CRISPR/Cas9-Mediated Gene Correction to Understand ALS

1
Department of Neurosurgery, Spine and Spinal Cord Institute, College of Medicine, Yonsei University, Seoul 03722, Korea
2
Brain Korea 21 PLUS Project for Medical Science, College of Medicine, Yonsei University, Seoul 03722, Korea
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2020, 21(11), 3801; https://doi.org/10.3390/ijms21113801
Submission received: 18 April 2020
/
Revised: 21 May 2020
/
Accepted: 21 May 2020
/
Published: 27 May 2020
(This article belongs to the Special Issue Disease Modeling Using Human Induced Pluripotent Stem Cells 2.0)
Abstract
:Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease caused by the death of motor neurons in the spinal cord and brainstem. ALS has a diverse genetic origin; at least 20 genes have been shown to be related to ALS. Most familial and sporadic cases of ALS are caused by variants of the SOD1, C9orf72, FUS, and TARDBP genes. Genome editing using clustered regularly interspaced short palindromic repeats/CRISPR-associated system 9 (CRISPR/Cas9) can provide insights into the underlying genetics and pathophysiology of ALS. By correcting common mutations associated with ALS in animal models and patient-derived induced pluripotent stem cells (iPSCs), CRISPR/Cas9 has been used to verify the effects of ALS-associated mutations and observe phenotype differences between patient-derived and gene-corrected iPSCs. This technology has also been used to create mutations to investigate the pathophysiology of ALS. Here, we review recent studies that have used CRISPR/Cas9 to understand the genetic underpinnings of ALS.
1. Introduction
Amyotrophic lateral sclerosis (ALS) is the most common form of motor neuron disease (MND). The term “amyotrophic” derives from “a”, which means no; “myo”, which means muscle; and “trophic”, which refers to nourishment. Furthermore, “lateral” refers to the lateral corticospinal tracts and “sclerosis” refers to scarring of lateral region [1]. Taken together, “amyotrophic lateral sclerosis” denotes a lack of nourishment that leads to muscle wasting in the spinal cord, where descending motor pathways carry motor signals from the brain down to the target muscle or organ. ALS, which is diagnosed using an electromyography (EMG) test, is characterized by the loss of motor neurons and muscle weakness, which results in respiratory failure and death within 3–5 years of onset [2].
A study in 1993 showed that the SOD1 gene was associated with some cases of ALS [3]. Since this seminal study, the genetic etiology of ALS has expanded dramatically to include more than 20 different genes (ALSoDs) [4]. The onset of ALS usually occurs in adulthood, but juvenile-onset ALS has been observed in patients with mutations in the FUS gene [5]. There is no definitive cure for ALS, although Riluzole increases patient survival by three months; the United States Food and Drug Administration (FDA) has also recently approved a new treatment, Radicut [6,7,8,9].
Clustered regularly interspaced short palindromic repeats/CRISPR-associated system 9 (CRISPR/Cas9) genome-editing technology has advanced the field of genome engineering [10]. CRISPR/Cas9 has been adapted from a natural genome-editing system of bacteria. In the bacterial immune system, bacteriophages recognize foreign genetic elements that occur as a result of infection and destroy these foreign DNA fragments. The CRISPR/Cas9 system works similar to cut and replace DNA fragments. Although other genome editing tools exist, such as zinc finger nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs), CRISPR/Cas9 is simple and inexpensive to use compared with prior tools [11,12]. In this review, we focus on recent studies that have used CRISPR/Cas9-mediated gene correction and disease modeling to study ALS. In particular, we briefly summarize and discuss prominent mutations in the SOD1, C9orf72, FUS, and TARDBP genes and their roles in ALS, as revealed by CRISPR/Cas9 technology.
2. Amyotrophic Lateral Sclerosis (ALS)
ALS, also known as Lou Gehrig’s disease, is the most common MND. ALS is characterized by muscle weakness and the loss of motor neurons in the spinal cord and brainstem. Clinical symptoms are anatomically divided into two categories based on whether symptoms start in the limbs or bulbar region. Limb onset displays symptoms in the upper (arm or hand) or lower limbs (leg or foot), and bulbar onset patients have symptoms including weakness of muscle controls in speech and swallowing [13,14,15]. The age of onset is typically 55–60 years old, and the worldwide prevalence is approximately 1.68 per 100,000 individuals [16,17]. One study estimates that the proportion of individuals affected by ALS will increase by 69% by 2040, primarily owing to a rapidly aging population [18]. The average survival of patients with ALS is 2–5 years. Approximately 20% of patients survive for five years, eventually succumbing to respiratory failure, whereas 5% of patients may live for 20 years [19,20].
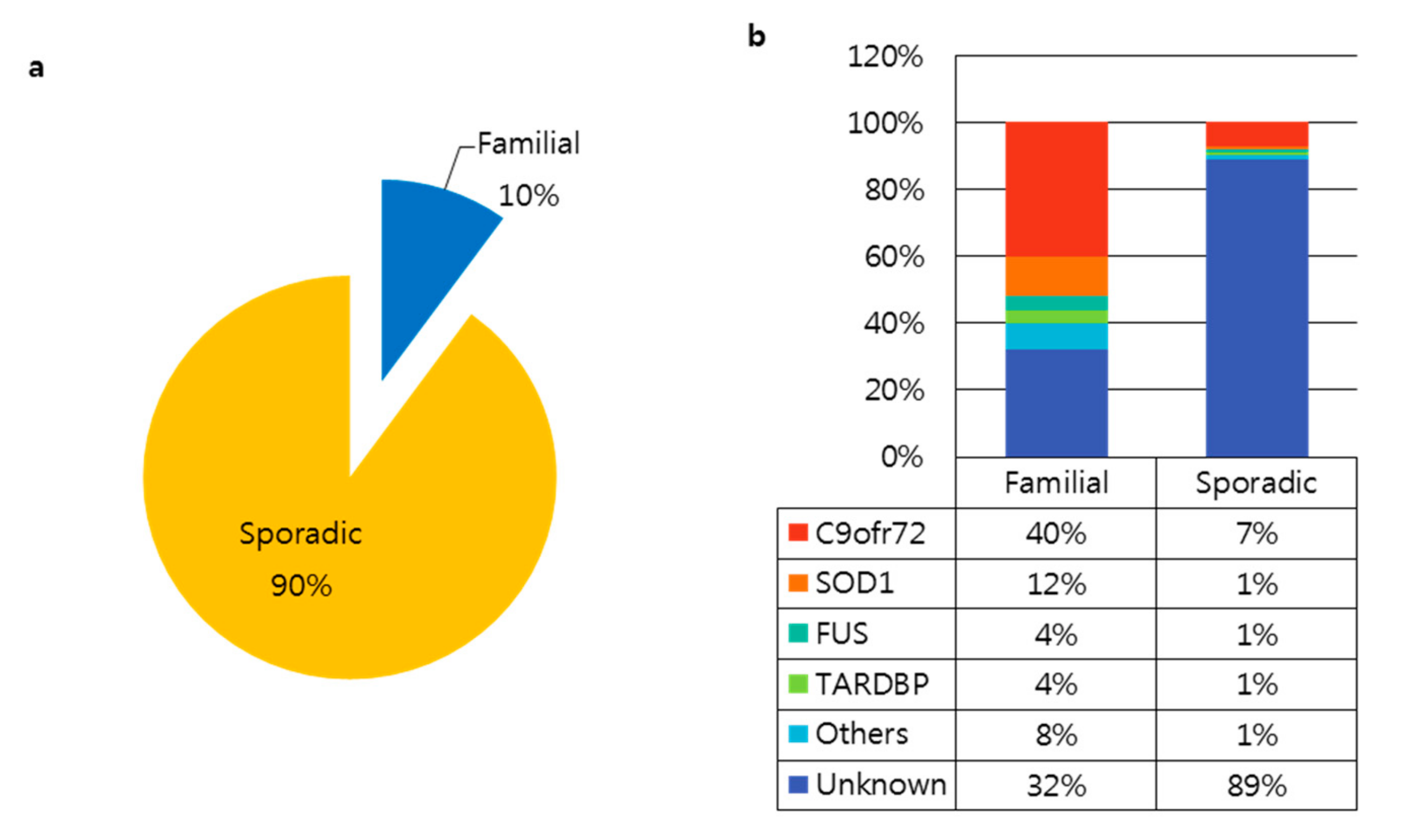
There are two types of ALS, familial (fALS, 10% of cases) and sporadic (sALS, 90% of cases) (Figure 1a). Since the discovery of superoxide dismutase 1 (SOD1) in 1993, over 20 genes have been linked to ALS. According to the Amyotrophic Lateral Sclerosis online Database (ALSoD), over 100 genes may be linked to ALS [3,21,22]. Of these, the most common genetic causes of ALS involve mutations in the SOD1, C9orf72, FUS, and TARDBP genes (Figure 1b). The Genome Aggregation Database (gnomAD) provides whole-genome or exome sequences of various diseases. We explored variants in SOD1, C9orf72, FUS, and TARDBP in the gnomAD v2.1.1 database as well as the Human Genome Variation Society (HGVS) database from the National Institutes of Health (NIH) to gather more information about each variant, including protein changes and dbSNPs (Table 1). Gene variants are classified into five categories according to their clinical significance: pathogenic, likely pathogenic, uncertain significance, likely benign, and benign [23]. There are 36 ALS-related variants in SOD1, 11 variants in FUS, and 14 variants in TARDBP with a pathogenic clinical significance. Variants in SOD1, C9orf72, FUS, and TARDBP account for up to 60% of familial ALS and 10% of sporadic ALS; the cause of the remaining cases remains unclear. The most common genetic cause of ALS is the (G4C2)n repeat expansion in C9orf72 [2,24].
A recent study, Project MinE, has begun to analyze the whole-genome sequence data of at least 15,000 patients with ALS worldwide [26]. Whole-genome sequencing is a promising tool to discover unknown genetic variants that contribute to disease. In a pilot analysis of 1169 patients with ALS, several rare genetic variations were found [27]. Several mutations were genetically associated with cellular pathology, which is characterized by motor neuron degeneration due to an interplay of axonal dysfunction, oxidative stress, mitochondrial dysfunction, microglia activation, and protein aggregation [13,28].
3. Targeted Genome Editing Using CRISPR/Cas9
CRISPR (clustered regularly interspaced short palindromic repeats)/CRISPR-associated system (Cas) is an immune system derived from prokaryotes that can be used to target and edit specific sequences. CRISPR sequences are located in prokaryotes such as bacteria and archaea. New foreign DNA fragments derived from infection are integrated into host CRISPR sequences and transcribed into CRISPR RNA. These RNA sequences are used as a guide to recognize foreign genetic elements derived from previously infected prokaryotes. The Cas protein is an enzyme that cleaves DNA fragments to match the CRISPR RNA [29,30].
To use CRISPR/Cas, a researcher designs small guide RNA (sgRNA) near the proto-spacer adjacent motif (PAM) sequences for Cas nuclease to bind target DNA sequences recognized by sgRNA. The Cas protein-coding gene and sgRNA plasmids are usually transfected via electroporation or lipid-based transfection agents. Other CRISPR/Cas9 delivery methods include ribonucleoprotein (RNP) complex and packaging into lentivirus or adeno-associated virus (AAV). Once in the nucleus, Cas nuclease binding leads to a double-strand break (DSB) in DNA that is recovered by the cell’s own repair system. These DSBs are commonly repaired by non-homologous end joining (NHEJ) or homology-directed repair (HDR) in the presence of a donor template. The difference between NHEJ and HDR is that the NHEJ mechanism generates insertions and deletions (indel) at a cleavage site, resulting in the knockout of a protein coding gene, whereas the HDR mechanism induces insertions or replaces DNA fragments with a donor template [31,32].
Cas9 from Streptococcus pyogenes (SpCas9) is widely used for the CRISPR/Cas system. Various other subtypes of the Cas protein, including saCas9, c2c2, RCas, and dCas, have also been identified and developed for applications in genome editing [33]. Each Cas protein has a unique PAM sequence and different protein sizes for different applications. These proteins have specific cleavage targets in DNA and RNA, and generate single-strand nicks (i.e., Cas9n) that activate or inhibit transcription (i.e., CRISPRa or CRISPRi) [34,35].
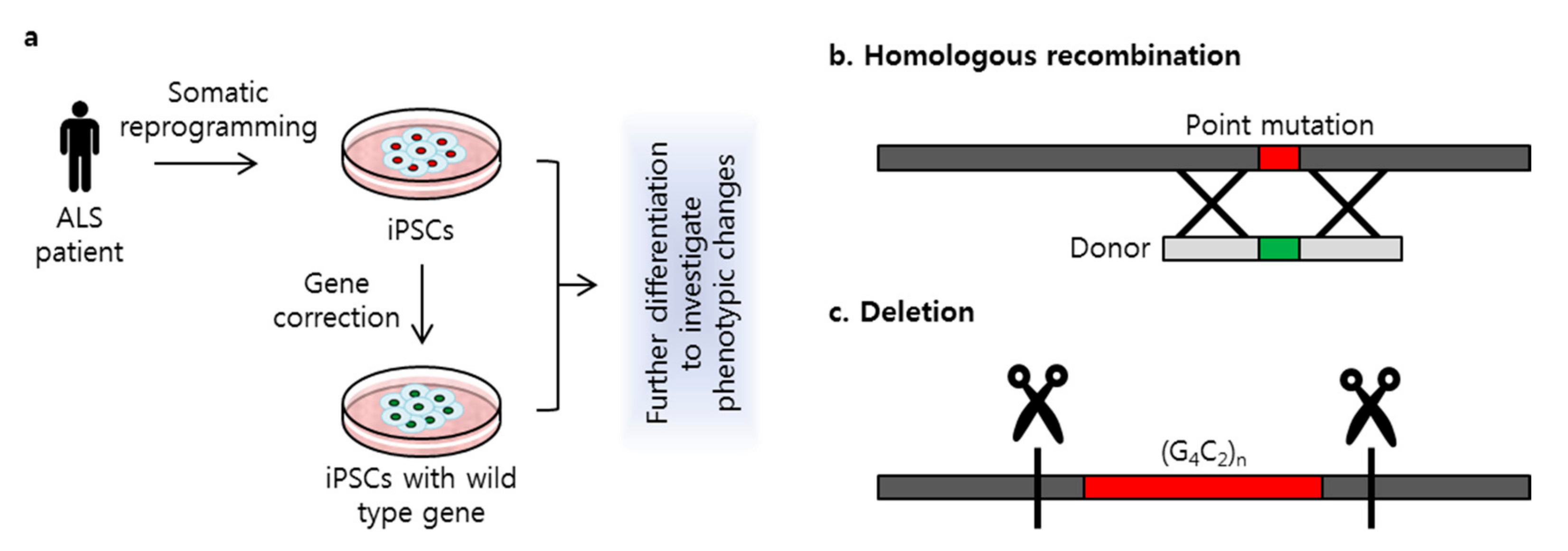
Genome editing using CRISPR/Cas9 is used in disease research to study genetic modifications in various cell types. Many years ago, mutagenesis or mutant transfection was widely used to investigate the genetic effects of mutations on disease [31,36]. Since the development of reprogramming technology, somatic cells have been used for induced pluripotent stem cell (iPSC) generation [37]. These iPSCs have the potential to differentiate into various cell types, such as neurons, liver cells, myoblasts, and cardiomyocytes. Patient-derived iPSCs provide an unlimited source of cells with which to study and treat disease [38,39,40,41]. For genetic disorders in particular, pathologic effects of mutations can be investigated in iPSCs using CRISPR/Cas9 to correct mutations (Figure 2a). The correction of specific mutations related to ALS can thus provide important insights into both verifying the pathologic effects of these mutations and identifying targets for therapeutic interventions.
In both fALS and sALS, the majority of causative genetic mutations are found in the SOD1, C9orf72, FUS, and TARDBP genes. Most pathogenic variants are the result of missense mutations, although repeat expansion in the intron region of the C9orf72 gene has been identified as a known cause of ALS. Patient-derived iPSCs are an important disease model to study ALS. One drawback to patient-derived iPSCs, however, is the phenotypic variation, which makes it difficult to determine which genetic mutations are relevant to ALS. Here, we review recent studies that have applied CRISPR/Cas9 to patient-derived iPSCs to study genetic mutations in ALS. These methods have included the generation of isogenic cell lines with the same genetic background as the iPSCs to understand the genetic etiology and pathophysiology of ALS (Table 2).
3.1. Superoxide Dismutase 1 (SOD1)
Mutant SOD1 was the first gene to be linked to ALS. SOD1 is located on chromosome 21q22.1 and contains five exons. More than 100 SOD1 mutations have been identified, 30 of which are associated with ALS, according to the genomAD and ClinVar online databases [42]. Mutations in SOD1 are reported in ~12% of fALS and ~1% of sALS cases [25,43]. SOD1 is an antioxidant enzyme that binds to copper and zinc to break down superoxide radicals, preventing cell damage. Mutations in SOD1 lead to neuronal excitability, endoplasmic reticulum stress, mitochondrial dysfunction, oxidative stress, impaired protein transport, accumulation of misfolded SOD1 protein, and inflammation, all of which may increase cell death [44,45].
A recent study [46] conducted a targeted gene correction of the SOD1 A272C mutation in patient-derived iPSCs. Fibroblasts harboring the A272C mutation were obtained from a patient with ALS and reprogrammed to generate iPSCs. Guide RNA was designed to include a PAM sequence that targeted the A272C mutation. A donor template to replace this point mutation was co-transfected with Cas9 nuclease and sgRNA via electroporation (Figure 2b). Because ALS is an MND, patient-derived iPSCs and the gene-corrected isogenic cell line were differentiated into motor neurons to compare neuronal differentiation capacity. RNA sequencing revealed significantly altered gene expression between the patient-derived motor neurons and the gene-corrected isogenic motor neurons. Gene ontology analysis revealed that expression was altered in RNA related to nervous system activity, signal transduction, and endoplasmic reticulum homeostasis. A similar study [47] applied this CRISPR/Cas9 gene-correction strategy to the SOD1 E100G mutation. Targeted gene correction was performed in iPSCs derived from a patient with the E100G mutation, which then were differentiated into motor neurons. Correction of the SOD1 E100G mutation in an isogenic cell line increased motor neuron proportion, soma size, and neurite length and decreased cell death. Transcriptional changes detected by RNA sequencing identified several up- or down-regulated pathways, including those related to AP1, oxidative phosphorylation, and ion channel transport. The functional gene network showed that these pathways share functional interactions and cascades. On the basis of these results, the authors hypothesized that activated ERK and JNK signaling is critical to neurodegeneration in mutant SOD1 motor neurons. Treatment with small-molecule inhibitors that target ERK, MAPK, JNK, WNT, TP53, and CDK kinases decreased motor neuron degeneration, suggesting that these pathways are critical in SOD1-linked ALS. A study by Imamura et al. [48] used CRISPR/Cas9-mediated gene corrected cell lines in drug screening assay as a control. iPSCs were generated from one ALS patient carrying the L144FVX mutation in the SOD1 gene and mutation was corrected by inserting wild type SOD1 sequences with homologous arms. The corrected cell line showed reversal of SOD1-associated pathologic phenotype, including increased motor neurons survival, degradation of misfolded SOD1 protein, and reserved internal ATP level. On the other hand, high-throughput screening discovered that more than half of hit targeted the Src/c-Abl signaling pathway in motor neurons differentiated from iPSCs. The treatment of bosutinib, one of the directly inhibited Src/c-Abl, showed therapeutic effects on motor neurons’ survival, decreased phosphorylation of Src/c-Abl, promoted autophagy activity, adecreased misfolded mutant SOD1, and rescue of ATP, as did gene correction of L144FVX. In this study, the authors revealed the potential application of the gene corrected isogenic cell line used as a control to validate the therapeutic effects of drugs on ALS disease.
3.2. C9orf72
The hexanucleotide (G4C2) repeat expansion in C9orf72 is the most common genetic cause of ALS and is also common in frontotemporal dementia (FTD). This repeat expansion is responsible for up to 40% of fALS and 7% of sALS cases [25,43,49]. The repeat is located in intron1 of the C9orf72 gene. The number of repeats is highly variable within an individual, ranging from hundreds to thousands of repeats; less than 30 repeats is considered normal [50]. Mutant C9orf72 has several pathologic neurodegenerative phenotypes, including reduced protein expression levels, promoter hypermethylation, excitotoxicity, RNA foci formation, and generation of dipeptide repeats (DPRs). Transcription of this repeat expansion produces RNA with abnormal secondary structures that aggregate to form intranuclear foci, which results in the disruption of transcription and cytoplasmic transport [51,52]. Repeat-associated non-AUG (RAN) translation produces toxic DPR proteins [53]. Poly GR(glycine arginine), one of the dipeptide chains, is accumulated in patients with C9orf72-associated ALS and FTD [54,55].
Mutant C9orf72 genes with 800–1050 repeats from patients with ALS are also associated with RNA foci formation and hypermethylation [56]. CRISPR/Cas9 genome editing technology with two sgRNAs has been designed to excise repeat expansions via the cleavage and removal of the repeat region (Figure 2c). Deletion of the repeat expansion results in decreased RNA foci formation and reduces methylation in repeat knockout iPSC-derived neurons. In contrast, deletion of the normal allele did not reverse the methylation level, which indicates that the hexanucleotide (G4C2) expansion is the cause of hypermethylation within the GC-rich region of C9orf72. Another study that used iPSC lines with up to 638, 760, and 960 repeats in C9orf72 showed that the C9orf72 mutation increased AMPA receptor expression, leading to enhanced excitotoxicity via intracellular Ca2+ overload [57], as well as RNA foci formation and DPR toxicity, but no differences in neuron excitability in iPSC-derived motor neurons were observed. A genome-editing approach that deleted the repeat expansion was able to rescue the AMPA-induced excitotoxicity and reduced the RNA foci formation and DPR expression. RNA sequencing also showed that deletion of the repeat expansion altered the gene expression in genes related to RNA processing, protein targeting, nuclear transport, synaptic transmission, and the regulation of phosphate metabolism [58].
Unlike the hSOD1 G93A transgenic mouse model, deletion of C9orf72 in the mouse does not produce an ALS-like phenotype [59]. As an alternative, Drosophila expressing 58 repeats of G4C2 or poly GR overexpression has been used to investigate the involvement of the Ku80 protein in ALS neurodegeneration [60,61]. Studies have confirmed that Ku80 protein levels are increased in both this Drosophila model and in C9orf72 iPSC-derived motor neurons with 70, 590, 1000, or 1100 repeats in each cell line. Ku80 is an essential DNA repair protein. The elevated expression of Ku80 leads to an increase in downstream pro-apoptotic proteins such as PUMA, BAX, and cleaved caspase-3. The application of genome editing using two sgRNAs reduced Ku80 levels and prevented nuclear RNA foci formation in iPSC-derived motor neurons with the corrected repeat expansion. Using CRISPR/Cas9, one copy of Ku80 in the C9orf72 iPSC was deleted, which resulted in the reduction of Ku80 expression as well as PUMA and cleaved caspase-3 levels. These studies, therefore, demonstrate that the CRISPR/Cas9 approach can be applied to the C9orf72 repeat expansion to validate pathologic phenotypes in patient-derived iPSCs and identify gene-based therapeutic approaches.
3.3. Fused in Sarcoma (FUS)
The fused in sarcoma (FUS) gene encodes an RNA-binding protein that is located primarily in the nucleus and composed of seven domains: QGSY-rich, Gly-rich, RNA recognition motif (RRM), Arg-Gly-Gly1 (RGG1), zinc finger (ZnF), RGG2, and nuclear localization signal (NLS) [62]. The majority of disease-causing FUS mutations are heterozygous with an autosomal-dominant inheritance pattern [63]. In addition, most FUS mutations affect the C-terminal nuclear localization signal. The known functions of FUS include DNA damage repair, transcription, and splicing [64]. The effect of mutant FUS in neurodegeneration and other biological processes is unknown; however, one hallmark of mutant FUS is cytoplasmic mislocalization, which may be related to a toxic gain of function in the cytoplasm [65]. Previous findings have suggested that mutant FUS may impact DNA repair, energy metabolism, and axonal transport. Recent studies have used CRISPR/Cas9 gene editing to study the impact of mutant FUS on ALS pathologic phenotypes in patient-derived iPSCs. A study by Wang et al. [46] first demonstrated CRISPR/Cas9-mediated gene correction of the FUS G1566A missense mutation in ALS patient-derived iPSCs. Another study used the CRISPR/Cas9 system to correct the recessive FUS H517Q mutation to the wild-type sequence in patient-derived iPSCs. Both mutant and corrected iPSCs were differentiated into Isl1/Tuj1-positive motor neurons to investigate the expression of the MAPK family. These results showed that both p38 and ERK kinase are activated in mutant FUS, indicating that the activation of MAPK signaling is a key pathway in FUS-mediated neurodegeneration in ALS [47]. Motor neurons derived from patients with ALS with the FUS mutation typically show cytoplasmic mislocalization [64]. Motor neurons carrying the FUS P525L and R521H mutations also exhibit mislocalization of FUS, compared with no aggregation in healthy control cells.
Electrophysiological changes and defects in axonal transport have also been observed in motor neurons with mutant FUS. The movement of endoplasmic reticulum vesicles is also significantly reduced, which results in impaired axonal transport [66]. To confirm whether these observed pathologic phenotypes were the result of the FUS mutation, CRISPR/Cas9-mediated gene correction was applied to iPSCs derived from patients with the FUS R521H mutation. Isogenic cell lines with the same genetic background as the mutant iPSCs were also established. Mitochondrial transport experiments showed that gene correction rescued the number of total and moving mitochondria as well as cytoplasmic FUS localization. Moreover, these experiments found that an HDAC6 inhibitor is an effective drug for targeting axonal transport impairment. This study thus demonstrates that gene correction can mediate the investigation of FUS-related disease pathologic phenotypes and may have the potential to be used for therapeutic discovery and validation.
Oxidative stress promotes genome damage and neurodegeneration results in motor neuron death, which is consistent with increased markers of oxidative stress in patients with ALS [67,68]. In healthy neurons, FUS is localized predominantly to the nucleus and promotes genome repair by recruiting PARP-1 dependent XRCC1/DNA ligase IIIα (LigIII) in response to DNA damage response (DDR) signaling [69]. ALS patient-derived motor neurons with FUS P525L and R521H mutations exhibit an approximately threefold increase in the cytoplasmic accumulation of FUS as well as a cytotoxic response to H2O2 and >50% decreased efficiency of nick ligation owing to the delay of DNA break repair [70]. Gene-corrected isogenic cell lines have been established using CRISPR/Cas9 to investigate the direct attribution of FUS mutations to DNA ligation impairment. Correction of these FUS mutations rescued DNA ligation defects, as assessed by ligation assay, and DNA integrity was also restored, indicating that mutant FUS fails to correct DNA ligation defects caused by oxidative stress. This study suggests that LigIII function may be a therapeutic target in patients with FUS mutations.
Energy metabolism is a critical component in neurodegeneration as well as genome repair, because most biological processes, including neuronal firing and axonal transport, are energy demanding [71,72]. The elevation of glycogen stores observed in patients with ALS implies a defect in mitochondrial function and a failure in glucose matabolism, which has been hypothesized to cause the hallmark motor neuron degeneration in ALS. Mislocalization caused by mutant FUS increased enzymatic interactions in glucose metabolism [73], indicating that energy metabolism may be involved in FUS-related ALS degeneration. Substrate-tracing experiments using radiolabeled 13C6—glucose and 13C3—lactate reveal the involvement of oxidative metabolism, but not the glycolytic pathway in motor neurons differentiated from iPSCs. In motor neurons carrying an FUS mutation, however, there was no evidence of altered mitochondrial morphology, such as intermembrane space and cristae patterns, as measured by transmission electron microscopy (TEM), whereas FUS overexpression was associated with a defect in the mitochondrial structure. Radioactive tracer analysis using 13C was performed to investigate FUS-related changes in glucose and/or lactate metabolism in FUS-mutant versus gene-corrected motor neurons. Although lactate was revealed as a main energy source in motor neurons, there was no altered contribution of glucose or lactate to glycolytic or TCA metabolites after gene correction, indicating that there is no metabolic dysfunction in FUS-related ALS phenotypes [5]. The use of CRISPR/Cas9 genome editing in patient-derived iPSCs also reveals that mutant FUS does not alter metabolic pathways, providing further evidence that metabolic dysfunction is not involved in FUS-related ALS.
3.4. TAR-Binding Protein Gene (TARDBP)
The TARDBP gene is located on chromosome 1p36.22 and contains six exons encoding TAR-DNA binding protein TDP-43. It is a DNA/RNA binding protein and functions in a variety of RNA metabolism such as splicing, stability, and trafficking. Mutations in TARDBP gene were first reported by Sreedharan et al. in 2008 [74]. Since then, many mutations have been identified through genetic analysis in ALS patients, and most mutations are conserved within exon 6 of the TARDBP gene, which encodes the carboxy-terminal glycine-rich domain [75]. TDP-43 protein undergoes post-translational modification including ubiquitination, phosphorylation, acetylation, cysteine oxidation, and PARylation. Ubiquitinated and hyper-phosphorylated TDP-43 forms cytoplasmic inclusion bodies—a pathological hallmark of ALS. Mutations in the C-terminal region hasten aggregations, mislocalization to the cytoplasm, and cytotoxicity [76].
In the nervous system, brain-derived neurotrophic factor (BDNF) is essential for neuron survival, differentiation, and synaptic plasticity. It is produced and regulated to the secretory pathway in neurons by binding of Sortilin (encoded by sort1 gene) to the pro-domain of BDNF. TDP-43 regulates the splicing of mRNA encoding Sortilin. Loss of TDP-43 leads to the generation of isoform owing to additional exon 17b and soluble Sortilin protein. Therefore, a study by Tann et al. hypothesized that TDP-43 dysfunction caused by mutations in TARDBP gene altered BDNF secretion and synaptic plasticity [77]. In a mouse model, the authors confirmed that hippocampal CA1-specific knockout of TDP-43 increased exon 17b mRNA of sort1 mRNA and reduced dendrite complexity and spine. To investigate the effect mutations could have in human neurons, CRISPR/Cas9 was used to correct M337V in three iPSCs lines from a patient. Corrected iPSCs were differentiated into excitatory cortico/hippocampal-like neurons. All isogenic lines showed a similar mRNA level of TARDBP and SORT1, but a decreased level of exon 17b mRNA of sort1 compared with cells bearing M337V. Lentivirus expressing human pro-BDNF was transduced to neurons and to assess BDNF secretion. M337V displayed a decreased level of BDNF, and significantly increased in all isogenic gene corrected neurons. This study indicated that TDP-43 mutation is associated to Sortilin isoform and BDNF secretion, and the gene correction strategy demonstrated that M337V mutation in TARDBP is a direct cause of abnormal BDNF secretion.
4. CRISPR/Cas9-Mediated In Vivo Therapeutic Approach
A transgenic mouse model that overexpresses the human SOD1 G93A mutation is widely used in studies of ALS. This mouse model expresses a similar pathologic phenotype to human ALS, including short survival and loss of motor behavior function. Recent studies using this mouse model [78,79] have reported that CRISPR/Cas9 disturbs the expression of mutant SOD1, which may be a promising potential therapeutic strategy. These studies have hypothesized that the reduced expression of mutant protein reverses the disease phenotype.

{kind=link}
{kind=link}
Table 2.
Studies of clustered regularly interspaced short palindromic repeats/CRISPR-associated system 9 (CRISPR/Cas9)-mediated genome editing in amyotrophic lateral sclerosis. N/R, not recorded; ALS, amyotrophic lateral sclerosis; iPSC, induced pluripotent stem cell; FTD, frontotemporal dementia.
Table 2.
Studies of clustered regularly interspaced short palindromic repeats/CRISPR-associated system 9 (CRISPR/Cas9)-mediated genome editing in amyotrophic lateral sclerosis. N/R, not recorded; ALS, amyotrophic lateral sclerosis; iPSC, induced pluripotent stem cell; FTD, frontotemporal dementia.
| Study | Gene | Variants | Model | Gene Editing Efficiency |
|---|---|---|---|---|
| Homologous recombination | ||||
| Wang et al., 2017 [46] | SOD1 | A272C | Human ALS patient-derived iPSCs | 20% |
| Bhinge et al., 2017 [47] | SOD1 | E100G | Human ALS patient-derived iPSCs | 0.5% (one of ~ 180 colonies) |
| Imamura et al., 2017 [48] | SOD1 | L144FVX | Human ALS patient-derived iPSCs | Selected with puromycin |
| Guo et al., 2017 [66] | FUS | R521H | Human ALS patient-derived iPSCs | Selected with puromycin |
| Wang et al., 2017 [46] | FUS | G1566A | Human ALS patient-derived iPSCs | 1% |
| Bhinge et al., 2017 [47] | FUS | H517Q | Human ALS patient-derived iPSCs | 0.5% (one of ~ 180 colonies) |
| Wang et al., 2018 [70] | FUS | R521H, P525L | Human ALS patient-derived iPSCs | Selected with puromycin |
| Vandoorne et al., 2019 [5] | FUS | R521H, P525L | Human ALS patient-derived iPSCs | Selected with puromycin |
| Tann et al., 2019 [77] | TARDBP | M337V | Human ALS patient-derived iPSCs | FACS sorting |
| Deletion | ||||
| Pribadi et al., 2016 [56] | C9orf72 | GGGGCC | Human FTD/ALS patient-derived iPSCs | 11.1% (66 of 593 colonies) |
| Selvaraj et al., 2018 [58] | C9orf72 | GGGGCC | Human ALS patient-derived iPSCs | 0.6% ~ 4.5% |
| Lopez-Gonzalez et al., 2019 [61] | C9orf72 | GGGGCC | C9orf72 patient-derived iPSCs | N/R |
| Duan et al., 2019 [79] | SOD1 | G93A | hSOD1 G93A transgenic mouse | N/R |
| Indel formation | ||||
| Gaj et al., 2017 [78] | SOD1 | G93A | hSOD1 G93A transgenic mouse | N/R |
Several sgRNAs that do not overlap with the G93A mutation have been designed for application to other SOD1 mutations. For in vivo genome editing such as that using animal models, AAV is the safest delivery vehicle. Because AAV has a limited capacity to harbor the plasmid, however, Cas9 nuclease from Staphylococcus aureus (SaCas9) is often used, thanks to its smaller size than spCas9 [80]. The plasmid, consisting of SaCas9 under the CMV promoter and sgRNA under the U6 promoter, is packaged into AAV. In a study of the hSOD1 G93A mouse model, this AAV was injected via the facial vein at postnatal day 0–1. Four weeks after injection, Western blot and histological analysis confirmed reduced expression of the SOD1 protein and an increased number of motor neurons in the lumbar spinal cord. Furthermore, these findings showed that genome editing improved motor function, reduced muscle atrophy, delayed disease onset, and increased survival compared with a control model.
Taken together, studies using CRISPR/Cas9-mediated in vivo genome editing of missense mutations in SOD1 show that this technique can be used not only to identify the underlying molecular mechanism but also to reverse the pathologic phenotype in patient-derived disease models, and thus may play a potential role in gene therapy for ALS.
5. Conclusions and Future
In this review, we discussed recent studies that have used CRISPR/Cas9-mediated gene correction to investigate the pathophysiology of ALS using patient-derived iPSCs. Although CRISPR/Cas9 technology is simple and easy to use across several research applications, there remain limitations with this approach. One critical concern is off-target effects, which could occur if the specificities of Cas9 and sgRNA are reduced such that they recognize and cleave non-target DNA sites [81]. Another limitation is the low (<1%) efficiency of gene correction through the HDR mechanism. Techniques such as timed delivery with cell cycle synchronization [82,83] and suppression of NHEJ key molecules [84] are needed to enhance HDR genome engineering rates.
As mutant SOD1 was revealed as a genetic cause of ALS in 1993, over 20 genes associated with ALS have been identified; however, more than 80% of patients with ALS do not exhibit known genetic variants [85]. Clinical differences between patients may imply that multiple mechanisms are involved with various genetic causes. According to ALSoD, over 100 genes are listed as ALS-associated genes. Recently, Vildan et al. analyzed SOD1, C9orf72, FUS, TARDBP, and UBQLN2 to find common genetic alterations in 30 patients with ALS [86]. In addition, many European countries and the United States have joined Project MinE to analyze the genes of 15,000 patients with ALS and 7500 controls. The aim of this project is to better understand the genetic background of ALS. Online open access resources also exist to obtain information about gene variants in various diseases, including ClinVar and Miner. The application of genetic information, advanced methods in genome engineering with CRISPR/Cas9, and in vitro disease modeling using iPSCs is likely to facilitate the discovery of disease-causing mutations and further investigate the underlying pathophysiology of various diseases. It is the hope that this strategy will ultimately lead to a thorough investigation of the various mechanisms underlying ALS to identify treatments and a cure for ALS.
Author Contributions
Y.Y. and Y.H. wrote and critically reviewed the manuscript. All authors approved the final manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Hulisz, D. Amyotrophic lateral sclerosis: Disease state overview. Am. J. Manag. Care 2018, 24, S320–S326. [Google Scholar]
- Oskarsson, B.; Gendron, T.F.; Staff, N.P. Amyotrophic lateral sclerosis: An update for 2018. Mayo Clin. Proc. 2018, 93, 1617–1628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X.; et al. Mutations in cu/zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Wroe, R.; Wai-Ling Butler, A.; Andersen, P.M.; Powell, J.F.; Al-Chalabi, A. Alsod: The amyotrophic lateral sclerosis online database. Amyotroph. Lateral Scler. Off. Publ. World Fed. Neurol. Res. Group Mot. Neuron Dis. 2008, 9, 249–250. [Google Scholar] [CrossRef]
- Vandoorne, T.; Veys, K.; Guo, W.; Sicart, A.; Vints, K.; Swijsen, A.; Moisse, M.; Eelen, G.; Gounko, N.V.; Fumagalli, L.; et al. Differentiation but not als mutations in fus rewires motor neuron metabolism. Nat. Commun. 2019, 10, 4147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothstein, J.D. Edaravone: A new drug approved for als. Cell 2017, 171, 725. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.G.; Appel, S.H. Introduction to supplement: The current status of treatment for als. Amyotroph. Lateral Scler. Front. Degener. 2017, 18, 1–4. [Google Scholar] [CrossRef]
- Bensimon, G.; Lacomblez, L.; Meininger, V. A controlled trial of riluzole in amyotrophic lateral sclerosis. Als/riluzole study group. N. Engl. J. Med. 1994, 330, 585–591. [Google Scholar] [CrossRef]
- Jaiswal, M.K. Riluzole and edaravone: A tale of two amyotrophic lateral sclerosis drugs. Med. Res. Rev. 2019, 39, 733–748. [Google Scholar] [CrossRef] [PubMed]
- You, L.; Tong, R.; Li, M.; Liu, Y.; Xue, J.; Lu, Y. Advancements and obstacles of crispr-cas9 technology in translational research. Mol. Ther. Methods Clin. Dev. 2019, 13, 359–370. [Google Scholar] [CrossRef] [Green Version]
- Maeder, M.L.; Gersbach, C.A. Genome-editing technologies for gene and cell therapy. Mol. Ther. J. Am. Soc. Gene Ther. 2016, 24, 430–446. [Google Scholar] [CrossRef] [Green Version]
- Gaj, T.; Gersbach, C.A.; Barbas, C.F., 3rd. Zfn, talen, and crispr/cas-based methods for genome engineering. Trends Biotechnol. 2013, 31, 397–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ragagnin, A.M.G.; Shadfar, S.; Vidal, M.; Jamali, M.S.; Atkin, J.D. Motor neuron susceptibility in als/ftd. Front. Neurosci. 2019, 13, 532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Statland, J.M.; Barohn, R.J.; McVey, A.L.; Katz, J.S.; Dimachkie, M.M. Patterns of weakness, classification of motor neuron disease, and clinical diagnosis of sporadic amyotrophic lateral sclerosis. Neurol. Clin. 2015, 33, 735–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grad, L.I.; Rouleau, G.A.; Ravits, J.; Cashman, N.R. Clinical spectrum of amyotrophic lateral sclerosis (als). Cold Spring Harb. Perspect. Med. 2017, 7, a024117. [Google Scholar] [CrossRef] [Green Version]
- Marin, B.; Boumédiene, F.; Logroscino, G.; Couratier, P.; Babron, M.C.; Leutenegger, A.L.; Copetti, M.; Preux, P.M.; Beghi, E. Variation in worldwide incidence of amyotrophic lateral sclerosis: A meta-analysis. Int. J. Epidemiol. 2017, 46, 57–74. [Google Scholar] [CrossRef] [Green Version]
- Mehta, P.; Kaye, W.; Raymond, J.; Wu, R.; Larson, T.; Punjani, R.; Heller, D.; Cohen, J.; Peters, T.; Muravov, O.; et al. Prevalence of amyotrophic lateral sclerosis—United states, 2014. Mmwr. Morb. Mortal. Wkly. Rep. 2018, 67, 216–218. [Google Scholar] [CrossRef] [Green Version]
- Arthur, K.C.; Calvo, A.; Price, T.R.; Geiger, J.T.; Chiò, A.; Traynor, B.J. Projected increase in amyotrophic lateral sclerosis from 2015 to 2040. Nat. Commun. 2016, 7, 12408. [Google Scholar] [CrossRef] [Green Version]
- Brown, R.H.; Al-Chalabi, A. Amyotrophic lateral sclerosis. N. Engl. J. Med. 2017, 377, 162–172. [Google Scholar] [CrossRef] [Green Version]
- Mehta, P.; Horton, D.K.; Kasarskis, E.J.; Tessaro, E.; Eisenberg, M.S.; Laird, S.; Iskander, J. Cdc grand rounds: National amyotrophic lateral sclerosis (als) registry impact, challenges, and future directions. Mmwr. Morb. Mortal. Wkly. Rep. 2017, 66, 1379–1382. [Google Scholar] [CrossRef] [Green Version]
- Abel, O.; Shatunov, A.; Jones, A.R.; Andersen, P.M.; Powell, J.F.; Al-Chalabi, A. Development of a smartphone app for a genetics website: The amyotrophic lateral sclerosis online genetics database (alsod). Jmir Mhealth Uhealth 2013, 1, e18. [Google Scholar] [CrossRef] [PubMed]
- Abel, O.; Powell, J.F.; Andersen, P.M.; Al-Chalabi, A. Alsod: A user-friendly online bioinformatics tool for amyotrophic lateral sclerosis genetics. Hum. Mutat. 2012, 33, 1345–1351. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the american college of medical genetics and genomics and the association for molecular pathology. Genet. Med. Off. J. Am. Coll. Med. Genet. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Mejzini, R.; Flynn, L.L.; Pitout, I.L.; Fletcher, S.; Wilton, S.D.; Akkari, P.A. Als genetics, mechanisms, and therapeutics: Where are we now? Front. Neurosci. 2019, 13, 1310. [Google Scholar] [CrossRef] [Green Version]
- Scarrott, J.M.; Herranz-Martín, S.; Alrafiah, A.R.; Shaw, P.J.; Azzouz, M. Current developments in gene therapy for amyotrophic lateral sclerosis. Expert Opin. Biol. Ther. 2015, 15, 935–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Spek, R.A.A.; Van Rheenen, W.; Pulit, S.L.; Kenna, K.P.; Van den Berg, L.H.; Veldink, J.H. The project mine databrowser: Bringing large-scale whole-genome sequencing in als to researchers and the public. Amyotroph. Lateral Scler. Front. Degener. 2019, 20, 432–440. [Google Scholar] [CrossRef] [Green Version]
- Rheenen, W.V.; Pulit, S.L.; Dekker, A.M.; Khleifat, A.A.; Brands, W.J.; Iacoangeli, A.; Kenna, K.P.; Kooyman, M.; McLaughlin, R.L.; Middelkoop, B.; et al. Project mine: Study design and pilot analyses of a large-scale whole genome sequencing study in amyotrophic lateral sclerosis. J. bioRxiv 2017, 152553. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Sayana, P.; Zhang, X.; Le, W. Genetics of amyotrophic lateral sclerosis: An update. Mol. Neurodegener. 2013, 8, 28. [Google Scholar] [CrossRef] [Green Version]
- Charpentier, E.; Marraffini, L.A. Harnessing crispr-cas9 immunity for genetic engineering. Curr. Opin. Microbiol. 2014, 19, 114–119. [Google Scholar] [CrossRef] [Green Version]
- Hryhorowicz, M.; Lipiński, D.; Zeyland, J.; Słomski, R. Crispr/cas9 immune system as a tool for genome engineering. Arch. Immunol. Et Ther. Exp. 2017, 65, 233–240. [Google Scholar] [CrossRef] [Green Version]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the crispr-cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Chung, J.H.; Kim, H.M.; Kim, D.W.; Kim, H. Designed nucleases for targeted genome editing. Plant. Biotechnol. J. 2016, 14, 448–462. [Google Scholar] [CrossRef] [PubMed]
- Nakade, S.; Yamamoto, T.; Sakuma, T. Cas9, cpf1 and c2c1/2/3-what’s next? Bioengineered 2017, 8, 265–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, K.; Cai, D.; Wang, Z.; He, Z.; Chen, S. Development of an efficient genome editing tool in bacillus licheniformis using crispr-cas9 nickase. Appl. Environ. Microbiol. 2018, 84, e02608-17. [Google Scholar] [CrossRef] [Green Version]
- Dominguez, A.A.; Lim, W.A.; Qi, L.S. Beyond editing: Repurposing crispr-cas9 for precision genome regulation and interrogation. Nat. Rev. Mol. Cell Biol. 2016, 17, 5–15. [Google Scholar] [CrossRef] [Green Version]
- Pearson, M.M.; Himpsl, S.D.; Mobley, H.L.T. Insertional mutagenesis protocol for constructing single or sequential mutations. Methods Mol. Biol. (CliftonN.J.) 2019, 2021, 61–76. [Google Scholar]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Qian, K.; Du, Z.; Cao, J.; Petersen, A.; Liu, H.; Blackbourn, L.W.T.; Huang, C.L.; Errigo, A.; Yin, Y.; et al. Modeling als with ipscs reveals that mutant sod1 misregulates neurofilament balance in motor neurons. Cell Stem Cell 2014, 14, 796–809. [Google Scholar] [CrossRef] [Green Version]
- Kiskinis, E.; Sandoe, J.; Williams, L.A.; Boulting, G.L.; Moccia, R.; Wainger, B.J.; Han, S.; Peng, T.; Thams, S.; Mikkilineni, S.; et al. Pathways disrupted in human als motor neurons identified through genetic correction of mutant sod1. Cell Stem Cell 2014, 14, 781–795. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Chen, J.; Liu, W.; Li, X.; Chen, Q.; Liu, T.; Gao, S.; Deng, M. The fused in sarcoma protein forms cytoplasmic aggregates in motor neurons derived from integration-free induced pluripotent stem cells generated from a patient with familial amyotrophic lateral sclerosis carrying the fus-p525l mutation. Neurogenetics 2015, 16, 223–231. [Google Scholar] [CrossRef]
- Fujimori, K.; Ishikawa, M.; Otomo, A.; Atsuta, N.; Nakamura, R.; Akiyama, T.; Hadano, S.; Aoki, M.; Saya, H.; Sobue, G.; et al. Modeling sporadic als in ipsc-derived motor neurons identifies a potential therapeutic agent. Nat. Med. 2018, 24, 1579–1589. [Google Scholar] [CrossRef]
- Yamashita, S.; Ando, Y. Genotype-phenotype relationship in hereditary amyotrophic lateral sclerosis. Transl. Neurodegener. 2015, 4, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, Z.Y.; Zhou, Z.R.; Che, C.H.; Liu, C.Y.; He, R.L.; Huang, H.P. Genetic epidemiology of amyotrophic lateral sclerosis: A systematic review and meta-analysis. J. Neurol. Neurosurg. Psychiatry 2017, 88, 540–549. [Google Scholar] [CrossRef]
- Kaur, S.J.; McKeown, S.R.; Rashid, S. Mutant sod1 mediated pathogenesis of amyotrophic lateral sclerosis. Gene 2016, 577, 109–118. [Google Scholar] [CrossRef]
- Kato, S.; Kato, M.; Abe, Y.; Matsumura, T.; Nishino, T.; Aoki, M.; Itoyama, Y.; Asayama, K.; Awaya, A.; Hirano, A.; et al. Redox system expression in the motor neurons in amyotrophic lateral sclerosis (als): Immunohistochemical studies on sporadic als, superoxide dismutase 1 (sod1)-mutated familial als, and sod1-mutated als animal models. Acta Neuropathol. 2005, 110, 101–112. [Google Scholar] [CrossRef]
- Wang, L.; Yi, F.; Fu, L.; Yang, J.; Wang, S.; Wang, Z.; Suzuki, K.; Sun, L.; Xu, X.; Yu, Y.; et al. Crispr/cas9-mediated targeted gene correction in amyotrophic lateral sclerosis patient ipscs. Protein Cell 2017, 8, 365–378. [Google Scholar] [CrossRef]
- Bhinge, A.; Namboori, S.C.; Zhang, X.; VanDongen, A.M.J.; Stanton, L.W. Genetic correction of sod1 mutant ipscs reveals erk and jnk activated ap1 as a driver of neurodegeneration in amyotrophic lateral sclerosis. Stem Cell Rep. 2017, 8, 856–869. [Google Scholar] [CrossRef] [Green Version]
- Imamura, K.; Izumi, Y.; Watanabe, A.; Tsukita, K.; Woltjen, K.; Yamamoto, T.; Hotta, A.; Kondo, T.; Kitaoka, S.; Ohta, A.; et al. The src/c-abl pathway is a potential therapeutic target in amyotrophic lateral sclerosis. Sci. Transl. Med. 2017, 9, eaaf3962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majounie, E.; Renton, A.E.; Mok, K.; Dopper, E.G.; Waite, A.; Rollinson, S.; Chiò, A.; Restagno, G.; Nicolaou, N.; Simon-Sanchez, J.; et al. Frequency of the c9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: A cross-sectional study. Lancet Neurol. 2012, 11, 323–330. [Google Scholar] [CrossRef]
- Gami, P.; Murray, C.; Schottlaender, L.; Bettencourt, C.; De Pablo Fernandez, E.; Mudanohwo, E.; Mizielinska, S.; Polke, J.M.; Holton, J.L.; Isaacs, A.M.; et al. A 30-unit hexanucleotide repeat expansion in c9orf72 induces pathological lesions with dipeptide-repeat proteins and rna foci, but not tdp-43 inclusions and clinical disease. Acta Neuropathol. 2015, 130, 599–601. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Donnelly, C.J.; Haeusler, A.R.; Grima, J.C.; Machamer, J.B.; Steinwald, P.; Daley, E.L.; Miller, S.J.; Cunningham, K.M.; Vidensky, S.; et al. The c9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 2015, 525, 56–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagier-Tourenne, C.; Baughn, M.; Rigo, F.; Sun, S.; Liu, P.; Li, H.R.; Jiang, J.; Watt, A.T.; Chun, S.; Katz, M.; et al. Targeted degradation of sense and antisense c9orf72 rna foci as therapy for als and frontotemporal degeneration. Proc. Natl. Acad. Sci. USA 2013, 110, E4530–E4539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westergard, T.; McAvoy, K.; Russell, K.; Wen, X.; Pang, Y.; Morris, B.; Pasinelli, P.; Trotti, D.; Haeusler, A. Repeat-associated non-aug translation in c9orf72-als/ftd is driven by neuronal excitation and stress. Embo Mol. Med. 2019, 11, e9423. [Google Scholar] [CrossRef] [PubMed]
- Saberi, S.; Stauffer, J.E.; Jiang, J.; Garcia, S.D.; Taylor, A.E.; Schulte, D.; Ohkubo, T.; Schloffman, C.L.; Maldonado, M.; Baughn, M.; et al. Sense-encoded poly-gr dipeptide repeat proteins correlate to neurodegeneration and uniquely co-localize with tdp-43 in dendrites of repeat-expanded c9orf72 amyotrophic lateral sclerosis. Acta Neuropathol. 2018, 135, 459–474. [Google Scholar] [CrossRef]
- Sakae, N.; Bieniek, K.F.; Zhang, Y.J.; Ross, K.; Gendron, T.F.; Murray, M.E.; Rademakers, R.; Petrucelli, L.; Dickson, D.W. Poly-gr dipeptide repeat polymers correlate with neurodegeneration and clinicopathological subtypes in c9orf72-related brain disease. Acta Neuropathol. Commun. 2018, 6, 63. [Google Scholar] [CrossRef]
- Pribadi, M.; Yang, Z.; Kim, T.S.; Swartz, E.W.; Huang, A.Y.; Chen, J.A.; Dokuru, D.; Baek, J.; Gao, F.; Fua, A.T.; et al. Crispr-cas9 targeted deletion of the c9orf72 repeat expansion mutation corrects cellular phenotypes in patient-derived ips cells. J. bioRxiv 2016, 051193. [Google Scholar] [CrossRef] [Green Version]
- Joshi, D.C.; Tewari, B.P.; Singh, M.; Joshi, P.G.; Joshi, N.B. Ampa receptor activation causes preferential mitochondrial ca2+ load and oxidative stress in motor neurons. Brain Res. 2015, 1616, 1–9. [Google Scholar] [CrossRef]
- Selvaraj, B.T.; Livesey, M.R.; Zhao, C.; Gregory, J.M.; James, O.T.; Cleary, E.M.; Chouhan, A.K.; Gane, A.B.; Perkins, E.M.; Dando, O.; et al. C9orf72 repeat expansion causes vulnerability of motor neurons to ca(2+)-permeable ampa receptor-mediated excitotoxicity. Nat. Commun. 2018, 9, 347. [Google Scholar] [CrossRef] [Green Version]
- Koppers, M.; Blokhuis, A.M.; Westeneng, H.J.; Terpstra, M.L.; Zundel, C.A.; Vieira de Sá, R.; Schellevis, R.D.; Waite, A.J.; Blake, D.J.; Veldink, J.H.; et al. C9orf72 ablation in mice does not cause motor neuron degeneration or motor deficits. Ann. Neurol. 2015, 78, 426–438. [Google Scholar] [CrossRef] [Green Version]
- Freibaum, B.D.; Lu, Y.; Lopez-Gonzalez, R.; Kim, N.C.; Almeida, S.; Lee, K.H.; Badders, N.; Valentine, M.; Miller, B.L.; Wong, P.C.; et al. Ggggcc repeat expansion in c9orf72 compromises nucleocytoplasmic transport. Nature 2015, 525, 129–133. [Google Scholar] [CrossRef]
- Lopez-Gonzalez, R.; Yang, D.; Pribadi, M.; Kim, T.S.; Krishnan, G.; Choi, S.Y.; Lee, S.; Coppola, G.; Gao, F.B. Partial inhibition of the overactivated ku80-dependent DNA repair pathway rescues neurodegeneration in c9orf72-als/ftd. Proc. Natl. Acad. Sci. USA 2019, 116, 9628–9633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shang, Y.; Huang, E.J. Mechanisms of fus mutations in familial amyotrophic lateral sclerosis. Brain Res. 2016, 1647, 65–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, H.; Skelt, L.; Notaro, A.; Highley, J.R.; Fox, A.H.; La Bella, V.; Buchman, V.L.; Shelkovnikova, T.A. Als-linked fus mutations confer loss and gain of function in the nucleus by promoting excessive formation of dysfunctional paraspeckles. Acta Neuropathol. Commun. 2019, 7, 7. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Lyashchenko, A.K.; Lu, L.; Nasrabady, S.E.; Elmaleh, M.; Mendelsohn, M.; Nemes, A.; Tapia, J.C.; Mentis, G.Z.; Shneider, N.A. Als-associated mutant fus induces selective motor neuron degeneration through toxic gain of function. Nat. Commun. 2016, 7, 10465. [Google Scholar] [CrossRef] [PubMed]
- Nakaya, T.; Maragkakis, M. Amyotrophic lateral sclerosis associated fus mutation shortens mitochondria and induces neurotoxicity. Sci. Rep. 2018, 8, 15575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, W.; Naujock, M.; Fumagalli, L.; Vandoorne, T.; Baatsen, P.; Boon, R.; Ordovás, L.; Patel, A.; Welters, M.; Vanwelden, T.; et al. Hdac6 inhibition reverses axonal transport defects in motor neurons derived from fus-als patients. Nat. Commun. 2017, 8, 861. [Google Scholar] [CrossRef] [PubMed]
- Barber, S.C.; Shaw, P.J. Oxidative stress in als: Key role in motor neuron injury and therapeutic target. Free Radic. Biol. Med. 2010, 48, 629–641. [Google Scholar] [CrossRef]
- Formella, I.; Svahn, A.J.; Radford, R.A.W.; Don, E.K.; Cole, N.J.; Hogan, A.; Lee, A.; Chung, R.S.; Morsch, M. Real-time visualization of oxidative stress-mediated neurodegeneration of individual spinal motor neurons in vivo. Redox Biol. 2018, 19, 226–234. [Google Scholar] [CrossRef]
- Wang, H.; Dharmalingam, P.; Vasquez, V.; Mitra, J.; Boldogh, I.; Rao, K.S.; Kent, T.A.; Mitra, S.; Hegde, M.L. Chronic oxidative damage together with genome repair deficiency in the neurons is a double whammy for neurodegeneration: Is damage response signaling a potential therapeutic target? Mech. Ageing Dev. 2017, 161, 163–176. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Guo, W.; Mitra, J.; Hegde, P.M.; Vandoorne, T.; Eckelmann, B.J.; Mitra, S.; Tomkinson, A.E.; Van Den Bosch, L.; Hegde, M.L. Mutant fus causes DNA ligation defects to inhibit oxidative damage repair in amyotrophic lateral sclerosis. Nat. Commun. 2018, 9, 3683. [Google Scholar] [CrossRef] [Green Version]
- Naumann, M.; Pal, A.; Goswami, A.; Lojewski, X.; Japtok, J.; Vehlow, A.; Naujock, M.; Günther, R.; Jin, M.; Stanslowsky, N.; et al. Impaired DNA damage response signaling by fus-nls mutations leads to neurodegeneration and fus aggregate formation. Nat. Commun. 2018, 9, 335. [Google Scholar] [CrossRef] [PubMed]
- Naujock, M.; Stanslowsky, N.; Bufler, S.; Naumann, M.; Reinhardt, P.; Sterneckert, J.; Kefalakes, E.; Kassebaum, C.; Bursch, F.; Lojewski, X.; et al. 4-aminopyridine induced activity rescues hypoexcitable motor neurons from amyotrophic lateral sclerosis patient-derived induced pluripotent stem cells. Stem Cells (Dayt. Ohio) 2016, 34, 1563–1575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Jiang, X.; Chen, G.; Xu, J. Interaction of amyotrophic lateral sclerosis/frontotemporal lobar degeneration-associated fused-in-sarcoma with proteins involved in metabolic and protein degradation pathways. Neurobiol. Aging 2015, 36, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Sreedharan, J.; Blair, I.P.; Tripathi, V.B.; Hu, X.; Vance, C.; Rogelj, B.; Ackerley, S.; Durnall, J.C.; Williams, K.L.; Buratti, E.; et al. Tdp-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 2008, 319, 1668–1672. [Google Scholar] [CrossRef]
- Prasad, A.; Bharathi, V.; Sivalingam, V.; Girdhar, A.; Patel, B.K. Molecular mechanisms of tdp-43 misfolding and pathology in amyotrophic lateral sclerosis. Front. Mol. Neurosci. 2019, 12, 25. [Google Scholar] [CrossRef]
- Johnson, B.S.; Snead, D.; Lee, J.J.; McCaffery, J.M.; Shorter, J.; Gitler, A.D. Tdp-43 is intrinsically aggregation-prone, and amyotrophic lateral sclerosis-linked mutations accelerate aggregation and increase toxicity. J. Biol. Chem. 2009, 284, 20329–20339. [Google Scholar] [CrossRef] [Green Version]
- Tann, J.Y.; Wong, L.W.; Sajikumar, S.; Ibáñez, C.F. Abnormal tdp-43 function impairs activity-dependent bdnf secretion, synaptic plasticity, and cognitive behavior through altered sortilin splicing. Embo J. 2019, 38. [Google Scholar] [CrossRef]
- Gaj, T.; Ojala, D.S.; Ekman, F.K.; Byrne, L.C.; Limsirichai, P.; Schaffer, D.V. In vivo genome editing improves motor function and extends survival in a mouse model of als. Sci. Adv. 2017, 3, eaar3952. [Google Scholar] [CrossRef] [Green Version]
- Duan, W.; Guo, M.; Yi, L.; Liu, Y.; Li, Z.; Ma, Y.; Zhang, G.; Liu, Y.; Bu, H.; Song, X.; et al. The deletion of mutant sod1 via crispr/cas9/sgrna prolongs survival in an amyotrophic lateral sclerosis mouse model. Gene Ther. 2019. [Google Scholar] [CrossRef]
- Ran, F.A.; Cong, L.; Yan, W.X.; Scott, D.A.; Gootenberg, J.S.; Kriz, A.J.; Zetsche, B.; Shalem, O.; Wu, X.; Makarova, K.S.; et al. In vivo genome editing using staphylococcus aureus cas9. Nature 2015, 520, 186–191. [Google Scholar] [CrossRef]
- Chen, S.J. Minimizing off-target effects in crispr-cas9 genome editing. Cell Biol. Toxicol. 2019, 35, 399–401. [Google Scholar] [CrossRef] [Green Version]
- Yang, D.; Scavuzzo, M.A.; Chmielowiec, J.; Sharp, R.; Bajic, A.; Borowiak, M. Enrichment of g2/m cell cycle phase in human pluripotent stem cells enhances hdr-mediated gene repair with customizable endonucleases. Sci. Rep. 2016, 6, 21264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.; Staahl, B.T.; Alla, R.K.; Doudna, J.A. Enhanced homology-directed human genome engineering by controlled timing of crispr/cas9 delivery. eLife 2014, 3, e04766. [Google Scholar] [CrossRef] [PubMed]
- Chu, V.T.; Weber, T.; Wefers, B.; Wurst, W.; Sander, S.; Rajewsky, K.; Kühn, R. Increasing the efficiency of homology-directed repair for crispr-cas9-induced precise gene editing in mammalian cells. Nat. Biotechnol. 2015, 33, 543–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corcia, P.; Couratier, P.; Blasco, H.; Andres, C.R.; Beltran, S.; Meininger, V.; Vourc’h, P. Genetics of amyotrophic lateral sclerosis. Rev. Neurol. 2017, 173, 254–262. [Google Scholar] [CrossRef]
- Vildan, C.; Sule, D.; Turker, B.; Hilmi, U.; Sibel, K.B. Genetic alterations of c9orf72, sod1, tardbp, fus, and ubqln2 genes in patients with amyotrophic lateral sclerosis. Cogent Med. 2019, 6, 1. [Google Scholar] [CrossRef]
Figure 1.
Genetic causes of amyotrophic lateral sclerosis (ALS). (a) Prevalence of familial and sporadic ALS cases. (b) Proportion of causative genes in familial and sporadic ALS [25].
Figure 1.
Genetic causes of amyotrophic lateral sclerosis (ALS). (a) Prevalence of familial and sporadic ALS cases. (b) Proportion of causative genes in familial and sporadic ALS [25].

Figure 2.
Strategy of clustered regularly interspaced short palindromic repeats/CRISPR-associated system 9 (CRISPR/Cas9)-mediated gene correction. (a) Patient-derived induced pluripotent stem cells (iPSCs) were used to perform gene correction. Isogenic cell line was generated to investigate phenotypic changes comparing iPSCs with genetic mutation; (b) A point mutation was target specific corrected using a donor via homology-directed repair (HDR) mechanism; (c) Repeat expansion in C9orf72 was deleted with two sgRNAs.
Figure 2.
Strategy of clustered regularly interspaced short palindromic repeats/CRISPR-associated system 9 (CRISPR/Cas9)-mediated gene correction. (a) Patient-derived induced pluripotent stem cells (iPSCs) were used to perform gene correction. Isogenic cell line was generated to investigate phenotypic changes comparing iPSCs with genetic mutation; (b) A point mutation was target specific corrected using a donor via homology-directed repair (HDR) mechanism; (c) Repeat expansion in C9orf72 was deleted with two sgRNAs.

Table 1.
Amyotrophic lateral sclerosis (ALS)-associated pathogenic variants in SOD1, C9orf72, FUS, and TARDBP (Genome Aggregation Database (gnomAD) v2.1.1).
Table 1.
Amyotrophic lateral sclerosis (ALS)-associated pathogenic variants in SOD1, C9orf72, FUS, and TARDBP (Genome Aggregation Database (gnomAD) v2.1.1).
| Nucleotide | Protein | dbSNP | Mutation Type |
|---|---|---|---|
| SOD1 | |||
| NM_000454.4:c.14C >T | NP_000445.1:p.Ala5Val | rs121912442 | Missense |
| NM_000454.4:c.20G >T | NP_000445.1:p.Cys7Phe | rs121912448 | Missense |
| NM_000454.4:c.13G >A | NP_000445.1:p.Ala5Thr | rs121912444 | Missense |
| NM_000454.4:c.37G >C | NP_000445.1:p.Gly13Arg | rs121912456 | Missense |
| NM_000454.4:c.49G >A | NP_000445.1:p.Gly17Ser | rs121912453 | Missense |
| NM_000454.4:c.64G >A | NP_000445.1:p.Glu22Lys | rs121912450 | Missense |
| NM_000454.4:c.115C >G | NP_000445.1:p.Leu39Val | rs121912432 | Missense |
| NM_000454.4:c.112G >C | NP_000445.1:p.Gly38Arg | rs121912431 | Missense |
| NM_000454.4:c.112G >A | NP_000445.1:p.Gly38Arg | rs121912431 | Missense |
| NM_000454.4:c.131A >G | NP_000445.1:p.His44Arg | rs121912435 | Missense |
| NM_000454.4:c.125G >A | NP_000445.1:p.Gly42Asp | rs121912434 | Missense |
| NM_000454.4:c.140A >G | NP_000445.1:p.His47Arg | rs121912443 | Missense |
| NM_000454.4:c.124G >A | NP_000445.1:p.Gly42Ser | rs121912433 | Missense |
| NM_000454.4:c.137T >G | NP_000445.1:p.Phe46Cys | rs121912457 | Missense |
| NM_000454.4:c.217G >A | NP_000445.1:p.Gly73Ser | rs121912455 | Missense |
| NM_000454.4:c.242A >G | NP_000445.1:p.His81Arg | rs121912458 | Missense |
| NM_000454.4:c.256G >C | NP_000445.1:p.Gly86Arg | rs121912436 | Missense |
| NM_000454.4:c.253T >G | NP_000445.1:p.Leu85Val | rs121912452 | Missense |
| NM_000454.4:c.281G >C | NP_000445.1:p.Gly94Ala | rs121912438 | Missense |
| NM_000454.4:c.280G >T | NP_000445.1:p.Gly94Cys | rs121912437 | Missense |
| NM_000454.4:c.280G >C | NP_000445.1:p.Gly94Arg | rs121912437 | Missense |
| NM_000454.4:c.289G >A | NP_000445.1:p.Asp97Asn | rs121912459 | Missense |
| NM_000454.4:c.302A >G | NP_000445.1:p.Glu101Gly | rs121912439 | Missense |
| NM_000454.4:c.319C >G | NP_000445.1:p.Leu107Val | rs121912440 | Missense |
| LRG_652t1:c.317C >T | LRG_652p1:p.Ser106Leu | Missense | |
| NM_000454.4:c.313A >T | NP_000445.1:p.Ile105Phe | rs121912445 | Missense |
| NM_000454.4:c.341T >C | NP_000445.1:p.Ile114Thr | rs121912441 | Missense |
| NM_000454.4:c.338T >C | NP_000445.1:p.Ile113Thr | rs74315452 | Missense |
| NM_000454.4:c.358-10T >G | Intron variant | ||
| NM_000454.4:c.358-11A >G | Intron variant | ||
| NM_000454.4:c.380T >A | NP_000445.1:p.Leu127Ter | rs121912454 | Stop gain |
| NM_000454.4:c.404G >A | NP_000445.1:p.Ser135Asn | rs121912451 | Missense |
| NM_000454.4:c.436G >A | NP_000445.1:p.Ala146Thr | rs121912447 | Missense |
| NM_000454.4:c.435G >C | NP_000445.1:p.Leu145Phe | rs1482760341 | Missense |
| NM_000454.4:c.434T >C | NP_000445.1:p.Leu145Ser | rs121912446 | Missense |
| NM_000454.4:c.455T >C | NP_000445.1:p.Ile152Thr | rs121912449 | Missense |
| C9orf72 | |||
| NM_001256054.2:c. − 45 + 163GGGGCC[>24] | rs143561967 | Intron variant | |
| FUS | |||
| NM_004960.3:c.616G >A | NP_004951.1:p.Gly206Ser | rs387906628 | Missense |
| NM_004960.3:c.646C >T | NP_004951.1:p.Arg216Cys | rs267606832 | Missense |
| NM_004960.3:c.1507_1508AG [3] | NP_004951.1:p.Gly504fs | Frameshift | |
| NM_004960.3:c.1483C >T | NP_004951.1:p.Arg495Ter | rs387906627 | Stop gain |
| NM_004960.3:c.1520G >A | NP_004951.1:p.Gly507Asp | rs267606831 | Missense |
| NM_004960.3:c.1570A >T | NP_004951.1:p.Arg524Trp | rs267606833 | Missense |
| NM_004960.3:c.1562G >A | NP_004951.1:p.Arg521His | rs121909671 | Missense |
| NM_004960.3:c.1561C >T | NP_004951.1:p.Arg521Cys | rs121909668 | Missense |
| NM_004960.3:c.1561C >G | NP_004951.1:p.Arg521Gly | rs121909668 | Missense |
| NM_004960.3:c.1553G >A | NP_004951.1:p.Arg518Lys | rs121909669 | Missense |
| NM_004960.3:c.1551C >G | NP_004951.1:p.His517Gln | rs121909667 | Missense |
| TARDBP | |||
| NM_007375.3:c.800A >G | NP_031401.1:p.Asn267Ser | rs80356718 | Missense |
| NM_007375.3:c.869G >C | NP_031401.1:p.Gly290Ala | rs121908395 | Missense |
| NM_007375.3:c.881G >T | NP_031401.1:p.Gly294Val | rs80356721 | Missense |
| NM_007375.3:c.881G >C | NP_031401.1:p.Gly294Ala | rs80356721 | Missense |
| NM_007375.3:c.883G >A | NP_031401.1:p.Gly295Ser | rs80356723 | Missense |
| NM_007375.3:c.892G >A | NP_031401.1:p.Gly298Ser | rs4884357 | Missense |
| NM_007375.3:c.943G >A | NP_031401.1:p.Ala315Thr | rs80356726 | Missense |
| NM_007375.3:c.991C >A | NP_031401.1:p.Gln331Lys | rs80356727 | Missense |
| NM_007375.3:c.1009A >G | NP_031401.1:p.Met337Val | rs80356730 | Missense |
| NM_007375.3:c.1028A >G | NP_031401.1:p.Gln343Arg | rs80356731 | Missense |
| NM_007375.3:c.1042G >T | NP_031401.1:p.Gly348Cys | rs80356733 | Missense |
| NM_007375.3:c.1055A >G | NP_031401.1:p.Asn352Ser | rs80356734 | Missense |
| NM_007375.3:c.1144G >A | NP_031401.1:p.Ala382Thr | rs367543041 | Missense |
| NM_007375.3:c.1153T >G | NP_031401.1:p.Trp385Gly | rs797044595 | Missense |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Yun, Y.; Ha, Y. CRISPR/Cas9-Mediated Gene Correction to Understand ALS. Int. J. Mol. Sci. 2020, 21, 3801. https://doi.org/10.3390/ijms21113801
AMA Style
Yun Y, Ha Y. CRISPR/Cas9-Mediated Gene Correction to Understand ALS. International Journal of Molecular Sciences. 2020; 21(11):3801. https://doi.org/10.3390/ijms21113801
Chicago/Turabian StyleYun, Yeomin, and Yoon Ha. 2020. "CRISPR/Cas9-Mediated Gene Correction to Understand ALS" International Journal of Molecular Sciences 21, no. 11: 3801. https://doi.org/10.3390/ijms21113801
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.