The Mechanisms Underlying the Cytotoxic Effects of Copper Via Differentiated Embryonic Chondrocyte Gene 1


Abstract
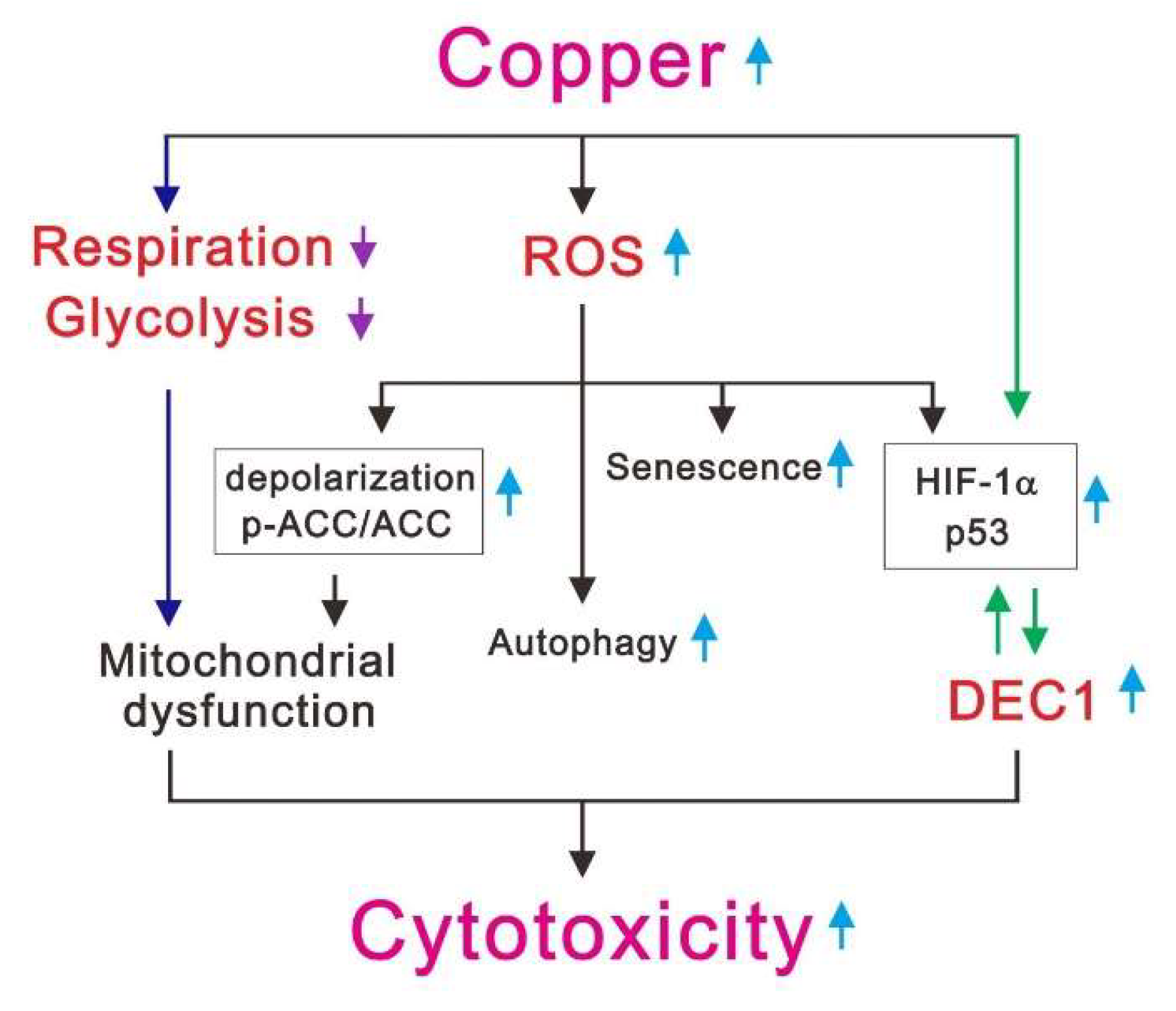
:1. Introduction
2. Results
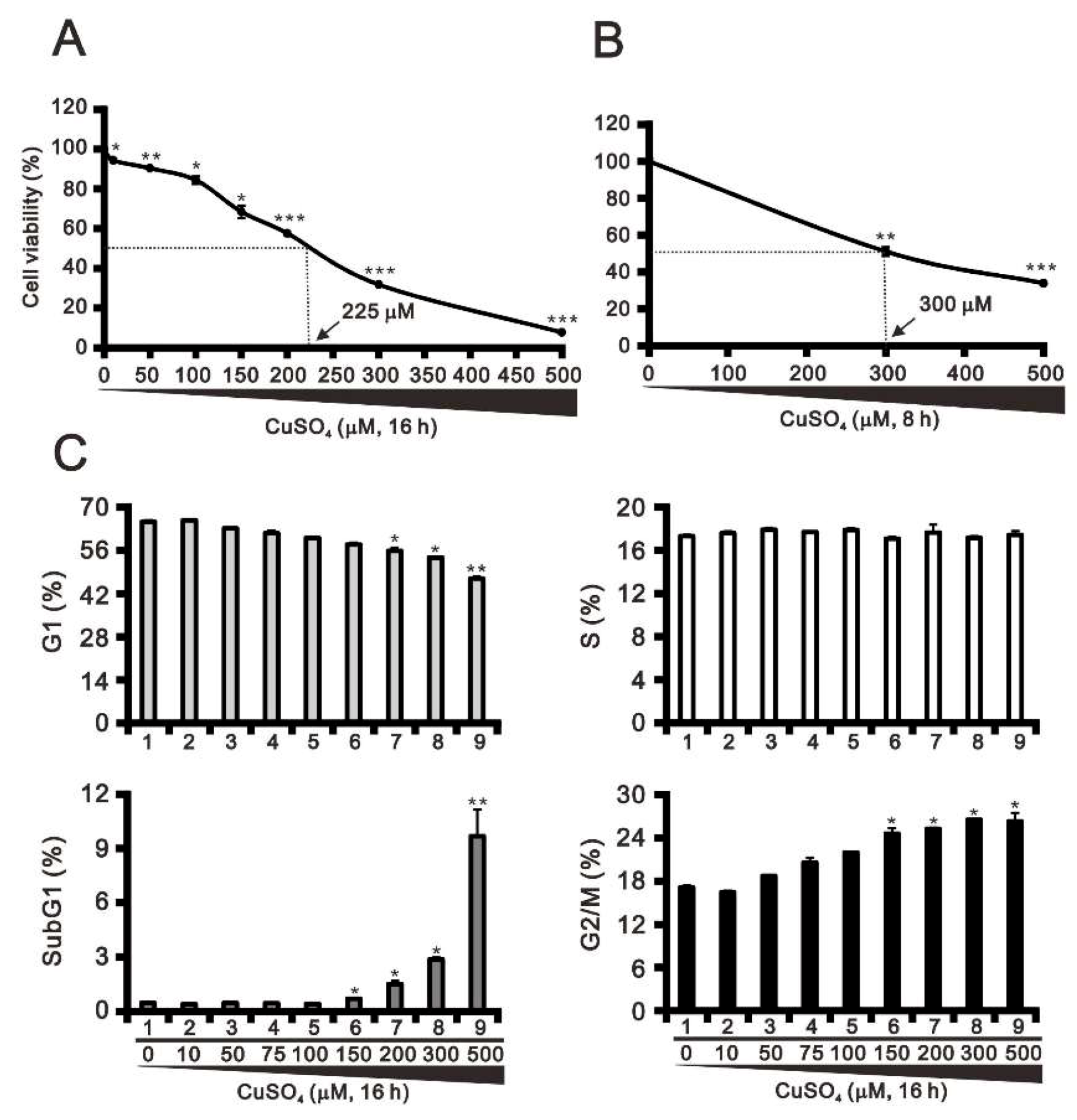
2.1. The Cytotoxic Effects of Copper Sulfate in HeLa Cells
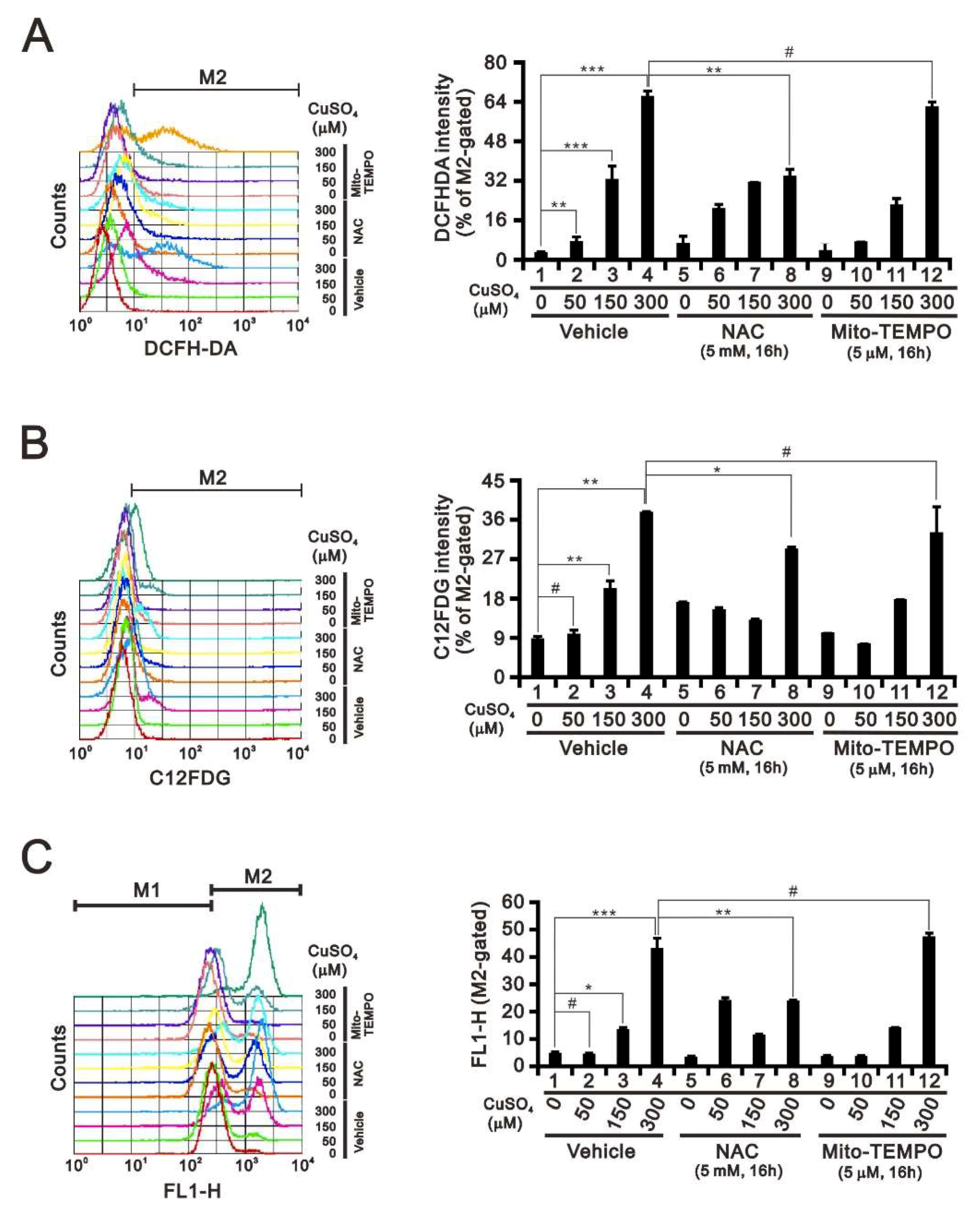
2.2. Effects of Copper Sulfate on ROS, Senescence, and Mitochondrial Membrane Potential in HeLa Cells
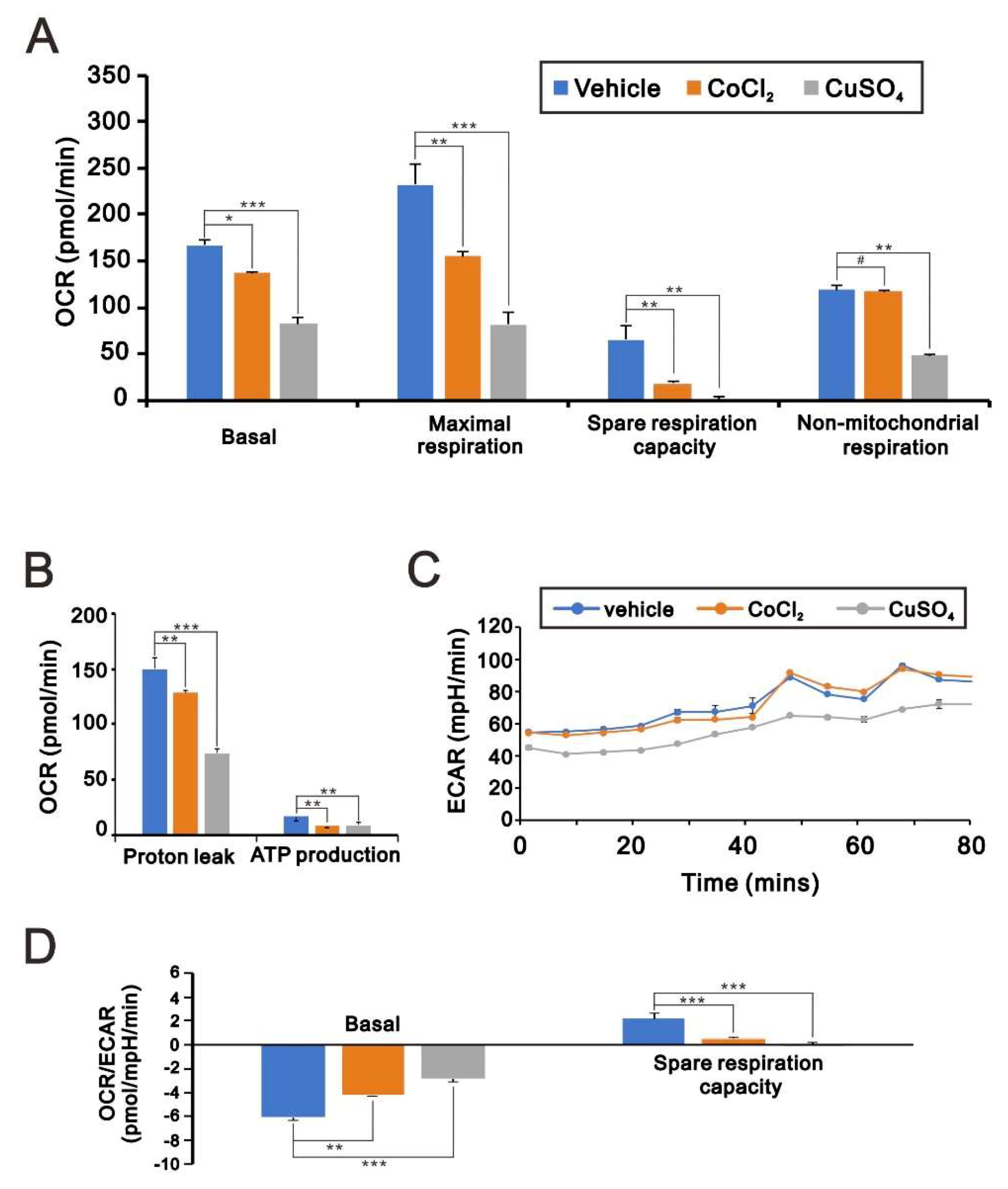
2.3. Effects of Copper Sulfate on Mitochondrial Bioenergetics in HeLa Cells
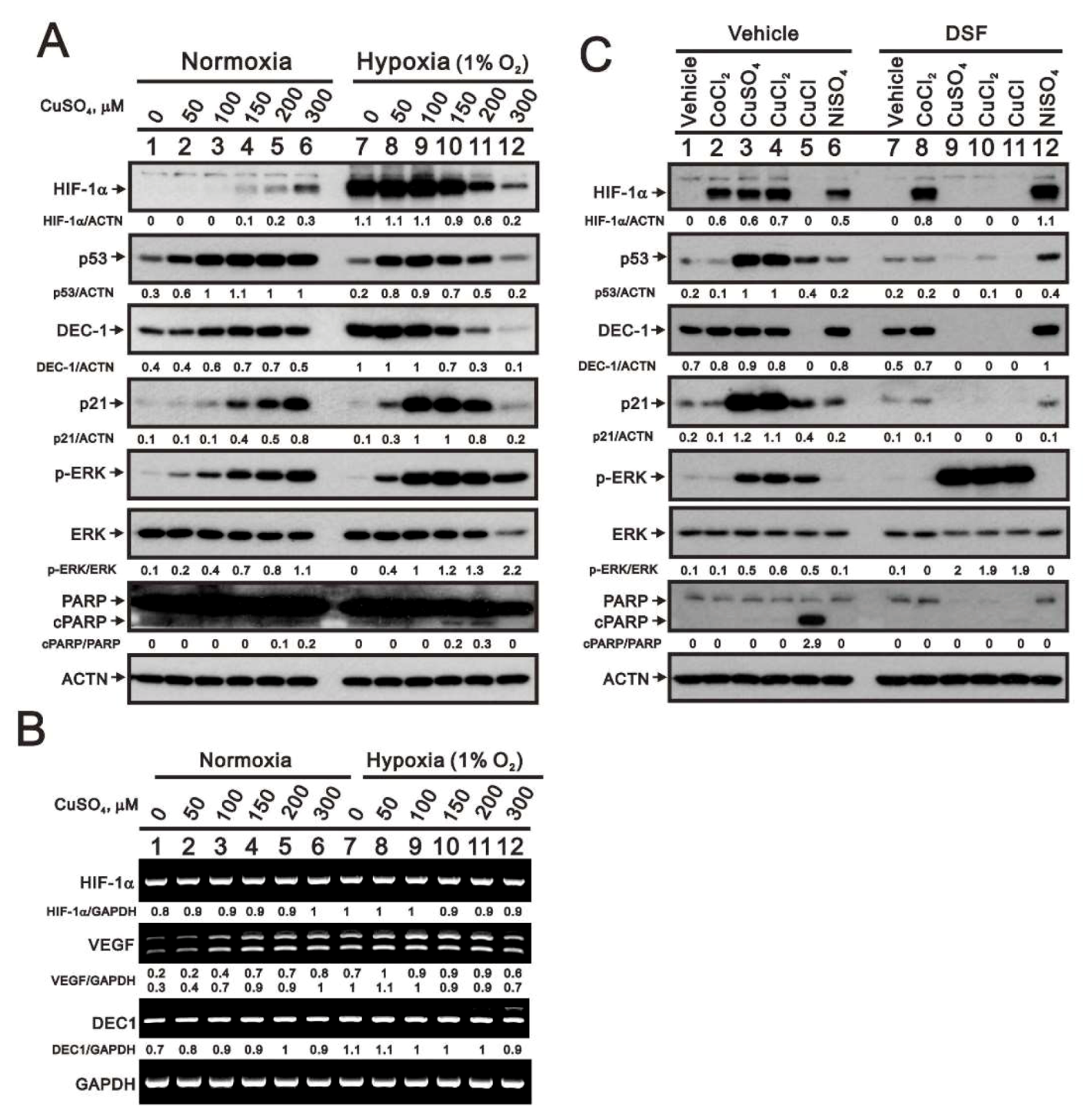
2.4. The Effects of Copper-Containing Compounds on the Stability of HIF-1α Protein in HeLa Cells
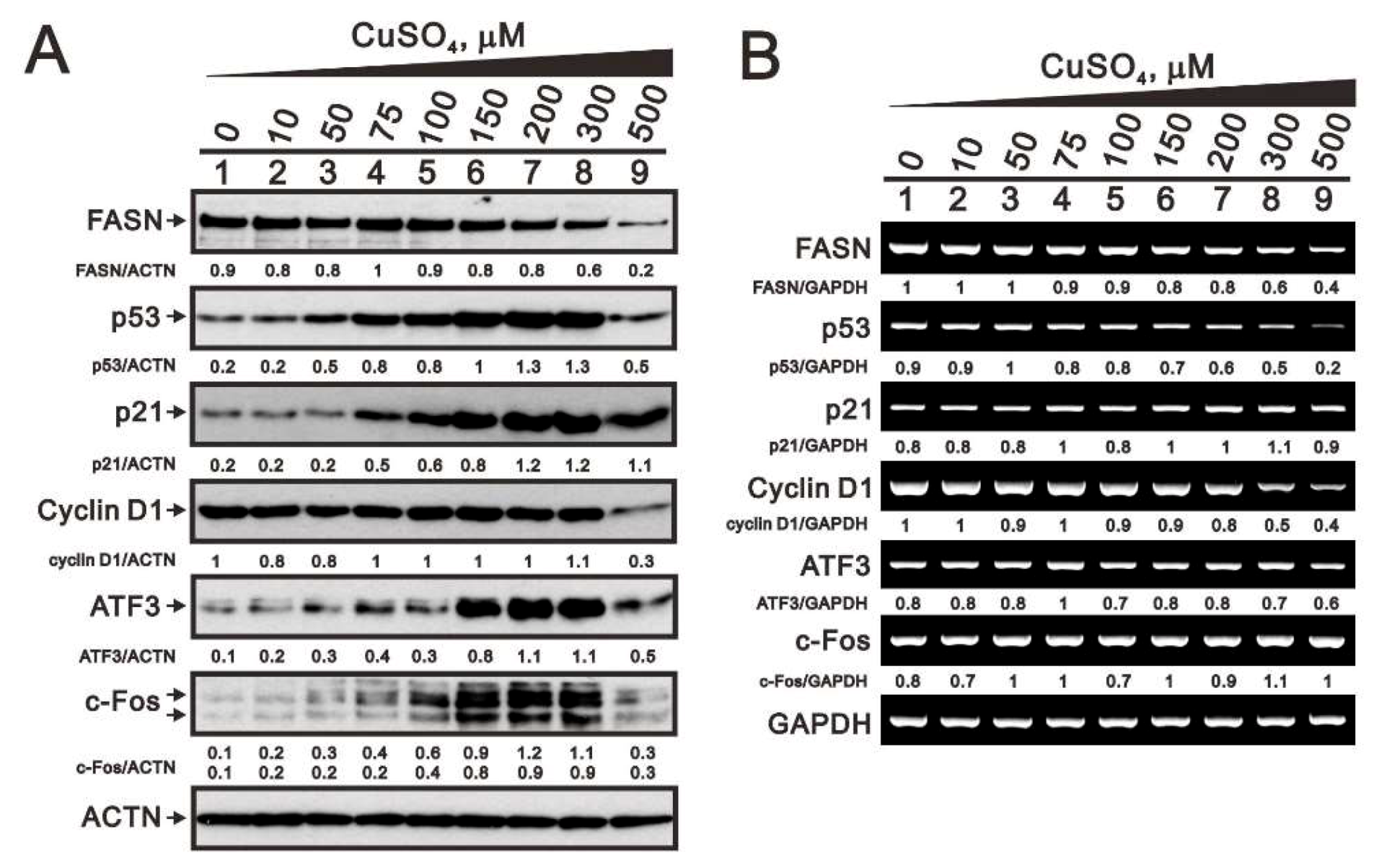
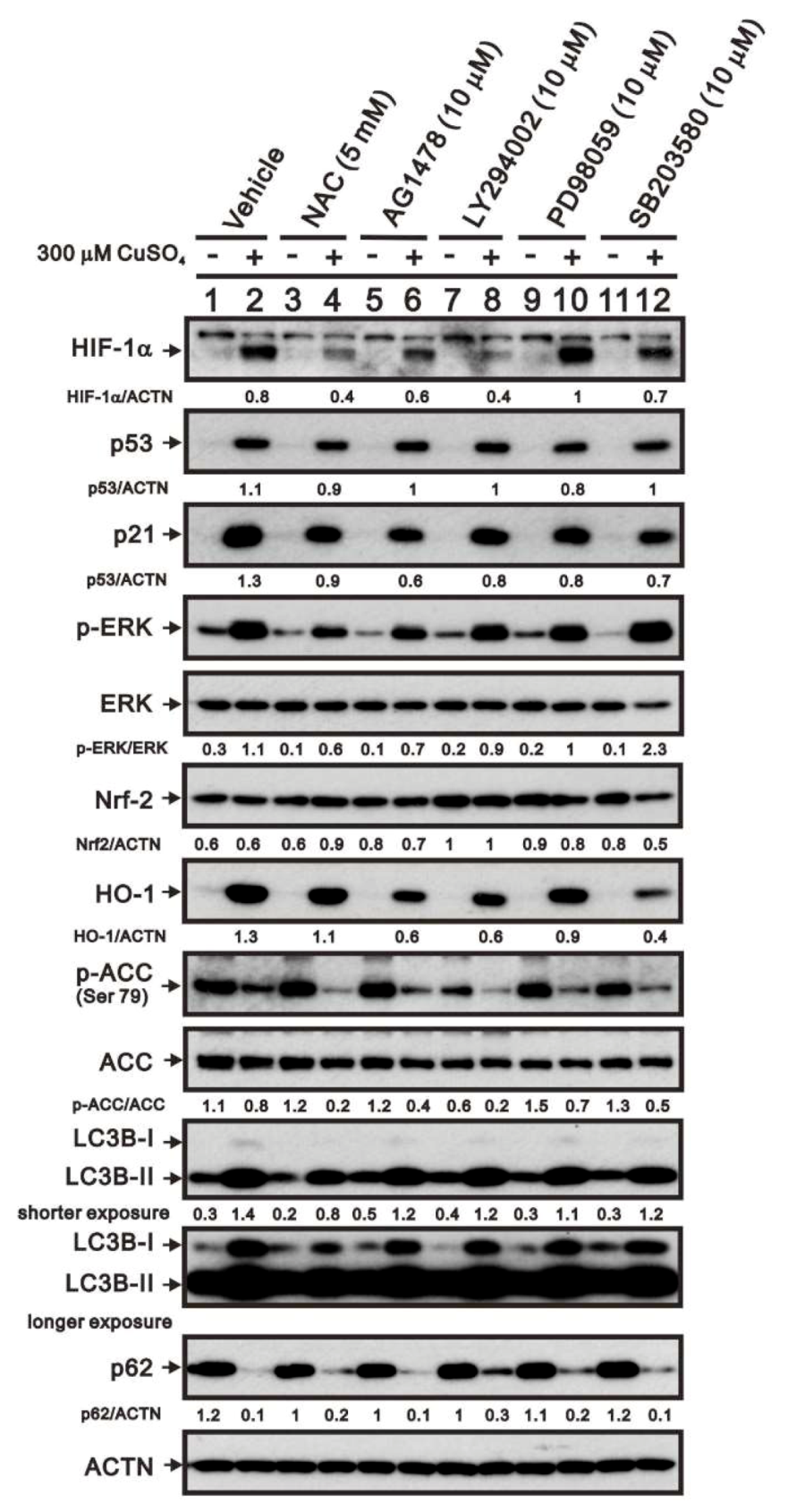
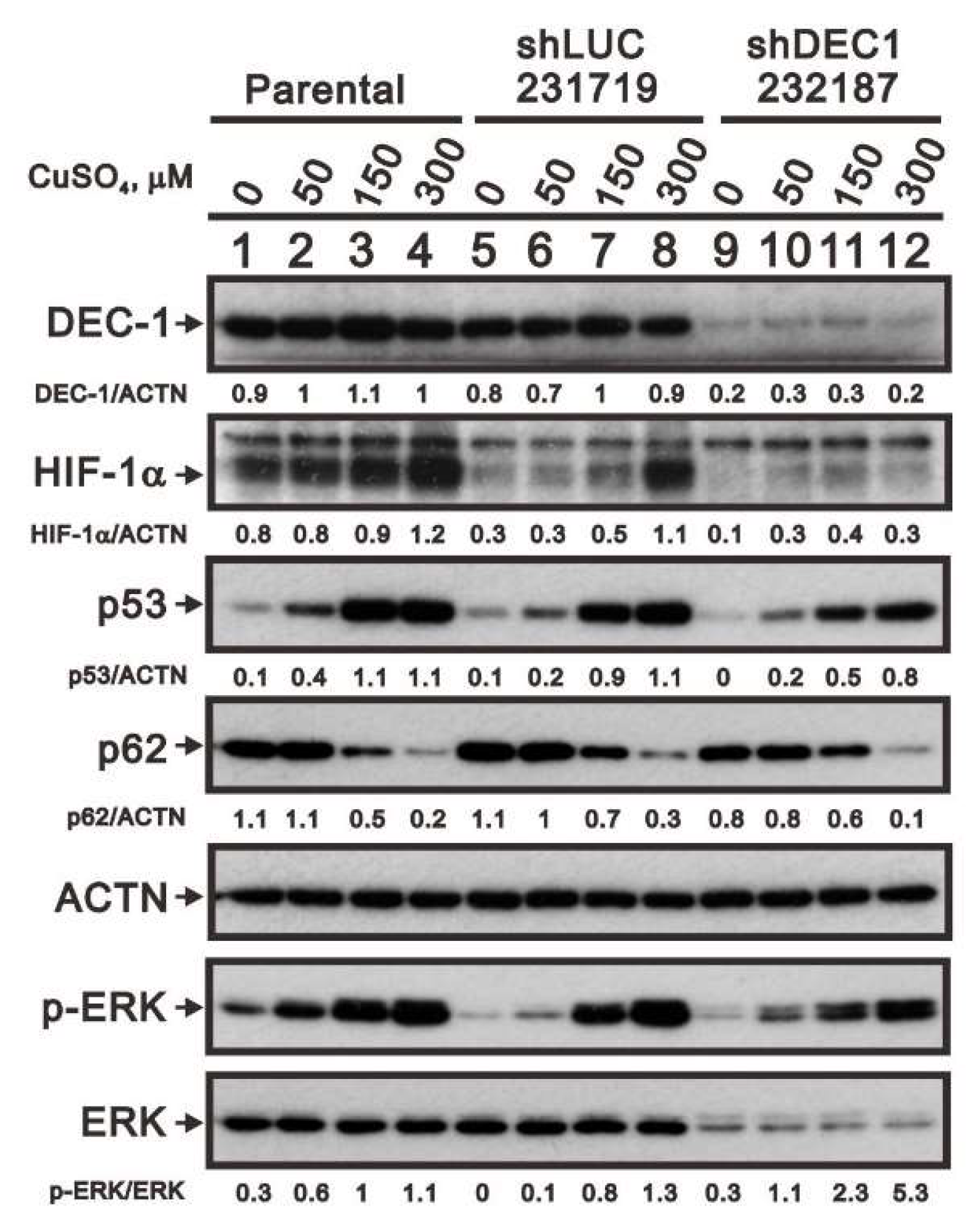
2.5. Mechanism of Copper Sulfate-Induced Protein Expression in HeLa Cells
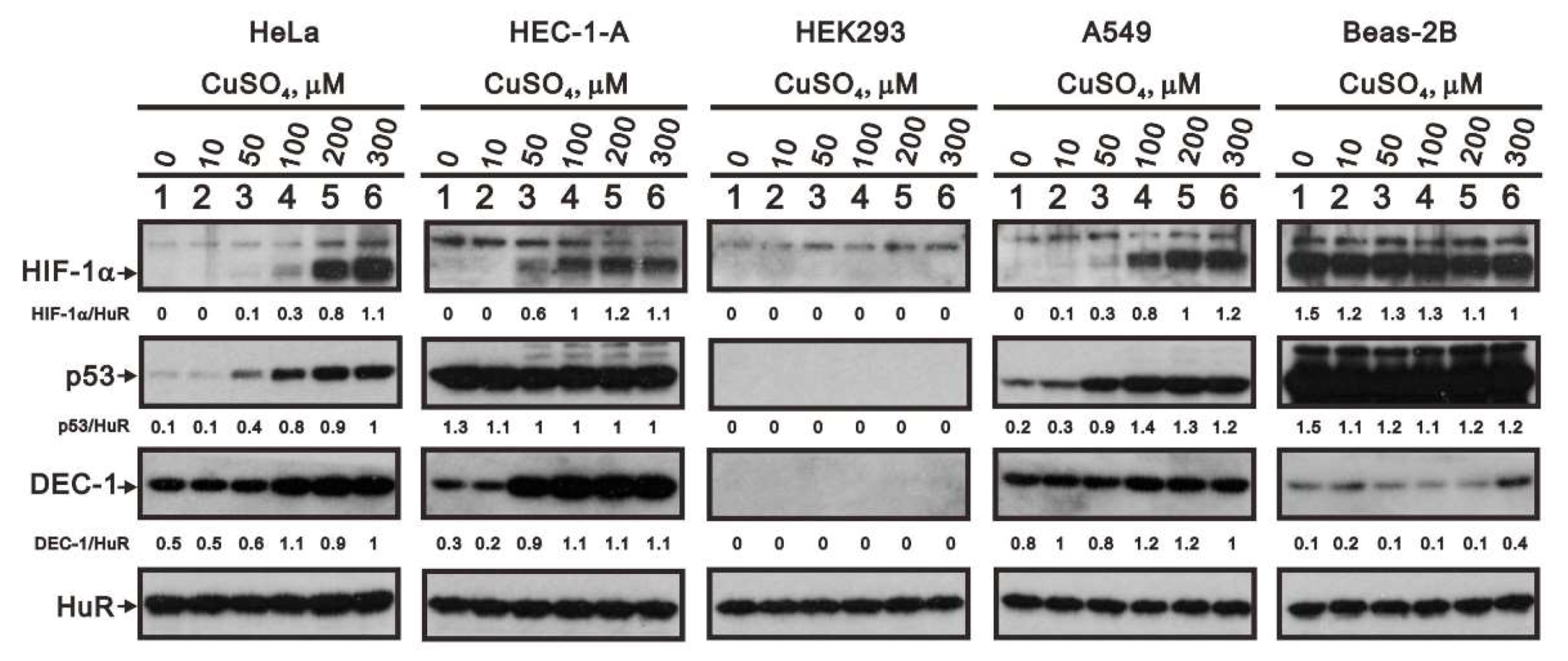
2.6. The Cell Type-Dependent Effects of Copper Sulfate on the Stability of HIF-1α
3. Discussion
4. Methods and Materials
4.1. Cell Culture and Reagents
4.2. Cell Survival Analysis
4.3. Fluorescence-Activated Cell Sorting (FACS), Cell Cycle Profiles, ROS, Senescence, and Mitochondrial Membrane Potential Analyses
4.4. Western Blotting
4.5. Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
4.6. Detection of the Oxygen Consumption Rate (OCR) and Extracellular Acidification Rate (ECAR)
4.7. DEC-1 mRNA Interference
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ACC | AMP-activated protein kinase |
| ACTN | α-actinin |
| AMPK | AMP-activated protein kinase |
| C12FDG | 5-dodecanoylaminofluorescein di-β-D-galactopyranoside |
| DCFH-DA | 2′,7-dichlorofluorescein diacetate |
| DEC1 | Differentiated embryonic chondrocyte gene 1 |
| DMEM | Dulbecco’s modified Eagle’s medium |
| DSF | Disulfiram |
| FACS | Fluorescence-activated cell sorting |
| FASN | Fatty acid synthase |
| FBS | Fetal bovine serum |
| FCCP | Carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone |
| H2O2 | Hydrogen peroxide |
| HIF-1α | Hypoxia-inducible factor 1 alpha |
| HO-1 | Heme oxygenase 1 |
| JC-1 | 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimi-dazolyl carbo-cyanine iodide |
| LC3B | Microtubule-associated protein 1 light chain 3B |
| MTT | Thiazolyl blue tetrazlium bromide |
| NAC | N-acetyl cysteine |
| OCR | Oxygen consumption rate |
| cPARP | Cleaved Poly-ADP-ribose polymerase |
| PBS | Phosphate buffered saline |
| pVHL | Von Hippel-Lindau tumor suppressor protein |
| PI | Propidium iodide |
| ROS | Reactive oxygen species |
| RPMI | Roswell Park Memorial Institute |
| RT-PCR | Reverse transcription-polymerase chain reaction |
| SOD | Superoxide dismutase |
| VEGF | Vascular endothelial growth factor |
References
- Harris, Z.L.; Gitlin, J.D. Genetic and molecular basis for copper toxicity. Am. J. Clin. Nutr. 1996, 63, 836S–841S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Festa, R.A.; Thiele, D.J. Copper: An essential metal in biology. Curr. Biol. 2011, 21, R877–R883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaplan, J.H.; Maryon, E.B. How Mammalian Cells Acquire Copper: An Essential but Potentially Toxic Metal. Biophys. J. 2016, 110, 7–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prohaska, J.R.; Gybina, A.A. Intracellular copper transport in mammals. J. Nutr. 2004, 134, 1003–1006. [Google Scholar] [CrossRef]
- Guengerich, F.P. Introduction to Metals in Biology 2018: Copper homeostasis and utilization in redox enzymes. J. Biol. Chem. 2018, 293, 4603–4605. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Jiao, P.; Qi, M.; Frezza, M.; Dou, Q.P.; Yan, B. Turning tumor-promoting copper into an anti-cancer weapon via high-throughput chemistry. Curr. Med. Chem. 2010, 17, 2685–2698. [Google Scholar] [CrossRef]
- Nicco, C.; Batteux, F. ROS Modulator Molecules with Therapeutic Potential in Cancers Treatments. Molecules 2017, 23, 84. [Google Scholar] [CrossRef]
- Wang, J.; Yi, J. Cancer cell killing via ROS: To increase or decrease, that is the question. Cancer Biol. Ther. 2008, 7, 1875–1884. [Google Scholar] [CrossRef]
- Gupte, A.; Mumper, R.J. Elevated copper and oxidative stress in cancer cells as a target for cancer treatment. Cancer Treat. Rev. 2009, 35, 32–46. [Google Scholar] [CrossRef]
- Matos, L.; Gouveia, A.M.; Almeida, H. Resveratrol Attenuates Copper-Induced Senescence by Improving Cellular Proteostasis. Oxid. Med. Cell. Longev. 2017, 2017, 3793817. [Google Scholar] [CrossRef]
- Matos, L.; Gouveia, A.; Almeida, H. Copper ability to induce premature senescence in human fibroblasts. Age (Dordr.) 2012, 34, 783–794. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Hu, J.; Guan, F.; Song, L.; Fan, R.; Zhu, H.; Hu, X.; Shen, E.; Yang, B. Copper induces cellular senescence in human glioblastoma multiforme cells through downregulation of Bmi-1. Oncol. Rep. 2013, 29, 1805–1810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masaldan, S.; Clatworthy, S.A.S.; Gamell, C.; Smith, Z.M.; Francis, P.S.; Denoyer, D.; Meggyesy, P.M.; Fontaine, S.; Cater, M.A. Copper accumulation in senescent cells: Interplay between copper transporters and impaired autophagy. Redox Biol. 2018, 16, 322–331. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Ye, F.; Xue, W.; Zhou, Z.; Kang, Y.J. Copper regulation of hypoxia-inducible factor-1 activity. Mol. Pharmacol. 2009, 75, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Rigiracciolo, D.C.; Scarpelli, A.; Lappano, R.; Pisano, A.; Santolla, M.F.; De Marco, P.; Cirillo, F.; Cappello, A.R.; Dolce, V.; Belfiore, A.; et al. Copper activates HIF-1alpha/GPER/VEGF signalling in cancer cells. Oncotarget 2015, 6, 34158–34177. [Google Scholar] [CrossRef] [PubMed]
- Leary, S.C. Redox regulation of SCO protein function: Controlling copper at a mitochondrial crossroad. Antioxid. Redox Signal. 2010, 13, 1403–1416. [Google Scholar] [CrossRef] [PubMed]
- He, L.; He, T.; Farrar, S.; Ji, L.; Liu, T.; Ma, X. Antioxidants Maintain Cellular Redox Homeostasis by Elimination of Reactive Oxygen Species. Cell. Physiol. Biochem. 2017, 44, 532–553. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef]
- Jomova, K.; Valko, M. Advances in metal-induced oxidative stress and human disease. Toxicology 2011, 283, 65–87. [Google Scholar] [CrossRef]
- Deavall, D.G.; Martin, E.A.; Horner, J.M.; Roberts, R. Drug-induced oxidative stress and toxicity. J. Toxicol. 2012, 2012, 645460. [Google Scholar] [CrossRef]
- Iakovidis, I.; Delimaris, I.; Piperakis, S.M. Copper and its complexes in medicine: A biochemical approach. Mol. Biol. Int. 2011, 2011, 594529. [Google Scholar] [CrossRef] [PubMed]
- Athar, M.; Back, J.H.; Kopelovich, L.; Bickers, D.R.; Kim, A.L. Multiple molecular targets of resveratrol: Anti-carcinogenic mechanisms. Arch. Biochem. Biophys. 2009, 486, 95–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, Y.; Zhang, C.; Nawa, T.; Aso, T.; Tanaka, M.; Oshiro, S.; Ichijo, H.; Kitajima, S. Homocysteine-responsive ATF3 gene expression in human vascular endothelial cells: Activation of c-Jun NH(2)-terminal kinase and promoter response element. Blood 2000, 96, 2140–2148. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Gan, X.; Zhang, L.; Liu, B.; Zhu, Z.; Li, T.; Zhu, J.; Chen, J.; Yu, H. CoCl2 induces apoptosis via a ROS-dependent pathway and Drp1-mediated mitochondria fission in periodontal ligament stem cells. Am. J. Physiol. Cell Physiol. 2018, 315, C389–C397. [Google Scholar] [CrossRef]
- Gartner, E.M.; Griffith, K.A.; Pan, Q.; Brewer, G.J.; Henja, G.F.; Merajver, S.D.; Zalupski, M.M. A pilot trial of the anti-angiogenic copper lowering agent tetrathiomolybdate in combination with irinotecan, 5-flurouracil, and leucovorin for metastatic colorectal cancer. Investig. New Drugs 2009, 27, 159–165. [Google Scholar] [CrossRef]
- Khan, G.; Merajver, S. Copper chelation in cancer therapy using tetrathiomolybdate: An evolving paradigm. Expert Opin. Investig. Drugs 2009, 18, 541–548. [Google Scholar] [CrossRef]
- Chan, N.; Willis, A.; Kornhauser, N.; Ward, M.M.; Lee, S.B.; Nackos, E.; Seo, B.R.; Chuang, E.; Cigler, T.; Moore, A.; et al. Influencing the Tumor Microenvironment: A Phase II Study of Copper Depletion Using Tetrathiomolybdate in Patients with Breast Cancer at High Risk for Recurrence and in Preclinical Models of Lung Metastases. Clin. Cancer Res. 2017, 23, 666–676. [Google Scholar] [CrossRef]
- Chen, R.; Jiang, T.; She, Y.; Xu, J.; Li, C.; Zhou, S.; Shen, H.; Shi, H.; Liu, S. Effects of Cobalt Chloride, a Hypoxia-Mimetic Agent, on Autophagy and Atrophy in Skeletal C2C12 Myotubes. Biomed. Res. Int. 2017, 2017, 7097580. [Google Scholar] [CrossRef]
- Hudson, C.C.; Liu, M.; Chiang, G.G.; Otterness, D.M.; Loomis, D.C.; Kaper, F.; Giaccia, A.J.; Abraham, R.T. Regulation of hypoxia-inducible factor 1alpha expression and function by the mammalian target of rapamycin. Mol. Cell. Biol. 2002, 22, 7004–7014. [Google Scholar] [CrossRef]
- Hsieh Li, S.M.; Liu, S.T.; Chang, Y.L.; Ho, C.L.; Huang, S.M. Metformin causes cancer cell death through downregulation of p53-dependent differentiated embryo chondrocyte 1. J. Biomed. Sci. 2018, 25, 81. [Google Scholar] [CrossRef]
- Calderon-Aparicio, A.; Strasberg-Rieber, M.; Rieber, M. Disulfiram anti-cancer efficacy without copper overload is enhanced by extracellular H2O2 generation: Antagonism by tetrathiomolybdate. Oncotarget 2015, 6, 29771–29781. [Google Scholar] [CrossRef] [PubMed]
- Cen, D.; Gonzalez, R.I.; Buckmeier, J.A.; Kahlon, R.S.; Tohidian, N.B.; Meyskens, F.L., Jr. Disulfiram induces apoptosis in human melanoma cells: A redox-related process. Mol. Cancer Ther. 2002, 1, 197–204. [Google Scholar] [PubMed]
- Li, Y.; Fu, S.Y.; Wang, L.H.; Wang, F.Y.; Wang, N.N.; Cao, Q.; Wang, Y.T.; Yang, J.Y.; Wu, C.F. Copper improves the anti-angiogenic activity of disulfiram through the EGFR/Src/VEGF pathway in gliomas. Cancer Lett. 2015, 369, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Loo, T.W.; Clarke, D.M. Blockage of drug resistance in vitro by disulfiram, a drug used to treat alcoholism. J. Natl. Cancer Inst. 2000, 92, 898–902. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Cui, Q.C.; Yang, H.; Dou, Q.P. Disulfiram, a clinically used anti-alcoholism drug and copper-binding agent, induces apoptotic cell death in breast cancer cultures and xenografts via inhibition of the proteasome activity. Cancer Res. 2006, 66, 10425–10433. [Google Scholar] [CrossRef] [PubMed]
- Chua, Y.L.; Dufour, E.; Dassa, E.P.; Rustin, P.; Jacobs, H.T.; Taylor, C.T.; Hagen, T. Stabilization of hypoxia-inducible factor-1alpha protein in hypoxia occurs independently of mitochondrial reactive oxygen species production. J. Biol. Chem. 2010, 285, 31277–31284. [Google Scholar] [CrossRef]
- Gao, Y.H.; Li, C.X.; Shen, S.M.; Li, H.; Chen, G.Q.; Wei, Q.; Wang, L.S. Hypoxia-inducible factor 1alpha mediates the down-regulation of superoxide dismutase 2 in von Hippel-Lindau deficient renal clear cell carcinoma. Biochem. Biophys. Res. Commun. 2013, 435, 46–51. [Google Scholar] [CrossRef]
- Kato, Y.; Kawamoto, T.; Fujimoto, K.; Noshiro, M. DEC1/STRA13/SHARP2 and DEC2/SHARP1 coordinate physiological processes, including circadian rhythms in response to environmental stimuli. Curr. Top. Dev. Biol. 2014, 110, 339–372. [Google Scholar] [CrossRef]
- Sato, F.; Bhawal, U.K.; Yoshimura, T.; Muragaki, Y. DEC1 and DEC2 Crosstalk between Circadian Rhythm and Tumor Progression. J. Cancer 2016, 7, 153–159. [Google Scholar] [CrossRef] [Green Version]
- Ivanova, A.V.; Ivanov, S.V.; Danilkovitch-Miagkova, A.; Lerman, M.I. Regulation of STRA13 by the von Hippel-Lindau tumor suppressor protein, hypoxia, and the UBC9/ubiquitin proteasome degradation pathway. J. Biol. Chem. 2001, 276, 15306–15315. [Google Scholar] [CrossRef]
- Kurihara, T.; Kubota, Y.; Ozawa, Y.; Takubo, K.; Noda, K.; Simon, M.C.; Johnson, R.S.; Suematsu, M.; Tsubota, K.; Ishida, S.; et al. von Hippel-Lindau protein regulates transition from the fetal to the adult circulatory system in retina. Development 2010, 137, 1563–1571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, Y.; Xing, X.; Li, S.; Bi, H.; Yang, C.; Zhao, F.; Liu, Y.; Ao, X.; Chang, A.K.; Wu, H. SUMOylation of DEC1 protein regulates its transcriptional activity and enhances its stability. PLoS ONE 2011, 6, e23046. [Google Scholar] [CrossRef] [PubMed]
- Murakami, K.; Wu, Y.; Imaizumi, T.; Aoki, Y.; Liu, Q.; Yan, X.; Seino, H.; Yoshizawa, T.; Morohashi, S.; Kato, Y.; et al. DEC1 promotes hypoxia-induced epithelial-mesenchymal transition (EMT) in human hepatocellular carcinoma cells. Biomed. Res. 2017, 38, 221–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, W.; Shi, X.; Lu, S.; Wu, L.; Wang, Y. Hypoxia-induced overexpression of DEC1 is regulated by HIF-1alpha in hepatocellular carcinoma. Oncol. Rep. 2013, 30, 2957–2962. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Lai, X.; Hu, L.; Lei, C.; Lan, X.; Zhang, C.; Ma, Y.; Zheng, L.; Bai, Y.Y.; Lin, F.; et al. Over-expression of DEC1 inhibits myogenic differentiation by modulating MyoG activity in bovine satellite cell. J. Cell. Physiol. 2018, 233, 9365–9374. [Google Scholar] [CrossRef]
- Ivanov, S.V.; Salnikow, K.; Ivanova, A.V.; Bai, L.; Lerman, M.I. Hypoxic repression of STAT1 and its downstream genes by a pVHL/HIF-1 target DEC1/STRA13. Oncogene 2007, 26, 802–812. [Google Scholar] [CrossRef]
- Qian, Y.; Zhang, J.; Yan, B.; Chen, X. DEC1, a basic helix-loop-helix transcription factor and a novel target gene of the p53 family, mediates p53-dependent premature senescence. J. Biol. Chem. 2008, 283, 2896–2905. [Google Scholar] [CrossRef]
- Qian, Y.; Jung, Y.S.; Chen, X. Differentiated embryo-chondrocyte expressed gene 1 regulates p53-dependent cell survival versus cell death through macrophage inhibitory cytokine-1. Proc. Natl. Acad. Sci. USA 2012, 109, 11300–11305. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Li, Q.Q. Embryo-chondrocyte expressed gene 1, downregulating hypoxia-inducible factor 1alpha, is another marker of lung tumor hypoxia. Acta Pharmacol. Sin. 2007, 28, 549–558. [Google Scholar] [CrossRef]
- Chen, Y.C.; Chan, J.Y.; Chiu, Y.L.; Liu, S.T.; Lozano, G.; Wang, S.L.; Ho, C.L.; Huang, S.M. Grail as a molecular determinant for the functions of the tumor suppressor p53 in tumorigenesis. Cell Death Differ. 2013, 20, 732–743. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Primer Sequence (5′→3′) |
|---|---|
| cyclin D1 | Forward: 5′-ATGGAACACCAGCTCC-3′ Reverse: 5′-TCAGATGTCCACGTCCCGC-3′ |
| DEC1 | Forward: 5′-GTACCCTGCCCACATGTACC-3′ Reverse: 5′-GCTTGGCCAGATACTGAAGC-3′ |
| GAPDH | Forward: 5′-CTTCATTGACCTCAACTAC-3′ Reverse: 5′-GCCATCCACAGTCTTCTG-3′ |
| p21 | Forward: 5′-CTGAGCCGCGACTGTGATGCG-3′ Reverse: 5′-GGTCTGCCGCCGTTTTCGACC-3′ |
| p53 | Forward: 5′-CTCTGACTGTACCACCATCCACTA-3′ Reverse: 5′-GAGTTCCAAGGCCTCATTCAGCTC-3′ |
| ATF3 | Forward: 5′-ATGGGTGCCCCGACGTTG-3′ Reverse: 5′-AGAGGCCTCAATCCATGG-3′ |
| c-Fos | Forward: 5′-GACTACGAGGCGTCATCCTC-3′ Reverse: 5′-GCTCTGGTCTGCGATGGGGCC-3′ |
| VEGF | Forward: 5′-GGACATCTTCCAGGAGTACC-3′ Reverse: 5′-GTTCCCGAAACCCTGAGGG-3′ |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, S.-Y.; Liu, S.-T.; Lin, W.-R.; Lin, C.-K.; Huang, S.-M. The Mechanisms Underlying the Cytotoxic Effects of Copper Via Differentiated Embryonic Chondrocyte Gene 1. Int. J. Mol. Sci. 2019, 20, 5225. https://doi.org/10.3390/ijms20205225
Chen S-Y, Liu S-T, Lin W-R, Lin C-K, Huang S-M. The Mechanisms Underlying the Cytotoxic Effects of Copper Via Differentiated Embryonic Chondrocyte Gene 1. International Journal of Molecular Sciences. 2019; 20(20):5225. https://doi.org/10.3390/ijms20205225
Chicago/Turabian StyleChen, Ssu-Yu, Shu-Ting Liu, Wun-Rong Lin, Chi-Kang Lin, and Shih-Ming Huang. 2019. "The Mechanisms Underlying the Cytotoxic Effects of Copper Via Differentiated Embryonic Chondrocyte Gene 1" International Journal of Molecular Sciences 20, no. 20: 5225. https://doi.org/10.3390/ijms20205225