Behavioral, Biochemical and Electrophysiological Changes in Spared Nerve Injury Model of Neuropathic Pain


Abstract
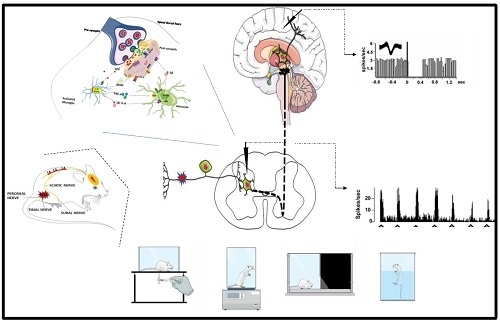
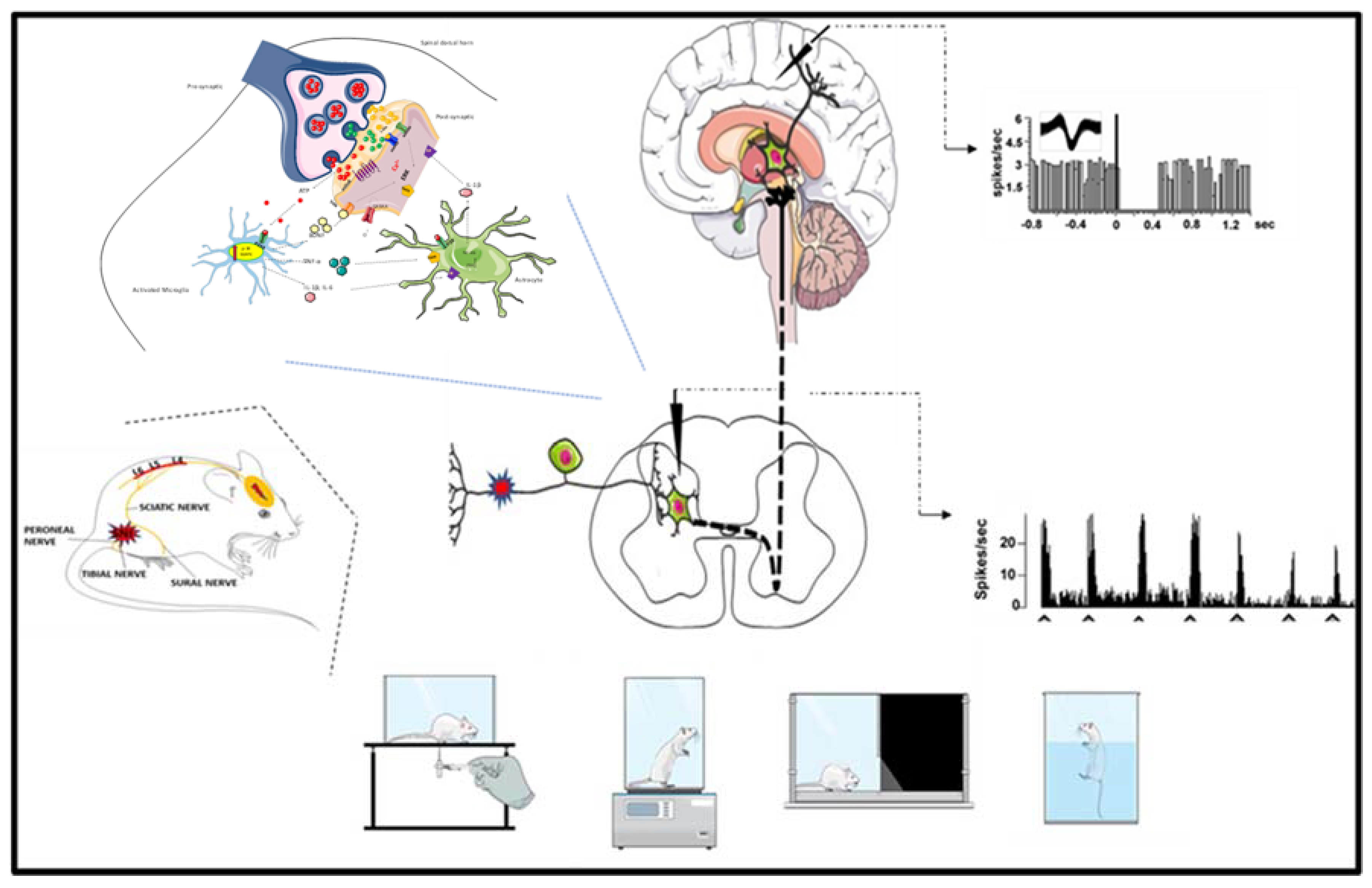
:
1. Introduction
- The surgical procedure is relatively easier than other methods and offers high reproducibility
- SNI permits behavioural testing of the non-injured sural nerve territory (adjacent to the denervated areas).
- As unilateral injury, the SNI-induced behavioural signs or biochemical markers can be compared with to contralateral side.
- SNI does not affect daily life activity, such as food intake, drinking and locomotion or circadian patterns [12]. Autonomy is not observed, unlike other models (i.e. Sciatic Nerve Transection).
- As compared with the chronic constriction injury (CCI), SNI induces more intense and prolonged (up to one year) mechanical sensitivity [10,12]. The long-lasting pain behaviours allows, similarly to the clinical situations, to perform chronic treatments (several weeks or months) after onset of symptoms and clear diagnosis (therapeutic treatment).
- SNI produces a low local inflammation that is present in the CCI or partial sciatic nerve ligation (PSNL) and SNL models [8].
- Sciatic nerve injuries in humans are rare to due the deep anatomical location within the lower extremity.
2. Surgical Procedure
3. Behavioral Symptoms Associated with the Spared Nerve Injury
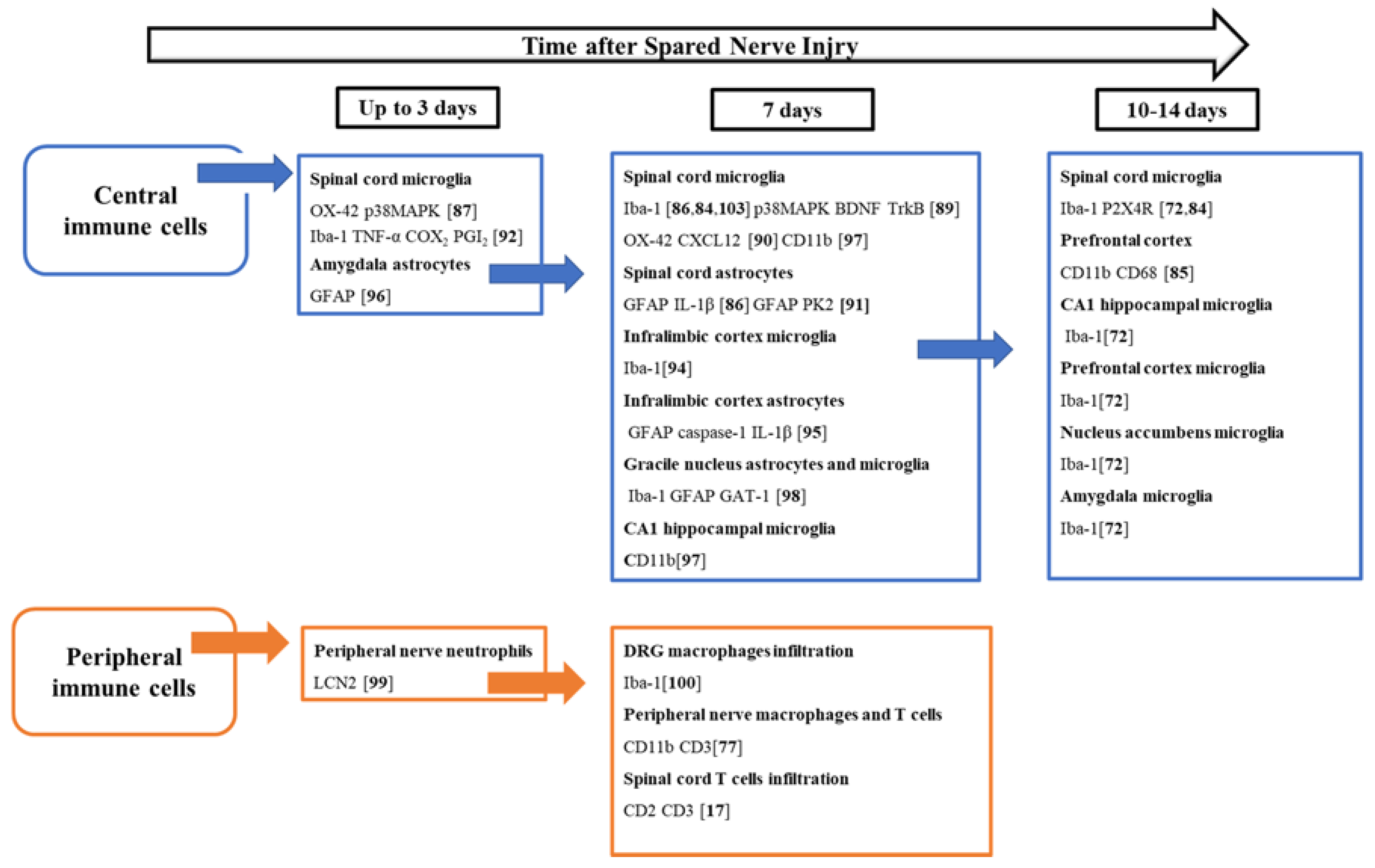
4. Role of Glia and Immune Cells in the SNI Model
5. Sex Differences in SNI-Associated Pain Signaling
6. Electrophysiological Characterization of SNI Model
7. Cutting-Edge Techniques in SNI Model: Focus on Optogenetic and DREADD
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Woolf, C.J.; Mannion, R.J. Neuropathic pain: Aetiology, symptoms, mechanisms, and management. Lancet 1999, 353, 1959–1964. [Google Scholar] [CrossRef]
- Campbell, J.N.; Meyer, R.A. Mechanisms of neuropathic pain. Neuron 2006, 52, 77–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennett, G.J.; Xie, Y.K. A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain 1988, 33, 87–107. [Google Scholar] [CrossRef]
- Kim, S.H.; Chung, J.M. An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain 1992, 50, 355–363. [Google Scholar] [CrossRef]
- Kumar, A.; Kaur, H.; Singh, A. Neuropathic Pain models caused by damage to central or peripheral nervous system. Pharmacol. Rep. 2018, 70, 206–216. [Google Scholar] [CrossRef] [PubMed]
- Seltzer, Z.; Dubner, R.; Shir, Y. A novel behavioral model of neuropathic pain disorders produced in rats by partial sciatic nerve injury. Pain 1990, 43, 205–218. [Google Scholar] [CrossRef]
- Shields, S.D.; Eckert, W.A., III; Basbaum, A.I. Spared nerve injury model of neuropathic pain in the mouse: A behavioral and anatomic analysis. J. Pain 2003, 4, 465–470. [Google Scholar] [CrossRef]
- Challa, S.R. Surgical animal models of neuropathic pain: Pros and Cons. Int. J. Neurosci. 2015, 125, 170–174. [Google Scholar] [CrossRef]
- Decosterd, I.; Woolf, C.J. Spared nerve injury: An animal model of persistent peripheral neuropathic pain. Pain 2000, 87, 149–158. [Google Scholar] [CrossRef]
- D’Aniello, A.; Luongo, L.; Romano, R.; Iannotta, M.; Marabese, I.; Boccella, S.; Belardo, C.; de Novellis, V.; Arra, C.; Barbieri, A.; et al. D-Aspartic acid ameliorates painful and neuropsychiatric changes and reduces β-amyloid Aβ(1-42) peptide in a long lasting model of neuropathic pain. Neurosci. Lett. 2017, 651, 151–158. [Google Scholar] [CrossRef]
- Langford, D.J.; Bailey, A.L.; Chanda, M.L.; Clarke, S.E.; Drummond, T.E.; Echols, S.; Glick, S.; Ingrao, J.; Klassen-Ross, T.; LaCroix-Fralish, M.L. Coding of facial expressions of pain in the laboratory mouse. Nat. Methods 2010, 7, 447. [Google Scholar] [CrossRef]
- Urban, R.; Scherrer, G.; Goulding, E.H.; Tecott, L.H.; Basbaum, A.I. Behavioral indices of ongoing pain are largely unchanged in male mice with tissue or nerve injury-induced mechanical hypersensitivity. Pain 2011, 152, 990–1000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urban, D.J.; Roth, B.L. DREADDs (designer receptors exclusively activated by designer drugs): Chemogenetic tools with therapeutic utility. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 399–417. [Google Scholar] [CrossRef] [PubMed]
- Apkarian, A.V.; Bushnell, M.C.; Treede, R.D.; Zubieta, J.K. Human brain mechanisms of pain perception and regulation in health and disease. Eur. J. Pain 2005, 9, 463–484. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Larrea, L.; Bastuji, H. Pain and consciousness. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2018, 87 Pt B, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Chao, T.H.; Chen, J.H.; Yen, C.T. Plasticity changes in forebrain activity and functional connectivity during neuropathic pain development in rats with sciatic spared nerve injury. Mol. Brain 2018, 11, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costigan, M.; Moss, A.; Latremoliere, A.; Johnston, C.; Verma-Gandhu, M.; Herbert, T.A.; Barrett, L.; Brenner, G.J.; Vardeh, D.; Woolf, C.J.; et al. T-cell infiltration and signaling in the adult dorsal spinal cord is a major contributor to neuropathic pain-like hypersensitivity. J. Neurosci. 2009, 29, 14415–14422. [Google Scholar] [CrossRef]
- Guida, F.; Boccella, S.; Belardo, C.; Iannotta, M.; Piscitelli, F.; De Filippis, F.; Paino, S.; Ricciardi, F.; Siniscalco, D.; Marabese, I.; et al. Altered gut microbiota and endocannabinoid system tone in vitamin D deficiency-mediated chronic pain. Brain Behav. Immun. 2019, 85, 128–141. [Google Scholar] [CrossRef]
- Guo, R.; Chen, L.H.; Xing, C.; Liu, T. Pain regulation by gut microbiota: Molecular mechanisms and therapeutic potential. Br. J. Anaesth. 2019, 123, 637–654. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Fang, X.; Zhan, G.; Huang, N.; Li, S.; Bi, J.; Jiang, R.; Yang, L.; Miao, L.; Zhu, B.; et al. Key role of gut microbiota in anhedonia-like phenotype in rodents with neuropathic pain. Transl. Psychiatry 2019, 9, 57. [Google Scholar] [CrossRef] [Green Version]
- Boccella, S.; Guida, F.; Palazzo, E.; Marabese, I.; de Novellis, V.; Maione, S.; Luongo, L. Spared Nerve Injury as a Long-Lasting Model of Neuropathic Pain. Methods Mol. Biol. 2018, 1727, 373–378. [Google Scholar] [PubMed]
- Cichon, J.; Sun, L.; Yang, G. Spared Nerve Injury Model of Neuropathic Pain in Mice. Bio. Protoc. 2018, 8, e2777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erichsen, H.K.; Blackburn-Munro, G. Pharmacological characterisation of the spared nerve injury model of neuropathic pain. Pain 2002, 98, 151–161. [Google Scholar] [CrossRef]
- Bourquin, A.F.; Süveges, M.; Pertin, M.; Gilliard, N.; Sardy, S.; Davison, A.C.; Spahn, D.R.; Decosterd, I. Assessment and analysis of mechanical allodynia-like behavior induced by spared nerve injury (SNI) in the mouse. Pain 2006, 122, e1–e14. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.H.; Won, R.; Baik, E.J.; Lee, S.H.; Moon, C.H. An animal model of neuropathic pain employing injury to the sciatic nerve branches. Neuroreport 2000, 11, 657–661. [Google Scholar] [CrossRef] [PubMed]
- Baron, R.; Binder, A.; Wasner, G. Neuropathic pain: Diagnosis, pathophysiological mechanisms, and treatment. Lancet Neurol. 2010, 9, 807–819. [Google Scholar] [CrossRef]
- Gonçalves, L.; Silva, R.; Pinto-Ribeiro, F.; Pêgo, J.M.; Bessa, J.M.; Pertovaara, A.; Sousa, N.; Almeida, A. Neuropathic pain is associated with depressive behaviour and induces neuroplasticity in the amygdala of the rat. Exp. Neurol. 2008, 213, 48–56. [Google Scholar] [CrossRef] [Green Version]
- Norman, G.J.; Karelina, K.; Zhang, N.; Walton, J.C.; Morris, J.S.; Devries, A.C. Stress and IL-1beta contribute to the development of depressive-like behavior following peripheral nerve injury. Mol. Psychiatry 2010, 15, 404–414. [Google Scholar] [CrossRef] [Green Version]
- Chambers, C.T.; Mogil, J.S. Ontogeny and phylogeny of facial expression of pain. Pain 2015, 156, 798–799. [Google Scholar] [CrossRef] [Green Version]
- Maletic, V.; Raison, C.L. Neurobiology of depression, fibromyalgia and neuropathic pain. Front. Biosci. 2009, 14, 5291–5338. [Google Scholar] [CrossRef]
- Kremer, M.; Becker, L.J.; Barrot, M.; Yalcin, I. How to study anxiety and depression in rodent models of chronic pain? Eur. J. Neurosci. 2020. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.G.; Chen, J. Preclinical research on pain comorbidity with affective disorders and cognitive deficits: Challenges and perspectives. Prog. Neurobiol. 2014, 116, 13–32. [Google Scholar] [CrossRef] [PubMed]
- Guida, F.; Luongo, L.; Marmo, F.; Romano, R.; Iannotta, M.; Napolitano, F.; Belardo, C.; Marabese, I.; D’Aniello, A.; De Gregorio, D.; et al. Palmitoylethanolamide reduces pain-related behaviors and restores glutamatergic synapses homeostasis in the medial prefrontal cortex of neuropathic mice. Mol. Brain 2015, 8, 47. [Google Scholar] [CrossRef]
- Leite-Almeida, H.; Almeida-Torres, L.; Mesquita, A.R.; Pertovaara, A.; Sousa, N.; Cerqueira, J.J.; Almeida, A. The impact of age on emotional and cognitive behaviours triggered by experimental neuropathy in rats. Pain 2009, 144, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Mutso, A.A.; Radzicki, D.; Baliki, M.N.; Huang, L.; Banisadr, G.; Centeno, M.V.; Radulovic, J.; Martina, M.; Miller, R.J.; Apkarian, A.V. Abnormalities in hippocampal functioning with persistent pain. J. Neurosci 2012, 32, 5747–5756. [Google Scholar] [CrossRef] [Green Version]
- Palazzo, E.; Luongo, L.; Guida, F.; Marabese, I.; Romano, R.; Iannotta, M.; Rossi, F.; D’Aniello, A.; Stella, L.; Marmo, F.; et al. D-Aspartate drinking solution alleviates pain and cognitive impairment in neuropathic mice. Amino Acids 2016, 48, 1553–1567. [Google Scholar] [CrossRef]
- Palazzo, E.; Romano, R.; Luongo, L.; Boccella, S.; De Gregorio, D.; Giordano, M.E.; Rossi, F.; Marabese, I.; Scafuro, M.A.; de Novellis, V.; et al. MMPIP, an mGluR7-selective negative allosteric modulator, alleviates pain and normalizes affective and cognitive behavior in neuropathic mice. Pain 2015, 156, 1060–1073. [Google Scholar] [CrossRef]
- Seminowicz, D.A.; Laferriere, A.L.; Millecamps, M.; Yu, J.S.; Coderre, T.J.; Bushnell, M.C. MRI structural brain changes associated with sensory and emotional function in a rat model of long-term neuropathic pain. Neuroimage 2009, 47, 1007–1014. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Goffer, Y.; Xu, D.; Tukey, D.S.; Shamir, D.B.; Eberle, S.E.; Zou, A.H.; Blanck, T.J.; Ziff, E.B. A single subanesthetic dose of ketamine relieves depression-like behaviors induced by neuropathic pain in rats. Anesthesiology 2011, 115, 812–821. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Wei, H.; Sagalajev, B.; Koivisto, A.; Pertovaara, A. Amygdaloid administration of tetrapentylammonium attenuates development of pain and anxiety-like behavior following peripheral nerve injury. Pharmacol. Rep. 2019, 71, 54–60. [Google Scholar] [CrossRef]
- Avila-Martin, G.; Galan-Arriero, I.; Ferrer-Donato, A.; Busquets, X.; Gomez-Soriano, J.; Escribá, P.V.; Taylor, J. Oral 2-hydroxyoleic acid inhibits reflex hypersensitivity and open-field-induced anxiety after spared nerve injury. Eur. J. Pain 2015, 19, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Leite-Almeida, H.; Cerqueira, J.J.; Wei, H.; Ribeiro-Costa, N.; Anjos-Martins, H.; Sousa, N.; Pertovaara, A.; Almeida, A. Differential effects of left/right neuropathy on rats’ anxiety and cognitive behavior. Pain 2012, 153, 2218–2225. [Google Scholar] [CrossRef] [Green Version]
- Gong, X.; Chen, Y.; Chang, J.; Huang, Y.; Cai, M.; Zhang, M. Environmental enrichment reduces adolescent anxiety- and depression-like behaviors of rats subjected to infant nerve injury. J. Neuroinflamm. 2018, 15, 262. [Google Scholar] [CrossRef] [PubMed]
- Sang, K.; Bao, C.; Xin, Y.; Hu, S.; Gao, X.; Wang, Y.; Bodner, M.; Zhou, Y.D.; Dong, X.W. Plastic change of prefrontal cortex mediates anxiety-like behaviors associated with chronic pain in neuropathic rats. Mol. Pain 2018, 14. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, C.S.; Khan, S.A.; Xu, S.; Cha, M.; Masri, R.; Seminowicz, D.A. Behavioral, metabolic and functional brain changes in a rat model of chronic neuropathic pain: A longitudinal MRI study. Neuroimage 2015, 107, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Descalzi, G.; Mitsi, V.; Purushothaman, I.; Gaspari, S.; Avrampou, K.; Loh, Y.E.; Shen, L.; Zachariou, V. Neuropathic pain promotes adaptive changes in gene expression in brain networks involved in stress and depression. Sci. Signal. 2017, 10, eaaj1549. [Google Scholar] [CrossRef] [Green Version]
- De Gregorio, D.; McLaughlin, R.J.; Posa, L.; Ochoa-Sanchez, R.; Enns, J.; Lopez-Canul, M.; Aboud, M.; Maione, S.; Comai, S.; Gobbi, G. Cannabidiol modulates serotonergic transmission and reverses both allodynia and anxiety-like behavior in a model of neuropathic pain. Pain 2019, 160, 136–150. [Google Scholar] [CrossRef]
- Zhang, Z.J.; Jiang, B.C.; Gao, Y.J. Chemokines in neuron-glial cell interaction and pathogenesis of neuropathic pain. Cell Mol. Life Sci. 2017, 74, 3275–3291. [Google Scholar] [CrossRef]
- Sieberg, C.B.; Taras, C.; Gomaa, A.; Nickerson, C.; Wong, C.; Ward, C.; Baskozos, G.; Bennett DL, H.; Ramirez, J.D.; Themistocleous, A.C.; et al. Neuropathic pain drives anxiety behavior in mice, results consistent with anxiety levels in diabetic neuropathy patients. Pain Rep. 2018, 3, e651. [Google Scholar] [CrossRef]
- Zhou, W.; Jin, Y.; Meng, Q.; Zhu, X.; Bai, T.; Tian, Y.; Mao, Y.; Wang, L.; Xie, W.; Zhong, H.; et al. A neural circuit for comorbid depressive symptoms in chronic pain. Nat. Neurosci. 2019, 22, 1649–1658. [Google Scholar] [CrossRef]
- Angoa-Pérez, M.; Kane, M.J.; Briggs, D.I.; Francescutti, D.M.; Kuhn, D.M. Marble burying and nestlet shredding as tests of repetitive, compulsive-like behaviors in mice. J. Vis. Exp. 2013, 82, e50978. [Google Scholar] [CrossRef] [PubMed]
- Pitzer, C.; La Porta, C.; Treede, R.D.; Tappe-Theodor, A. Inflammatory and neuropathic pain conditions do not primarily evoke anxiety-like behaviours in C57BL/6 mice. Eur. J. Pain 2019, 23, 285–306. [Google Scholar] [CrossRef] [PubMed]
- Humo, M.; Lu, H.; Yalcin, I. The molecular neurobiology of chronic pain-induced depression. Cell Tissue Res. 2019, 377, 21–43. [Google Scholar] [CrossRef] [PubMed]
- Yalcin, I.; Megat, S.; Barthas, F.; Waltisperger, E.; Kremer, M.; Salvat, E.; Barrot, M. The sciatic nerve cuffing model of neuropathic pain in mice. J. Vis. Exp. 2014, 89, e51608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goffer, Y.; Xu, D.; Eberle, S.E.; D’Amour, J.; Lee, M.; Tukey, D.; Froemke, R.C.; Ziff, E.B.; Wang, J. Calcium-permeable AMPA receptors in the nucleus accumbens regulate depression-like behaviors in the chronic neuropathic pain state. J. Neurosci. 2013, 33, 19034–19044. [Google Scholar] [CrossRef] [Green Version]
- Pan, W.; Zhang, G.F.; Li, H.H.; Ji, M.H.; Zhou, Z.Q.; Li, K.Y.; Yang, J.J. Ketamine differentially restores diverse alterations of neuroligins in brain regions in a rat model of neuropathic pain-induced depression. Neuroreport 2018, 29, 863–869. [Google Scholar] [CrossRef]
- Xie, Z.M.; Wang, X.M.; Xu, N.; Wang, J.; Pan, W.; Tang, X.H.; Zhou, Z.Q.; Hashimoto, K.; Yang, J.J. Alterations in the inflammatory cytokines and brain-derived neurotrophic factor contribute to depression-like phenotype after spared nerve injury: Improvement by ketamine. Sci. Rep. 2017, 7, 3124. [Google Scholar] [CrossRef]
- Zhang, X.M.; Wang, L.Z.; He, B.; Xiang, Y.K.; Fan, L.X.; Wang, Q.; Tao, L. The gap junction inhibitor INI-0602 attenuates mechanical allodynia and depression-like behaviors induced by spared nerve injury in rats. Neuroreport 2019, 30, 369–377. [Google Scholar] [CrossRef]
- Fu, B.; Wen, S.N.; Wang, B.; Wang, K.; Zhang, J.Y.; Weng, X.C.; Liu, S.J. Gabapentin regulates dopaminergic neuron firing and theta oscillation in the ventral tegmental area to reverse depression-like behavior in chronic neuropathic pain state. J. Pain Res. 2018, 11, 2247–2256. [Google Scholar] [CrossRef] [Green Version]
- Fang, X.; Zhan, G.; Zhang, J.; Xu, H.; Zhu, B.; Hu, Y.; Yang, C.; Luo, A. Abnormalities in Inflammatory Cytokines Confer Susceptible to Chronic Neuropathic Pain-related Anhedonia in a Rat Model of Spared Nerve Injury. Clin. Psychopharmacol. Neurosci. 2019, 17, 189–199. [Google Scholar] [CrossRef] [Green Version]
- Thompson, S.J.; Pitcher, M.H.; Stone, L.S.; Tarum, F.; Niu, G.; Chen, X.; Kiesewetter, D.O.; Schweinhardt, P.; Bushnell, M.C. Chronic neuropathic pain reduces opioid receptor availability with associated anhedonia in rat. Pain 2018, 159, 1856–1866. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Wei, H.; Pertovaara, A.; Wang, J.; Carlson, S. Anxiety- and activity-related effects of paracetamol on healthy and neuropathic rats. Pharmacol. Res. Perspect. 2018, 6, e00367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, W.; Dantzer, R.; Budac, D.P.; Walker, A.K.; Mao-Ying, Q.L.; Lee, A.W.; Heijnen, C.J.; Kavelaars, A. Peripheral indoleamine 2,3-dioxygenase 1 is required for comorbid depression-like behavior but does not contribute to neuropathic pain in mice. Brain Behav. Immun. 2015, 46, 147–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moriarty, O.; McGuire, B.E.; Finn, D.P. The effect of pain on cognitive function: A review of clinical and preclinical research. Prog. Neurobiol. 2011, 93, 385–404. [Google Scholar] [CrossRef] [Green Version]
- Cardoso-Cruz, H.; Dourado, M.; Monteiro, C.; Galhardo, V. Blockade of dopamine D2 receptors disrupts intrahippocampal connectivity and enhances pain-related working memory deficits in neuropathic pain rats. Eur. J. Pain 2018, 22, 1002–1015. [Google Scholar] [CrossRef]
- Cardoso-Cruz, H.; Paiva, P.; Monteiro, C.; Galhardo, V. Bidirectional optogenetic modulation of prefrontal-hippocampal connectivity in pain-related working memory deficits. Sci. Rep. 2019, 9, 10980. [Google Scholar] [CrossRef]
- Grégoire, S.; Millecamps, M.; Naso, L.; Do Carmo, S.; Cuello, A.C.; Szyf, M.; Stone, L.S. Therapeutic benefits of the methyl donor S-adenosylmethionine on nerve injury-induced mechanical hypersensitivity and cognitive impairment in mice. Pain 2017, 158, 802–810. [Google Scholar] [CrossRef]
- Paul, C.M.; Magda, G.; Abel, S. Spatial memory: Theoretical basis and comparative review on experimental methods in rodents. Behav. Brain Res. 2009, 203, 151–164. [Google Scholar] [CrossRef]
- Low, L.A.; Millecamps, M.; Seminowicz, D.A.; Naso, L.; Thompson, S.J.; Stone, L.S.; Bushnell, M.C. Nerve injury causes long-term attentional deficits in rats. Neurosci. Lett. 2012, 529, 103–107. [Google Scholar] [CrossRef]
- Higgins, G.A.; Silenieks, L.B.; Van Niekerk, A.; Desnoyer, J.; Patrick, A.; Lau, W.; Thevarkunnel, S. Enduring attentional deficits in rats treated with a peripheral nerve injury. Behav. Brain Res. 2015, 286, 347–355. [Google Scholar] [CrossRef]
- Wang, J.; Liu, Y.; Zhou, L.J.; Wu, Y.; Li, F.; Shen, K.F.; Pang, R.P.; Wei, X.H.; Li, Y.Y.; Liu, X.G. Magnesium L-threonate prevents and restores memory deficits associated with neuropathic pain by inhibition of TNF-α. Pain Physician 2013, 16, E563–E575. [Google Scholar] [PubMed]
- Gui, W.S.; Wei, X.; Mai, C.L.; Murugan, M.; Wu, L.J.; Xin, W.J.; Zhou, L.J.; Liu, X.G. Interleukin-1β overproduction is a common cause for neuropathic pain, memory deficit, and depression following peripheral nerve injury in rodents. Mol. Pain 2016, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mai, C.L.; Wei, X.; Gui, W.S.; Xu, Y.N.; Zhang, J.; Lin, Z.J.; Tan, Z.; Meng, Y.T.; Li, Y.Y.; Zhou, L.J.; et al. Differential regulation of GSK-3β in spinal dorsal horn and in hippocampus mediated by interleukin-1beta contributes to pain hypersensitivity and memory deficits following peripheral nerve injury. Mol. Pain 2019, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boccella, S.; Cristiano, C.; Romano, R.; Iannotta, M.; Belardo, C.; Farina, A.; Guida, F.; Piscitelli, F.; Palazzo, E.; Mazzitelli, M.; et al. Ultra-micronized palmitoylethanolamide rescues the cognitive decline-associated loss of neural plasticity in the neuropathic mouse entorhinal cortex-dentate gyrus pathway. Neurobiol. Dis. 2019, 121, 106–119. [Google Scholar] [CrossRef] [PubMed]
- Albuquerque, B.; Häussler, A.; Vannoni, E.; Wolfer, D.P.; Tegeder, I. Learning and memory with neuropathic pain: Impact of old age and progranulin deficiency. Front. Behav. Neurosci. 2013, 7, 174. [Google Scholar] [CrossRef] [Green Version]
- Shiers, S.; Mwirigi, J.; Pradhan, G.; Kume, M.; Black, B.; Barragan-Iglesias, P.; Moy, J.K.; Dussor, G.; Pancrazio, J.J.; Kroener, S.; et al. Reversal of peripheral nerve injury-induced neuropathic pain and cognitive dysfunction via genetic and tomivosertib targeting of MNK. Neuropsychopharmacology 2020, 45, 524–533. [Google Scholar] [CrossRef]
- Karl, F.; Grießhammer, A.; Üçeyler, N.; Sommer, C. Differential Impact of miR-21 on Pain and Associated Affective and Cognitive Behavior after Spared Nerve Injury in B7-H1 ko Mouse. Front. Mol. Neurosci. 2017, 10, 219. [Google Scholar] [CrossRef]
- Xu, N.; Tang, X.H.; Pan, W.; Xie, Z.M.; Zhang, G.F.; Ji, M.H.; Yang, J.J.; Zhou, M.T.; Zhou, Z.Q. Spared Nerve Injury Increases the Expression of Microglia M1 Markers in the Prefrontal Cortex of Rats and Provokes Depression-Like Behaviors. Front. Neurosci. 2017, 11, 209. [Google Scholar] [CrossRef]
- Becker, S.; Navratilova, E.; Nees, F.; Van Damme, S. Emotional and motivational pain processing: Current state of knowledge and perspectives in translational research. Pain Res. Manag. 2018, 2018, 5457870. [Google Scholar] [CrossRef]
- Bushnell, M.C.; Čeko, M.; Low, L.A. Cognitive and emotional control of pain and its disruption in chronic pain. Nat. Rev. Neurosci. 2013, 14, 502–511. [Google Scholar] [CrossRef] [Green Version]
- Moss, A.; Beggs, S.; Vega-Avelaira, D.; Costigan, M.; Hathway, G.J.; Salter, M.W.; Fitzgerald, M. Spinal microglia and neuropathic pain in young rats. Pain 2007, 128, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Kettenmann, H.; Hanisch, U.K.; Noda, M.; Verkhratsky, A. Physiology of microglia. Physiol. Rev. 2011, 91, 461–553. [Google Scholar] [CrossRef]
- McMahon, S.B.; Malcangio, M. Current challenges in glia-pain biology. Neuron 2009, 64, 46–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, T.T.; Wu, J.R.; Chen, Z.Y.; Liu, Z.X.; Miao, B. Effects of dexmedetomidine on P2X4Rs, p38-MAPK and BDNF in spinal microglia in rats with spared nerve injury. Brain Res. 2014, 1568, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wei, H.; Piirainen, S.; Chen, Z.; Kalso, E.; Pertovaara, A.; Tian, L. Spinal versus brain microglial and macrophage activation traits determine the differential neuroinflammatory responses and analgesic effect of minocycline in chronic neuropathic pain. Brain Behav. Immun. 2016, 58, 107–117. [Google Scholar] [CrossRef] [Green Version]
- Luongo, L.; Palazzo, E.; Tambaro, S.; Giordano, C.; Gatta, L.; Scafuro, M.A.; Rossi, F.S.; Lazzari, P.; Pani, L.; de Novellis, V.; et al. 1-(2’,4’-dichlorophenyl)-6-methyl-N-cyclohexylamine-1,4-dihydroindeno [1,2-c]pyrazole-3-carboxamide, a novel CB2 agonist, alleviates neuropathic pain through functional microglial changes in mice. Neurobiol. Dis. 2010, 37, 177–185. [Google Scholar] [CrossRef]
- Wen, Y.R.; Suter, M.R.; Kawasaki, Y.; Huang, J.; Pertin, M.; Kohno, T.; Berde, C.B.; Decosterd, I.; Ji, R.R. Nerve conduction blockade in the sciatic nerve prevents but does not reverse the activation of p38 mitogen-activated protein kinase in spinal microglia in the rat spared nerve injury model. Anesthesiology 2007, 107, 312–321. [Google Scholar] [CrossRef] [Green Version]
- Shao, J.; Xu, R.; Li, M.; Zhao, Q.; Ren, X.; Li, Z.; Cao, J.; Zang, W. Glucocorticoid receptor inhibit the activity of NF-κB through p38 signaling pathway in spinal cord in the spared nerve injury rats. Life Sci. 2018, 208, 268–275. [Google Scholar] [CrossRef]
- Zhou, L.J.; Yang, T.; Wei, X.; Liu, Y.; Xin, W.J.; Chen, Y.; Pang, R.P.; Zang, Y.; Li, Y.Y.; Liu, X.G. Brain-derived neurotrophic factor contributes to spinal long-term potentiation and mechanical hypersensitivity by activation of spinal microglia in rat. Brain Behav. Immun. 2011, 25, 322–334. [Google Scholar] [CrossRef]
- Bai, L.; Wang, X.; Li, Z.; Kong, C.; Zhao, Y.; Qian, J.L.; Kan, Q.; Zhang, W.; Xu, J.T. Upregulation of Chemokine CXCL12 in the Dorsal Root Ganglia and Spinal Cord Contributes to the Development and Maintenance of Neuropathic Pain Following Spared Nerve Injury in Rats. Neurosci. Bull. 2016, 32, 27–40. [Google Scholar] [CrossRef] [Green Version]
- Guida, F.; Lattanzi, R.; Boccella, S.; Maftei, D.; Romano, R.; Marconi, V.; Balboni, G.; Salvadori, S.; Scafuro, M.A.; de Novellis, V.; et al. PC1, a non-peptide PKR1-preferring antagonist, reduces pain behavior and spinal neuronal sensitization in neuropathic mice. Pharmacol. Res. 2015, 91, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Kanda, H.; Kobayashi, K.; Yamanaka, H.; Okubo, M.; Noguchi, K. Microglial TNFα Induces COX2 and PGI2 Synthase Expression in Spinal Endothelial Cells during Neuropathic Pain. eNeuro 2017, 4. [Google Scholar] [CrossRef] [PubMed]
- Kiyoyuki, Y.; Taniguchi, W.; Okubo, M.; Yamanaka, H.; Kobayashi, K.; Nishio, N.; Nakatsuka, T.; Noguchi, K. Leukotriene enhances NMDA-induced inward currents in dorsal horn neurons of the rat spinal cord after peripheral nerve injury. Mol. Pain 2015, 11, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu Sin Chung, P.; Panigada, T.; Cardis, R.; Decosterd, I.; Gosselin, R.D. Peripheral nerve injury induces a transitory microglial reaction in the rat infralimbic cortex. Neurosci. Lett. 2017, 655, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Giordano, C.; Cristino, L.; Luongo, L.; Siniscalco, D.; Petrosino, S.; Piscitelli, F.; Marabese, I.; Gatta, L.; Rossi, F.; Imperatore, R.; et al. TRPV1-dependent and -independent alterations in the limbic cortex of neuropathic mice: Impact on glial caspases and pain perception. Cereb. Cortex 2012, 22, 2495–2518. [Google Scholar] [CrossRef] [Green Version]
- Marcello, L.; Cavaliere, C.; Colangelo, A.M.; Bianco, M.R.; Cirillo, G.; Alberghina, L.; Papa, M. Remodelling of supraspinal neuroglial network in neuropathic pain is featured by a reactive gliosis of the nociceptive amygdala. Eur. J. Pain 2013, 17, 799–810. [Google Scholar] [CrossRef]
- Liu, Y.; Zhou, L.J.; Wang, J.; Li, D.; Ren, W.J.; Peng, J.; Wei, X.; Xu, T.; Xin, W.J.; Pang, R.P.; et al. TNF-α Differentially Regulates Synaptic Plasticity in the Hippocampus and Spinal Cord by Microglia-Dependent Mechanisms after Peripheral Nerve Injury. J. Neurosci. 2017, 37, 871–881. [Google Scholar] [CrossRef] [Green Version]
- Gosselin, R.D.; Bebber, D.; Decosterd, I. Upregulation of the GABA transporter GAT-1 in the gracile nucleus in the spared nerve injury model of neuropathic pain. Neurosci. Lett. 2010, 480, 132–137. [Google Scholar] [CrossRef] [Green Version]
- Jeon, S.; Jha, M.K.; Ock, J.; Seo, J.; Jin, M.; Cho, H.; Lee, W.-H.; Suk, K. Role of lipocalin-2-chemokine axis in the development of neuropathic pain following peripheral nerve injury. J. Biol. Chem. 2013, 288, 24116–24127. [Google Scholar] [CrossRef] [Green Version]
- Vega-Avelaira, D.; Géranton, S.M.; Fitzgerald, M. Differential regulation of immune responses and macrophage/neuron interactions in the dorsal root ganglion in young and adult rats following nerve injury. Mol. Pain 2009, 5, 70. [Google Scholar] [CrossRef] [Green Version]
- Vicuña, L.; Strochlic, D.E.; Latremoliere, A.; Bali, K.K.; Simonetti, M.; Husainie, D.; Prokosch, S.; Riva, P.; Griffin, R.S.; Njoo, C. The serine protease inhibitor SerpinA3N attenuates neuropathic pain by inhibiting T cell–derived leukocyte elastase. Nat. Med. 2015, 21, 518–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gattlen, C.; Clarke, C.B.; Piller, N.; Kirschmann, G.; Pertin, M.; Decosterd, I.; Gosselin, R.-D.; Suter, M.R. Spinal cord T-cell infiltration in the rat spared nerve injury model: A time course study. Int. J. Mol. Sci. 2016, 17, 352. [Google Scholar] [CrossRef]
- Sorge, R.E.; Mapplebeck, J.C.; Rosen, S.; Beggs, S.; Taves, S.; Alexander, J.K.; Martin, L.J.; Austin, J.S.; Sotocinal, S.G.; Chen, D.; et al. Different immune cells mediate mechanical pain hypersensitivity in male and female mice. Nat. Neurosci. 2015, 18, 1081–1083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boccella, S.; Guida, F.; De Logu, F.; De Gregorio, D.; Mazzitelli, M.; Belardo, C.; Iannotta, M.; Serra, N.; Nassini, R.; de Novellis, V. Ketones and pain: Unexplored role of hydroxyl carboxylic acid receptor type 2 in the pathophysiology of neuropathic pain. FASEB J. 2019, 33, 1062–1073. [Google Scholar] [CrossRef] [PubMed]
- Brings, V.E.; Zylka, M.J. Sex, drugs and pain control. Nat. Neurosci. 2015, 18, 1059–1060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coraggio, V.; Guida, F.; Boccella, S.; Scafuro, M.; Paino, S.; Romano, D.; Maione, S.; Luongo, L. Neuroimmune-Driven Neuropathic Pain Establishment: A Focus on Gender Differences. Int. J. Mol. Sci. 2018, 19, 281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorge, R.E.; Totsch, S.K. Sex Differences in Pain. J. Neurosci. Res. 2017, 95, 1271–1281. [Google Scholar] [CrossRef]
- Inyang, K.E.; Szabo-Pardi, T.; Wentworth, E.; McDougal, T.A.; Dussor, G.; Burton, M.D.; Price, T.J. The antidiabetic drug metformin prevents and reverses neuropathic pain and spinal cord microglial activation in male but not female mice. Pharmacol. Res. 2019, 139, 1–16. [Google Scholar] [CrossRef]
- Gong, N.; Hagopian, G.; Holmes, T.C.; Luo, Z.D.; Xu, X. Functional Reorganization of Local Circuit Connectivity in Superficial Spinal Dorsal Horn with Neuropathic Pain States. eNeuro 2019, 6. [Google Scholar] [CrossRef] [Green Version]
- Doolen, S.; Blake, C.B.; Smith, B.N.; Taylor, B.K. Peripheral nerve injury increases glutamate-evoked calcium mobilization in adult spinal cord neurons. Mol. Pain 2012, 8, 56. [Google Scholar] [CrossRef] [Green Version]
- Inquimbert, P.; Moll, M.; Latremoliere, A.; Tong, C.K.; Whang, J.; Sheehan, G.F.; Smith, B.M.; Korb, E.; Athié, M.C.P.; Babaniyi, O.; et al. NMDA Receptor Activation Underlies the Loss of Spinal Dorsal Horn Neurons and the Transition to Persistent Pain after Peripheral Nerve Injury. Cell Rep. 2018, 23, 2678–2689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, L.; Cai, J.; Guo, X.Y.; Meng, X.L.; Xing, G.G. The antiallodynic action of pregabalin may depend on the suppression of spinal neuronal hyperexcitability in rats with spared nerve injury. Pain Res. Manag. 2014, 19, 205–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singer, T.; Seymour, B.; O’doherty, J.; Kaube, H.; Dolan, R.J.; Frith, C.D. Empathy for pain involves the affective but not sensory components of pain. Science 2004, 303, 1157–1162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Novellis, V.; Vita, D.; Gatta, L.; Luongo, L.; Bellini, G.; De Chiaro, M.; Marabese, I.; Siniscalco, D.; Boccella, S.; Piscitelli, F.; et al. The blockade of the transient receptor potential vanilloid type 1 and fatty acid amide hydrolase decreases symptoms and central sequelae in the medial prefrontal cortex of neuropathic rats. Mol. Pain 2011, 7, 7. [Google Scholar] [CrossRef] [Green Version]
- Fields, H.L.; Barbaro, N.M.; Heinricher, M.M. Brain stem neuronal circuitry underlying the antinociceptive action of opiates. Prog. Brain Res. 1988, 77, 245–257. [Google Scholar]
- Fields, H.L.; Heinricher, M.M.; Mason, P. Neurotransmitters in nociceptive modulatory circuits. Annu. Rev. Neurosci. 1991, 14, 219–245. [Google Scholar] [CrossRef]
- Palazzo, E.; Guida, F.; Gatta, L.; Luongo, L.; Boccella, S.; Bellini, G.; Marabese, I.; de Novellis, V.; Rossi, F.; Maione, S. EP1 receptor within the ventrolateral periaqueductal grey controls thermonociception and rostral ventromedial medulla cell activity in healthy and neuropathic rat. Mol. Pain 2011, 7, 82. [Google Scholar] [CrossRef] [Green Version]
- Fu, B.; Weng, X.C.; Wang, J.; Huang, T.; Wang, B.; Lin, S.D.; Liu, S.J. The impact of electrophysiology of central dopaminergic neurons and the depression-state induced by chronic neuropathic pain. Zhongguo Ying Yong Sheng Li Xue Za Zhi 2016, 32, 403–407. [Google Scholar]
- Huang, S.; Borgland, S.L.; Zamponi, G.W. Peripheral nerve injury-induced alterations in VTA neuron firing properties. Mol. Brain 2019, 12, 89. [Google Scholar] [CrossRef] [Green Version]
- Sagheddu, C.; Aroni, S.; De Felice, M.; Lecca, S.; Luchicchi, A.; Melis, M.; Muntoni, A.L.; Romano, R.; Palazzo, E.; Guida, F.; et al. Enhanced serotonin and mesolimbic dopamine transmissions in a rat model of neuropathic pain. Neuropharmacology 2015, 97, 383–393. [Google Scholar] [CrossRef]
- Jarrin, S.; Finn, D.P. Optogenetics and its application in pain and anxiety research. Neurosci. Biobehav. Rev. 2019, 105, 200–211. [Google Scholar] [CrossRef] [PubMed]
- Deisseroth, K. Optogenetics. Nat. Methods 2011, 8, 26–29. [Google Scholar] [CrossRef] [PubMed]
- Tye, K.M.; Deisseroth, K. Optogenetic investigation of neural circuits underlying brain disease in animal models. Nat. Rev. Neurosci. 2012, 13, 251–266. [Google Scholar] [CrossRef]
- Williams, S.C.; Deisseroth, K. Optogenetics. Proc. Natl. Acad. Sci. USA 2013, 110, 16287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Häusser, M. Optogenetics: The age of light. Nat. Methods 2014, 11, 1012. [Google Scholar] [CrossRef]
- Iyer, S.M.; Montgomery, K.L.; Towne, C.; Lee, S.Y.; Ramakrishnan, C.; Deisseroth, K.; Delp, S.L. Virally mediated optogenetic excitation and inhibition of pain in freely moving nontransgenic mice. Nat. Biotechnol. 2014, 32, 274–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iyer, S.M.; Vesuna, S.; Ramakrishnan, C.; Huynh, K.; Young, S.; Berndt, A.; Lee, S.Y.; Gorini, C.J.; Deisseroth, K.; Delp, S.L. Optogenetic and chemogenetic strategies for sustained inhibition of pain. Sci. Rep. 2016, 6, 30570. [Google Scholar] [CrossRef]
- Montgomery, K.L.; Yeh, A.J.; Ho, J.S.; Tsao, V.; Iyer, S.M.; Grosenick, L.; Ferenczi, E.A.; Tanabe, Y.; Deisseroth, K.; Delp, S.L. Wirelessly powered, fully internal optogenetics for brain, spinal and peripheral circuits in mice. Nat. Methods 2015, 12, 969–974. [Google Scholar] [CrossRef] [Green Version]
- Meda, K.S.; Patel, T.; Braz, J.M.; Malik, R.; Turner, M.L.; Seifikar, H.; Basbaum, A.I.; Sohal, V.S. Microcircuit mechanisms through which mediodorsal thalamic input to anterior cingulate cortex exacerbates pain-related aversion. Neuron 2019, 102, 944–959.e3. [Google Scholar] [CrossRef]
- Chang, Y.T.; Chen, W.H.; Shih, H.C.; Min, M.Y.; Shyu, B.C.; Chen, C.C. Anterior nucleus of paraventricular thalamus mediates chronic mechanical hyperalgesia. Pain 2019, 160, 1208–1223. [Google Scholar] [CrossRef]
- Lee, M.; Manders, T.R.; Eberle, S.E.; Su, C.; D’Amour, J.; Yang, R.; Lin, H.Y.; Deisseroth, K.; Froemke, R.C.; Wang, J. Activation of corticostriatal circuitry relieves chronic neuropathic pain. J. Neurosci. 2015, 35, 5247–5259. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Martinez, E.; Lin, H.H.; Yang, R.; Dale, J.A.; Liu, K.; Huang, D.; Wang, J. Inhibition of the Prefrontal Projection to the Nucleus Accumbens Enhances Pain Sensitivity and Affect. Front. Cell Neurosci. 2018, 12, 240. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Gadotti, V.M.; Chen, L.; Souza, I.A.; Stemkowski, P.L.; Zamponi, G.W. Role of Prelimbic GABAergic Circuits in Sensory and Emotional Aspects of Neuropathic Pain. Cell Rep. 2015, 12, 752–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gadotti, V.M.; Zhang, Z.; Huang, J.; Zamponi, G.W. Analgesic effects of optogenetic inhibition of basolateral amygdala inputs into the prefrontal cortex in nerve injured female mice. Mol. Brain 2019, 12, 105. [Google Scholar] [CrossRef] [PubMed]
- Daou, I.; Beaudry, H.; Ase, A.R.; Wieskopf, J.S.; Ribeiro-da-Silva, A.; Mogil, J.S.; Séguéla, P. Optogenetic silencing of Nav1. 8-positive afferents alleviates inflammatory and neuropathic pain. eNeuro 2016, 3. [Google Scholar] [CrossRef] [PubMed]
- Cowie, A.M.; Moehring, F.; O’Hara, C.; Stucky, C.L. Optogenetic inhibition of CGRPα sensory neurons reveals their distinct roles in neuropathic and incisional pain. J. Neurosci. 2018, 38, 5807–5825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nam, Y.; Kim, J.-H.; Kim, J.-H.; Jha, M.K.; Jung, J.Y.; Lee, M.-G.; Choi, I.-S.; Jang, I.-S.; Lim, D.G.; Hwang, S.-H. Reversible induction of pain hypersensitivity following optogenetic stimulation of spinal astrocytes. Cell Rep. 2016, 17, 3049–3061. [Google Scholar] [CrossRef] [Green Version]
- Whissell, P.D.; Tohyama, S.; Martin, L.J. The Use of DREADDs to Deconstruct Behavior. Front. Genet. 2016, 7, 70. [Google Scholar] [CrossRef] [Green Version]
- Roth, B.L. DREADDs for Neuroscientists. Neuron 2016, 89, 683–694. [Google Scholar] [CrossRef] [Green Version]
- Lee, G.H.; Kim, S.S. Therapeutic Strategies for Neuropathic Pain: Potential Application of Pharmacosynthetics and Optogenetics. Mediat. Inflamm. 2016, 2016, 5808215. [Google Scholar] [CrossRef] [Green Version]
- Cichon, J.; Blanck TJ, J.; Gan, W.B.; Yang, G. Activation of cortical somatostatin interneurons prevents the development of neuropathic pain. Nat. Neurosci. 2017, 20, 1122–1132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, N.; Miller, C.; Fields, H.L. Cortico-Accumbens Regulation of Approach-Avoidance Behavior Is Modified by Experience and Chronic Pain. Cell Rep. 2017, 19, 1522–1531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, H.; Fatima, M.; Li, A.; Lee, H.; Cai, W.; Horwitz, L.; Hor, C.C.; Zaher, N.; Cin, M.; Slade, H. Identification of a spinal circuit for mechanical and persistent spontaneous itch. Neuron 2019, 103, 1135–1149.e6. [Google Scholar] [CrossRef] [PubMed]
- Guettier, J.-M.; Gautam, D.; Scarselli, M.; de Azua, I.R.; Li, J.H.; Rosemond, E.; Ma, X.; Gonzalez, F.J.; Armbruster, B.N.; Lu, H. A chemical-genetic approach to study G protein regulation of β cell function in vivo. Proc. Natl. Acad. Sci. USA 2009, 106, 19197–19202. [Google Scholar] [CrossRef] [Green Version]
- Mogil, J.S. Animal models of pain: Progress and challenges. Nat. Rev. Neurosci. 2009, 10, 283–294. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Surgery | Specie and Sex | Effect | Duration | Methods | References |
|---|---|---|---|---|---|
| SNI | Male rat | Mechanical allodynia and hyperalgesia Thermal hyperalgesia | from 24 h to 7 months | Von Frey Pin prick Acetone/Ethyl Cloride test Hargreaves’ test | [10,24] |
| Crush injury of tibial and common peroneal nerves | from 4 days to 7 weeks | ||||
| Spared common peroneal | Male rats | Mechanical and cold allodynia | From day 4 to 10 weeks | [25] | |
| Spared tibial | Mechanical allodynia No cold allodynia | only at 14 days | |||
| Spared common peroneal and sural | Mechanical and cold allodynia | ||||
| Common peroneal, tibial and sural nerves injured | Mechanical and cold allodynia | ||||
| SNI | Male and female mice | Mechanical Allodynia | From 3 to 28 days | Von Frey | [24] |
| Male mice | Until 12 months | Dynamic Plantar Aesthesiometer | [10] | ||
| Spared tibial nerve | Male mouse | Mechanical allodynia | From 3 to 14 days | Von Frey | [7] |
| Spared sural and common peroneal | Male and female mice | No mechanical allodynia Mechanical allodynia | From 3 to 28 days | Von Frey | [7,24] |
| SNI | Male rats | Anxiety-like behavior | 14 days | Light dark box | [40] |
| Anxiety-like behavior | 21 days | Open field, elevated plus maze | [41,62] | ||
| Anxiety-like behavior | 23 days | Open field, elevated plus maze | [47] | ||
| Anxiety-like behavior | 28 days | Open field, elevated plus maze | [34,42] | ||
| Anxiety-like behavior | 20–40 days | Open field, elevated plus maze | [43] | ||
| Anxiety-like behavior | 4–8 weeks | Open field, elevated plus maze | [44] | ||
| Anxiety-like behavior | 24 weeks | Elevated plus maze | [38] | ||
| Female rats | No anxiety-like behaviour | 8 weeks | Open field, elevated plus maze | [27] | |
| No anxiety-like behaviour | From 2 to 19 weeks | Open field, elevated plus maze | [45] | ||
| SNI | Male mice | Anxiety-like behavior | 12 days | Fear condition and extinction, black box emergency | [35] |
| Male mice | Anxiety-like behavior | 14 days | Open field, elevated plus maze, marble burying | [37,46] | |
| Anxiety-like behavior | 28 days | Open field, elevated plus maze | [48] | ||
| Male mice | Anxiety-like behavior | 30 days | Light dark box, Marble burying | [36] | |
| Male and female mice | Anxiety-like behavior | 4–7 weeks | Elevated plus maze, light dark box, holeboard | [49] | |
| Male mice | Anxiety-like behavior | 6 weeks | Open field, elevated plus maze | [50] | |
| Male mice | No anxiety-like behavior | From day 3 to week 7 | Elevated zero maze, marble, burying | [12] | |
| Male and female mice | No anxiety-like behavior | From 3 to 97 days | Elevated plus maze, hole-board | [52] | |
| Male rats | Depression-like behavior | 14 days | Forced swim, sucrose preference | [55,78] | |
| Male rats | Depression-like behavior | 13–16 and 20–23 days | Forced swim, sucrose preference | [57] | |
| Male rats | Depression-like behavior | 14 and 18 days | Forced swim, sucrose reference | [56] | |
| Male rats | Depression-like behavior | 14 and 56 days | Forced swim, sucrose preference | [39] | |
| Male rats | Depression-like behavior | day 25 | Forced swim, sucrose preference, tail suspension | [58] | |
| Male rats | Depression-like behavior | day 28 | Forced swim | [34] | |
| Male rats | Depression-like behavior | 12 and 19 day | Sucrose preference | [60] | |
| Male rats | Depression-like behavior | 42, 56 days | Forced swim, sucrose preference | [59] | |
| Male rats | Depression-like behavior | 42 days | Sucrose preference | [43] | |
| Male rats | Depression-like behavior | 7 weeks | Forced swim | [27] | |
| Male rats | Depression-like behavior | 11 weeks | Sucrose preference | [61] | |
| Male rats | No depression-like behavior | 3 weeks | Sucrose preference | [62] | |
| SNI | Male mice | Depression-like behavior | 3 days to 7 weeks | Forced swim, sucrose preference | [12] |
| Male mice | Depression-like behavior | 7 days | Forced swim | [28,63] | |
| Male mice | Depression-like behavior | 14 days | Tail suspension | [37] | |
| Male mice | Depression-like behavior | 30 days | Forced swim, Tail suspension | [33,36] | |
| Male mice | Depression-like behavior | 6 weeks | Tai suspension, sucrose preference | [50] | |
| Male mice | Depression-like behavior | 9 weeks | Forced swim | [46] | |
| Male mice | Depression-like behavior | 1 year | Tail suspension | [10] | |
| Male mice | No depression-like behavior | 3 to 97 days | Forced swim | [52] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guida, F.; De Gregorio, D.; Palazzo, E.; Ricciardi, F.; Boccella, S.; Belardo, C.; Iannotta, M.; Infantino, R.; Formato, F.; Marabese, I.; et al. Behavioral, Biochemical and Electrophysiological Changes in Spared Nerve Injury Model of Neuropathic Pain. Int. J. Mol. Sci. 2020, 21, 3396. https://doi.org/10.3390/ijms21093396
Guida F, De Gregorio D, Palazzo E, Ricciardi F, Boccella S, Belardo C, Iannotta M, Infantino R, Formato F, Marabese I, et al. Behavioral, Biochemical and Electrophysiological Changes in Spared Nerve Injury Model of Neuropathic Pain. International Journal of Molecular Sciences. 2020; 21(9):3396. https://doi.org/10.3390/ijms21093396
Chicago/Turabian StyleGuida, Francesca, Danilo De Gregorio, Enza Palazzo, Flavia Ricciardi, Serena Boccella, Carmela Belardo, Monica Iannotta, Rosmara Infantino, Federica Formato, Ida Marabese, and et al. 2020. "Behavioral, Biochemical and Electrophysiological Changes in Spared Nerve Injury Model of Neuropathic Pain" International Journal of Molecular Sciences 21, no. 9: 3396. https://doi.org/10.3390/ijms21093396