Single Nucleotide Polymorphisms as Practical Molecular Tools to Support European Chestnut Agrobiodiversity Management


Abstract
:
1. Introduction
2. Results
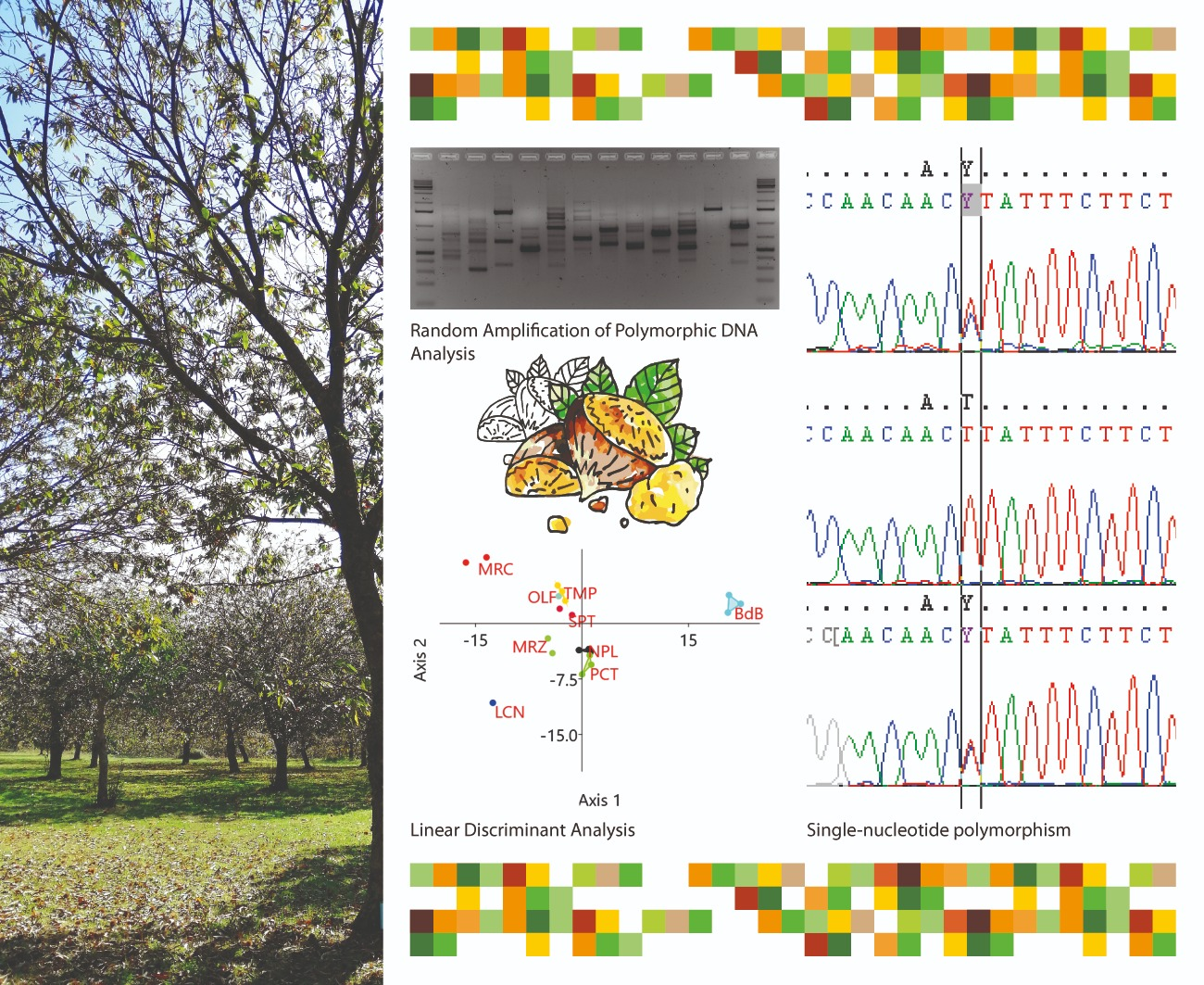
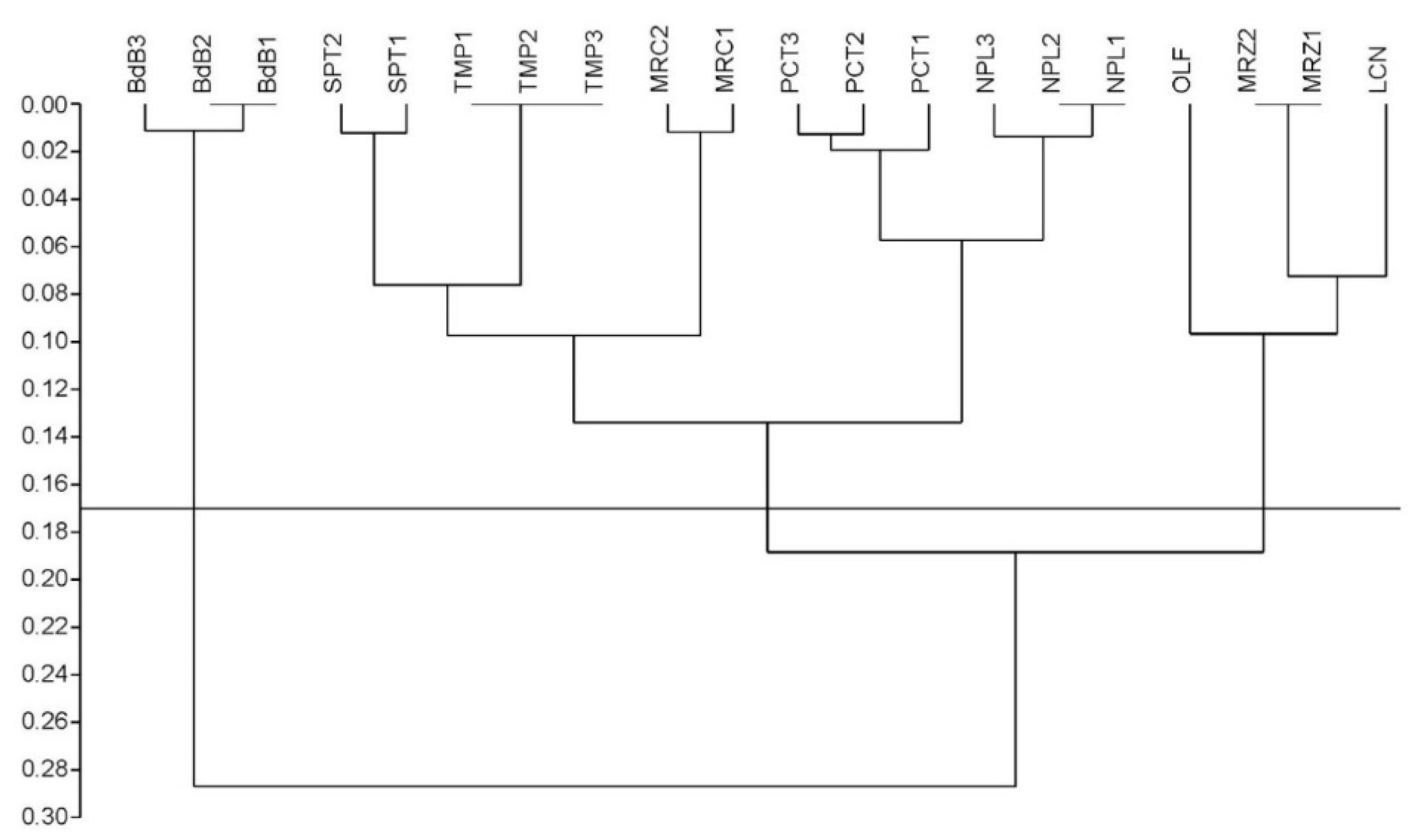
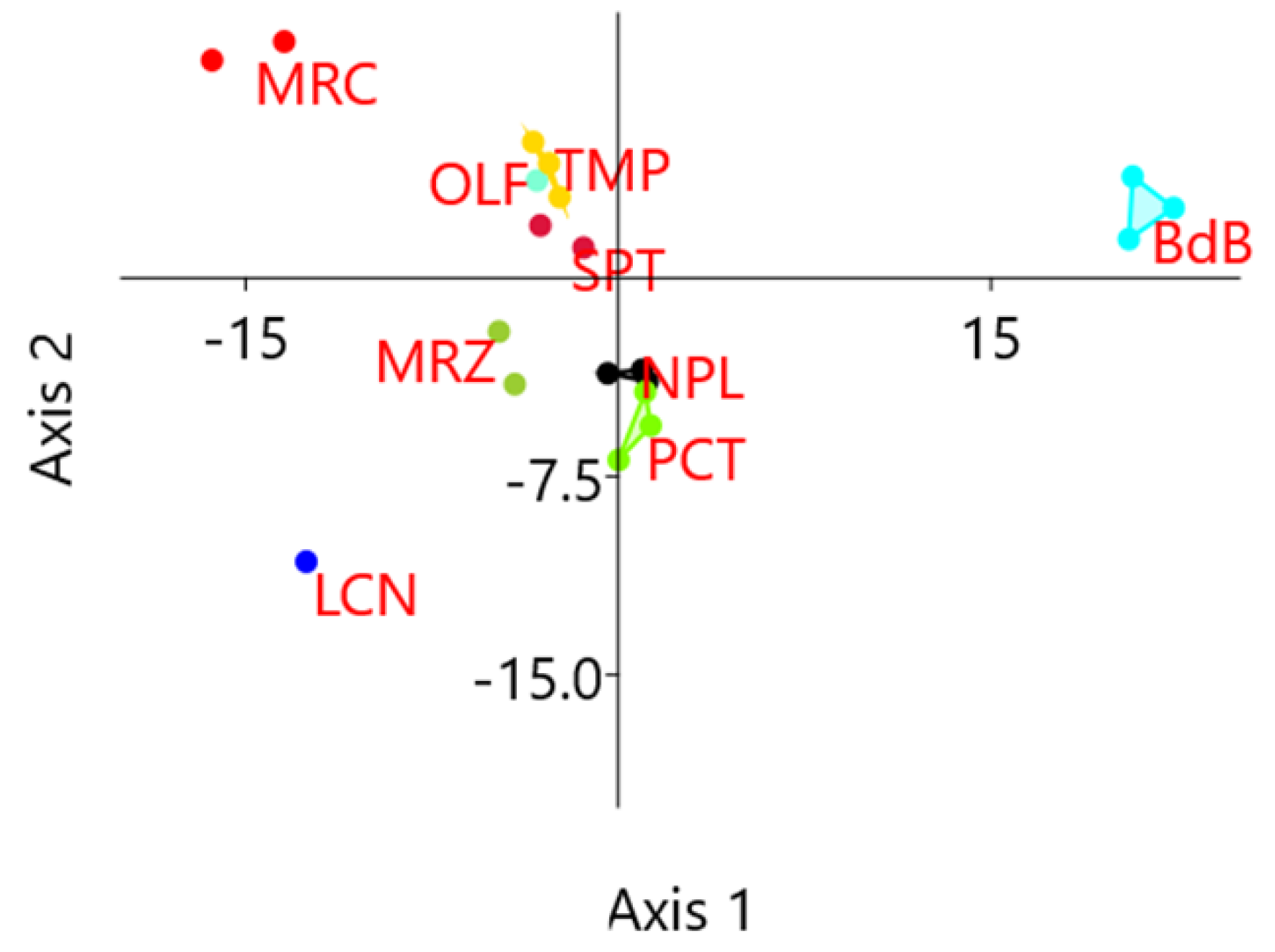
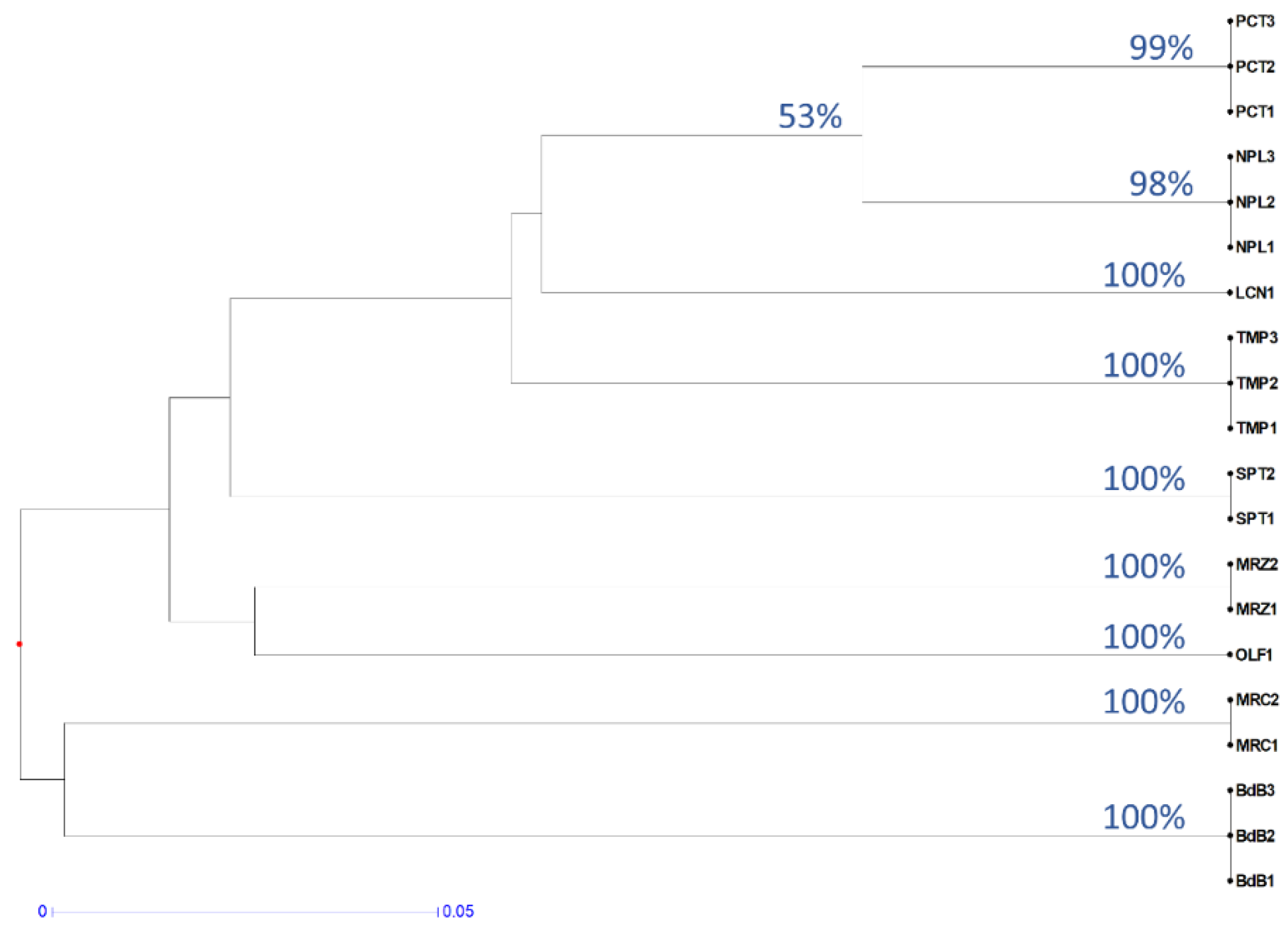
2.1. Preliminary Assessment of Genetic Diversity in Chestnut Cultivars by RAPD Molecular Markers
2.2. SNP Mining in C. sativa
2.2.1. Selection of Target Loci from C. Crenata
2.2.2. High-Resolution Melting (HRM) Screening to Select Polymorphic C. sativa DNA Fragments for Subsequent Resequencing
2.2.3. Target Resequencing of Polymorphic Fragments Selected by HRM Analysis
2.3. KASP Assay Selection for Varietal Identification
3. Discussion
4. Materials and Methods
4.1. Plant Material and DNA isoLation
4.2. Random Amplified Polymorphic DNA (RAPD) Analysis and Clustering
4.3. High-Resolution Melting (HRM) Analysis
4.4. Target Resequencing
4.5. Kompetitive Allele-Specific PCR (KASP) Assays
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| EST | Expressed Sequence Tag |
| FRET | Fluorescence Resonance Energy Transfer |
| HRM | High-Resolution Melting |
| PGI | Protected Geographical Indication |
| ISSR | Inter Simple Sequence Repeat |
| KASP | Kompetitive Allele-Specific PCR |
| LDA | Linear Discriminant Analysis |
| MAF | Minor Allele Frequency |
| MAS | Molecular Marker-Assisted Selection |
| NCBI | National Center for Biotechnology Information |
| NGS | Next Generation Sequencing |
| RAPD | Random Amplified Polymorphic DNA |
| SNP | Single Nucleotide Polymorphism |
| SSR | Simple Sequence Repeats |
| UPGMA | Unweighted Pair Group Method with Arithmetic mean |
| UPOV | International Union for the Protection of New Varieties of Plants |
References
- FAOSTAT. Available online: http://www.fao.org/faostat/en/#home (accessed on 5 June 2020).
- Beccaro, G.L.; Alma, A.; Boni, I.; Botta, R.; Bussone, M.; Corgnati, M.; Ebone, A.; Gonthier, P.; Locatelli, G.; Malacarne, E.; et al. Chestnut R&D Centre: The trategic project of Piemonte to support the whole chestnut supply chain. L’italia. E Mont. 2020, 75, 39–48. [Google Scholar] [CrossRef]
- Pereira-Lorenzo, S.; Ballester, A.; Corredoira, E.; Vieitez, A.M.; Agnanostakis, S.; Costa, R.; Bounous, G.; Botta, R.; Beccaro, G.L.; Kubisiak, T.L.; et al. Fruit Breeding; Badenes, M.L.H., Byrne, D., Eds.; Springe: Boston, MA, USA, 2012; pp. 729–769. [Google Scholar]
- De Biaggi, M.; Beccaro, G.; Casey, J.; Riqué, P.H.; Conedera, M.; Gomes-Laranjo, J.; Fulbright, D.W.; Nishio, S.; Serdar, Ü.; Zou, F.; et al. The Chestnut Handbook; CRC Press: Boca Raton, FL, USA, 2019; pp. 35–52. [Google Scholar]
- Roces-Díaz, J.V.; Jiménez-Alfaro, B.; Chytrý, M.; Díaz-Varela, E.R.; Álvarez-Álvarez, P. Glacial refugia and mid-Holocene expansion delineate the current distribution of Castanea sativa in Europe. Palaeogeogr. Palaeoclim. Palaeoecol. 2018, 491, 152–160. [Google Scholar] [CrossRef] [Green Version]
- Vella, F.M.; De Masi, L.; Calandrelli, R.; Morana, A.; Laratta, B. Valorization of the agro-forestry wastes from Italian chestnut cultivars for the recovery of bioactive compounds. Eur. Food Res. Technol. 2019, 245, 2679–2686. [Google Scholar] [CrossRef]
- Galderisi, U.; Cipollaro, M.; Di Bernardo, G.; De Masi, L.; Galano, G.; Cascino, A. Molecular typing of Italian sweet chestnut cultivars by random amplified polymorphic DNA analysis. J. Hortic. Sci. Biotechnol. 1998, 73, 259–263. [Google Scholar] [CrossRef]
- Grassi, G. Germoplasma e biodiversità del castagno da frutto in Campania. In Il Castagno in Campania—Problematiche e Prospettive Della Filiera; Cristinzio, G., Testa, A., Eds.; Società Editrice Imago Media: Dragoni (CE), Italy, 2006; pp. 62–73. [Google Scholar]
- NCBI—Nucleotide Database. Available online: https://www.ncbi.nlm.nih.gov/nucleotide/ (accessed on 5 June 2020).
- European and Japanese Chestnut RNASeq|HWG. Available online: https://www.hardwoodgenomics.org/content/european-and-japanese-chestnut-rnaseq (accessed on 5 June 2020).
- Santos, C.; Nelson, C.D.; Zhebentyayeva, T.; Machado, H.; Gomes-Laranjo, J.; Costa, R.L. First interspecific genetic linkage map for Castanea sativa × Castanea crenata revealed QTLs for resistance to Phytophthora cinnamomi. PLoS ONE 2017, 12, e0184381. [Google Scholar] [CrossRef] [Green Version]
- Serrazina, S.; Santos, C.; Machado, H.; Pesquita, C.; Vicentini, R.; Pais, M.S.; Sebastiana, M.; Costa, R. Castanea root transcriptome in response to Phytophthora cinnamomi challenge. Tree Genet. Genomes 2015, 11, 1–19. [Google Scholar] [CrossRef]
- Acquadro, A.; Torello Marinoni, D.; Sartor, C.; Dini, F.; Macchio, M.; Botta, R. Transcriptome characterization and expression profiling in chestnut cultivars resistant or susceptible to the gall wasp Dryocosmus kuriphilus. Mol. Genet. Genom. 2020, 295, 107–120. [Google Scholar] [CrossRef]
- Casasoli, M.; Mattioni, C.; Cherubini, M.; Villani, F. A genetic linkage map of European chestnut (Castanea sativa Mill.) based on RAPD, ISSR and isozyme markers. Appl. Genet. 2001, 102, 1190–1199. [Google Scholar] [CrossRef]
- Martín, M.A.; Mattioni, C.; Cherubini, M.; Taurchini, D.; Villani, F. Genetic characterisation of traditional chestnut varieties in Italy using microsatellites (simple sequence repeats) markers. Ann. Appl. Biol. 2010, 157, 37–44. [Google Scholar] [CrossRef]
- Mellano, M.G.; Beccaro, G.L.; Donno, D.; Marinoni, D.T.; Boccacci, P.; Canterino, S.; Cerutti, A.K.; Bounous, G. Castanea spp. biodiversity conservation: Collection and characterization of the genetic diversity of an endangered species. Genet. Resour. Crop Evol. 2012, 59, 1727–1741. [Google Scholar] [CrossRef]
- Marinoni, D.; Akkak, A.; Bounous, G.; Edwards, K.J.; Botta, R. Development and characterization of microsatellite markers in Castanea sativa (Mill.). Mol. Breed. 2003, 11, 127–136. [Google Scholar] [CrossRef]
- Buck, E.J.; Hadonou, M.; James, C.J.; Blakesley, D.; Russell, K. Isolation and characterization of polymorphic microsatellites in European chestnut (Castanea sativa Mill.). Mol. Ecol. Notes 2003, 3, 239–241. [Google Scholar] [CrossRef]
- Sebastiani, F.; Carnevale, S.; Vendramin, G.G. A new set of mono- and dinucleotide chloroplast microsatellites in Fagaceae. Mol. Ecol. Notes 2004, 4, 259–261. [Google Scholar] [CrossRef]
- Míguez-Soto, B.; Fernández-Cruz, J.; Fernández-López, J. Mediterranean and Northern Iberian gene pools of wild Castanea sativa Mill. are two differentiated ecotypes originated under natural divergent selection. PLoS ONE 2019, 14, e0211315. [Google Scholar] [CrossRef] [Green Version]
- Jarman, R.; Mattioni, C.; Russell, K.; Chambers, F.M.; Bartlett, D.; Angela Martin, M.; Cherubini, M.; Villani, F.; Webb, J. DNA analysis of Castanea sativa (sweet chestnut) in Britain and Ireland: Elucidating European origins and genepool diversity. PLoS ONE 2019, 14, e0222936. [Google Scholar] [CrossRef]
- Nishio, S.; Terakami, S.; Matsumoto, T.; Yamamoto, T.; Takada, N.; Kato, H.; Katayose, Y.; Saito, T. Identification of QTLs for agronomic traits in the Japanese chestnut (Castanea crenata Sieb. et Zucc.) breeding. Hortic. J. 2018, 87, 43–54. [Google Scholar] [CrossRef] [Green Version]
- Bouffartigue, C.; Debille, S.; Fabreguette, O.; Cabrer, A.R.; Pereira-Lorenzo, S.; Flutre, T.; Harvengt, L. Genetic diversity and structure of sweet chestnut (Castanea sativa Mill.) in France: At the intersection between Spain and Italy. bioRxiv 2019, 792259. [Google Scholar] [CrossRef] [Green Version]
- Bianco, L.; Cestaro, A.; Sargent, D.J.; Banchi, E.; Derdak, S.; Di Guardo, M.; Salvi, S.; Jansen, J.; Viola, R.; Gut, I.; et al. Development and Validation of a 20K Single Nucleotide Polymorphism (SNP) Whole Genome Genotyping Array for Apple (Malus × domestica Borkh). PLoS ONE 2014, 9, e110377. [Google Scholar] [CrossRef] [Green Version]
- Chagné, D.; Crowhurst, R.N.; Troggio, M.; Davey, M.W.; Gilmore, B.; Lawley, C.; Vanderzande, S.; Hellens, R.P.; Kumar, S.; Cestaro, A.; et al. Genome-wide SNP detection, validation, and development of an 8K SNP array for apple. PLoS ONE 2012, 7, e31745. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Liu, S.M.; Fu, H.W.; Huang, J.Z.; Shu, Q.Y. High-resolution melting-based TILLING of γ ray-induced mutations in rice. J. Zhejiang Univ. Sci. B 2018, 19, 620–629. [Google Scholar] [CrossRef]
- Gady, A.L.F.; Hermans, F.W.K.; Van De Wal, M.H.B.J.; Van Loo, E.N.; Visser, R.G.F.; Bachem, C.W.B. Implementation of two high through-put techniques in a novel application: Detecting point mutations in large EMS mutated plant populations. Plant Methods 2009, 5, 13. [Google Scholar] [CrossRef] [Green Version]
- Semagn, K.; Babu, R.; Hearne, S.; Olsen, M. Single nucleotide polymorphism genotyping using Kompetitive Allele Specific PCR (KASP): Overview of the technology and its application in crop improvement. Mol. Breed. 2014, 33, 1–14. [Google Scholar] [CrossRef]
- Hammer, Ø.; Harper, D.A.T.; Ryan, P.D. Past: Paleontological statistics software package for education and data analysis. Palaeontol. Electron. 2001, 4, 1–9. [Google Scholar]
- Staton, M.; Zhebentyayeva, T.; Olukolu, B.; Fang, G.C.; Nelson, D.; Carlson, J.E.; Abbott, A.G. Substantial genome synteny preservation among woody angiosperm species: Comparative genomics of Chinese chestnut (Castanea mollissima) and plant reference genomes. BMC Genom. 2015, 16, 744. [Google Scholar] [CrossRef] [Green Version]
- Kang, M.J.; Shin, A.Y.; Shin, Y.; Lee, S.A.; Lee, H.R.; Kim, T.D.; Choi, M.; Koo, N.; Kim, Y.M.; Kyeong, D.; et al. Identification of transcriptome-wide, nut weight-associated SNPs in Castanea crenata. Sci. Rep. 2019, 9, 13161. [Google Scholar] [CrossRef] [Green Version]
- Nishio, S.; Yamamoto, T.; Terakami, S.; Sawamura, Y.; Takada, N.; Saito, T. Genetic diversity of Japanese chestnut cultivars assessed by SSR markers. Breed. Sci. 2011, 61, 109–120. [Google Scholar] [CrossRef] [Green Version]
- Vondras, A.M.; Minio, A.; Blanco-Ulate, B.; Figueroa-Balderas, R.; Penn, M.A.; Zhou, Y.; Seymour, D.; Ye, Z.; Liang, D.; Espinoza, L.K.; et al. The genomic diversification of grapevine clones. BMC Genom. 2019, 20, 972. [Google Scholar] [CrossRef] [Green Version]
- De Masi, L.; Minasi, P.; Boscaino, F.; Galano, G.; Castaldo, D.; Laratta, B. A preliminary study on DNA typing of Calabrian chestnut by RAPD analysis. Essenze Deriv. Agrum. 2002, 7, 3–9. [Google Scholar]
- De Masi, L.; Castaldo, D.; Minasi, P.; Laratta, B. Caratterizzazione e Certificazione di Qualità di fico e Castagno Calabresi Attraverso Moderne Tecniche di Biologia Molecolare; Monografia.; Stazione Sperimentale per l’Industria delle Essenze e dei Derivati dagli Agrumi: Reggio Calabria, Italy, 2004. [Google Scholar]
- De Masi, L.; Castaldo, D.; Galano, G.; Minasi, P.; Laratta, B. Genotyping of fig (Ficus carica L) via RAPD markers. J. Sci. Food Agric. 2005, 85, 2235–2242. [Google Scholar] [CrossRef]
- Cautela, D.; Balestrieri, M.L.; Savini, S.; Sannino, A.; Ferrari, G.; Servillo, L.; De Masi, L.; Pastore, A.; Castaldo, D. The ancient neapolitan sweet lime and the calabrian lemoncetta locrese belong to the same citrus species. Molecules 2020, 25, 113. [Google Scholar] [CrossRef] [Green Version]
- Williams, J.G.K.; Kubelik, A.R.; Livak, K.J.; Rafalski, J.A.; Tingey, S.V. DNA polymorphisms amplified by arbitrary primers are useful as genetic markers. Nucleic Acids Res. 1990. [Google Scholar] [CrossRef] [Green Version]
- Jamali, S.H.; Cockram, J.; Hickey, L.T. Insights into deployment of DNA markers in plant variety protection and registration. Appl. Genet. 2019, 132, 1911–1929. [Google Scholar] [CrossRef] [PubMed]
- Mammadov, J.; Aggarwal, R.; Buyyarapu, R.; Kumpatla, S. SNP markers and their impact on plant breeding. Int. J. Plant Genom. 2012, 2012, 728398. [Google Scholar] [CrossRef]
- Pereira-Lorenzo, S.; Ramos-Cabrer, A.M.; Barreneche, T.; Mattioni, C.; Villani, F.; Díaz-Hernández, M.B.; Martín, L.M.; Martín, Á. Database of European chestnut cultivars and definition of a core collection using simple sequence repeats. Tree Genet. Genomes 2017, 13, 114. [Google Scholar] [CrossRef]
- Pereira-Lorenzo, S.; Ramos-Cabrer, A.M.; Barreneche, T.; Mattioni, C.; Villani, F.; Díaz-Hernández, B.; Martín, L.M.; Robles-Loma, A.; Cáceres, Y.; Martín, A. Instant domestication process of European chestnut cultivars. Ann. Appl. Biol. 2019, 174, 74–85. [Google Scholar] [CrossRef] [Green Version]
- Van Inghelandt, D.; Melchinger, A.E.; Lebreton, C.; Stich, B. Population structure and genetic diversity in a commercial maize breeding program assessed with SSR and SNP markers. Appl. Genet. 2010, 120, 1289–1299. [Google Scholar] [CrossRef] [Green Version]
- Martín, M.A.; Mattioni, C.; Cherubini, M.; Villani, F.; Martín, L.M. A comparative study of European chestnut varieties in relation to adaptive markers. Agrofor. Syst. 2017, 91, 97–109. [Google Scholar] [CrossRef]
- Dini, F.; Sartor, C.; Botta, R. Detection of a hypersensitive reaction in the chestnut hybrid “Bouche de Bétizac” infested by Dryocosmus kuriphilus Yasumatsu. Plant Physiol. Biochem. 2012, 60, 67–73. [Google Scholar] [CrossRef]
- PAST. Available online: https://folk.uio.no/ohammer/past/ (accessed on 5 June 2020).
- Sokal, R.R.; Michener, C.D. A Statistical Methods for Evaluating Relationships. Univ. Kans. Sci. Bull. 1958, 38, 1409–1448. [Google Scholar]
- Wittwer, C.T.; Reed, G.H.; Gundry, C.N.; Vandersteen, J.G.; Pryor, R.J. High-resolution genotyping by amplicon melting analysis using LCGreen. Clin. Chem. 2003, 49, 853–860. [Google Scholar] [CrossRef] [Green Version]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gene Ontology Resource. Available online: http://geneontology.org/ (accessed on 5 June 2020).
- Ensembl Variations. Available online: https://m.ensembl.org/info/genome/variation/prediction/predicted_data.html (accessed on 5 June 2020).
- DARwin–Dissimilarity Analysis and Representation for Windows. Available online: https://darwin.cirad.fr/index.php (accessed on 5 June 2020).




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SNP Code | Description | AA Change | Variant Type | Impact | Functional Description | Ref Sequence_ID |
|---|---|---|---|---|---|---|
| A3079 | caC/caT | His/His | synonymous | LOW | uncharacterized protein LOC115978171 | Mad_contig_257281 |
| A4081 | Aca/Gca | Thr/Ala | missense | MODERATE | two-component response regulator-like APRR9 | BdB_contig_30280 |
| A5096 | gtA/gtC | Val/Val | synonymous | LOW | uncharacterized membrane protein At3g27390 | BdB_contig_81679 |
| A7075 | ccA/ccC | Pro/Pro | synonymous | LOW | DExH-box ATP-dependent RNA helicase DExH7 | BdB_contig_45109 |
| A8045 | caC/caT | His/His | synonymous | LOW | NAC domain-containing protein 35 | BdB_contig_49951 |
| A8095 | cTg/cAg | Leu/Gln | missense | MODERATE | ||
| A8121 | Ttt/Ctt | Phe/Leu | missense | MODERATE | ||
| A9081 | ctA/ctT | Leu/Leu | synonymous | LOW | Fanconi anemia group J protein homolog | BdB_contig_161049 |
| B0033 | gcA/gcG | Ala/Ala | synonymous | LOW | meiotic recombination protein DMC1 homolog | BdB_contig_344176 |
| B0042 | caT/caC | His/His | synonymous | LOW | ||
| B0081 | agG/agA | Arg/Arg | synonymous | LOW | ||
| B1077 | acA/acG | Thr/Thr | synonymous | LOW | putative E3 ubiquitin-protein ligase RF298 | BdB_contig_20383 |
| B2081, B2082 | AGa/RRa | Arg/Glu | missense | MODERATE | probable protein phosphatase 2C 42 | Mad_contig_4982354 |
| B3127 | ctT/ctC | Leu/Leu | synonymous | LOW | nuclear pore complex protein NUP58 | XM_024070927 |
| B5060 | gaA/gaG | Glu/Glu | missense | MODERATE | trihelix transcription factor GT-2-like | BdB_contig_32135 |
| C0114 | agT/agC | Ser/Ser | synonymous | LOW | uncharacterized protein LOC115981485 | Mad_contig_4969272 |
| C1083 | aAg/aGa | Lys/Arg | missense | MODERATE | intermediate cleaving peptidase 55, mitochondrial | BdB_contig_2198 |
| C1115 | atT/atA | Ile/Ile | synonymous | LOW | ||
| C2122 | aCt/aAt | - | intergenic | MODIFIER | protein PHR1-LIKE 1-like | XM_024065219 |
| C3057 | ggC/ggT | Gly/Gly | synonymous | LOW | pentatricopeptide repeat-containing protein | BdB_contig_73204 |
| C4088 | Acc/Gcc | Thr/Ala | missense | MODERATE | uncharacterized protein LOC115978856 | BdB_contig_23188 |
| C4092 | tTc/tCt | Phe/Ser | missense | MODERATE | ||
| C5126 | ccA/ccC | Pro/Pro | synonymous | LOW | peptidyl-prolyl cis-trans isomerase E | XM_024025215 |
| C7034, C7035 | cRY/cRY | His/Arg | missense | MODERATE | 26S proteasome non-ATPase regulatory subunit 10 | XM_024044673 |
| C7040 | aAa/aTa | Lys/Ile | missense | MODERATE | ||
| C9098 | aCt/aAt | Thr/Asn | missense | MODERATE | dentin sialophospho | BdB_contig_9459 |
| D3098 | ggG/ggT | Gly/Gly | synonymous | LOW | Ribosomal protein | BdB_contig_47152 |
| D3104 | tcG/tcA | Ser/Ser | synonymous | LOW | ||
| E0104 | Ggt/Agt | Gly/Ser | missense | MODERATE | midasin | XM_031113223 |
| E1081 | Agg/Ggg | Arg/Gly | missense | MODERATE | probable E3 ubiquitin-protein ligase RHC1A | Mad_contig_9295 |
| E1091 | gCt/gGt | Ala/Gly | missense | MODERATE | zinc finger protein ZAT3-like | Mad_contig_9295 |
| E2081 | ttT/ttC | Phe/Phe | synonymous | LOW | peroxidase 19 | BdB_contig_168068 |
| E2115 | cAt/cGt | His/Arg | missense | MODERATE | ||
| F0090 | gAa/gGa | Glu/Leu | missense | MODERATE | formin-like protein 5 | BdB_contig_78166 |
| F0115 | Tat/Cat | Tyr/His | missense | MODERATE | ||
| F0116 | ctG/ctT | Leu/Leu | synonymous | LOW | ||
| F1081 | tcT/tcA | Ser/Ser | synonymous | LOW | protein-tyrosine-phosphatase IBR5 | BdB_contig_59988 |
| F3045 | gaC/gaT | Asp/Asp | synonymous | LOW | uncharacterized aarF domain-containing protein kinase | BdB_contig_105211 |
| F3081 | Tct/Cct | Ser/Pro | missense | MODERATE | ||
| G0115 | Act/Gct | Ala/Thr | missense | MODERATE | probable E3 ubiquitin-protein ligase RHC1A | XM_024060281 |
| G1038 | atT/atC | Ile/Ile | synonymous | LOW | protease Do-like 7 | BdB_contig_20564 |
| G3081 | ggC/ggT | Gly/Gly | synonymous | LOW | phospholipase A-2-activating protein | BdB_contig_6253 |
| G3111 | tgT/tgC | Cys/Cys | synonymous | LOW | ||
| G4023 | gCg/gTg | Ala/Val | missense | MODERATE | uncharacterized protein LOC112036137 | Mad_contig_4962484 |
| G4120 | tcT/tcC | Ser/Ser | synonymous | LOW | ||
| G5075 | ggG/ggA | Gly/Gly | synonymous | LOW | zinc finger protein ZAT3-like | XM_031086265 |
| G5083 | Tcc/Gcc | Ser/Ala | missense | MODERATE | ||
| G5135 | ccA/ccG | Pro/Pro | synonymous | LOW | ||
| H0106 | tTt/tAt | Phe/Tyr | missense | MODERATE | AP2/ERF and B3 domain-containing RAV1-like | BdB_contig_11865 |
| H2081 | gcC/gcT | Ala/Ala | synonymous | LOW | importin-5-like | BdB_contig_11365 |
| H2132 | tcG/tcA | Ser/Ser | synonymous | LOW | ||
| H4081 | gcC/gcT | Ala/Ala | synonymous | LOW | ATP synthase subunit beta, mitochondrial | BdB_contig_1478 |
| I0081 | acG/acA | Thr/Thr | synonymous | LOW | mediator of RNA polymerase II transcription subunit 4 | BdB_contig_35713 |
| I2081 | gGt/gCt | Gly/Ala | missense | MODERATE | WUSCHEL-related homeobox 11-like | XM_031069633 |
| J0109 | cAt/cGt | His/Arg | missense | MODERATE | probable receptor-like serine/threonine-protein kinase | BdB_contig_35898 |
| J1105 | tcG/tcC | Ser/Ser | synonymous | LOW | zinc finger CCCH domain-containing protein 12 | BdB_contig_42954 |
| J1108 | tcT/tcG | Ser/Ser | synonymous | LOW | ||
| J2081 | aAc/aGc | Asn/Ser | missense | MODERATE | E3 ubiquitin-protein ligase | BdB_contig_13323 |
| K0044 | ggC/ggT | Gly/Gly | synonymous | LOW | nuclear-pore anchor | BdB_contig_7400 |
| K0049 | aCa/aTa | Thr/Ile | missense | MODERATE | ||
| K0081 | Atg/Gtg/Ttg | Met/Val/Leu | missense | MODERATE | ||
| K1126 | atA/atG | Ile/Met | missense | MODERATE | serrate RNA effector molecule-like | BdB_contig_6695 |
| K4081 | gaC/gaT | Asp/Asp | synonymous | LOW | O-fucosyltransferase 20 | BdB_contig_67709 |
| L0115 | gCg/gGg | Ala/Gly | missense | MODERATE | protein DOWNY MILDEW RESISTANCE 6 | BdB_contig_72305 |
| L1081 | ccG/ccA | Pro/Pro | synonymous | LOW | serine/threonine-protein kinase-like protein | Mad_contig_4983178 |
| L1117 | cgA/cgT | Arg/Arg | synonymous | LOW | ||
| L2056 | cAt/cGt | His/Arg | missense | MODERATE | uncharacterized protein LOC112025178 | BdB_contig_51854 |
| L4081 | ctC/ctT | Leu/Leu | synonymous | LOW | GTP cyclohydrolase 1 | BdB_contig_65823 |
| Clone | A3079 | A5096 | A7075 | A8095 | A9081 | B0042 | B1077 | B2082 | B3127 | B5060 | C0114 | C1083 | C2122 | C3057 | C4088 | C4092 | C9098 | D3098 | D3104 | E0104 | E1091 | E2115 | F1081 | F3045 | G0115 | G4120 | G5083 | H0106 | J0109 | J1108 | J1105 | K0081 | K0049 | K1126 | L0115 | L1117 | L4081 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BdB1 | Y | M | T | W | T | Y | R | R | A | G | T | C | C | C | G | Y | A | G | C | C | S | Y | W | R | R | C | M | W | R | G | G | G | T | C | S | A | A |
| BdB2 | Y | M | T | W | T | Y | R | R | A | G | T | C | C | C | G | Y | A | G | C | C | S | Y | W | R | R | C | M | W | R | G | G | G | T | C | S | A | A |
| BdB3 | Y | M | T | W | T | Y | R | R | A | G | T | C | C | C | G | Y | A | G | C | C | S | Y | W | R | R | C | M | W | R | G | G | G | T | C | S | A | A |
| LCN1 | T | A | T | T | A | C | G | A | A | A | T | Y | C | Y | G | C | A | G | C | Y | C | ? | A | R | R | Y | M | W | A | G | C | G | C | C | G | A | A |
| MRZ1 | T | M | T | T | A | C | ? | R | A | A | Y | Y | C | C | G | C | A | T | Y | C | C | T | A | G | A | T | M | W | A | G | C | R | Y | C | G | W | A |
| MRZ2 | T | M | T | T | A | C | G | R | A | A | Y | Y | C | C | G | C | A | T | Y | C | C | T | A | G | A | T | M | W | A | G | C | R | Y | C | G | W | A |
| MRC1 | Y | M | K | T | T | T | G | G | A | A | Y | C | C | Y | G | C | M | G | C | C | S | Y | W | G | G | Y | M | W | A | K | G | G | Y | Y | G | T | G |
| MRC2 | Y | M | K | T | T | T | G | G | A | A | Y | C | C | Y | G | C | M | G | C | C | S | Y | W | G | G | Y | M | W | A | K | G | G | Y | Y | G | T | G |
| NPL1 | T | A | T | T | W | Y | G | R | A | A | T | T | M | C | G | Y | A | G | C | Y | C | T | A | R | R | Y | A | A | A | G | G | R | Y | Y | G | A | A |
| NPL2 | T | A | T | T | W | Y | G | R | A | A | T | ? | M | C | G | Y | A | G | C | Y | C | T | A | R | R | Y | A | A | A | G | G | R | Y | Y | G | A | A |
| NPL3 | T | A | T | T | W | Y | G | R | A | A | T | T | M | C | G | Y | A | G | C | Y | C | T | A | R | R | Y | A | A | A | G | G | R | Y | Y | G | A | A |
| OLF1 | C | C | T | T | A | Y | G | G | A | A | T | Y | C | Y | R | Y | A | G | C | C | C | Y | A | G | A | T | M | A | R | G | G | G | Y | C | G | A | A |
| PCT1 | T | M | T | T | A | Y | G | R | A | A | T | T | M | C | G | Y | A | G | C | Y | C | T | A | R | R | Y | ? | W | A | K | C | R | Y | ? | G | W | A |
| PCT2 | T | M | T | T | A | Y | G | R | A | A | T | T | M | C | G | Y | A | G | C | Y | C | T | A | R | R | Y | A | W | A | K | C | R | Y | Y | G | W | A |
| PCT3 | T | M | T | T | A | Y | G | R | A | A | T | T | M | C | G | Y | A | G | C | Y | C | T | A | R | R | Y | A | W | A | K | C | R | Y | Y | G | W | A |
| SPT1 | T | A | T | T | A | Y | ? | G | R | A | Y | C | C | C | G | Y | A | G | Y | C | C | C | W | G | R | Y | A | A | A | K | G | G | C | T | G | W | R |
| SPT2 | T | A | T | T | A | Y | G | G | R | A | Y | C | C | C | G | ? | A | G | Y | C | C | C | W | G | R | Y | A | A | A | K | G | G | C | T | G | W | R |
| TMP1 | T | A | K | T | W | Y | G | R | A | A | T | Y | M | Y | G | Y | A | G | C | Y | C | Y | W | R | G | Y | A | A | R | K | G | G | C | Y | S | A | A |
| TMP2 | T | A | K | T | W | Y | ? | R | A | A | T | Y | M | Y | G | Y | A | G | C | Y | C | Y | W | R | G | Y | A | A | R | K | G | G | C | Y | S | A | A |
| TMP3 | T | A | K | T | W | Y | ? | R | A | A | T | Y | M | Y | G | Y | A | G | C | Y | C | Y | W | R | G | Y | A | ? | R | K | G | G | C | Y | S | A | A |
| Cultivar | C1083 | G0115 | A5096 |
|---|---|---|---|
| Bouche de Bétizac | C | R | M |
| Lucente | Y | R | A |
| Marzatica | Y | A | M |
| Mercogliana | C | G | M |
| Napoletana | T | R | A |
| Olefarella | Y | A | C |
| Paccuta | T | R | M |
| San Pietro | C | R | A |
| Tempestiva | Y | G | A |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nunziata, A.; Ruggieri, V.; Petriccione, M.; De Masi, L. Single Nucleotide Polymorphisms as Practical Molecular Tools to Support European Chestnut Agrobiodiversity Management. Int. J. Mol. Sci. 2020, 21, 4805. https://doi.org/10.3390/ijms21134805
Nunziata A, Ruggieri V, Petriccione M, De Masi L. Single Nucleotide Polymorphisms as Practical Molecular Tools to Support European Chestnut Agrobiodiversity Management. International Journal of Molecular Sciences. 2020; 21(13):4805. https://doi.org/10.3390/ijms21134805
Chicago/Turabian StyleNunziata, Angelina, Valentino Ruggieri, Milena Petriccione, and Luigi De Masi. 2020. "Single Nucleotide Polymorphisms as Practical Molecular Tools to Support European Chestnut Agrobiodiversity Management" International Journal of Molecular Sciences 21, no. 13: 4805. https://doi.org/10.3390/ijms21134805