Pioglitazone and Deoxyribonucleoside Combination Treatment Increases Mitochondrial Respiratory Capacity in m.3243A>G MELAS Cybrid Cells

 , ,
, ,
Abstract
:1. Introduction
2. Results
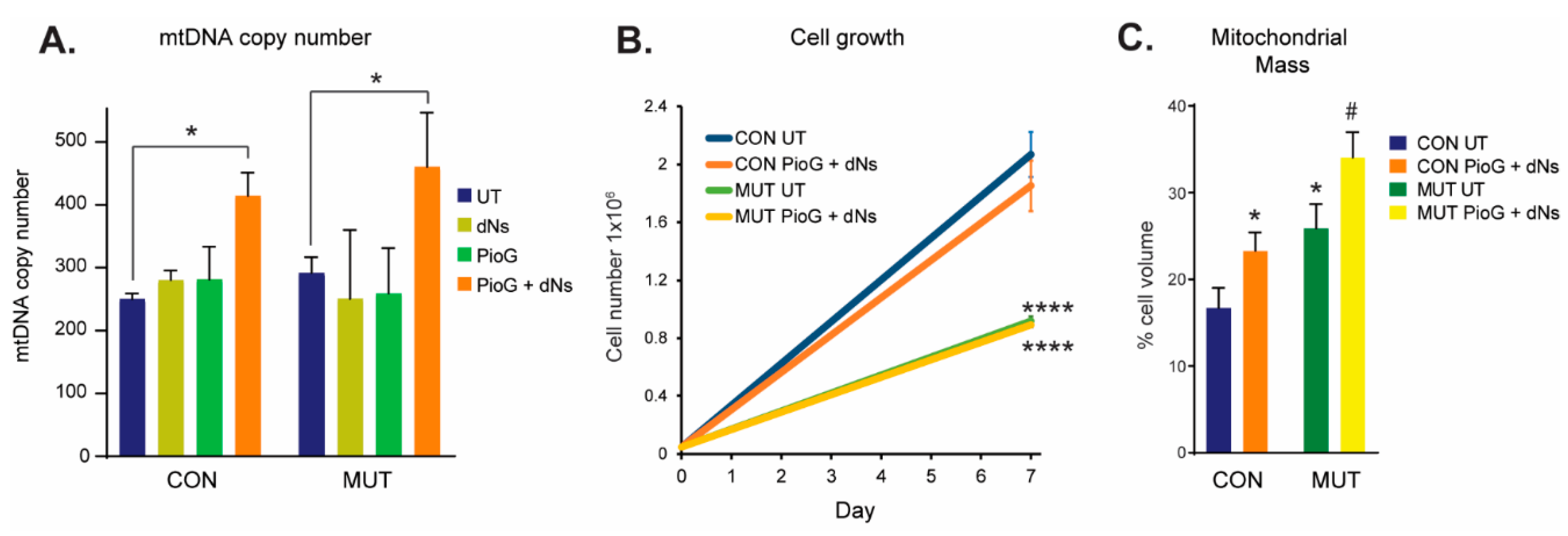
2.1. Combination Treatment with Pioglitazone (PioG) and Deoxyribonucleosides (dNs) Increases mtDNA Copy Number and Mitochondrial Mass in Both Control and m.3243A>G Mutant Cybrids
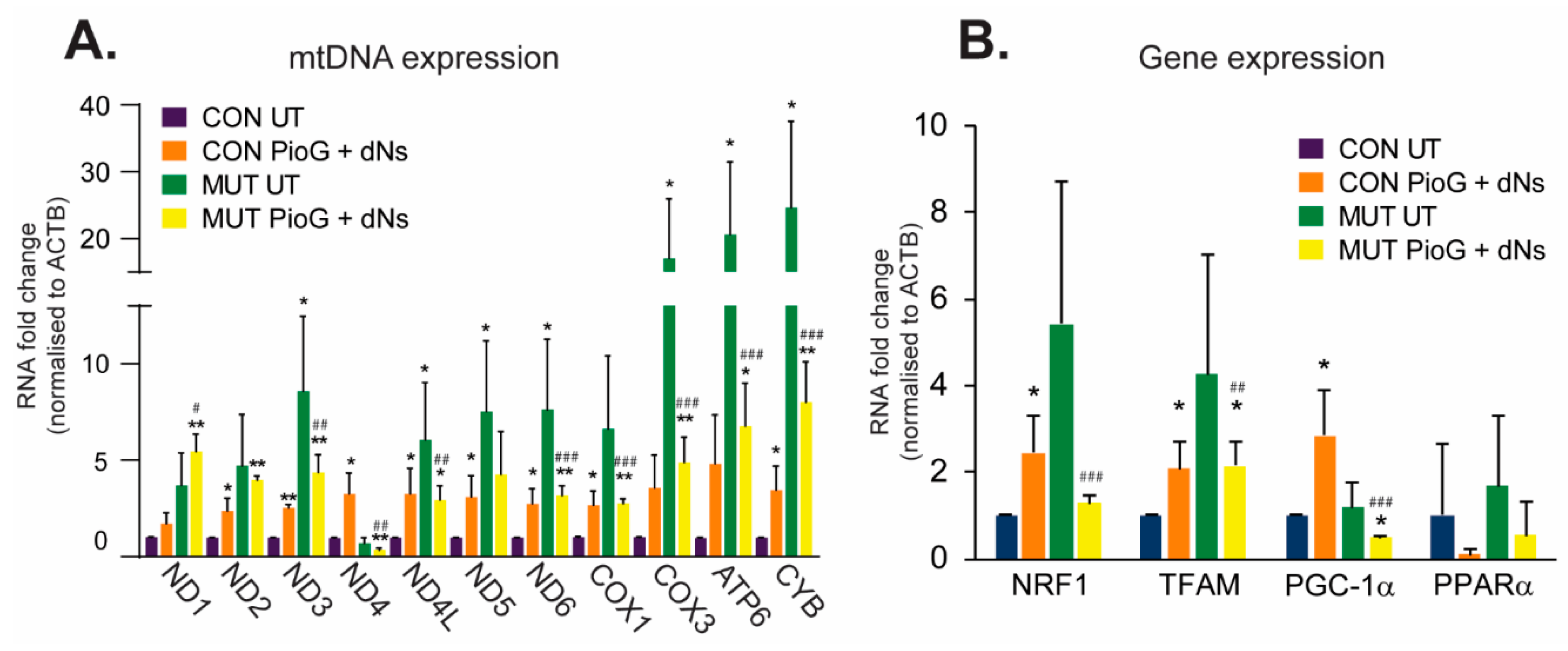
2.2. PioG + dNs Treatment Increases mtDNA Transcript Levels in Control Cybrids, but Not in MUT Cybrids
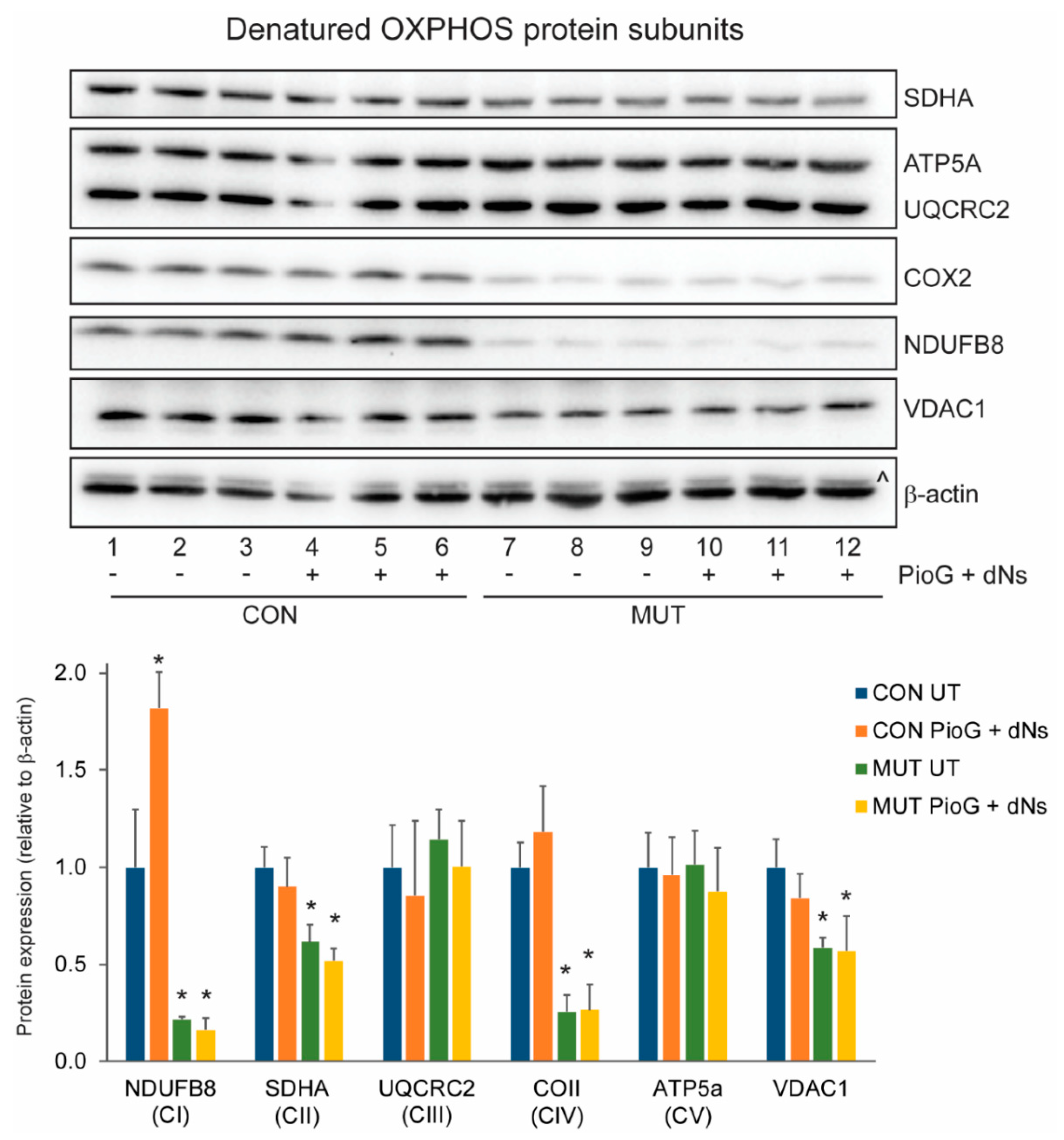
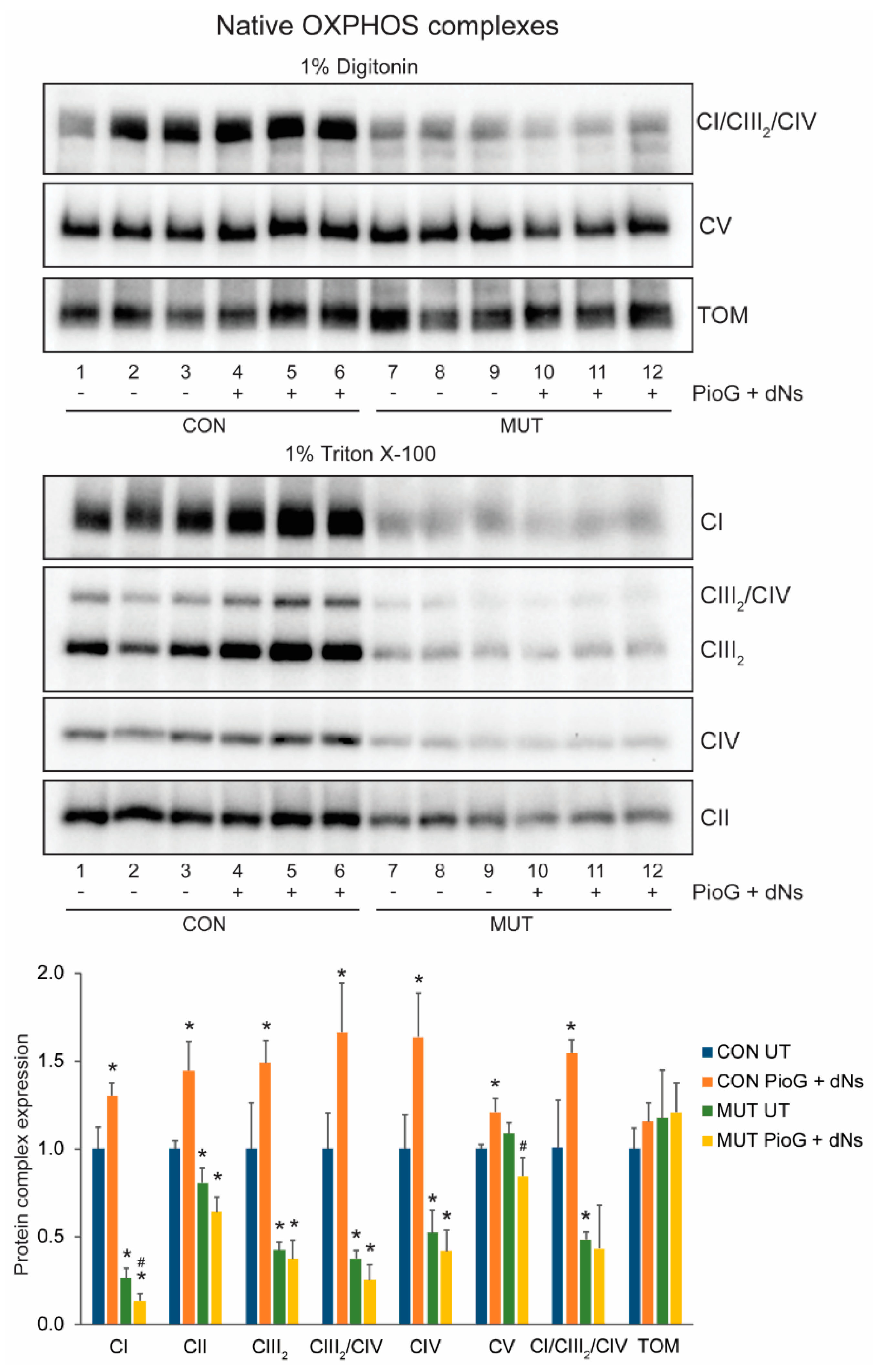
2.3. PioG + dNs Treatment Increased Oxidative Phosphorylation Protein Steady-State Levels in Control Cybrids, but Not in MUT Cybrids
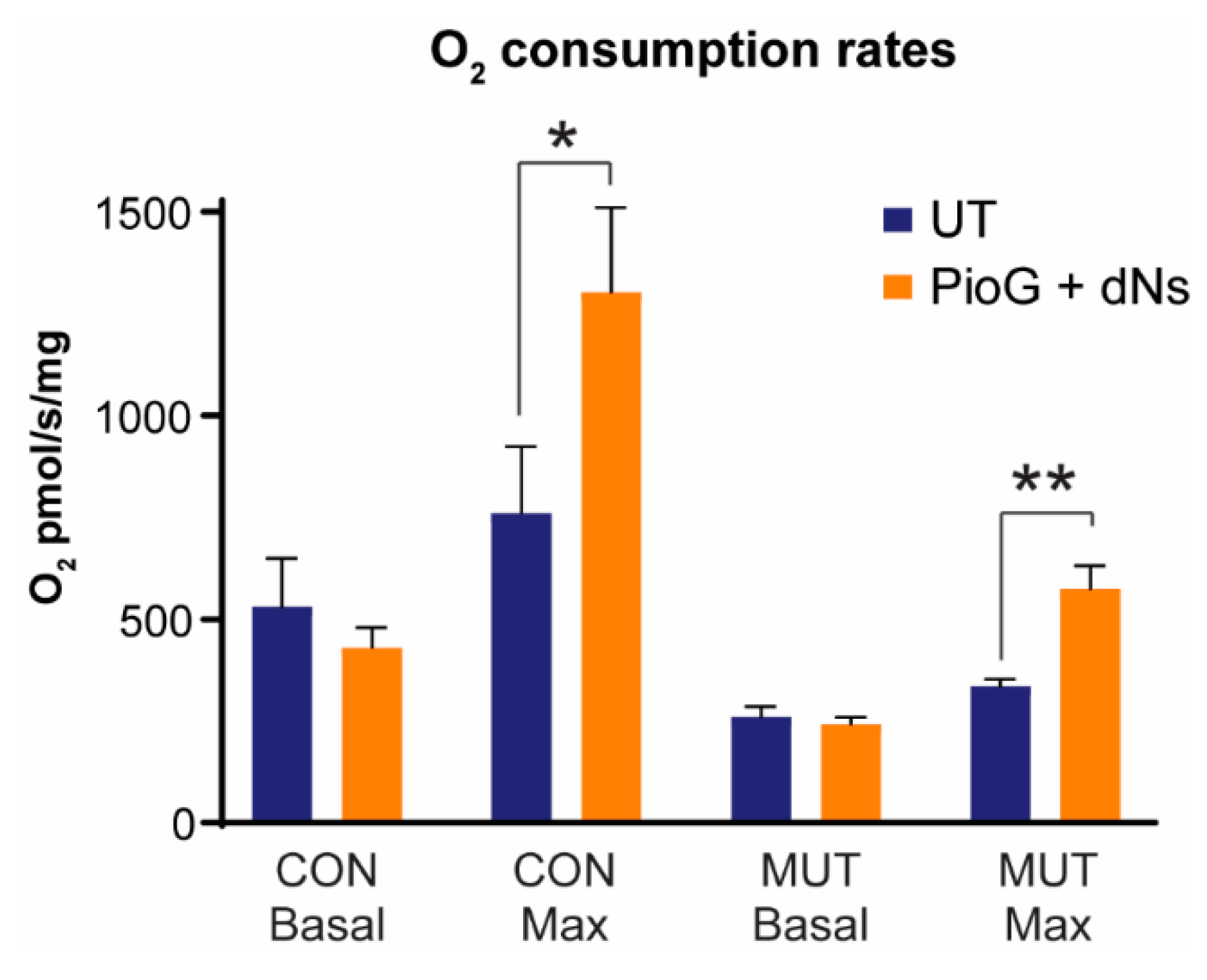
2.4. Treatment with PioG + dNs Increases Mitochondrial Respiratory Capacity in both Control Cybrids and MUT Cybrids
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Cell Lines and Culture Conditions
4.3. Mitochondrial DNA (mtDNA) Copy Number Quantitation
4.4. Quantification of m.3243A>G MT-TL1 Mutant Load
4.5. MtDNA Haplogroup Determination
4.6. Assessment of Mitochondrial Mass
4.7. Real-Time Quantitative RT-PCR Analysis
4.8. Denaturing Gel Electrophoresis
4.9. Native Gel Electrophoresis
4.10. Western Blotting
4.11. Measurement of Oxygen Consumption Rates
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Gorman, G.S.; Schaefer, A.M.; Ng, Y.; Gomez, N.; Blakely, E.L.; Alston, C.L.; Feeney, C.; Horvath, R.; Yu-Wai-Man, P.; Chinnery, P.F.; et al. Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Ann. Neurol. 2015, 77, 753–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parikh, S.; Goldstein, A.; Koenig, M.K.; Scaglia, F.; Enns, G.M.; Saneto, R.; Anselm, I.; Cohen, B.H.; Falk, M.J.; Greene, C.; et al. Diagnosis and management of mitochondrial disease: A consensus statement from the Mitochondrial Medicine Society. Genet. Med. 2015, 17, 689–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komaki, H.; Nishigaki, Y.; Fuku, N.; Hosoya, H.; Murayama, K.; Ohtake, A.; Goto, Y.; Wakamoto, H.; Koga, Y.; Tanaka, M. Pyruvate therapy for Leigh syndrome due to cytochrome c oxidase deficiency. Biochim. Biophys. Acta 2010, 1800, 313–315. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, G.; Majamaa, K.; Turnbull, D.M.; Thorburn, D.; Chinnery, P.F. Treatment for mitochondrial disorders. Cochrane Database Syst. Rev. 2012. [Google Scholar] [CrossRef] [PubMed]
- Weissig, V. Drug Development for the Therapy of Mitochondrial Diseases. Trends Mol. Med. 2019, 26, 40–57. [Google Scholar] [CrossRef]
- Janssen, M.C.H.; Koene, S.; de Laat, P.; Hemelaar, P.; Pickkers, P.; Spaans, E.; Beukema, R.; Beyrath, J.; Groothuis, J.; Verhaak, C.; et al. The KHENERGY Study: Safety and Efficacy of KH176 in Mitochondrial m.3243A>G Spectrum Disorders. Clin. Pharmacol. Ther. 2019, 105, 101–111. [Google Scholar] [CrossRef] [Green Version]
- Zesiewicz, T.; Salemi, J.L.; Perlman, S.; Sullivan, K.L.; Shaw, J.D.; Huang, Y.; Isaacs, C.; Gooch, C.; Lynch, D.R.; Klein, M.B. Double-blind, randomized and controlled trial of EPI-743 in Friedreich’s ataxia. Neurodegener. Dis. Manag. 2018, 8, 233–242. [Google Scholar] [CrossRef]
- Lyseng-Williamson, K.A. Idebenone: A Review in Leber’s Hereditary Optic Neuropathy. Drugs 2016, 76, 805–813. [Google Scholar] [CrossRef]
- Olsen, R.K.J.; Olpin, S.E.; Andresen, B.S.; Miedzybrodzka, Z.H.; Pourfarzam, M.; Merinero, B.; Frerman, F.E.; Beresford, M.W.; Dean, J.C.S.; Cornelius, N.; et al. ETFDH mutations as a major cause of riboflavin-responsive multiple acyl-CoA dehydrogenation deficiency. Brain 2007, 130, 2045–2054. [Google Scholar] [CrossRef]
- Barshop, B.A.; Naviaux, R.K.; McGowan, K.A.; Levine, F.; Nyhan, W.L.; Loupis-Geller, A.; Haas, R.H. Chronic treatment of mitochondrial disease patients with dichloroacetate. Mol. Genet. Metab. 2004, 83, 138–149. [Google Scholar] [CrossRef]
- Fujii, T.; Nozaki, F.; Saito, K.; Hayashi, A.; Nishigaki, Y.; Murayama, K.; Tanaka, M.; Koga, Y.; Hiejima, I.; Kumada, T. Efficacy of pyruvate therapy in patients with mitochondrial disease: A semi-quantitative clinical evaluation study. Mol. Genet. Metab. 2014, 112, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Murayama, K.; Shimura, M.; Liu, Z.; Okazaki, Y.; Ohtake, A. Recent topics: The diagnosis, molecular genesis, and treatment of mitochondrial diseases. J. Hum. Genet. 2019, 64, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Szeto, H.H. First-in-class cardiolipin-protective compound as a therapeutic agent to restore mitochondrial bioenergetics. Br. J. Pharmacol. 2014, 171, 2029–2050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cameron, R.B.; Beeson, C.C.; Schnellmann, R.G. Development of Therapeutics That Induce Mitochondrial Biogenesis for the Treatment of Acute and Chronic Degenerative Diseases. J. Med. Chem. 2016, 59, 10411–10434. [Google Scholar] [CrossRef] [Green Version]
- Scarpulla, R.C. Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochimica et Biophysica Acta (BBA)-Mol. Cell Res. 2011, 1813, 1269–1278. [Google Scholar] [CrossRef] [Green Version]
- Jornayvaz, F.R.; Shulman, G.I. Regulation of mitochondrial biogenesis. Essays Biochem. 2010, 47, 69–84. [Google Scholar]
- Palikaras, K.; Tavernarakis, N. Mitochondrial homeostasis: The interplay between mitophagy and mitochondrial biogenesis. Exp. Gerontol. 2014, 56, 182–188. [Google Scholar] [CrossRef]
- Gillies, P.S.; Dunn, C.J. Pioglitazone. Drugs 2000, 60, 333–343. [Google Scholar] [CrossRef]
- Bogacka, I.; Xie, H.; Bray, G.A.; Smith, S.R. Pioglitazone induces mitochondrial biogenesis in human subcutaneous adipose tissue In Vivo. Diabetes 2005, 54, 1392–1399. [Google Scholar] [CrossRef] [Green Version]
- Bolten, C.W.; Blanner, P.M.; McDonald, W.G.; Staten, N.R.; Mazzarella, R.A.; Arhancet, G.B.; Meier, M.F.; Weiss, D.J.; Sullivan, P.M.; Hromockyj, A.E.; et al. Insulin sensitizing pharmacology of thiazolidinediones correlates with mitochondrial gene expression rather than activation of PPAR gamma. Gene Regul. Syst. Biol. 2007, 1, 73–82. [Google Scholar]
- Medeiros, D.M. Assessing mitochondria biogenesis. Methods 2008, 46, 288–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cámara, Y.; González-Vioque, E.; Scarpelli, M.; Torres-Torronteras, J.; Caballero, A.; Hirano, M.; Martí, R. Administration of deoxyribonucleosides or inhibition of their catabolism as a pharmacological approach for mitochondrial DNA depletion syndrome. Hum. Mol. Genet. 2013, 23, 2459–2467. [Google Scholar] [CrossRef] [PubMed]
- El-Hattab, A.W.; Zarante, A.M.; Almannai, M.; Scaglia, F. Therapies for mitochondrial diseases and current clinical trials. Mol. Genet. Metab. 2017, 122, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, A.M.; McFarland, R.; Blakely, E.L.; He, L.; Whittaker, R.G.; Taylor, R.W.; Chinnery, P.F.; Turnbull, D.M. Prevalence of mitochondrial DNA disease in adults. Ann. Neurol. 2008, 63, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Manwaring, N.; Jones, M.M.; Wang, J.J.; Rochtchina, E.; Howard, C.; Mitchell, P.; Sue, C.M. Population prevalence of the MELAS A3243G mutation. Mitochondrion 2007, 7, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Goto, Y.; Horai, S.; Matsuoka, T.; Koga, Y.; Nihei, K.; Kobayashi, M.; Nonaka, I. Mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes (MELAS): A correlative study of the clinical features and mitochondrial DNA mutation. Neurology 1992, 42, 545–550. [Google Scholar] [CrossRef]
- Ciafaloni, E.; Ricci, E.; Shanske, S.; Moraes, C.T.; Silvestri, G.; Hirano, M.; Simonetti, S.; Angelini, C.; Donati, M.A.; Garcia, C.; et al. MELAS: Clinical features, biochemistry, and molecular genetics. Ann. Neurol. 1992, 31, 391–398. [Google Scholar] [CrossRef]
- Koga, Y.; Akita, Y.; Takane, N.; Sato, Y.; Kato, H. Heterogeneous presentation in A3243G mutation in the mitochondrial tRNA(Leu(UUR)) gene. Arch. Dis. Child. 2000, 82, 407–411. [Google Scholar] [CrossRef]
- Ghosh, S.; Patel, N.; Rahn, D.; McAllister, J.; Sadeghi, S.; Horwitz, G.; Berry, D.; Wang, K.X.; Swerdlow, R.H. The thiazolidinedione pioglitazone alters mitochondrial function in human neuron-like cells. Mol. Pharmacol. 2007, 71, 1695–1702. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Vioque, E.; Torres-Torronteras, J.; Andreu, A.L.; Marti, R. Limited dCTP availability accounts for mitochondrial DNA depletion in mitochondrial neurogastrointestinal encephalomyopathy (MNGIE). PLoS Genet. 2011, 7, e1002035. [Google Scholar] [CrossRef]
- Fornuskova, D.; Brantova, O.; Tesarova, M.; Stiburek, L.; Honzik, T.; Wenchich, L.; Tietzeova, E.; Hansikova, H.; Zeman, J. The impact of mitochondrial tRNA mutations on the amount of ATP synthase differs in the brain compared to other tissues. Biochimica et Biophysica Acta (BBA)-Mol. Basis Dis. 2008, 1782, 317–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chomyn, A.; Martinuzzi, A.; Yoneda, M.; Daga, A.; Hurko, O.; Johns, D.; Lai, S.T.; Nonaka, I.; Angelini, C.; Attardi, G. MELAS mutation in mtDNA binding site for transcription termination factor causes defects in protein synthesis and in respiration but no change in levels of upstream and downstream mature transcripts. Proc. Natl. Acad. Sci. USA 1992, 89, 4221–4225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koopman, W.J.; Beyrath, J.; Fung, C.-W.; Koene, S.; Rodenburg, R.J.; Willems, P.H.; Smeitink, J.A. Mitochondrial disorders in children: Toward development of small-molecule treatment strategies. EMBO Mol. Med. 2016, 8, 311–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picard, M.; Zhang, J.; Hancock, S.; Derbeneva, O.; Golhar, R.; Golik, P.; O’Hearn, S.; Levy, S.; Potluri, P.; Lvova, M.; et al. Progressive increase in mtDNA 3243A > G heteroplasmy causes abrupt transcriptional reprogramming. Proc. Natl. Acad. Sci. USA 2014, 111, E4033–E4042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garone, C.; Viscomi, C. Towards a therapy for mitochondrial disease: An update. Biochem. Soc. Trans. 2018, 46, 1247–1261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, S.W.; Haydar, G.; Taniane, C.; Farrell, G.; Arias, I.M.; Lippincott-Schwartz, J.; Fu, D. AMPK Activation Prevents and Reverses Drug-Induced Mitochondrial and Hepatocyte Injury by Promoting Mitochondrial Fusion and Function. PLoS ONE 2016, 11, e0165638. [Google Scholar] [CrossRef]
- Morishita, M.; Kawamoto, T.; Hara, H.; Onishi, Y.; Ueha, T.; Minoda, M.; Katayama, E.; Takemori, T.; Fukase, N.; Kurosaka, M.; et al. AICAR induces mitochondrial apoptosis in human osteosarcoma cells through an AMPK-dependent pathway. Int. J. Oncol. 2017, 50, 23–30. [Google Scholar] [CrossRef] [Green Version]
- Musi, N.; Goodyear, L.J. Targetting the AMP-Activated Protein Kinase for the Treatment of Type 2 Diabetes. Curr. Drug Targets Immune Endocr. Metab. Disord. 2002, 2, 119–127. [Google Scholar] [CrossRef]
- Goodyear, L.J. The Exercise Pill- Too Good to Be True? N. Engl. J. Med. 2008, 359, 1842–1844. [Google Scholar] [CrossRef]
- Yamaguchi, S.; Li, H.; Purevsuren, J.; Yamada, K.; Furui, M.; Takahashi, T.; Mushimoto, Y.; Kobayashi, H.; Hasegawa, Y.; Taketani, T.; et al. Bezafibrate can be a new treatment option for mitochondrial fatty acid oxidation disorders: Evaluation by in vitro probe acylcarnitine assay. Mol. Genet. Metab. 2012, 107, 87–91. [Google Scholar] [CrossRef]
- Orngreen, M.C.; Madsen, K.L.; Preisler, N.; Andersen, G.; Vissing, J.; Laforet, P. Bezafibrate in skeletal muscle fatty acid oxidation disorders: A randomized clinical trial. Neurology 2014, 82, 607–613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanchet, J.; Longpre, F.; Bureau, G.; Morissette, M.; DiPaolo, T.; Bronchti, G.; Martinoli, M.G. Resveratrol, a red wine polyphenol, protects dopaminergic neurons in MPTP-treated mice. Prog. Neuropsychopharmacol. Biol. Psychiatry 2008, 32, 1243–1250. [Google Scholar] [CrossRef] [PubMed]
- Csiszar, A.; Labinskyy, N.; Pinto, J.T.; Ballabh, P.; Zhang, H.; Losonczy, G.; Pearson, K.; de Cabo, R.; Pacher, P.; Zhang, C.; et al. Resveratrol induces mitochondrial biogenesis in endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H13–H20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, K.; Tao, Y.; Zhang, J.; Wang, J.; Ye, F.; Dan, G.; Zhao, Y.; Cai, Y.; Zhao, J.; Wu, Q.; et al. Resveratrol Regulates Mitochondrial Biogenesis and Fission/Fusion to Attenuate Rotenone-Induced Neurotoxicity. Oxid. Med. Cell. Longev. 2016, 2016, 6705621. [Google Scholar] [CrossRef] [Green Version]
- Patel, K.R.; Scott, E.; Brown, V.A.; Gescher, A.J.; Steward, W.P.; Brown, K. Clinical trials of resveratrol. Ann. N. Y. Acad. Sci. 2011, 1215, 161–169. [Google Scholar] [CrossRef]
- McMillan, R.P.; Stewart, S.; Budnick, J.A.; Caswell, C.C.; Hulver, M.W.; Mukherjee, K.; Srivastava, S. Quantitative Variation in m.3243A > G Mutation Produce Discrete Changes in Energy Metabolism. Sci. Rep. 2019, 9, 5752. [Google Scholar] [CrossRef] [Green Version]
- Hämäläinen, R.H.; Manninen, T.; Koivumäki, H.; Kislin, M.; Otonkoski, T.; Suomalainen, A. Tissue- and cell-type–specific manifestations of heteroplasmic mtDNA 3243A>G mutation in human induced pluripotent stem cell-derived disease model. Proc. Natl. Acad. Sci. USA 2013, 110, E3622–E3630. [Google Scholar] [CrossRef] [Green Version]
- Sasarman, F.; Antonicka, H.; Shoubridge, E.A. The A3243G tRNALeu(UUR) MELAS mutation causes amino acid misincorporation and a combined respiratory chain assembly defect partially suppressed by overexpression of EFTu and EFG2. Hum. Mol. Genet. 2008, 17, 3697–3707. [Google Scholar] [CrossRef] [Green Version]
- Guarás, A.; Perales-Clemente, E.; Calvo, E.; Acín-Pérez, R.; Loureiro-Lopez, M.; Pujol, C.; Martínez-Carrascoso, I.; Nuñez, E.; García-Marqués, F.; Rodríguez-Hernández, M.A.; et al. The CoQH2/CoQ Ratio Serves as a Sensor of Respiratory Chain Efficiency. Cell Rep. 2016, 15, 197–209. [Google Scholar] [CrossRef] [Green Version]
- Brown, M.D.; Trounce, I.A.; Jun, A.S.; Allen, J.C.; Wallace, D.C. Functional analysis of lymphoblast and cybrid mitochondria containing the 3460, 11778, or 14484 Leber’s hereditary optic neuropathy mitochondrial DNA mutation. J. Biol. Chem. 2000, 275, 39831–39836. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.; Kelly, R.D.; Yeung, K.Y.; Cagnone, G.; McKenzie, M.; John, J.C. Analysis of Mitochondrial DNA in Induced Pluripotent and Embryonic Stem Cells. Methods Mol. Biol. 2015, 1330, 219–252. [Google Scholar]
- Lott, M.T.; Leipzig, J.N.; Derbeneva, O.; Xie, H.M.; Chalkia, D.; Sarmady, M.; Procaccio, V.; Wallace, D.C. mtDNA Variation and Analysis Using Mitomap and Mitomaster. Curr. Protoc. Bioinform. 2013, 44, 1.23.1–1.23.26. [Google Scholar] [CrossRef] [Green Version]
- McKenzie, M.; Lim, S.C.; Duchen, M.R. Simultaneous Measurement of Mitochondrial Calcium and Mitochondrial Membrane Potential in Live Cells by Fluorescent Microscopy. J. Vis. Exp. JoVE 2017, e55166. [Google Scholar] [CrossRef] [Green Version]
- Kelly, R.D.; Rodda, A.E.; Dickinson, A.; Mahmud, A.; Nefzger, C.M.; Lee, W.; Forsythe, J.S.; Polo, J.M.; Trounce, I.A.; McKenzie, M.; et al. Mitochondrial DNA haplotypes define gene expression patterns in pluripotent and differentiating embryonic stem cells. Stem Cells 2013, 31, 703–716. [Google Scholar] [CrossRef]
- McKenzie, M.; Lazarou, M.; Ryan, M.T. Chapter 18 Analysis of respiratory chain complex assembly with radiolabeled nuclear- and mitochondrial-encoded subunits. Methods Enzymol. 2009, 456, 321–339. [Google Scholar]
- McKenzie, M.; Lazarou, M.; Thorburn, D.R.; Ryan, M.T. Analysis of mitochondrial subunit assembly into respiratory chain complexes using Blue Native polyacrylamide gel electrophoresis. Anal. Biochem. 2007, 364, 128–137. [Google Scholar] [CrossRef]
- Johnston, A.J.; Hoogenraad, J.; Dougan, D.A.; Truscott, K.N.; Yano, M.; Mori, M.; Hoogenraad, N.J.; Ryan, M.T. Insertion and assembly of human tom7 into the preprotein translocase complex of the outer mitochondrial membrane. J. Biol. Chem. 2002, 277, 42197–42204. [Google Scholar] [CrossRef] [Green Version]
- Brand, M.D.; Nicholls, D.G. Assessing mitochondrial dysfunction in cells. Biochem. J. 2011, 435, 297–312. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Basal pmol O2/s/mg | Maximal pmol O2/s/mg | Proton Leak pmol O2/s/mg | Spare Respiratory Capacity | Cell Respiratory Control Ratio | |
|---|---|---|---|---|---|
| CON UT | 530 ± 120 | 761 ± 163 | 59 ± 3 | 231 ± 250 | 13 ± 3 |
| CON PioG + dNs | 431 ± 49 | 1303 ± 207 * | 65 ± 10 | 872 ± 200 * | 20 ± 3 * |
| MUT UT | 260 ± 26 * | 335 ± 18 * | 41 ± 11 * | 75 ± 13 | 8 ± 0.4 * |
| MUT PioG + dNs | 242 ± 17 | 575 ± 57 ### | 41 ± 5 | 332 ± 45 ### | 14 ± 1 ### |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burgin, H.J.; Lopez Sanchez, M.I.G.; Smith, C.M.; Trounce, I.A.; McKenzie, M. Pioglitazone and Deoxyribonucleoside Combination Treatment Increases Mitochondrial Respiratory Capacity in m.3243A>G MELAS Cybrid Cells. Int. J. Mol. Sci. 2020, 21, 2139. https://doi.org/10.3390/ijms21062139
Burgin HJ, Lopez Sanchez MIG, Smith CM, Trounce IA, McKenzie M. Pioglitazone and Deoxyribonucleoside Combination Treatment Increases Mitochondrial Respiratory Capacity in m.3243A>G MELAS Cybrid Cells. International Journal of Molecular Sciences. 2020; 21(6):2139. https://doi.org/10.3390/ijms21062139
Chicago/Turabian StyleBurgin, Harrison J., M. Isabel G. Lopez Sanchez, Craig M. Smith, Ian A. Trounce, and Matthew McKenzie. 2020. "Pioglitazone and Deoxyribonucleoside Combination Treatment Increases Mitochondrial Respiratory Capacity in m.3243A>G MELAS Cybrid Cells" International Journal of Molecular Sciences 21, no. 6: 2139. https://doi.org/10.3390/ijms21062139