261–267. [Google Scholar] [CrossRef]
- Koskinen, S. Long-term follow-up of health in blood donors with primary selective IgA deficiency. J. Clin. Immunol. 1996, 16, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Burgio, G.R.; Duse, M.; Monafo, V.; Ascione, A.; Nespoli, L. Selective IgA deficiency: Clinical and immunological evaluation of 50 pediatric patients. Eur. J. Pediatr. 1980, 133, 101–106. [Google Scholar] [CrossRef]
- Gualdi, G.; Lougaris, V.; Baronio, M.; Vitali, M.; Tampella, G.; Moratto, D.; Tanghetti, P.; Monari, P.; Calzavara-Pinton, P.; Plebani, A. Burden of Skin Disease in Selective IgA Deficiency and Common Variable Immunodeficiency. J. Investig. Allergol. Clin. Immunol. 2015, 25, 369–371. [Google Scholar]
- Mortaz, E.; Tabarsi, P.; Mansouri, D.; Khosravi, A.; Garssen, J.; Velayati, A.; Adcock, I.M. Cancers Related to Immunodeficiencies: Update and Perspectives. Front. Immunol. 2016, 7, 365. [Google Scholar] [CrossRef] [Green Version]
- Riaz, I.B.; Faridi, W.; Patnaik, M.M.; Abraham, R.S. A Systematic Review on Predisposition to Lymphoid (B and T cell) Neoplasias in Patients With Primary Immunodeficiencies and Immune Dysregulatory Disorders (Inborn Errors of Immunity). Front. Immunol. 2019, 10, 777. [Google Scholar] [CrossRef]
- Ludvigsson, J.F.; Neovius, M.; Ye, W.; Hammarström, L. IgA deficiency and risk of cancer: A population-based matched cohort study. J. Clin. Immunol. 2015, 35, 182–188. [Google Scholar] [CrossRef]
- Edwards, E.; Razvi, S.; Cunningham-Rundles, C. IgA deficiency: Clinical correlates and responses to pneumococcal vaccine. Clin. Immunol. 2004, 111, 93–97. [Google Scholar] [CrossRef]
- Kersey, J.H.; Shapiro, R.S.; Filipovich, A.H. Relationship of immunodeficiency to lymphoid malignancy. Pediatr. Infect. Dis. J. 1988, 7, S10–S12. [Google Scholar] [CrossRef] [PubMed]
- Mellemkjaer, L.; Hammarstrom, L.; Andersen, V.; Yuen, J.; Heilmann, C.; Barington, T.; Bjorkander, J.; Olsen, J.H. Cancer risk among patients with IgA deficiency or common variable immunodeficiency and their relatives: A combined Danish and Swedish study. Clin. Exp. Immunol. 2002, 130, 495–500. [Google Scholar] [CrossRef]
- Yazdani, R.; Habibi, S.; Sharifi, L.; Azizi, G.; Abolhassani, H.; Olbrich, P.; Aghamohammadi, A. Common Variable Immunodeficiency: Epidemiology, Pathogenesis, Clinical Manifestations, Diagnosis, Classification, and Management. J. Investig. Allergol. Clin. Immunol. 2020, 30, 14–34. [Google Scholar] [CrossRef] [Green Version]
- Cunningham-Rundles, C.; Maglione, P.J. Common variable immunodeficiency. J. Allergy Clin. Immunol. 2012, 129, 1425–1426. [Google Scholar] [CrossRef]
- Bonilla, F.A.; Barlan, I.; Chapel, H.; Costa-Carvalho, B.T.; Cunningham-Rundles, C.; de la Morena, M.T.; Espinosa-Rosales, F.J.; Hammarström, L.; Nonoyama, S.; Quinti, I.; et al. International Consensus Document (ICON): Common Variable Immunodeficiency Disorders. J. Allergy Clin. Immunol. Pract. 2016, 4, 38–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picard, C.; Bobby Gaspar, H.; Al-Herz, W.; Bousfiha, A.; Casanova, J.-L.; Chatila, T.; Crow, Y.J.; Cunningham-Rundles, C.; Etzioni, A.; Franco, J.L.; et al. International Union of Immunological Societies: 2017 Primary Immunodeficiency Diseases Committee Report on Inborn Errors of Immunity. J. Clin. Immunol. 2018, 38, 96–128. [Google Scholar] [CrossRef] [Green Version]
- Aggarwal, V.; Banday, A.Z.; Jindal, A.K.; Das, J.; Rawat, A. Recent advances in elucidating the genetics of common variable immunodeficiency. Genes Dis. 2020, 7, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Shillitoe, B.; Bangs, C.; Guzman, D.; Gennery, A.R.; Longhurst, H.J.; Slatter, M.; Edgar, D.M.; Thomas, M.; Worth, A.; Huissoon, A.; et al. The United Kingdom Primary Immune Deficiency (UKPID) registry 2012 to 2017. Clin. Exp. Immunol. 2018, 192, 284–291. [Google Scholar] [CrossRef] [Green Version]
- Saikia, B.; Gupta, S. Common Variable Immunodeficiency. Indian J. Pediatr. 2016, 83, 338–344. [Google Scholar] [CrossRef]
- Aghamohammadi, A.; Abolhassani, H.; Moazzami, K.; Parvaneh, N.; Rezaei, N. Correlation between common variable immunodeficiency clinical phenotypes and parental consanguinity in children and adults. J. Investig. Allergol. Clin. Immunol. 2010, 20, 372–379. [Google Scholar]
- Karaca, N.E.; Severcan, E.U.; Bilgin, B.G.; Azarsiz, E.; Akarcan, S.; Gunaydın, N.C.; Gulez, N.; Genel, F.; Aksu, G.; Kutukculer, N. Familial inheritance and screening of first-degree relatives in common variable immunodeficiency and immunoglobulin A deficiency patients. Int. J. Immunopathol. Pharmacol. 2018, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uzzan, M.; Ko, H.M.; Mehandru, S.; Cunningham-Rundles, C. Gastrointestinal Disorders Associated with Common Variable Immune Deficiency (CVID) and Chronic Granulomatous Disease (CGD). Curr. Gastroenterol. Rep. 2016, 18, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwal, S.; Cunningham-Rundles, C. Autoimmunity in common variable immunodeficiency. Curr. Allergy Asthma Rep. 2009, 9, 347–352. [Google Scholar] [CrossRef] [PubMed]
- Seidel, M.G.; Kindle, G.; Gathmann, B.; Quinti, I.; Buckland, M.; van Montfrans, J.; Scheible, R.; Rusch, S.; Gasteiger, L.M.; Grimbacher, B.; et al. The European Society for Immunodeficiencies (ESID) Registry Working Definitions for the Clinical Diagnosis of Inborn Errors of Immunity. J. Allergy Clin. Immunol. Pract. 2019, 7, 1763–1770. [Google Scholar] [CrossRef] [PubMed]
- Bousfiha, A.; Jeddane, L.; Picard, C.; Ailal, F.; Bobby, G.H.; Al-Herz, W.; Chatila, T.; Crow, Y.J.; Cunningham-Rundles, C.; Etzioni, A.; et al. The 2017 IUIS Phenotypic Classification for Primary Immunodeficiencies. J. Clin. Immunol. 2018, 38, 129–143. [Google Scholar] [CrossRef] [Green Version]
- Ahn, S.; Cunningham-Rundles, C. Role of B cells in common variable immune deficiency. Expert Rev. Clin. Immunol. 2009, 5, 557–564. [Google Scholar] [CrossRef] [Green Version]
- Warnatz, K.; Schlesier, M. Flowcytometric phenotyping of common variable immunodeficiency. Cytom. B Clin. Cytom. 2008, 74, 261–271. [Google Scholar] [CrossRef]
- Romberg, N.; Chamberlain, N.; Saadoun, D.; Gentile, M.; Kinnunen, T.; Ng, Y.S.; Virdee, M.; Menard, L.; Cantaert, T.; Morbach, H.; et al. CVID-associated TACI mutations affect autoreactive B cell selection and activation. J. Clin. Investig. 2013, 123, 4283–4293. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.E.; Knight, A.K.; Radigan, L.; Marron, T.U.; Zhang, L.; Sanchez-Ramón, S.; Cunningham-Rundles, C. Toll-like receptor 7 and 9 defects in common variable immunodeficiency. J. Allergy Clin. Immunol. 2009, 124, 349–356. [Google Scholar] [CrossRef] [Green Version]
- Romberg, N.; Lawrence, M.G. Birds of a feather: Common variable immune deficiencies. Ann. Allergy Asthma Immunol. Off. Publ. Am. Coll. Allergy Asthma Immunol. 2019, 123, 461–467. [Google Scholar] [CrossRef] [Green Version]
- Bryant, A.; Calver, N.C.; Toubi, E.; Webster, A.D.; Farrant, J. Classification of patients with common variable immunodeficiency by B cell secretion of IgM and IgG in response to anti-IgM and interleukin-2. Clin. Immunol. Immunopathol. 1990, 56, 239–248. [Google Scholar] [CrossRef]
- Van Schouwenburg, P.A.; IJspeert, H.; Pico-Knijnenburg, I.; Dalm, V.A.S.H.; van Hagen, P.M.; van Zessen, D.; Stubbs, A.P.; Patel, S.Y.; van der Burg, M. Identification of CVID Patients With Defects in Immune Repertoire Formation or Specification. Front. Immunol. 2018, 9, 2545. [Google Scholar] [CrossRef] [PubMed]
- Carsetti, R.; Rosado, M.M.; Donnanno, S.; Guazzi, V.; Soresina, A.; Meini, A.; Plebani, A.; Aiuti, F.; Quinti, I. The loss of IgM memory B cells correlates with clinical disease in common variable immunodeficiency. J. Allergy Clin. Immunol. 2005, 115, 412–417. [Google Scholar] [CrossRef] [PubMed]
- Warnatz, K.; Denz, A.; Dräger, R.; Braun, M.; Groth, C.; Wolff-Vorbeck, G.; Eibel, H.; Schlesier, M.; Peter, H.H. Severe deficiency of switched memory B cells (CD27(+)IgM(-)IgD(-)) in subgroups of patients with common variable immunodeficiency: A new approach to classify a heterogeneous disease. Blood 2002, 99, 1544–1551. [Google Scholar] [CrossRef] [Green Version]
- Piqueras, B.; Lavenu-Bombled, C.; Galicier, L.; Bergeron-van der Cruyssen, F.; Mouthon, L.; Chevret, S.; Debré, P.; Schmitt, C.; Oksenhendler, E. Common variable immunodeficiency patient classification based on impaired B cell memory differentiation correlates with clinical aspects. J. Clin. Immunol. 2003, 23, 385–400. [Google Scholar] [CrossRef] [PubMed]
- Ganjalikhani-Hakemi, M.; Yazdani, R.; Esmaeili, M.; Abolhassani, H.; Rae, W.; Azizi, G.; Dizaji, M.Z.; Shaghaghi, M.; Rezaei, A.; Abbasi-Rad, F.; et al. Role of Apoptosis in the Pathogenesis of Common Variable Immunodeficiency (CVID). Endocr. Metab. Immune Disord. Drug Targets 2017, 17, 332–340. [Google Scholar] [CrossRef]
- López-Gómez, A.; Clemente, A.; Cunill, V.; Pons, J.; Ferrer, J.M. IL-21 and anti-CD40 restore Bcl-2 family protein imbalance in vitro in low-survival CD27+ B cells from CVID patients. Cell Death Dis. 2018, 9, 1156. [Google Scholar] [CrossRef]
- Warnatz, K.; Salzer, U.; Rizzi, M.; Fischer, B.; Gutenberger, S.; Böhm, J.; Kienzler, A.-K.; Pan-Hammarström, Q.; Hammarström, L.; Rakhmanov, M.; et al. B-cell activating factor receptor deficiency is associated with an adult-onset antibody deficiency syndrome in humans. Proc. Natl. Acad. Sci. USA 2009, 106, 13945–13950. [Google Scholar] [CrossRef] [Green Version]
- Unger, S.; Seidl, M.; van Schouwenburg, P.; Rakhmanov, M.; Bulashevska, A.; Frede, N.; Grimbacher, B.; Pfeiffer, J.; Schrenk, K.; Munoz, L.; et al. The TH1 phenotype of follicular helper T cells indicates an IFN-γ-associated immune dysregulation in patients with CD21low common variable immunodeficiency. J. Allergy Clin. Immunol. 2018, 141, 730–740. [Google Scholar] [CrossRef] [Green Version]
- Thorarinsdottir, K.; Camponeschi, A.; Gjertsson, I.; Mårtensson, I.-L. CD21-/low B cells: A Snapshot of a Unique B Cell Subset in Health and Disease. Scand. J. Immunol. 2015, 82, 254–261. [Google Scholar] [CrossRef]
- Ochtrop, M.L.G.; Goldacker, S.; May, A.M.; Rizzi, M.; Draeger, R.; Hauschke, D.; Stehfest, C.; Warnatz, K.; Goebel, H.; Technau-Ihling, K.; et al. T and B lymphocyte abnormalities in bone marrow biopsies of common variable immunodeficiency. Blood 2011, 118, 309–318. [Google Scholar] [CrossRef] [Green Version]
- Bateman, E.A.L.; Ayers, L.; Sadler, R.; Lucas, M.; Roberts, C.; Woods, A.; Packwood, K.; Burden, J.; Harrison, D.; Kaenzig, N.; et al. T cell phenotypes in patients with common variable immunodeficiency disorders: Associations with clinical phenotypes in comparison with other groups with recurrent infections. Clin. Exp. Immunol. 2012, 170, 202–211. [Google Scholar] [CrossRef] [PubMed]
- Oraei, M.; Aghamohammadi, A.; Rezaei, N.; Bidad, K.; Gheflati, Z.; Amirkhani, A.; Abolhassani, H.; Massoud, A. Naive CD4+ T cells and recent thymic emigrants in common variable immunodeficiency. J. Investig. Allergol. Clin. Immunol. 2012, 22, 160–167. [Google Scholar] [PubMed]
- Azizi, G.; Rezaei, N.; Kiaee, F.; Tavakolinia, N.; Yazdani, R.; Mirshafiey, A.; Aghamohammadi, A. T-Cell Abnormalities in Common Variable Immunodeficiency. J. Investig. Allergol. Clin. Immunol. 2016, 26, 233–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Saos-Patrinos, C.; Loizon, S.; Blanco, P.; Viallard, J.-F.; Duluc, D. Functions of Tfh Cells in Common Variable Immunodeficiency. Front. Immunol. 2020, 11, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giovannetti, A.; Pierdominici, M.; Mazzetta, F.; Marziali, M.; Renzi, C.; Mileo, A.M.; De Felice, M.; Mora, B.; Esposito, A.; Carello, R.; et al. Unravelling the complexity of T cell abnormalities in common variable immunodeficiency. J. Immunol. 2007, 178, 3932–3943. [Google Scholar] [CrossRef]
- Vlkova, M.; Ticha, O.; Nechvatalova, J.; Kalina, T.; Litzman, J.; Mauri, C.; Blair, P.A. Regulatory B cells in CVID patients fail to suppress multifunctional IFN-γ+ TNF-α+ CD4+ T cells differentiation. Clin. Immunol. 2015, 160, 292–300. [Google Scholar] [CrossRef]
- Carbone, J.; Sarmiento, E.; Micheloud, D.; Rodríguez-Molina, J.; Fernández-Cruz, E. Elevated levels of activated CD4 T cells in common variable immunodeficiency: Association with clinical findings. Allergol. Immunopathol. 2006, 34, 131–135. [Google Scholar] [CrossRef]
- Viallard, J.-F.; Ruiz, C.; Guillet, M.; Pellegrin, J.-L.; Moreau, J.-F. Perturbations of the CD8(+) T-cell repertoire in CVID patients with complications. Results Immunol. 2013, 3, 122–128. [Google Scholar] [CrossRef] [Green Version]
- Rezaei, N.; Haji-Molla-Hoseini, M.; Aghamohammadi, A.; Pourfathollah, A.A.; Moghtadaie, M.; Pourpak, Z. Increased serum levels of soluble CD30 in patients with common variable immunodeficiency and its clinical implications. J. Clin. Immunol. 2008, 28, 78–84. [Google Scholar] [CrossRef]
- North, M.E.; Ivory, K.; Funauchi, M.; Webster, A.D.; Lane, A.C.; Farrant, J. Intracellular cytokine production by human CD4+ and CD8+ T cells from normal and immunodeficient donors using directly conjugated anti-cytokine antibodies and three-colour flow cytometry. Clin. Exp. Immunol. 1996, 105, 517–522. [Google Scholar] [CrossRef] [PubMed]
- North, M.E.; Webster, A.D.; Farrant, J. Primary defect in CD8+ lymphocytes in the antibody deficiency disease (common variable immunodeficiency): Abnormalities in intracellular production of interferon-gamma (IFN-gamma) in CD28+ (‘cytotoxic’) and CD28- (‘suppressor’) CD8+ subsets. Clin. Exp. Immunol. 1998, 111, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Cambronero, R.; Sewell, W.A.; North, M.E.; Webster, A.D.; Farrant, J. Up-regulation of IL-12 in monocytes: A fundamental defect in common variable immunodeficiency. J. Immunol. 2000, 164, 488–494. [Google Scholar] [CrossRef]
- Bayry, J.; Lacroix-Desmazes, S.; Kazatchkine, M.D.; Galicier, L.; Lepelletier, Y.; Webster, D.; Lévy, Y.; Eibl, M.M.; Oksenhendler, E.; Hermine, O.; et al. Common variable immunodeficiency is associated with defective functions of dendritic cells. Blood 2004, 104, 2441–2443. [Google Scholar] [CrossRef] [PubMed]
- Cunningham-Rundles, C.; Radigan, L. Deficient IL-12 and dendritic cell function in common variable immune deficiency. Clin. Immunol. 2005, 115, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, R.R.; Silva, S.P.; Silva, S.L.; Melo, A.C.; Pedro, E.; Barbosa, M.P.; Pereira-Santos, M.C.; Victorino, R.M.M.; Sousa, A.E. Primary B-cell deficiencies reveal a link between human IL-17-producing CD4 T-cell homeostasis and B-cell differentiation. PLoS ONE 2011, 6, e22848. [Google Scholar] [CrossRef] [PubMed]
- Kutukculer, N.; Azarsiz, E.; Aksu, G.; Karaca, N.E. CD4+CD25+Foxp3+ T regulatory cells, Th1 (CCR5, IL-2, IFN-γ) and Th2 (CCR4, IL-4, Il-13) type chemokine receptors and intracellular cytokines in children with common variable immunodeficiency. Int. J. Immunopathol. Pharmacol. 2016, 29, 241–251. [Google Scholar] [CrossRef] [Green Version]
- Arandi, N.; Mirshafiey, A.; Abolhassani, H.; Jeddi-Tehrani, M.; Edalat, R.; Sadeghi, B.; Shaghaghi, M.; Aghamohammadi, A. Frequency and expression of inhibitory markers of CD4(+) CD25(+) FOXP3(+) regulatory T cells in patients with common variable immunodeficiency. Scand. J. Immunol. 2013, 77, 405–412. [Google Scholar] [CrossRef]
- Genre, J.; Errante, P.R.; Kokron, C.M.; Toledo-Barros, M.; Câmara, N.O.S.; Rizzo, L.V. Reduced frequency of CD4(+)CD25(HIGH)FOXP3(+) cells and diminished FOXP3 expression in patients with Common Variable Immunodeficiency: A link to autoimmunity? Clin. Immunol. 2009, 132, 215–221. [Google Scholar] [CrossRef]
- Yu, G.P.; Chiang, D.; Song, S.J.; Hoyte, E.G.; Huang, J.; Vanishsarn, C.; Nadeau, K.C. Regulatory T cell dysfunction in subjects with common variable immunodeficiency complicated by autoimmune disease. Clin. Immunol. 2009, 131, 240–253. [Google Scholar] [CrossRef] [Green Version]
- Aukrust, P.; Lien, E.; Kristoffersen, A.K.; Müller, F.; Haug, C.J.; Espevik, T.; Frøland, S.S. Persistent activation of the tumor necrosis factor system in a subgroup of patients with common variable immunodeficiency-possible immunologic and clinical consequences. Blood 1996, 87, 674–681. [Google Scholar] [CrossRef] [Green Version]
- Kutukculer, N.; Azarsiz, E.; Karaca, N.E.; Ulusoy, E.; Koturoglu, G.; Aksu, G. A Clinical and Laboratory Approach to the Evaluation of Innate Immunity in Pediatric CVID Patients. Front. Immunol. 2015, 6, 145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebbo, M.; Gérard, L.; Carpentier, S.; Vély, F.; Cypowyj, S.; Farnarier, C.; Vince, N.; Malphettes, M.; Fieschi, C.; Oksenhendler, E.; et al. Low Circulating Natural Killer Cell Counts are Associated With Severe Disease in Patients With Common Variable Immunodeficiency. EBioMedicine 2016, 6, 222–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abolhassani, H.; Hammarström, L.; Cunningham-Rundles, C. Current genetic landscape in common variable immune deficiency. Blood 2020, 135, 656–667. [Google Scholar] [CrossRef] [PubMed]
- Ameratunga, R.; Woon, S.-T. Perspective: Evolving Concepts in the Diagnosis and Understanding of Common Variable Immunodeficiency Disorders (CVID). Clin. Rev. Allergy Immunol. 2019, 59, 109–121. [Google Scholar] [CrossRef]
- Abolhassani, H.; Aghamohammadi, A.; Fang, M.; Rezaei, N.; Jiang, C.; Liu, X.; Pan-Hammarström, Q.; Hammarström, L. Clinical implications of systematic phenotyping and exome sequencing in patients with primary antibody deficiency. Genet. Med. Off. J. Am. Coll. Med. Genet. 2019, 21, 243–251. [Google Scholar] [CrossRef] [Green Version]
- Tuijnenburg, P.; Lango, A.H.; Burns, S.O.; Greene, D.; Jansen, M.H.; Staples, E.; Stephens, J.; Carss, K.J.; Biasci, D.; Baxendale, H.; et al. Loss-of-function nuclear factor κB subunit 1 (NFKB1) variants are the most common monogenic cause of common variable immunodeficiency in Europeans. J. Allergy Clin. Immunol. 2018, 142, 1285–1296. [Google Scholar] [CrossRef] [Green Version]
- Christiansen, M.; Offersen, R.; Jensen, J.M.B.; Petersen, M.S.; Larsen, C.S.; Mogensen, T.H. Identification of Novel Genetic Variants in CVID Patients With Autoimmunity, Autoinflammation, or Malignancy. Front. Immunol. 2019, 10, 3022. [Google Scholar] [CrossRef] [Green Version]
- Tangye, S.G.; Al-Herz, W.; Bousfiha, A.; Chatila, T.; Cunningham-Rundles, C.; Etzioni, A.; Franco, J.L.; Holland, S.M.; Klein, C.; Morio, T.; et al. Human Inborn Errors of Immunity: 2019 Update on the Classification from the International Union of Immunological Societies Expert Committee. J. Clin. Immunol. 2020, 40, 24–64. [Google Scholar] [CrossRef] [Green Version]
- Vorechovský, I.; Cullen, M.; Carrington, M.; Hammarström, L.; Webster, A.D. Fine mapping of IGAD1 in IgA deficiency and common variable immunodeficiency: Identification and characterization of haplotypes shared by affected members of 101 multiple-case families. J. Immunol. 2000, 164, 4408–4416. [Google Scholar] [CrossRef] [Green Version]
- Olerup, O.; Smith, C.I.; Hammarström, L. Different amino acids at position 57 of the HLA-DQ beta chain associated with susceptibility and resistance to IgA deficiency. Nature 1990, 347, 289–290. [Google Scholar] [CrossRef] [PubMed]
- Fiedorová, K.; Radvanský, M.; Bosák, J.; Grombiříková, H.; Němcová, E.; Králíčková, P.; Černochová, M.; Kotásková, I.; Lexa, M.; Litzman, J.; et al. Bacterial but Not Fungal Gut Microbiota Alterations Are Associated With Common Variable Immunodeficiency (CVID) Phenotype. Front. Immunol. 2019, 10, 1914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jørgensen, S.F.; Trøseid, M.; Kummen, M.; Anmarkrud, J.A.; Michelsen, A.E.; Osnes, L.T.; Holm, K.; Høivik, M.L.; Rashidi, A.; Dahl, C.P.; et al. Altered gut microbiota profile in common variable immunodeficiency associates with levels of lipopolysaccharide and markers of systemic immune activation. Mucosal Immunol. 2016, 9, 1455–1465. [Google Scholar] [CrossRef] [PubMed]
- Berbers, R.-M.; Nierkens, S.; van Laar, J.M.; Bogaert, D.; Leavis, H.L. Microbial Dysbiosis in Common Variable Immune Deficiencies: Evidence, Causes, and Consequences. Trends Immunol. 2017, 38, 206–216. [Google Scholar] [CrossRef]
- Yazdani, R.; Seify, R.; Ganjalikhani-Hakemi, M.; Abolhassani, H.; Eskandari, N.; Golsaz-Shirazi, F.; Ansaripour, B.; Salehi, E.; Azizi, G.; Rezaei, N.; et al. Comparison of various classifications for patients with common variable immunodeficiency (CVID) using measurement of B-cell subsets. Allergol. Immunopathol. 2017, 45, 183–192. [Google Scholar] [CrossRef]
- Cunningham-Rundles, C. Common variable immune deficiency: Dissection of the variable. Immunol. Rev. 2019, 287, 145–161. [Google Scholar] [CrossRef]
- Chapel, H.; Lucas, M.; Lee, M.; Bjorkander, J.; Webster, D.; Grimbacher, B.; Fieschi, C.; Thon, V.; Abedi, M.R.; Hammarstrom, L. Common variable immunodeficiency disorders: Division into distinct clinical phenotypes. Blood 2008, 112, 277–286. [Google Scholar] [CrossRef] [Green Version]
- Patuzzo, G.; Barbieri, A.; Tinazzi, E.; Veneri, D.; Argentino, G.; Moretta, F.; Puccetti, A.; Lunardi, C. Autoimmunity and infection in common variable immunodeficiency (CVID). Autoimmun. Rev. 2016, 15, 877–882. [Google Scholar] [CrossRef]
- Yazdani, R.; Abolhassani, H.; Asgardoon, M.; Shaghaghi, M.; Modaresi, M.; Azizi, G.; Aghamohammadi, A. Infectious and Noninfectious Pulmonary Complications in Patients With Primary Immunodeficiency Disorders. J. Investig. Allergol. Clin. Immunol. 2017, 27, 213–224. [Google Scholar] [CrossRef] [Green Version]
- Quinti, I.; Soresina, A.; Guerra, A.; Rondelli, R.; Spadaro, G.; Agostini, C.; Milito, C.; Trombetta, A.C.; Visentini, M.; Martini, H.; et al. Effectiveness of immunoglobulin replacement therapy on clinical outcome in patients with primary antibody deficiencies: Results from a multicenter prospective cohort study. J. Clin. Immunol. 2011, 31, 315–322. [Google Scholar] [CrossRef]
- Pasteur, M.C.; Bilton, D.; Hill, A.T. British Thoracic Society Bronchiectasis non-CF Guideline Group British Thoracic Society guideline for non-CF bronchiectasis. Thorax 2010, 65 (Suppl. 1), i1–i58. [Google Scholar] [CrossRef] [Green Version]
- Verma, N.; Grimbacher, B.; Hurst, J.R. Lung disease in primary antibody deficiency. Lancet Respir. Med. 2015, 3, 651–660. [Google Scholar] [CrossRef]
- Bang, T.J.; Richards, J.C.; Olson, A.L.; Groshong, S.D.; Gelfand, E.W.; Lynch, D.A. Pulmonary Manifestations of Common Variable Immunodeficiency. J. Thorac. Imaging 2018, 33, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Oksenhendler, E.; Gérard, L.; Fieschi, C.; Malphettes, M.; Mouillot, G.; Jaussaud, R.; Viallard, J.-F.; Gardembas, M.; Galicier, L.; Schleinitz, N.; et al. Infections in 252 patients with common variable immunodeficiency. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2008, 46, 1547–1554. [Google Scholar] [CrossRef] [PubMed]
- Malamut, G.; Verkarre, V.; Suarez, F.; Viallard, J.-F.; Lascaux, A.-S.; Cosnes, J.; Bouhnik, Y.; Lambotte, O.; Béchade, D.; Ziol, M.; et al. The enteropathy associated with common variable immunodeficiency: The delineated frontiers with celiac disease. Am. J. Gastroenterol. 2010, 105, 2262–2275. [Google Scholar] [CrossRef] [PubMed]
- Daniels, J.A.; Lederman, H.M.; Maitra, A.; Montgomery, E.A. Gastrointestinal tract pathology in patients with common variable immunodeficiency (CVID): A clinicopathologic study and review. Am. J. Surg. Pathol. 2007, 31, 1800–1812. [Google Scholar] [CrossRef]
- Ramírez-Vargas, N.; Arablin-Oropeza, S.E.; Mojica-Martínez, D.; Yamazaki-Nakashimada, M.A.; de la Luz García-Cruz, M.; Terán-Juárez, L.M.; Cortés-Grimaldo, R.M.; Torres-Lozano, C.; Madrigal-Beas, I.; Ortega-Cisneros, M.; et al. Clinical and immunological features of common variable immunodeficiency in Mexican patients. Allergol. Immunopathol. 2014, 42, 235–240. [Google Scholar] [CrossRef]
- Azizi, G.; Abolhassani, H.; Asgardoon, M.H.; Alinia, T.; Yazdani, R.; Mohammadi, J.; Rezaei, N.; Ochs, H.D.; Aghamohammadi, A. Autoimmunity in common variable immunodeficiency: Epidemiology, pathophysiology and management. Expert Rev. Clin. Immunol. 2017, 13, 101–115. [Google Scholar] [CrossRef]
- Resnick, E.S.; Moshier, E.L.; Godbold, J.H.; Cunningham-Rundles, C. Morbidity and mortality in common variable immune deficiency over 4 decades. Blood 2012, 119, 1650–1657. [Google Scholar] [CrossRef]
- Brandt, D.; Gershwin, M.E. Common variable immune deficiency and autoimmunity. Autoimmun. Rev. 2006, 5, 465–470. [Google Scholar] [CrossRef]
- Knight, A.K.; Cunningham-Rundles, C. Inflammatory and autoimmune complications of common variable immune deficiency. Autoimmun. Rev. 2006, 5, 156–159. [Google Scholar] [CrossRef]
- Xiao, X.; Miao, Q.; Chang, C.; Gershwin, M.E.; Ma, X. Common variable immunodeficiency and autoimmunity-an inconvenient truth. Autoimmun. Rev. 2014, 13, 858–864. [Google Scholar] [CrossRef] [PubMed]
- Karaca, N.E.; Aksu, G.; Yildiz, B.; Gulez, N.; Turk, B.; Dereli, T.; Kutukculer, N. Relapsing polychondritis in a child with common variable immunodeficiency. Int. J. Dermatol. 2009, 48, 525–528. [Google Scholar] [CrossRef]
- Todoric, K.; Koontz, J.B.; Mattox, D.; Tarrant, T.K. Autoimmunity in immunodeficiency. Curr. Allergy Asthma Rep. 2013, 13, 361–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Herrera, G.; Segura-Méndez, N.H.; O’Farril-Romanillos, P.; Nuñez-Nuñez, M.E.; Zarate-Hernández, M.C.; Mogica-Martínez, D.; Yamazaki-Nakashimada, M.A.; Staines-Boone, A.T.; Santos-Argumedo, L.; Berrón-Ruiz, L. Low percentages of regulatory T cells in common variable immunodeficiency (CVID) patients with autoimmune diseases and its association with increased numbers of CD4+CD45RO+ T and CD21low B cells. Allergol. Immunopathol. 2019, 47, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Kofod-Olsen, E.; Jørgensen, S.E.; Nissen, S.K.; Westh, L.; Møller, B.K.; Østergaard, L.; Larsen, C.S.; Mogensen, T.H. Altered fraction of regulatory B and T cells is correlated with autoimmune phenomena and splenomegaly in patients with CVID. Clin. Immunol. 2016, 162, 49–57. [Google Scholar] [CrossRef]
- Westh, L.; Mogensen, T.H.; Dalgaard, L.S.; Bernth Jensen, J.M.; Katzenstein, T.; Hansen, A.-B.E.; Larsen, O.D.; Terpling, S.; Nielsen, T.L.; Larsen, C.S. Identification and Characterization of a Nationwide Danish Adult Common Variable Immunodeficiency Cohort. Scand. J. Immunol. 2017, 85, 450–461. [Google Scholar] [CrossRef] [Green Version]
- Ardeniz, O.; Cunningham-Rundles, C. Granulomatous disease in common variable immunodeficiency. Clin. Immunol. 2009, 133, 198–207. [Google Scholar] [CrossRef]
- Boursiquot, J.-N.; Gérard, L.; Malphettes, M.; Fieschi, C.; Galicier, L.; Boutboul, D.; Borie, R.; Viallard, J.-F.; Soulas-Sprauel, P.; Berezne, A.; et al. Granulomatous disease in CVID: Retrospective analysis of clinical characteristics and treatment efficacy in a cohort of 59 patients. J. Clin. Immunol. 2013, 33, 84–95. [Google Scholar] [CrossRef]
- Fernández, P.E.R. Granulomatous lymphocytic interstitial lung disease. Immunol. Allergy Clin. N. Am. 2012, 32, 621–632. [Google Scholar] [CrossRef]
- Gathmann, B.; Mahlaoui, N.; Gérard, L.; Oksenhendler, E.; Warnatz, K.; Schulze, I.; Kindle, G.; Kuijpers, T.W.; van Beem, R.T.; Lic, D.G.; et al. Clinical picture and treatment of 2212 patients with common variable immunodeficiency. J. Allergy Clin. Immunol. 2014, 134, 116–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pensieri, M.V.; Pulvirenti, F.; Schiepatti, A.; Maimaris, S.; Lattanzio, S.; Quinti, I.; Klersy, C.; Corazza, G.R.; Biagi, F. The high mortality of patients with common variable immunodeficiency and small bowel villous atrophy. Scand. J. Gastroenterol. 2019, 54, 164–168. [Google Scholar] [CrossRef]
- Agarwal, S.; Smereka, P.; Harpaz, N.; Cunningham-Rundles, C.; Mayer, L. Characterization of immunologic defects in patients with common variable immunodeficiency (CVID) with intestinal disease. Inflamm. Bowel Dis. 2011, 17, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Resnick, E.S.; Cunningham-Rundles, C. The many faces of the clinical picture of common variable immune deficiency. Curr. Opin. Allergy Clin. Immunol. 2012, 12, 595–601. [Google Scholar] [CrossRef] [PubMed]
- Vajdic, C.M.; Mao, L.; van Leeuwen, M.T.; Kirkpatrick, P.; Grulich, A.E.; Riminton, S. Are antibody deficiency disorders associated with a narrower range of cancers than other forms of immunodeficiency? Blood 2010, 116, 1228–1234. [Google Scholar] [CrossRef]
- Aghamohammadi, A.; Mohammadi, J.; Parvaneh, N.; Rezaei, N.; Moin, M.; Espanol, T.; Hammarstrom, L. Progression of selective IgA deficiency to common variable immunodeficiency. Int. Arch. Allergy Immunol. 2008, 147, 87–92. [Google Scholar] [CrossRef]
- Schroeder, H.W.; Zhu, Z.B.; March, R.E.; Campbell, R.D.; Berney, S.M.; Nedospasov, S.A.; Turetskaya, R.L.; Atkinson, T.P.; Go, R.C.; Cooper, M.D.; et al. Susceptibility locus for IgA deficiency and common variable immunodeficiency in the HLA-DR3, -B8, -A1 haplotypes. Mol. Med. 1998, 4, 72–86. [Google Scholar] [CrossRef]
- Vorechovský, I.; Zetterquist, H.; Paganelli, R.; Koskinen, S.; Webster, A.D.; Björkander, J.; Smith, C.I.; Hammarström, L. Family and linkage study of selective IgA deficiency and common variable immunodeficiency. Clin. Immunol. Immunopathol. 1995, 77, 185–192. [Google Scholar] [CrossRef]
- De La Concha, E.G.; Fernandez-Arquero, M.; Martinez, A.; Vidal, F.; Vigil, P.; Conejero, L.; Garcia-Rodriguez, M.C.; Fontan, G. HLA class II homozygosity confers susceptibility to common variable immunodeficiency (CVID). Clin. Exp. Immunol. 1999, 116, 516–520. [Google Scholar] [CrossRef]
- Choung, R.S.; Larson, S.A.; Khaleghi, S.; Rubio-Tapia, A.; Ovsyannikova, I.G.; King, K.S.; Larson, J.J.; Lahr, B.D.; Poland, G.A.; Camilleri, M.J.; et al. Prevalence and Morbidity of Undiagnosed Celiac Disease From a Community-Based Study. Gastroenterology 2017, 152, 830–839. [Google Scholar] [CrossRef] [Green Version]
- Singh, P.; Arora, A.; Strand, T.A.; Leffler, D.A.; Catassi, C.; Green, P.H.; Kelly, C.P.; Ahuja, V.; Makharia, G.K. Global Prevalence of Celiac Disease: Systematic Review and Meta-analysis. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2018, 16, 823–836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, P.; Arora, S.; Lal, S.; Strand, T.A.; Makharia, G.K. Risk of Celiac Disease in the First- and Second-Degree Relatives of Patients With Celiac Disease: A Systematic Review and Meta-Analysis. Am. J. Gastroenterol. 2015, 110, 1539–1548. [Google Scholar] [CrossRef] [PubMed]
- Mårild, K.; Stephansson, O.; Grahnquist, L.; Cnattingius, S.; Söderman, G.; Ludvigsson, J.F. Down syndrome is associated with elevated risk of celiac disease: A nationwide case-control study. J. Pediatr. 2013, 163, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Oujamaa, I.; Sebbani, M.; Elmoumou, L.; Bourrahouate, A.; El Qadiry, R.; El Moussaoui, S.; Ait Sab, I.; Sbihi, M.; Ennazk, L.; El Mghari-Tabib, G.; et al. The Prevalence of Celiac Disease-Specific Auto-Antibodies in Type 1 Diabetes in a Moroccan Population. Int. J. Endocrinol. 2019, 2019, 7895207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagopian, W.; Lee, H.-S.; Liu, E.; Rewers, M.; She, J.-X.; Ziegler, A.-G.; Lernmark, Å.; Toppari, J.; Rich, S.S.; Krischer, J.P.; et al. Co-occurrence of Type 1 Diabetes and Celiac Disease Autoimmunity. Pediatrics 2017, 140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, E.; Rewers, M.; Eisenbarth, G.S. Genetic testing: Who should do the testing and what is the role of genetic testing in the setting of celiac disease? Gastroenterology 2005, 128, S33–S37. [Google Scholar] [CrossRef]
- Makharia, G.K.; Catassi, C. Celiac Disease in Asia. Gastroenterol. Clin. N. Am. 2019, 48, 101–113. [Google Scholar] [CrossRef]
- Ramakrishna, B.S.; Makharia, G.K.; Chetri, K.; Dutta, S.; Mathur, P.; Ahuja, V.; Amarchand, R.; Balamurugan, R.; Chowdhury, S.D.; Daniel, D.; et al. Prevalence of Adult Celiac Disease in India: Regional Variations and Associations. Am. J. Gastroenterol. 2016, 111, 115–123. [Google Scholar] [CrossRef]
- Catassi, C.; Kryszak, D.; Bhatti, B.; Sturgeon, C.; Helzlsouer, K.; Clipp, S.L.; Gelfond, D.; Puppa, E.; Sferruzza, A.; Fasano, A. Natural history of celiac disease autoimmunity in a USA cohort followed since 1974. Ann. Med. 2010, 42, 530–538. [Google Scholar] [CrossRef]
- Lohi, S.; Mustalahti, K.; Kaukinen, K.; Laurila, K.; Collin, P.; Rissanen, H.; Lohi, O.; Bravi, E.; Gasparin, M.; Reunanen, A.; et al. Increasing prevalence of coeliac disease over time. Aliment. Pharmacol. Ther. 2007, 26, 1217–1225. [Google Scholar] [CrossRef]
- Levinson-Castiel, R.; Eliakim, R.; Shinar, E.; Perets, T.-T.; Layfer, O.; Levhar, N.; Schvimer, M.; Marderfeld, L.; Ben-Horin, S.; Shamir, R. Rising prevalence of celiac disease is not universal and repeated testing is needed for population screening. United Eur. Gastroenterol. J. 2019, 7, 412–418. [Google Scholar] [CrossRef]
- Oxentenko, A.S.; Rubio-Tapia, A. Celiac Disease. Mayo Clin. Proc. 2019, 94, 2556–2571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Toma, A.; Volta, U.; Auricchio, R.; Castillejo, G.; Sanders, D.S.; Cellier, C.; Mulder, C.J.; Lundin, K.E.A. European Society for the Study of Coeliac Disease (ESsCD) guideline for coeliac disease and other gluten-related disorders. United Eur. Gastroenterol. J. 2019, 7, 583–613. [Google Scholar] [CrossRef] [PubMed]
- Balaban, D.V.; Popp, A.; Vasilescu, F.; Haidautu, D.; Purcarea, R.M.; Jinga, M. Diagnostic yield of endoscopic markers for celiac disease. J. Med. Life 2015, 8, 452–457. [Google Scholar] [PubMed]
- Husby, S.; Koletzko, S.; Korponay-Szabó, I.; Kurppa, K.; Mearin, M.L.; Ribes-Koninckx, C.; Shamir, R.; Troncone, R.; Auricchio, R.; Castillejo, G.; et al. European Society Paediatric Gastroenterology, Hepatology and Nutrition Guidelines for Diagnosing Coeliac Disease 2020. J. Pediatr. Gastroenterol. Nutr. 2020, 70, 141–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oberhuber, G.; Granditsch, G.; Vogelsang, H. The histopathology of coeliac disease: Time for a standardized report scheme for pathologists. Eur. J. Gastroenterol. Hepatol. 1999, 11, 1185–1194. [Google Scholar] [CrossRef]
- Gujral, N.; Freeman, H.J.; Thomson, A.B.R. Celiac disease: Prevalence, diagnosis, pathogenesis and treatment. World J. Gastroenterol. 2012, 18, 6036–6059. [Google Scholar] [CrossRef]
- McGowan, K.E.; Lyon, M.E.; Butzner, J.D. Celiac disease and IgA deficiency: Complications of serological testing approaches encountered in the clinic. Clin. Chem. 2008, 54, 1203–1209. [Google Scholar] [CrossRef] [Green Version]
- Pallav, K.; Xu, H.; Leffler, D.A.; Kabbani, T.; Kelly, C.P. Immunoglobulin A deficiency in celiac disease in the United States. J. Gastroenterol. Hepatol. 2016, 31, 133–137. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.; Shen, N.; Vyse, T.J.; Anand, V.; Gunnarson, I.; Sturfelt, G.; Rantapää-Dahlqvist, S.; Elvin, K.; Truedsson, L.; Andersson, B.A.; et al. Selective IgA deficiency in autoimmune diseases. Mol. Med. 2011, 17, 1383–1396. [Google Scholar] [CrossRef]
- Ludvigsson, J.F.; Neovius, M.; Hammarström, L. Association between IgA deficiency & other autoimmune conditions: A population-based matched cohort study. J. Clin. Immunol. 2014, 34, 444–451. [Google Scholar] [CrossRef] [PubMed]
- Lenhardt, A.; Plebani, A.; Marchetti, F.; Gerarduzzi, T.; Not, T.; Meini, A.; Villanacci, V.; Martelossi, S.; Ventura, A. Role of human-tissue transglutaminase IgG and anti-gliadin IgG antibodies in the diagnosis of coeliac disease in patients with selective immunoglobulin A deficiency. Dig. Liver Dis. Off. J. Ital. Soc. Gastroenterol. Ital. Assoc. Study Liver 2004, 36, 730–734. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Truedsson, L.; Elvin, K.; Andersson, B.A.; Rönnelid, J.; Mincheva-Nilsson, L.; Lindkvist, A.; Ludvigsson, J.F.; Hammarström, L.; Dahle, C. Serological assessment for celiac disease in IgA deficient adults. PLoS ONE 2014, 9, e93180. [Google Scholar] [CrossRef] [PubMed]
- Szajewska, H.; Shamir, R.; Mearin, L.; Ribes-Koninckx, C.; Catassi, C.; Domellöf, M.; Fewtrell, M.S.; Husby, S.; Papadopoulou, A.; Vandenplas, Y.; et al. Gluten Introduction and the Risk of Coeliac Disease: A Position Paper by the European Society for Pediatric Gastroenterology, Hepatology, and Nutrition. J. Pediatr. Gastroenterol. Nutr. 2016, 62, 507–513. [Google Scholar] [CrossRef] [Green Version]
- Mulder, S.J.; Mulder-Bos, G.C. Most probable origin of coeliac disease is low immune globulin A in the intestine caused by malfunction of Peyer’s patches. Med. Hypotheses 2006, 66, 757–762. [Google Scholar] [CrossRef]
- Prince, H.E.; Norman, G.L.; Binder, W.L. Immunoglobulin A (IgA) deficiency and alternative celiac disease-associated antibodies in sera submitted to a reference laboratory for endomysial IgA testing. Clin. Diagn. Lab. Immunol. 2000, 7, 192–196. [Google Scholar] [CrossRef] [Green Version]
- Dahlbom, I.; Olsson, M.; Forooz, N.K.; Sjöholm, A.G.; Truedsson, L.; Hansson, T. Immunoglobulin G (IgG) anti-tissue transglutaminase antibodies used as markers for IgA-deficient celiac disease patients. Clin. Diagn. Lab. Immunol. 2005, 12, 254–258. [Google Scholar] [CrossRef] [Green Version]
- Villalta, D.; Alessio, M.G.; Tampoia, M.; Tonutti, E.; Brusca, I.; Bagnasco, M.; Pesce, G.; Stella, S.; Bizzaro, N. Testing for IgG class antibodies in celiac disease patients with selective IgA deficiency. A comparison of the diagnostic accuracy of 9 IgG anti-tissue transglutaminase, 1 IgG anti-gliadin and 1 IgG anti-deaminated gliadin peptide antibody assays. Clin. Chim. Acta Int. J. Clin. Chem. 2007, 382, 95–99. [Google Scholar] [CrossRef]
- Valletta, E.; Fornaro, M.; Pecori, S.; Zanoni, G. Selective immunoglobulin A deficiency and celiac disease: let’s give serology a chance. J. Investig. Allergol. Clin. Immunol. 2011, 21, 242–244. [Google Scholar]
- Winkelstein, J.A.; Marino, M.C.; Ochs, H.; Fuleihan, R.; Scholl, P.R.; Geha, R.; Stiehm, E.R.; Conley, M.E. The X-linked hyper-IgM syndrome: Clinical and immunologic features of 79 patients. Medicine 2003, 82, 373–384. [Google Scholar] [CrossRef]
- Fuleihan, R.; Ramesh, N.; Loh, R.; Jabara, H.; Rosen, R.S.; Chatila, T.; Fu, S.M.; Stamenkovic, I.; Geha, R.S. Defective expression of the CD40 ligand in X chromosome-linked immunoglobulin deficiency with normal or elevated IgM. Proc. Natl. Acad. Sci. USA 1993, 90, 2170–2173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leven, E.A.; Maffucci, P.; Ochs, H.D.; Scholl, P.R.; Buckley, R.H.; Fuleihan, R.L.; Geha, R.S.; Cunningham, C.K.; Bonilla, F.A.; Conley, M.E.; et al. Hyper IgM Syndrome: A Report from the USIDNET Registry. J. Clin. Immunol. 2016, 36, 490–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heneghan, M.A.; Stevens, F.M.; Cryan, E.M.; Warner, R.H.; McCarthy, C.F. Celiac sprue and immunodeficiency states: A 25-year review. J. Clin. Gastroenterol. 1997, 25, 421–425. [Google Scholar] [CrossRef] [PubMed]
- Chow, M.A.; Lebwohl, B.; Reilly, N.R.; Green, P.H.R. Immunoglobulin A deficiency in celiac disease. J. Clin. Gastroenterol. 2012, 46, 850–854. [Google Scholar] [CrossRef] [Green Version]
- Rubio-Tapia, A.; Hill, I.D.; Kelly, C.P.; Calderwood, A.H.; Murray, J.A. American College of Gastroenterology ACG clinical guidelines: Diagnosis and management of celiac disease. Am. J. Gastroenterol. 2013, 108, 656–676. [Google Scholar] [CrossRef] [Green Version]
- Korponay-Szabó, I.R.; Dahlbom, I.; Laurila, K.; Koskinen, S.; Woolley, N.; Partanen, J.; Kovács, J.B.; Mäki, M.; Hansson, T. Elevation of IgG antibodies against tissue transglutaminase as a diagnostic tool for coeliac disease in selective IgA deficiency. Gut 2003, 52, 1567–1571. [Google Scholar] [CrossRef]
- Gidrewicz, D.; Trevenen, C.L.; Lyon, M.; Butzner, J.D. Normalization Time of Celiac Serology in Children on a Gluten-free Diet. J. Pediatr. Gastroenterol. Nutr. 2017, 64, 362–367. [Google Scholar] [CrossRef]
- Yuan, Q.; Bousvaros, A. Chapter 38—Gastrointestinal manifestations of primary immunodeficiency. In Pediatric Gastrointestinal and Liver Disease, 3rd ed.; Wyllie, R., Hyams, J.S., Kay, M., Eds.; W.B. Saunders: London, UK, 2006; pp. 601–616. ISBN 978-0-7216-3924-6. [Google Scholar]
- Pehlivanoğlu, B.; Ardeniz, Ö.; Hassoy, H.; Sezak, M.; Özdemir, H.; Ünal, N.G.; Onay, H.; Doğanavşargil, B. Gastrointestinal findings in 26 adults with common variable immunodeficiency: The fickle nature of the disease manifests in gastrointestinal biopsies. Turk. J. Gastroenterol. Off. J. Turk. Soc. Gastroenterol. 2019, 30, 789–800. [Google Scholar] [CrossRef]
- Luzi, G.; Zullo, A.; Iebba, F.; Rinaldi, V.; Sanchez, M.L.; Muscaritoli, M.; Aiuti, F. Duodenal pathology and clinical-immunological implications in common variable immunodeficiency patients. Am. J. Gastroenterol. 2003, 98, 118–121. [Google Scholar] [CrossRef]
- Hartono, S.; Ippoliti, M.R.; Mastroianni, M.; Torres, R.; Rider, N.L. Gastrointestinal Disorders Associated with Primary Immunodeficiency Diseases. Clin. Rev. Allergy Immunol. 2019, 57, 145–165. [Google Scholar] [CrossRef]
- Venhoff, N.; Emmerich, F.; Neagu, M.; Salzer, U.; Koehn, C.; Driever, S.; Kreisel, W.; Rizzi, M.; Effelsberg, N.M.; Kollert, F.; et al. The role of HLA DQ2 and DQ8 in dissecting celiac-like disease in common variable immunodeficiency. J. Clin. Immunol. 2013, 33, 909–916. [Google Scholar] [CrossRef] [PubMed]
- Giorgio, F.; Principi, M.; Losurdo, G.; Piscitelli, D.; Iannone, A.; Barone, M.; Amoruso, A.; Ierardi, E.; Di Leo, A. Seronegative Celiac Disease and Immunoglobulin Deficiency: Where to Look in the Submerged Iceberg? Nutrients 2015, 7, 7486–7504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Licinio, R.; Principi, M.; Amoruso, A.; Piscitelli, D.; Ierardi, E.; Di Leo, A. Celiac disease and common variable immunodeficiency: A familial inheritance? J. Gastrointest. Liver Dis. JGLD 2013, 22, 473. [Google Scholar]
- Viallard, J.-F.; Blanco, P.; André, M.; Etienne, G.; Liferman, F.; Neau, D.; Vidal, E.; Moreau, J.-F.; Pellegrin, J.-L. CD8+HLA-DR+ T lymphocytes are increased in common variable immunodeficiency patients with impaired memory B-cell differentiation. Clin. Immunol. 2006, 119, 51–58. [Google Scholar] [CrossRef]
- Jørgensen, S.F.; Reims, H.M.; Frydenlund, D.; Holm, K.; Paulsen, V.; Michelsen, A.E.; Jørgensen, K.K.; Osnes, L.T.; Bratlie, J.; Eide, T.J.; et al. A Cross-Sectional Study of the Prevalence of Gastrointestinal Symptoms and Pathology in Patients With Common Variable Immunodeficiency. Am. J. Gastroenterol. 2016, 111, 1467–1475. [Google Scholar] [CrossRef]
- Woodward, J.M.; Gkrania-Klotsas, E.; Cordero-Ng, A.Y.; Aravinthan, A.; Bandoh, B.N.; Liu, H.; Davies, S.; Zhang, H.; Stevenson, P.; Curran, M.D.; et al. The role of chronic norovirus infection in the enteropathy associated with common variable immunodeficiency. Am. J. Gastroenterol. 2015, 110, 320–327. [Google Scholar] [CrossRef]
- Pikkarainen, S.; Martelius, T.; Ristimäki, A.; Siitonen, S.; Seppänen, M.R.J.; Färkkilä, M. A High Prevalence of Gastrointestinal Manifestations in Common Variable Immunodeficiency. Am. J. Gastroenterol. 2019, 114, 648–655. [Google Scholar] [CrossRef] [Green Version]
- Jørgensen, S.F.; Reims, H.M.; Aukrust, P.; Lundin, K.E.A.; Fevang, B. CVID and Celiac Disease. Am. J. Gastroenterol. 2017, 112, 393. [Google Scholar] [CrossRef]
- Biagi, F.; Bianchi, P.I.; Zilli, A.; Marchese, A.; Luinetti, O.; Lougaris, V.; Plebani, A.; Villanacci, V.; Corazza, G.R. The significance of duodenal mucosal atrophy in patients with common variable immunodeficiency: A clinical and histopathologic study. Am. J. Clin. Pathol. 2012, 138, 185–189. [Google Scholar] [CrossRef] [Green Version]
- Steenholt, J.V.; Nielsen, C.; Baudewijn, L.; Staal, A.; Rasmussen, K.S.; Sabir, H.J.; Barington, T.; Husby, S.; Toft-Hansen, H. The composition of T cell subtypes in duodenal biopsies are altered in coeliac disease patients. PLoS ONE 2017, 12, e0170270. [Google Scholar] [CrossRef] [Green Version]
- Kelsen, J.R.; Russo, P.; Sullivan, K.E. Early-Onset Inflammatory Bowel Disease. Immunol. Allergy Clin. N. Am. 2019, 39, 63–79. [Google Scholar] [CrossRef] [PubMed]
- Geboes, K.; Van Eyken, P. Inflammatory bowel disease unclassified and indeterminate colitis: The role of the pathologist. J. Clin. Pathol. 2009, 62, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Tremaine, W.J. Diagnosis and treatment of indeterminate colitis. Gastroenterol. Hepatol. 2011, 7, 826–828. [Google Scholar]
- Ng, S.C.; Shi, H.Y.; Hamidi, N.; Underwood, F.E.; Tang, W.; Benchimol, E.I.; Panaccione, R.; Ghosh, S.; Wu, J.C.Y.; Chan, F.K.L.; et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: A systematic review of population-based studies. Lancet Lond. Engl. 2018, 390, 2769–2778. [Google Scholar] [CrossRef]
- Bach, J.-F. The effect of infections on susceptibility to autoimmune and allergic diseases. N. Engl. J. Med. 2002, 347, 911–920. [Google Scholar] [CrossRef]
- Benchimol, E.I.; Bernstein, C.N.; Bitton, A.; Carroll, M.W.; Singh, H.; Otley, A.R.; Vutcovici, M.; El-Matary, W.; Nguyen, G.C.; Griffiths, A.M.; et al. Trends in Epidemiology of Pediatric Inflammatory Bowel Disease in Canada: Distributed Network Analysis of Multiple Population-Based Provincial Health Administrative Databases. Am. J. Gastroenterol. 2017, 112, 1120–1134. [Google Scholar] [CrossRef] [Green Version]
- Chassaing, B.; Koren, O.; Goodrich, J.K.; Poole, A.C.; Srinivasan, S.; Ley, R.E.; Gewirtz, A.T. Dietary emulsifiers impact the mouse gut microbiota promoting colitis and metabolic syndrome. Nature 2015, 519, 92–96. [Google Scholar] [CrossRef] [Green Version]
- Lamb, C.A.; Kennedy, N.A.; Raine, T.; Hendy, P.A.; Smith, P.J.; Limdi, J.K.; Hayee, B.; Lomer, M.C.E.; Parkes, G.C.; Selinger, C.; et al. British Society of Gastroenterology consensus guidelines on the management of inflammatory bowel disease in adults. Gut 2019, 68, s1–s106. [Google Scholar] [CrossRef] [Green Version]
- Jostins, L.; Ripke, S.; Weersma, R.K.; Duerr, R.H.; McGovern, D.P.; Hui, K.Y.; Lee, J.C.; Schumm, L.P.; Sharma, Y.; Anderson, C.A.; et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012, 491, 119–124. [Google Scholar] [CrossRef] [Green Version]
- Feuerstein, J.D.; Moss, A.C.; Farraye, F.A. Ulcerative Colitis. Mayo Clin. Proc. 2019, 94, 1357–1373. [Google Scholar] [CrossRef] [Green Version]
- Ventham, N.T.; Kennedy, N.A.; Nimmo, E.R.; Satsangi, J. Beyond gene discovery in inflammatory bowel disease: The emerging role of epigenetics. Gastroenterology 2013, 145, 293–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moller, F.T.; Andersen, V.; Wohlfahrt, J.; Jess, T. Familial risk of inflammatory bowel disease: A population-based cohort study 1977-2011. Am. J. Gastroenterol. 2015, 110, 564–571. [Google Scholar] [CrossRef] [PubMed]
- UK IBD Genetics Consortium; Barrett, J.C.; Lee, J.C.; Lees, C.W.; Prescott, N.J.; Anderson, C.A.; Phillips, A.; Wesley, E.; Parnell, K.; Zhang, H. Genome-wide association study of ulcerative colitis identifies three new susceptibility loci, including the HNF4A region. Nat. Genet. 2009, 41, 1330–1334. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.C.; Bernstein, C.N.; Vatn, M.H.; Lakatos, P.L.; Loftus, E.V.; Tysk, C.; O’Morain, C.; Moum, B.; Colombel, J.-F. Epidemiology and Natural History Task Force of the International Organization of Inflammatory Bowel Disease (IOIBD) Geographical variability and environmental risk factors in inflammatory bowel disease. Gut 2013, 62, 630–649. [Google Scholar] [CrossRef]
- Loftus, E.V. Clinical epidemiology of inflammatory bowel disease: Incidence, prevalence, and environmental influences. Gastroenterology 2004, 126, 1504–1517. [Google Scholar] [CrossRef]
- Gradel, K.O.; Nielsen, H.L.; Schønheyder, H.C.; Ejlertsen, T.; Kristensen, B.; Nielsen, H. Increased short- and long-term risk of inflammatory bowel disease after salmonella or campylobacter gastroenteritis. Gastroenterology 2009, 137, 495–501. [Google Scholar] [CrossRef]
- Rodríguez, G.L.A.; González-Pérez, A.; Johansson, S.; Wallander, M.-A. Risk factors for inflammatory bowel disease in the general population. Aliment. Pharmacol. Ther. 2005, 22, 309–315. [Google Scholar] [CrossRef]
- Shaw, S.Y.; Blanchard, J.F.; Bernstein, C.N. Association between the use of antibiotics in the first year of life and pediatric inflammatory bowel disease. Am. J. Gastroenterol. 2010, 105, 2687–2692. [Google Scholar] [CrossRef]
- Ananthakrishnan, A.N. Epidemiology and risk factors for IBD. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 205–217. [Google Scholar] [CrossRef]
- Höie, O.; Wolters, F.; Riis, L.; Aamodt, G.; Solberg, C.; Bernklev, T.; Odes, S.; Mouzas, I.A.; Beltrami, M.; Langholz, E.; et al. Ulcerative colitis: Patient characteristics may predict 10-yr disease recurrence in a European-wide population-based cohort. Am. J. Gastroenterol. 2007, 102, 1692–1701. [Google Scholar] [CrossRef]
- Vavricka, S.R.; Brun, L.; Ballabeni, P.; Pittet, V.; Prinz-Vavricka, B.M.; Zeitz, J.; Rogler, G.; Schoepfer, A.M. Frequency and risk factors for extraintestinal manifestations in the Swiss inflammatory bowel disease cohort. Am. J. Gastroenterol. 2011, 106, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Rubin, D.T.; Ananthakrishnan, A.N.; Siegel, C.A.; Sauer, B.G.; Long, M.D. ACG Clinical Guideline: Ulcerative Colitis in Adults. Am. J. Gastroenterol. 2019, 114, 384–413. [Google Scholar] [CrossRef] [PubMed]
- Corte, C.; Fernandopulle, N.; Catuneanu, A.M.; Burger, D.; Cesarini, M.; White, L.; Keshav, S.; Travis, S. Association between the ulcerative colitis endoscopic index of severity (UCEIS) and outcomes in acute severe ulcerative colitis. J. Crohns Colitis 2015, 9, 376–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Lange, T.; Larsen, S.; Aabakken, L. Inter-observer agreement in the assessment of endoscopic findings in ulcerative colitis. BMC Gastroenterol. 2004, 4, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gajendran, M.; Loganathan, P.; Jimenez, G.; Catinella, A.P.; Ng, N.; Umapathy, C.; Ziade, N.; Hashash, J.G. A comprehensive review and update on ulcerative colitis. Dis. Mon. DM 2019, 65, 100851. [Google Scholar] [CrossRef]
- Maaser, C.; Sturm, A.; Vavricka, S.R.; Kucharzik, T.; Fiorino, G.; Annese, V.; Calabrese, E.; Baumgart, D.C.; Bettenworth, D.; Borralho-Nunes, P.; et al. ECCO-ESGAR Guideline for Diagnostic Assessment in IBD Part 1: Initial diagnosis, monitoring of known IBD, detection of complications. J. Crohns Colitis 2019, 13, 144–164. [Google Scholar] [CrossRef] [Green Version]
- Menees, S.B.; Powell, C.; Kurlander, J.; Goel, A.; Chey, W.D. A meta-analysis of the utility of C-reactive protein, erythrocyte sedimentation rate, fecal calprotectin, and fecal lactoferrin to exclude inflammatory bowel disease in adults with IBS. Am. J. Gastroenterol. 2015, 110, 444–454. [Google Scholar] [CrossRef]
- Sands, B.E. Biomarkers of Inflammation in Inflammatory Bowel Disease. Gastroenterology 2015, 149, 1275–1285. [Google Scholar] [CrossRef]
- Torres, J.; Bonovas, S.; Doherty, G.; Kucharzik, T.; Gisbert, J.P.; Raine, T.; Adamina, M.; Armuzzi, A.; Bachmann, O.; Bager, P.; et al. ECCO Guidelines on Therapeutics in Crohn’s Disease: Medical Treatment. J. Crohns Colitis 2020, 14, 4–22. [Google Scholar] [CrossRef]
- Ng, S.C.; Tang, W.; Ching, J.Y.; Wong, M.; Chow, C.M.; Hui, A.J.; Wong, T.C.; Leung, V.K.; Tsang, S.W.; Yu, H.H.; et al. Incidence and phenotype of inflammatory bowel disease based on results from the Asia-pacific Crohn’s and colitis epidemiology study. Gastroenterology 2013, 145, 158–165. [Google Scholar] [CrossRef]
- Gajendran, M.; Loganathan, P.; Catinella, A.P.; Hashash, J.G. A comprehensive review and update on Crohn’s disease. Dis. Mon. DM 2018, 64, 20–57. [Google Scholar] [CrossRef] [PubMed]
- Halme, L.; Paavola-Sakki, P.; Turunen, U.; Lappalainen, M.; Farkkila, M.; Kontula, K. Family and twin studies in inflammatory bowel disease. World J. Gastroenterol. 2006, 12, 3668–3672. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.Z.; van Sommeren, S.; Huang, H.; Ng, S.C.; Alberts, R.; Takahashi, A.; Ripke, S.; Lee, J.C.; Jostins, L.; Shah, T.; et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat. Genet. 2015, 47, 979–986. [Google Scholar] [CrossRef]
- McGovern, D.P.B.; Kugathasan, S.; Cho, J.H. Genetics of Inflammatory Bowel Diseases. Gastroenterology 2015, 149, 1163–1176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeissig, S.; Bürgel, N.; Günzel, D.; Richter, J.; Mankertz, J.; Wahnschaffe, U.; Kroesen, A.J.; Zeitz, M.; Fromm, M.; Schulzke, J.-D. Changes in expression and distribution of claudin 2, 5 and 8 lead to discontinuous tight junctions and barrier dysfunction in active Crohn’s disease. Gut 2007, 56, 61–72. [Google Scholar] [CrossRef]
- Mahid, S.S.; Minor, K.S.; Soto, R.E.; Hornung, C.A.; Galandiuk, S. Smoking and inflammatory bowel disease: A meta-analysis. Mayo Clin. Proc. 2006, 81, 1462–1471. [Google Scholar] [CrossRef]
- Ungaro, R.; Bernstein, C.N.; Gearry, R.; Hviid, A.; Kolho, K.-L.; Kronman, M.P.; Shaw, S.; Van Kruiningen, H.; Colombel, J.-F.; Atreja, A. Antibiotics associated with increased risk of new-onset Crohn’s disease but not ulcerative colitis: A meta-analysis. Am. J. Gastroenterol. 2014, 109, 1728–1738. [Google Scholar] [CrossRef]
- Ananthakrishnan, A.N.; Higuchi, L.M.; Huang, E.S.; Khalili, H.; Richter, J.M.; Fuchs, C.S.; Chan, A.T. Aspirin, nonsteroidal anti-inflammatory drug use, and risk for Crohn disease and ulcerative colitis: A cohort study. Ann. Intern. Med. 2012, 156, 350–359. [Google Scholar] [CrossRef]
- Ungaro, R.; Chang, H.L.; Côté-Daigneault, J.; Mehandru, S.; Atreja, A.; Colombel, J.-F. Statins Associated With Decreased Risk of New Onset Inflammatory Bowel Disease. Am. J. Gastroenterol. 2016, 111, 1416–1423. [Google Scholar] [CrossRef]
- Ananthakrishnan, A.N.; Khalili, H.; Konijeti, G.G.; Higuchi, L.M.; de Silva, P.; Fuchs, C.S.; Willett, W.C.; Richter, J.M.; Chan, A.T. Long-term intake of dietary fat and risk of ulcerative colitis and Crohn’s disease. Gut 2014, 63, 776–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gevers, D.; Kugathasan, S.; Denson, L.A.; Vázquez-Baeza, Y.; Van Treuren, W.; Ren, B.; Schwager, E.; Knights, D.; Song, S.J.; Yassour, M.; et al. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe 2014, 15, 382–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kostic, A.D.; Xavier, R.J.; Gevers, D. The microbiome in inflammatory bowel disease: Current status and the future ahead. Gastroenterology 2014, 146, 1489–1499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timm, S.; Svanes, C.; Janson, C.; Sigsgaard, T.; Johannessen, A.; Gislason, T.; Jogi, R.; Omenaas, E.; Forsberg, B.; Torén, K.; et al. Place of upbringing in early childhood as related to inflammatory bowel diseases in adulthood: A population-based cohort study in Northern Europe. Eur. J. Epidemiol. 2014, 29, 429–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castiglione, F.; Diaferia, M.; Morace, F.; Labianca, O.; Meucci, C.; Cuomo, A.; Panarese, A.; Romano, M.; Sorrentini, I.; D’Onofrio, C.; et al. Risk factors for inflammatory bowel diseases according to the “hygiene hypothesis”: A case-control, multi-centre, prospective study in Southern Italy. J. Crohns Colitis 2012, 6, 324–329. [Google Scholar] [CrossRef] [Green Version]
- Ng, S.C.; Tang, W.; Leong, R.W.; Chen, M.; Ko, Y.; Studd, C.; Niewiadomski, O.; Bell, S.; Kamm, M.A.; de Silva, H.J.; et al. Environmental risk factors in inflammatory bowel disease: A population-based case-control study in Asia-Pacific. Gut 2015, 64, 1063–1071. [Google Scholar] [CrossRef] [Green Version]
- Lichtenstein, G.R.; Loftus, E.V.; Isaacs, K.L.; Regueiro, M.D.; Gerson, L.B.; Sands, B.E. ACG Clinical Guideline: Management of Crohn’s Disease in Adults. Am. J. Gastroenterol. 2018, 113, 481–517. [Google Scholar] [CrossRef]
- Coremans, G.; Rutgeerts, P.; Geboes, K.; Van den Oord, J.; Ponette, E.; Vantrappen, G. The value of ileoscopy with biopsy in the diagnosis of intestinal Crohn’s disease. Gastrointest. Endosc. 1984, 30, 167–172. [Google Scholar] [CrossRef]
- Geboes, K.; Ectors, N.; D’Haens, G.; Rutgeerts, P. Is ileoscopy with biopsy worthwhile in patients presenting with symptoms of inflammatory bowel disease? Am. J. Gastroenterol. 1998, 93, 201–206. [Google Scholar] [CrossRef]
- Dubinsky, M.C.; Kugathasan, S.; Mei, L.; Picornell, Y.; Nebel, J.; Wrobel, I.; Quiros, A.; Silber, G.; Wahbeh, G.; Katzir, L.; et al. Increased immune reactivity predicts aggressive complicating Crohn’s disease in children. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2008, 6, 1105–1111. [Google Scholar] [CrossRef] [Green Version]
- Van Rheenen, P.F.; Van de Vijver, E.; Fidler, V. Faecal calprotectin for screening of patients with suspected inflammatory bowel disease: Diagnostic meta-analysis. BMJ 2010, 341, c3369. [Google Scholar] [CrossRef] [Green Version]
- Gardenbroek, T.J.; Tanis, P.J.; Buskens, C.J.; Bemelman, W.A. Surgery for Crohn’s disease: New developments. Dig. Surg. 2012, 29, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Neumann, P.-A.; Rijcken, E. Minimally invasive surgery for inflammatory bowel disease: Review of current developments and future perspectives. World J. Gastrointest. Pharmacol. Ther. 2016, 7, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Falchuk, K.R.; Falchuk, Z.M. Selective immunoglobulin a deficiency, ulcerative colitis, and gluten-sensitive enteropathy-a unique association. Gastroenterology 1975, 69, 503–506. [Google Scholar] [CrossRef]
- Marks, D.J.B.; Seymour, C.R.; Sewell, G.W.; Rahman, F.Z.; Smith, A.M.; McCartney, S.A.; Bloom, S.L. Inflammatory bowel diseases in patients with adaptive and complement immunodeficiency disorders. Inflamm. Bowel Dis. 2010, 16, 1984–1992. [Google Scholar] [CrossRef] [PubMed]
- Asada, Y.; Isomoto, H.; Shikuwa, S.; Wen, C.Y.; Fukuda, E.; Miyazato, M.; Okamoto, K.; Nakamura, T.; Nishiyama, H.; Mizuta, Y.; et al. Development of ulcerative colitis during the course of rheumatoid arthritis: Association with selective IgA deficiency. World J. Gastroenterol. 2006, 12, 5240–5243. [Google Scholar] [CrossRef]
- Hermaszewski, R.A.; Webster, A.D. Primary hypogammaglobulinaemia: A survey of clinical manifestations and complications. Q. J. Med. 1993, 86, 31–42. [Google Scholar]
- Cunningham-Rundles, C.; Bodian, C. Common variable immunodeficiency: Clinical and immunological features of 248 patients. Clin. Immunol. 1999, 92, 34–48. [Google Scholar] [CrossRef]
- Kainulainen, L.; Nikoskelainen, J.; Ruuskanen, O. Diagnostic findings in 95 Finnish patients with common variable immunodeficiency. J. Clin. Immunol. 2001, 21, 145–149. [Google Scholar] [CrossRef]
- Mannon, P.J.; Fuss, I.J.; Dill, S.; Friend, J.; Groden, C.; Hornung, R.; Yang, Z.; Yi, C.; Quezado, M.; Brown, M.; et al. Excess IL-12 but not IL-23 accompanies the inflammatory bowel disease associated with common variable immunodeficiency. Gastroenterology 2006, 131, 748–756. [Google Scholar] [CrossRef]
- Shulzhenko, N.; Dong, X.; Vyshenska, D.; Greer, R.L.; Gurung, M.; Vasquez-Perez, S.; Peremyslova, E.; Sosnovtsev, S.; Quezado, M.; Yao, M.; et al. CVID enteropathy is characterized by exceeding low mucosal IgA levels and interferon-driven inflammation possibly related to the presence of a pathobiont. Clin. Immunol. 2018, 197, 139–153. [Google Scholar] [CrossRef]
- Cols, M.; Rahman, A.; Maglione, P.J.; Garcia-Carmona, Y.; Simchoni, N.; Ko, H.-B.M.; Radigan, L.; Cerutti, A.; Blankenship, D.; Pascual, V.; et al. Expansion of inflammatory innate lymphoid cells in patients with common variable immune deficiency. J. Allergy Clin. Immunol. 2016, 137, 1206–1215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumgart, D.C.; Sandborn, W.J. Crohn’s disease. Lancet Lond. Engl. 2012, 380, 1590–1605. [Google Scholar] [CrossRef] [Green Version]
- Mannon, P.J.; Fuss, I.J.; Mayer, L.; Elson, C.O.; Sandborn, W.J.; Present, D.; Dolin, B.; Goodman, N.; Groden, C.; Hornung, R.L.; et al. Anti-interleukin-12 antibody for active Crohn’s disease. N. Engl. J. Med. 2004, 351, 2069–2079. [Google Scholar] [CrossRef]
- Sandborn, W.J.; Gasink, C.; Gao, L.-L.; Blank, M.A.; Johanns, J.; Guzzo, C.; Sands, B.E.; Hanauer, S.B.; Targan, S.; Rutgeerts, P.; et al. Ustekinumab induction and maintenance therapy in refractory Crohn’s disease. N. Engl. J. Med. 2012, 367, 1519–1528. [Google Scholar] [CrossRef] [PubMed]
- De Morales, R.J.G.; Muñoz, F.; Hernando, M. Successful Treatment of Common Variable Immunodeficiency-associated Inflammatory Bowel Disease With Ustekinumab. J. Crohns Colitis 2017, 11, 1154–1155. [Google Scholar] [CrossRef] [Green Version]
- Robertson, N.; Engelhardt, K.R.; Morgan, N.V.; Barge, D.; Cant, A.J.; Hughes, S.M.; Abinun, M.; Xu, Y.; Koref, M.S.; Arkwright, P.D.; et al. Astute Clinician Report: A Novel 10 bp Frameshift Deletion in Exon 2 of ICOS Causes a Combined Immunodeficiency Associated with an Enteritis and Hepatitis. J. Clin. Immunol. 2015, 35, 598–603. [Google Scholar] [CrossRef]
- Scott-Taylor, T.H.; Whiting, K.; Pettengell, R.; Webster, D.A. Enhanced formation of giant cells in common variable immunodeficiency: Relation to granulomatous disease. Clin. Immunol. 2017, 175, 1–9. [Google Scholar] [CrossRef]
- Sanges, M.; Spadaro, G.; Miniero, M.; Mattera, D.; Sollazzo, R.; D’Armiento, F.P.; De Palma, G.D.; Pecoraro, A.; Borrelli, F.; Genovese, A.; et al. Efficacy of subcutaneous immunoglobulins in primary immunodeficiency with Crohn’s-like phenotype: Report of a case. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 2641–2645. [Google Scholar]
- Agarwal, S.; Mayer, L. Pathogenesis and treatment of gastrointestinal disease in antibody deficiency syndromes. J. Allergy Clin. Immunol. 2009, 124, 658–664. [Google Scholar] [CrossRef] [Green Version]
- Kalha, I.; Sellin, J.H. Common variable immunodeficiency and the gastrointestinal tract. Curr. Gastroenterol. Rep. 2004, 6, 377–383. [Google Scholar] [CrossRef]
- Bosworth, B.P.; Sanders, A.; Maltz, C. Common variable immunodeficiency masquerading as Crohn’s ileocolitis. Inflamm. Bowel Dis. 2006, 12, 151–152. [Google Scholar] [CrossRef] [PubMed]
- Mudter, J.; Wirtz, S.; Weigmann, B.; Tiede, I.; Tubbe, I.; Kiesslich, R.; Galle, P.R.; Lehr, H.A.; Neurath, M.F. Crohn’s-like colitis in a patient with immunodeficiency associated with a defect in expression of inducible costimulator. Dig. Dis. Sci. 2006, 51, 711–717. [Google Scholar] [CrossRef] [PubMed]
- Vázquez-Morón, J.M.; Pallarés-Manrique, H.; Martín-Suárez, I.J.; Benítez-Rodríguez, B.; Ramos-Lora, M. Crohn’s-like disease in a patient with common variable immunodeficiency treated with azathioprine and adalimumab. Rev. Espanola Enfermedades Dig. Organo Of. Soc. Espanola Patol. Dig. 2013, 105, 299–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khodadad, A.; Aghamohammadi, A.; Parvaneh, N.; Rezaei, N.; Mahjoob, F.; Bashashati, M.; Movahedi, M.; Fazlollahi, M.R.; Zandieh, F.; Roohi, Z.; et al. Gastrointestinal manifestations in patients with common variable immunodeficiency. Dig. Dis. Sci. 2007, 52, 2977–2983. [Google Scholar] [CrossRef]
- Comunoglu, N.; Kara, S.; Kepil, N. Inflammatory bowel disease-like colitis pathology in a patient with common variable immune deficiency. BMJ Case Rep. 2015, 2015. [Google Scholar] [CrossRef] [Green Version]
- Washington, K.; Stenzel, T.T.; Buckley, R.H.; Gottfried, M.R. Gastrointestinal pathology in patients with common variable immunodeficiency and X-linked agammaglobulinemia. Am. J. Surg. Pathol. 1996, 20, 1240–1252. [Google Scholar] [CrossRef]
- Byrne, M.F.; Royston, D.; Patchett, S.E. Association of common variable immunodeficiency with atypical collagenous colitis. Eur. J. Gastroenterol. Hepatol. 2003, 15, 1051–1053. [Google Scholar] [CrossRef]
- Castellano, G.; Moreno, D.; Galvao, O.; Ballestín, C.; Colina, F.; Mollejo, M.; Morillas, J.D.; Solís-Herruzo, J.A. Malignant lymphoma of jejunum with common variable hypogammaglobulinemia and diffuse nodular hyperplasia of the small intestine. A case study and literature review. J. Clin. Gastroenterol. 1992, 15, 128–135. [Google Scholar] [CrossRef]
- Pecoraro, A.; Nappi, L.; Crescenzi, L.; D’Armiento, F.P.; Genovese, A.; Spadaro, G. Chronic Diarrhea in Common Variable Immunodeficiency: A Case Series and Review of the Literature. J. Clin. Immunol. 2018, 38, 67–76. [Google Scholar] [CrossRef]
- Elnachef, N.; McMorris, M.; Chey, W.D. Successful treatment of common variable immunodeficiency disorder-associated diarrhea with budesonide: A case report. Am. J. Gastroenterol. 2007, 102, 1322–1325. [Google Scholar] [CrossRef]
- Arieira, C.; de Dias, C.F.; Moreira, M.J.; Cotter, J. Common Variable Immunodeficiency-Associated Inflammatory Enteropathy: The New Era of Biological Therapy. GE Port. J. Gastroenterol. 2018, 25, 322–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chua, I.; Standish, R.; Lear, S.; Harbord, M.; Eren, E.; Raeiszadeh, M.; Workman, S.; Webster, D. Anti-tumour necrosis factor-alpha therapy for severe enteropathy in patients with common variable immunodeficiency (CVID). Clin. Exp. Immunol. 2007, 150, 306–311. [Google Scholar] [CrossRef]
- Nos, P.; Bastida, G.; Beltran, B.; Aguas, M.; Ponce, J. Crohn’s disease in common variable immunodeficiency: Treatment with antitumor necrosis factor alpha. Am. J. Gastroenterol. 2006, 101, 2165–2166. [Google Scholar] [CrossRef] [PubMed]
- Akazawa, Y.; Takeshima, F.; Yajima, H.; Imanishi, D.; Kanda, T.; Matsushima, K.; Minami, H.; Yamaguchi, N.; Ohnita, K.; Isomoto, H.; et al. Infliximab therapy for Crohn’s-like disease in common variable immunodeficiency complicated by massive intestinal hemorrhage: A case report. BMC Res. Notes 2014, 7, 382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saldaña, D.C.; Rubio, I.S. Immmunodeficiencies and autoimmune diseases: Common variable immunodeficiency and Crohn-like. Rev. Esp. Enferm. Dig. Organo Of. Soc. Esp. Patol. Dig. 2016, 108, 520–523. [Google Scholar] [CrossRef] [PubMed]
- Boland, B.S.; Riedl, M.A.; Valasek, M.A.; Crowe, S.E.; Sandborn, W.J. Vedolizumab in Patients With Common Variable Immune Deficiency and Gut Inflammation. Am. J. Gastroenterol. 2017, 112, 1621. [Google Scholar] [CrossRef]
- Aslam, A.; Misbah, S.A.; Talbot, K.; Chapel, H. Vitamin E deficiency induced neurological disease in common variable immunodeficiency: Two cases and a review of the literature of vitamin E deficiency. Clin. Immunol. 2004, 112, 24–29. [Google Scholar] [CrossRef]
- Aukrust, P.; Müller, F.; Ueland, T.; Svardal, A.M.; Berge, R.K.; Frøland, S.S. Decreased vitamin A levels in common variable immunodeficiency: Vitamin A supplementation in vivo enhances immunoglobulin production and downregulates inflammatory responses. Eur. J. Clin. Investig. 2000, 30, 252–259. [Google Scholar] [CrossRef]
- Teahon, K.; Webster, A.D.; Price, A.B.; Weston, J.; Bjarnason, I. Studies on the enteropathy associated with primary hypogammaglobulinaemia. Gut 1994, 35, 1244–1249. [Google Scholar] [CrossRef] [Green Version]





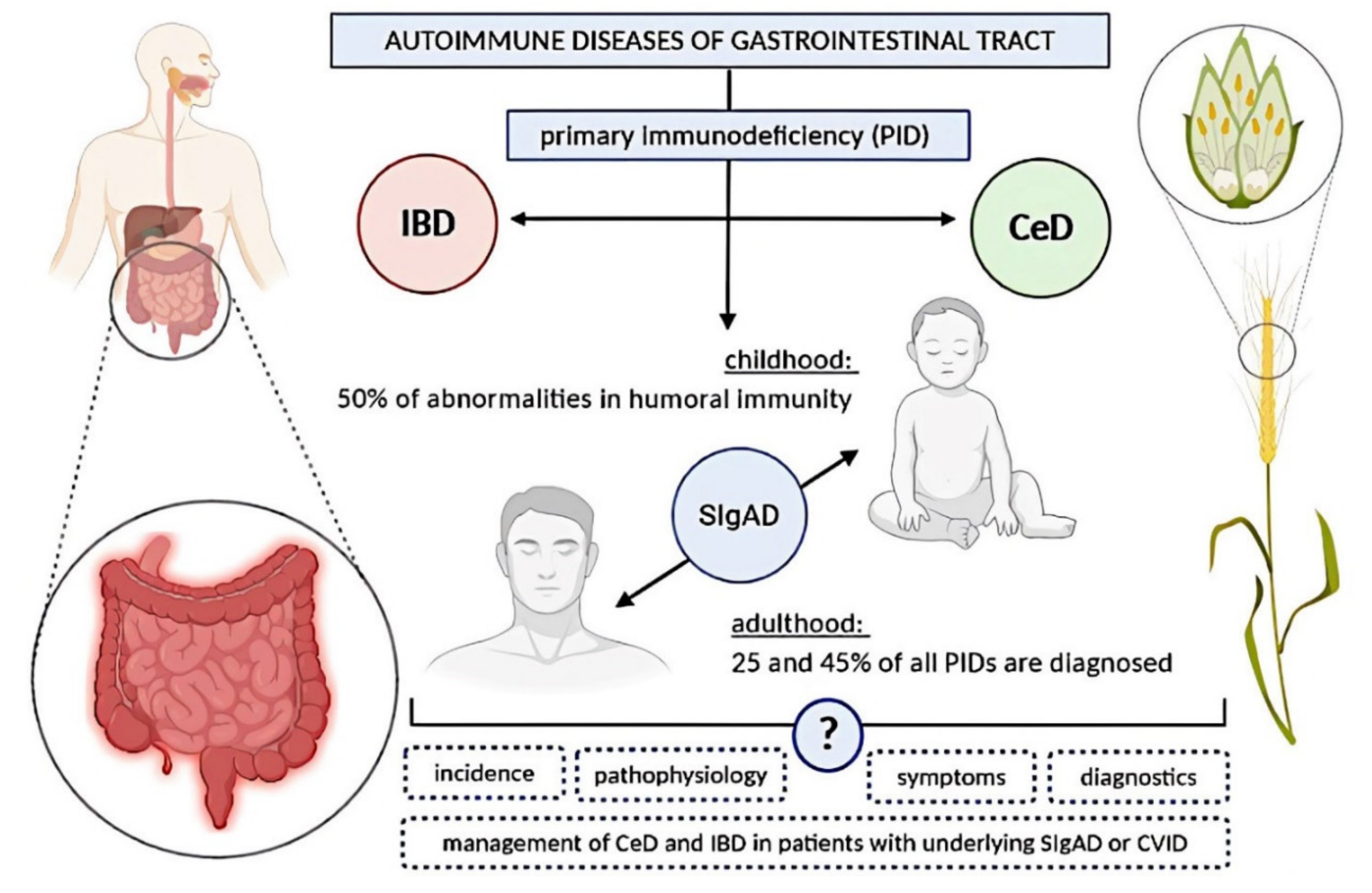{kind=link}
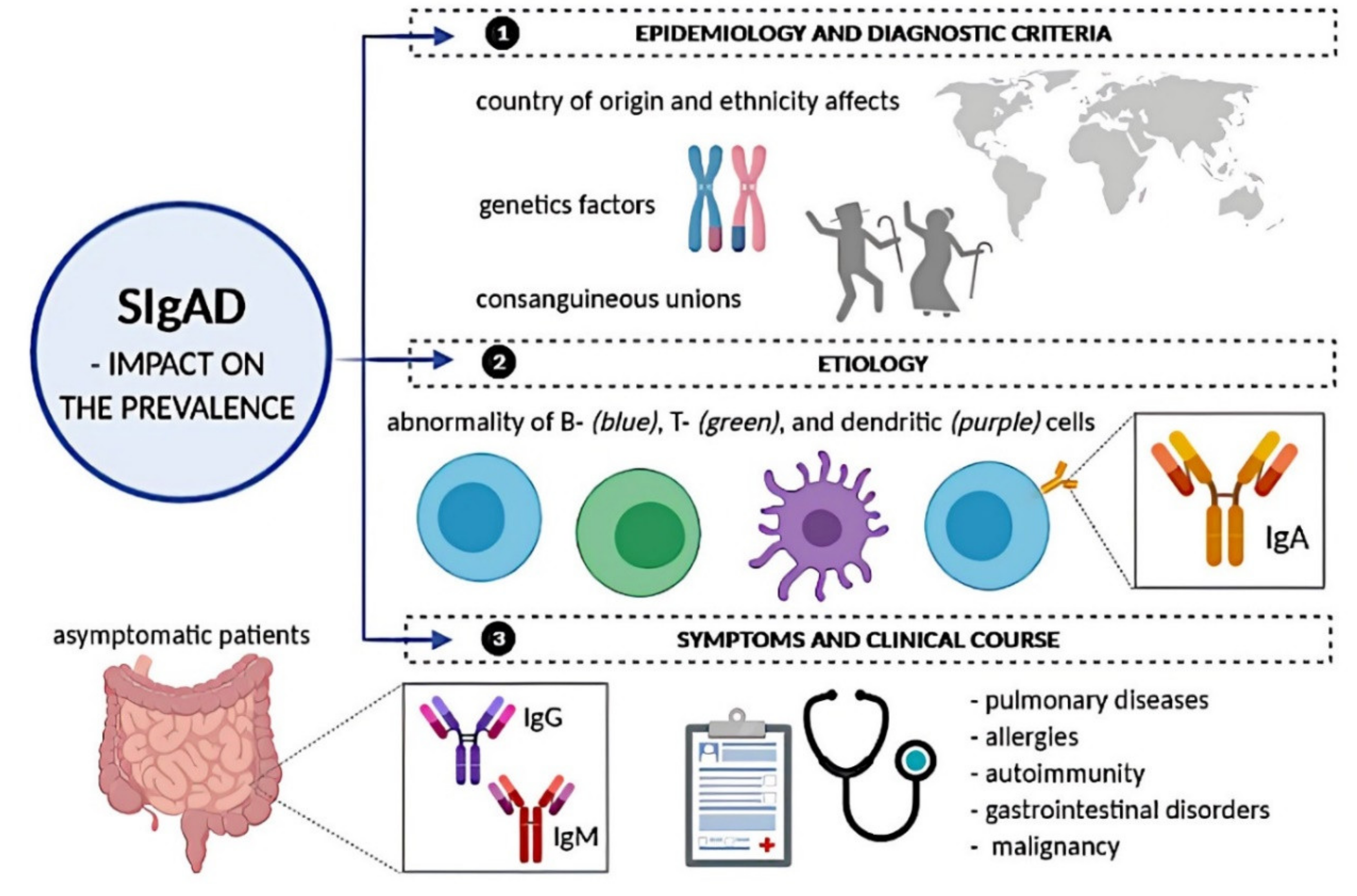{kind=link}
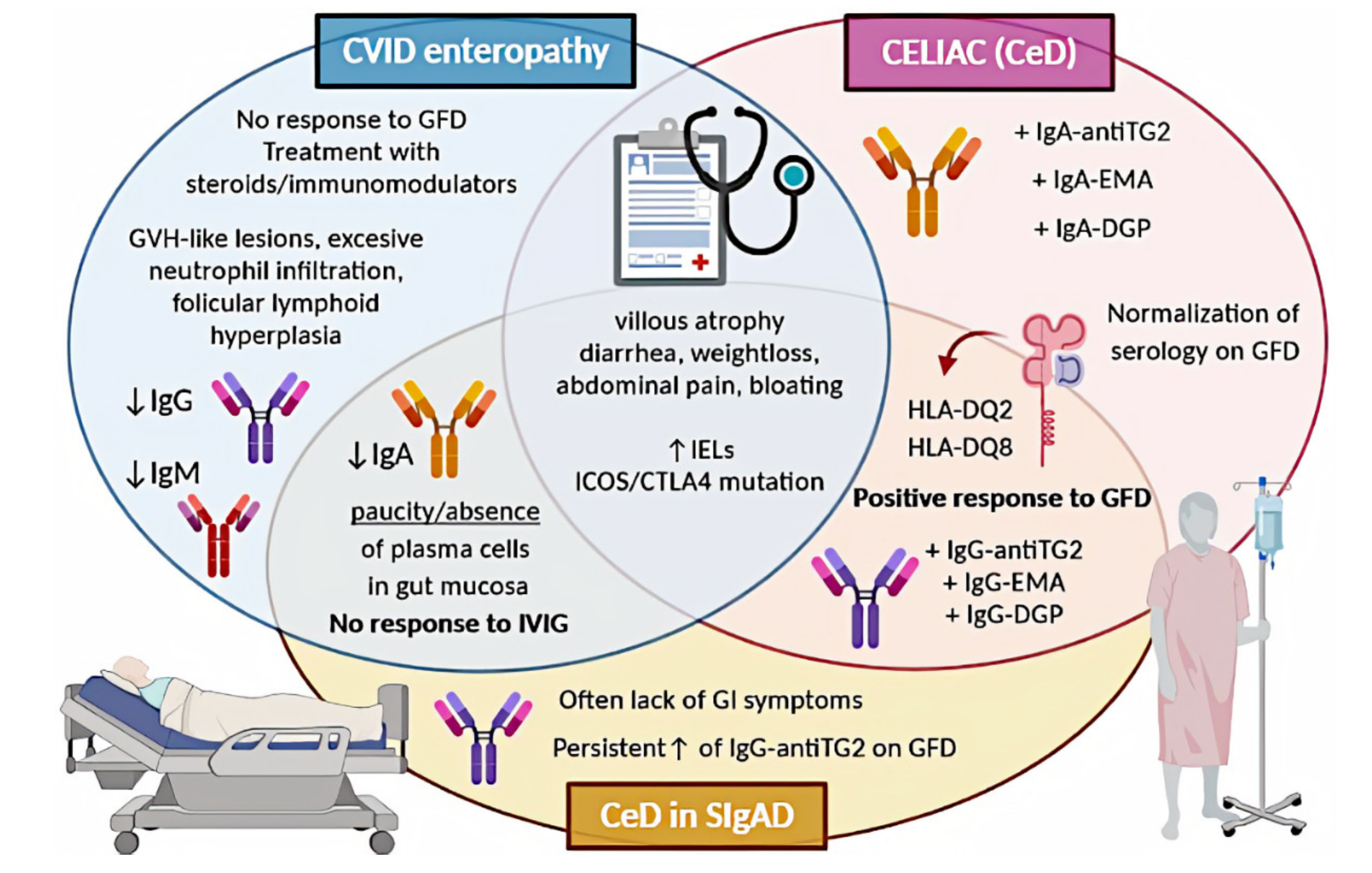{kind=link}
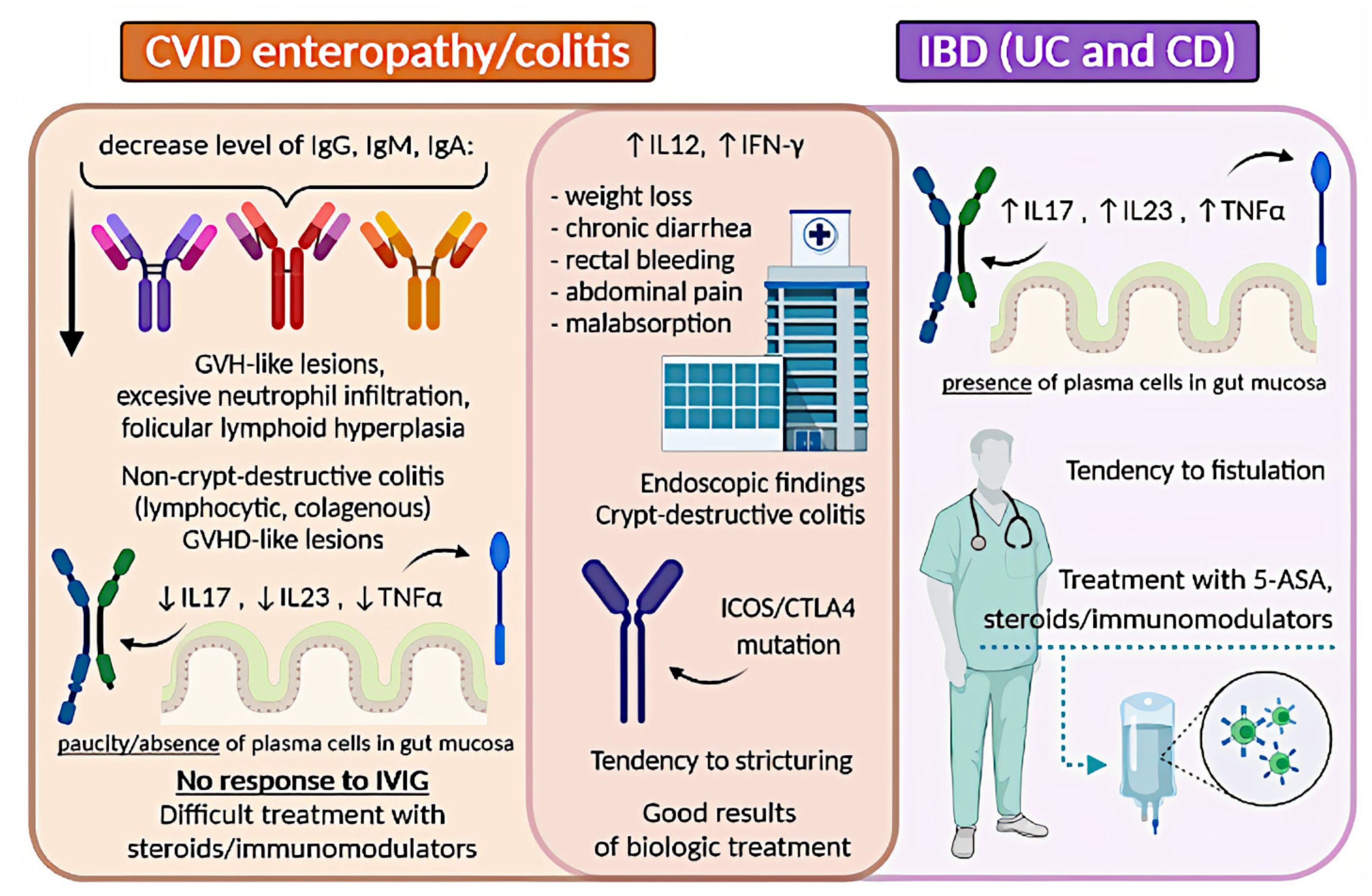{kind=link}
| Selective IgA Deficiency (SIgAD)—Diagnostic Criteria | |
|---|---|
| At least one of: | increased susceptibility to infection |
| autoimmune manifestations | |
| affected family member | |
| ALL of the following: | serum IgA level lower than 7 mg/dL (0.07 g/L) |
| normal serum levels of IgG and IgM | |
| patients older than 4 years | |
| normal IgG antibody response to vaccination | |
| excluded other causes of hypogammaglobulinemia | |
| excluded profound T-cell deficiency | |
| Common Variable Immunodeficiency (CVID)—Diagnostic Criteria | |
|---|---|
| At least one of: | increased susceptibility to infection |
| autoimmune manifestations | |
| granulomatous disease | |
| unexplained polyclonal lymphoproliferation | |
| affected family member with antibody deficiency | |
| ALL of the following: | serum IgG and IgA levels at least 2 SD below the mean for age with/without decreased serum IgM level |
| normal serum levels of IgG and IgM | |
| patients older than 4 years | |
| poor response to vaccination and/or low switched memory B-cells (<70% of age-related normal value) | |
| excluded other causes of hypogammaglobulinemia | |
| excluded profound T-cell deficiency | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malesza, I.J.; Malesza, M.; Krela-Kaźmierczak, I.; Zielińska, A.; Souto, E.B.; Dobrowolska, A.; Eder, P. Primary Humoral Immune Deficiencies: Overlooked Mimickers of Chronic Immune-Mediated Gastrointestinal Diseases in Adults. Int. J. Mol. Sci. 2020, 21, 5223. https://doi.org/10.3390/ijms21155223
Malesza IJ, Malesza M, Krela-Kaźmierczak I, Zielińska A, Souto EB, Dobrowolska A, Eder P. Primary Humoral Immune Deficiencies: Overlooked Mimickers of Chronic Immune-Mediated Gastrointestinal Diseases in Adults. International Journal of Molecular Sciences. 2020; 21(15):5223. https://doi.org/10.3390/ijms21155223
Chicago/Turabian StyleMalesza, Ida Judyta, Michał Malesza, Iwona Krela-Kaźmierczak, Aleksandra Zielińska, Eliana B. Souto, Agnieszka Dobrowolska, and Piotr Eder. 2020. "Primary Humoral Immune Deficiencies: Overlooked Mimickers of Chronic Immune-Mediated Gastrointestinal Diseases in Adults" International Journal of Molecular Sciences 21, no. 15: 5223. https://doi.org/10.3390/ijms21155223