Kainate Receptor-Mediated Depression of Glutamate Release Involves Protein Kinase A in the Cerebellum


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
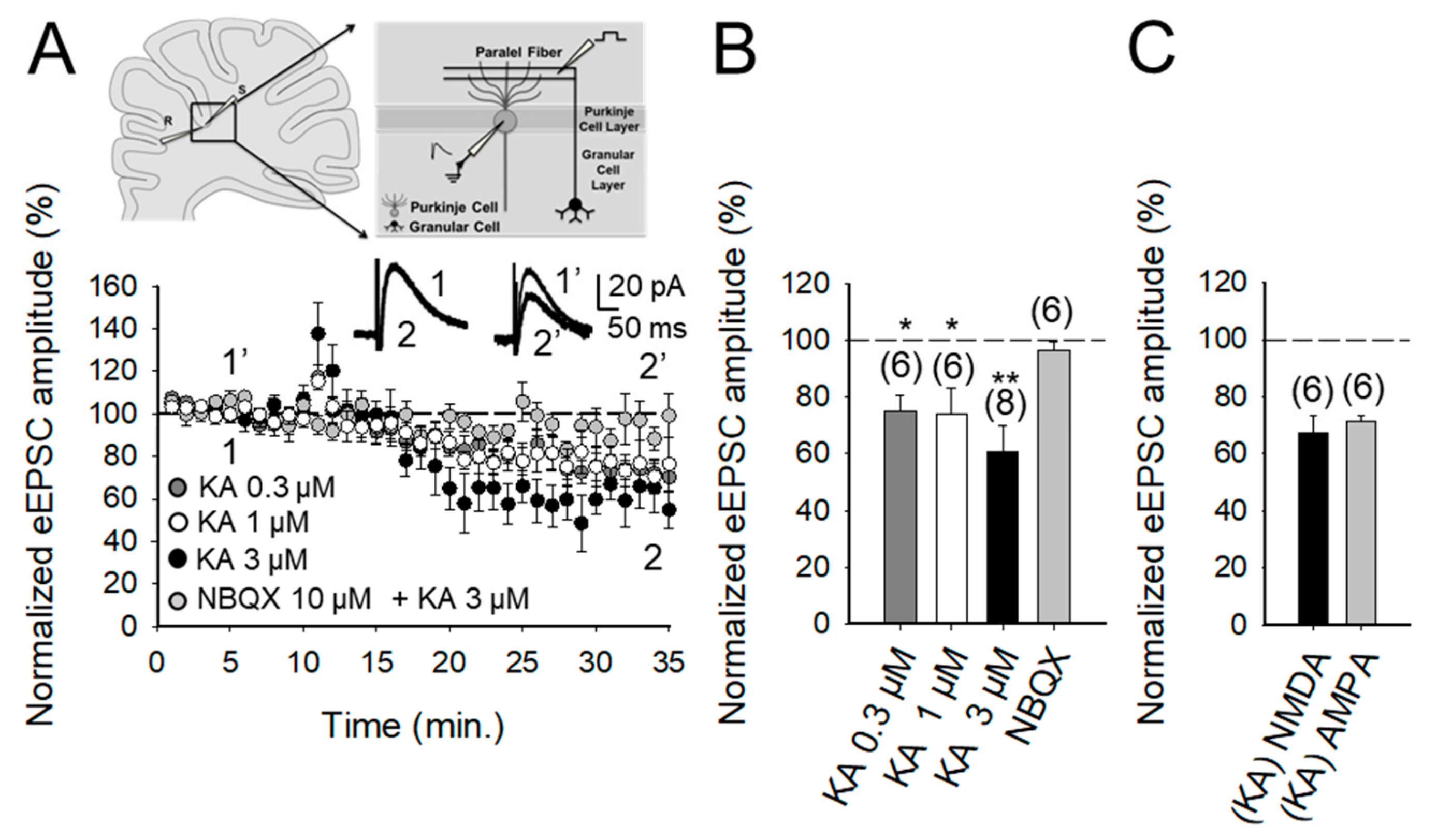
2.1. The Activation of Kainate Receptors by 3 μM KA Produces A Decrease in the Amplitude of NMDA Receptor-Mediated Postsynaptic Currents at PF-PuC Synapses
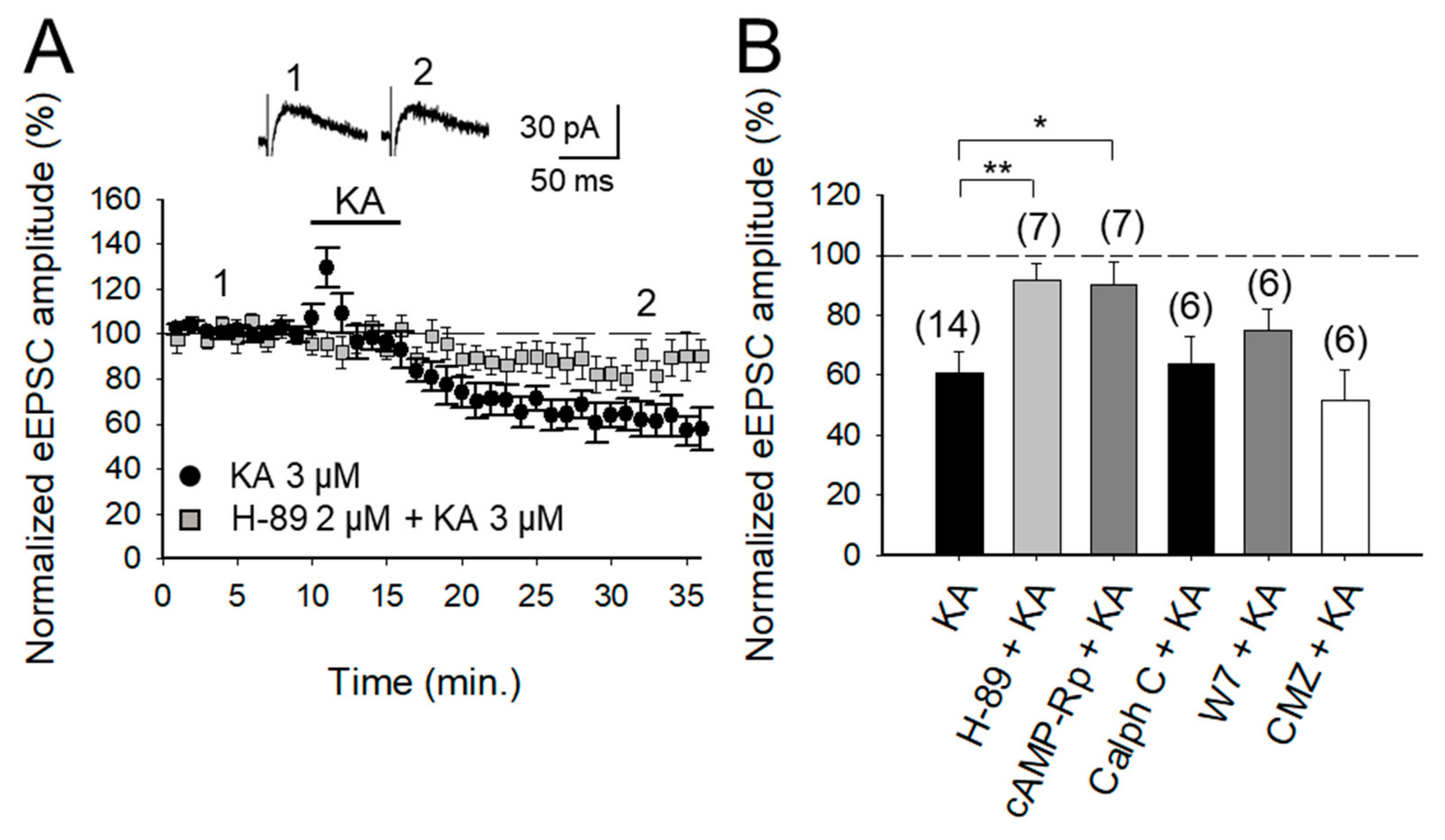
2.2. KAR-Mediated Depression of Glutamatergic Transmission at PF-PuC Is Contingent on cAMP-Dependent Signaling
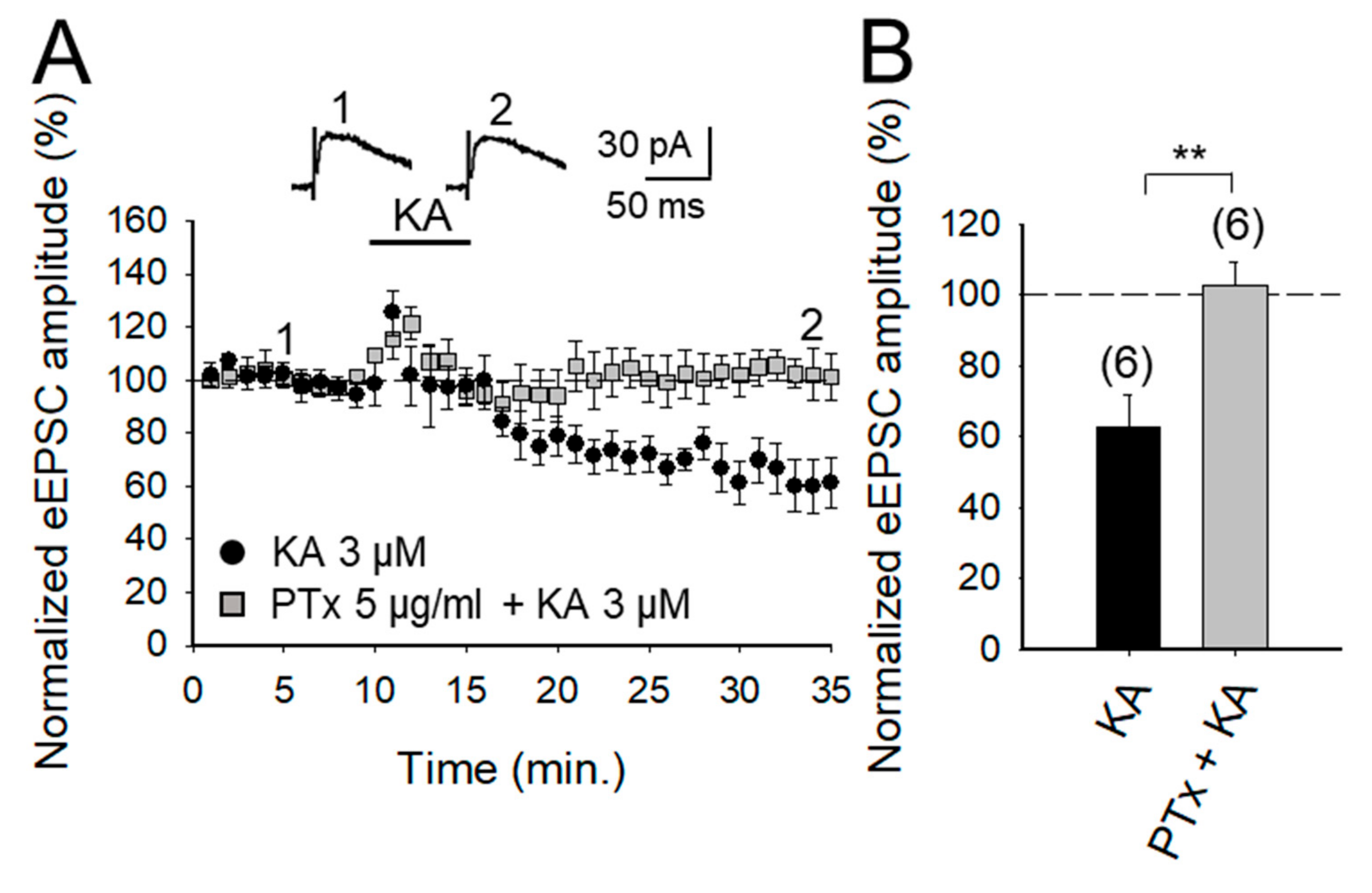
2.3. KAR-Mediated Depression of Glutamate Release Requires G Protein at PF-PuC Synapses
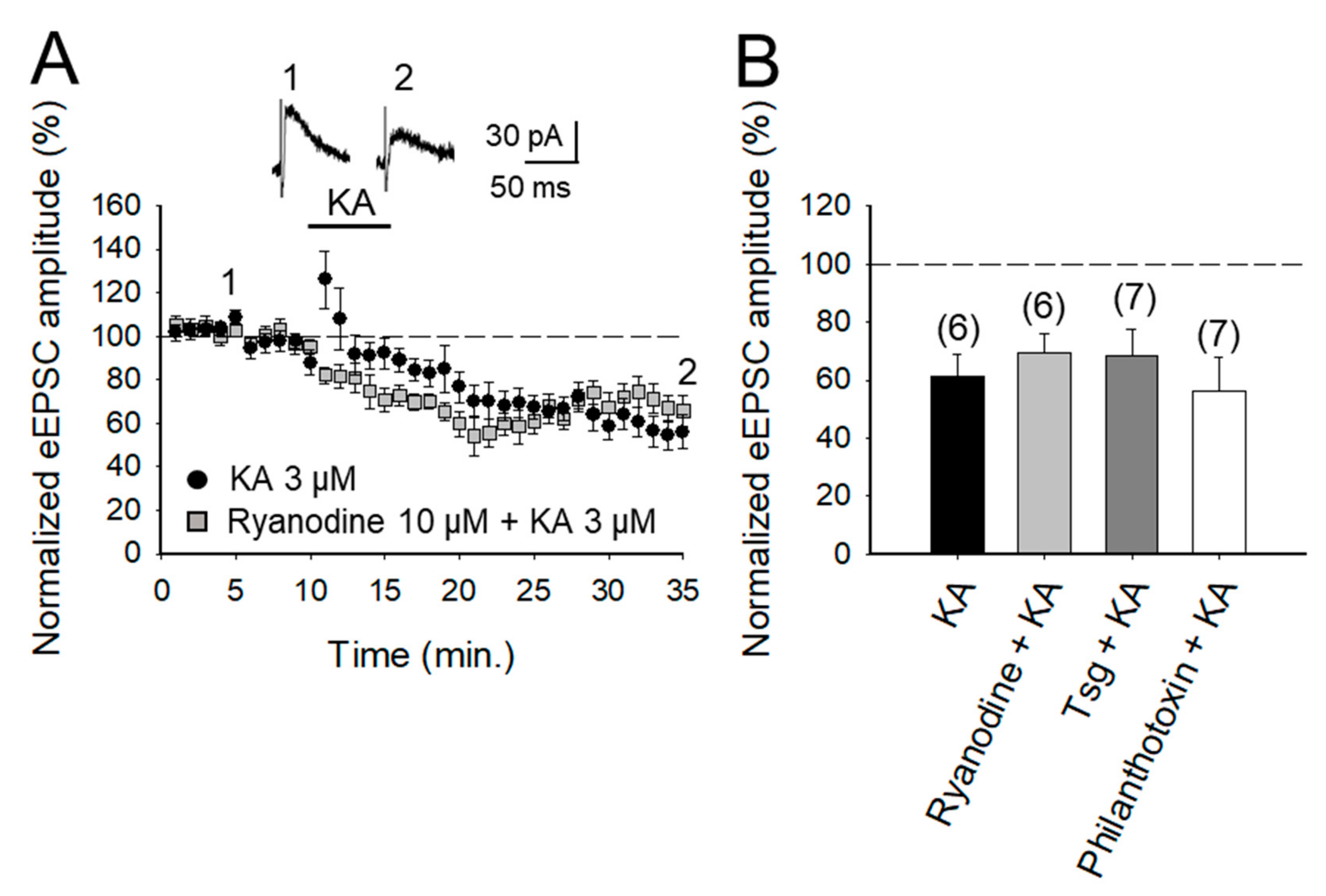
2.4. The Depression of Glutamate Release at PF-PuC Synapses is not Mediated by Calcium-Permeable KAR and does not Require Calcium Release from Intracellular Stores
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Slice Preparation
4.3. Electrophysiological Recordings
4.4. Data Analysis
4.5. Compounds
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lerma, J.; Paternain, A.V.; Rodríguez-Moreno, A.; López-García, J.C. Molecular physiology of kainate receptors. Physiol. Rev. 2001, 89, 971–998. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Moreno, A.; Sihra, T.S. Metabotropic actions of kainate receptors in the CNS. J. Neurochem. 2007, 103, 2121–2135. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Moreno, A.; Sihra, T.S. Kainate receptors with a metabotropic modus operandi. Trends Neurosci. 2007, 30, 630–637. [Google Scholar] [CrossRef] [PubMed]
- Jane, D.E.; Lodge, D.; Collingridge, G.L. Kainate receptors: Pharmacology, function and therapeutical potential. Neuropharmacology 2009, 56, 90–113. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Moreno, A.; Sihra, T.S. Metabotropic actions of kainate receptors in the control of glutamate release in the hippocampus. Adv. Exp. Med. Biol. 2011, 717, 39–48. [Google Scholar]
- Sihra, T.S.; Rodríguez-Moreno, A. Metabotropic actions of kainate receptors in the control of GABA release. Adv. Exp. Med. Biol. 2011, 717, 1–10. [Google Scholar]
- Shira, T.S.; Rodríguez-Moreno, A. Presynaptic kainate receptor-mediated bidirectional modulatory actions: Mechanisms. Neurochem. Int. 2013, 62, 982–987. [Google Scholar] [CrossRef]
- Valbuena, S.; Lerma, J. Non-canonical signalling, the hidden life of ligand-gated ion channels. Neuron 2016, 92, 316–329. [Google Scholar] [CrossRef]
- Negrete-Díaz, J.V.; Sihra, T.S.; Flores, G.; Rodríguez-Moreno, A. Non-canonical mechanisms of presynaptic kainate receptors controlling glutamate release. Front. Mol. Neurosci. 2018, 20, 128. [Google Scholar] [CrossRef]
- Lerma, J.; Marques, J.M. Kainate receptors in health and disease. Neuron 2013, 80, 292–311. [Google Scholar] [CrossRef]
- Smith, T.C.; Wang, L.Y.; Howe, J.R. Distinct kainate receptor phenotypes in immature and mature mouse cerebellar granule cells. J. Physiol. 1999, 517, 51–58. [Google Scholar] [CrossRef]
- Bettler, B.; Boulter, J.; Hermans-Borgmeyer, I.; O’Shea-Greenfield, A.; Deneris, E.S.; Moll, C. Cloning of a novel glutamate receptor subunit, GluR5: Expression in the nervous system during development. Neuron 1990, 5, 583–595. [Google Scholar] [CrossRef]
- Herb, A.; Burnashev, N.; Werner, P.; Sakmann, B.; Wisden, W.; Seeburg, P.H. The KA-2 subunit of excitatory amino acid receptors shows widespread expression in brain and forms ion channels with distantly related subunits. Neuron 1992, 8, 775–785. [Google Scholar] [CrossRef]
- Bahn, S.; Volk, B.; Wisden, W. Kainate receptor gene expression in the developing rat brain. J. Neurosci. 1994, 14, 5525–5547. [Google Scholar] [CrossRef]
- Petralia, R.S.; Wang, Y.X.; Wenthold, R.J. Histological and ultrastructural localization of the kainate receptor subunits, KA2 and GluR6/7, in the rat nervous system using selective antipeptide antibodies. J. Comp. Neurol. 1994, 349, 85–110. [Google Scholar] [CrossRef]
- Swanson, G.T.; Feldmeyer, D.; Kaneda, M.; Cull-Candy, S.G. Effect of RNA editing and subunit co-assembly single-channel properties of recombinant kainate receptors. J. Physiol. 1996, 492, 129–142. [Google Scholar] [CrossRef]
- Delaney, A.J.; Jahr, C.E. Kainate receptors differentially regulate release at two parallel fiber synapses. Neuron 2003, 36, 475–482. [Google Scholar] [CrossRef]
- Falcón-Moya, R.; Losada-Ruiz, P.; Sihra, T.S.; Rodríguez-Moreno, A. Cerebellar kainate receptor-mediated facilitation of glutamate release requires Ca2+-calmodulin and PKA. Front. Mol. Neurosci. 2018, 11, 195. [Google Scholar] [CrossRef]
- Lauri, S.E.; Bortolotto, Z.A.; Bleakman, D.; Ornstein, P.L.; Lodge, D.; Isaac, J.T.R. A critical role of a facilitatory kainate autoreceptor in mossy fiber LTP. Neuron 2001, 32, 697–709. [Google Scholar] [CrossRef]
- Lauri, S.E.; Delany, C.; Clarke, V.E.J.; Bortolotto, Z.A.; Ornstein, P.I.; Isaac, J.T. Synaptic activation of a presynaptic kainate receptor facilitates AMPA receptor-mediated synaptic transmission at hippocampal mossy fibre synapses. Neuropharmacology 2001, 41, 907–915. [Google Scholar] [CrossRef]
- Lauri, S.E.; Bortolotto, Z.A.; Nistico, R.; Bleakman, D.; Ornstein, P.L.; Lodge, D. A role for Ca2+ stores in kainate receptor-dependent synaptic facilitation and LTP at mossy fiber synapses in the hippocampus. Neuron 2003, 39, 327–341. [Google Scholar] [CrossRef]
- Schmitz, D.; Mellor, J.; Nicoll, R.A. Presynaptic kainate receptor mediation of frequency facilitation at hippocampal mossy fiber synapses. Science 2001, 291, 1972–1976. [Google Scholar] [CrossRef]
- Ji, Z.; Stäubli, U. Presynaptic kainate receptors play different physiological roles in mossy fiber and associational-commissural synapses in CA3 of hippocampus from adult rats. Neurosci. Lett. 2002, 331, 71–74. [Google Scholar] [CrossRef]
- Contractor, A.; Sailer, A.; Darstein, M.; Maron, C.; Xu, J.; Swanson, G. Loss of kainate receptor-mediated heterosynaptic facilitation at mossy-fiber synapses in KA2−/− mice. J. Neurosci. 2003, 23, 422–429. [Google Scholar] [CrossRef]
- Breustedt, J.; Schmitz, D. Assesing the role of GLUK5 and GLUK6 at hippocampal mossy fiber synapses. J. Neurosci. 2004, 24, 10093–10098. [Google Scholar] [CrossRef]
- Rodríguez-Moreno, A.; Sihra, T.S. Presynaptic kainate receptor facilitation of glutamate release involves protein kinase A in the rat hippocampus. J. Physiol. 2004, 557, 733–745. [Google Scholar] [CrossRef]
- Rodríguez-Moreno, A.; Sihra, T.S. Presynaptic kainate receptor-mediated facilitation of glutamate release involves Ca2+-calmodulin and PKA in cerebrocortical synaptosomes. FEBS Lett. 2013, 587, 788–792. [Google Scholar] [CrossRef]
- Campbell, S.L.; Mathew, S.S.; Hablitz, J.J. Pre-and postsynaptic effects of kainate on layer II/III pyramidal cells in rat neocortex. Neuropharmacology 2007, 53, 37–47. [Google Scholar] [CrossRef]
- Pinheiro, P.S.; Perrais, D.; Coussen, F.; Barhanin, J.; Bettler, B.; Mann, J.R. GluR7 is an essential subunit of presynaptic kainate autoreceptors at hippocampal mossy fiber synapses. Proc. Natl. Acad. Sci. USA 2007, 1004, 12181–12186. [Google Scholar] [CrossRef]
- Scott, R.; Lalic, T.; Kullmann, D.M.; Capogna, M.; Rusakov, D.A. Target-cell specificity of kainate autoreceptor and Ca2+ store-dependent short-term plasticity at hippocampal mossy fibers. J. Neurosci. 2008, 28, 13139–13149. [Google Scholar] [CrossRef]
- Fernandes, H.B.; Catches, J.S.; Petralia, R.S.; Copits, B.A.; Xu, J.; Russell, T.A. High-affinity kainate receptor subunits are necessary for ionotropic but not metabotropic signaling. Neuron 2009, 63, 818–829. [Google Scholar] [CrossRef]
- Jouhanneau, J.S.; Ball, S.M.; Molnár, E.; Isaac, J.T.R. Mechanisms of bi-directional modulation of thalamocortical transmission in barrel cortex by presynaptic kainite receptors. Neuropharmacology 2011, 60, 832–841. [Google Scholar] [CrossRef]
- Andrade-Talavera, Y.; Duque-Feria, P.; Negrete-Díaz, J.V.; Sihra, T.S.; Flores, G.; Rodríguez-Moreno, A. Presynaptic kainate receptor-mediated facilitation of glutamate release involves Ca2+-calmodulin at mossy fiber-CA3 synapses. J. Neurochem. 2012, 122, 891–899. [Google Scholar] [CrossRef]
- Andrade-Talavera, Y.; Duque-Feria, P.; Sihra, T.S.; Rodríguez-Moreno, A. Presynaptic kainate-receptor-mediated facilitation of glutamate release involves PKA and Ca2+ -calmodulin at thalamocortical synapses. J. Neurochem. 2013, 126, 565–578. [Google Scholar] [CrossRef]
- Manabe, T.; Willey, D.J.; Perkel, D.J.; Nicoll, R.A. Modulation of synaptic transmission and long-term potentiation: effects on paired pulse facilitation and EPSC variance in the CA1 region of the hippocampus. J. Neurophysiol. 1993, 70, 1451–1459. [Google Scholar] [CrossRef]
- Negrete-Díaz, J.V.; Sihra, T.S.; Delgado-García, J.M.; Rodríguez-Moreno, A. Kainate receptor-mediated inhibition of glutamate release involves protein kinase A in the mouse hippocampus. J. Neurophysiol. 2006, 96, 1829–1837. [Google Scholar] [CrossRef]
- Negrete-Díaz, J.V.; Duque-Feria, P.; Andrade-Talavera, Y.; Carrión, M.; Flores, G.; Rodríguez-Moreno, A. Kainate receptor-mediated depression of glutamatergic transmission involving protein kinase A in the lateral amygdala. J. Neurochem. 2012, 121, 36–43. [Google Scholar] [CrossRef]
- Lyon, L.; Borel, M.; Carrión, M.; Kew, J.N.; Corti, C.; Harrison, P.J.; Burnet, P.W.; Paulsen, O.; Rodríguez-Moreno, A. Hippocampal mossy fiber long-term depression in Grm2/3 double knockout mice. Synapse 2011, 65, 945–954. [Google Scholar] [CrossRef]
- Kamiya, H.; Umeda, K.; Ozawa, S.; Manabe, T. Presynaptic Ca2+ entry is unchanged during hippocampal mossy fiber long-term potentiation. J. Neurosci. 2002, 22, 10524–10528. [Google Scholar] [CrossRef]
- Kamiya, H.; Ozawa, S. Kainate receptor-mediated inhibition of presynaptic Ca2+ influx and EPSP in area CA1 of the rat hippocampus. J. Physiol. 1998, 509, 833–845. [Google Scholar] [CrossRef]
- Kamiya, H.; Ozawa, S. Kainate receptor-mediated presynaptic inhibition at the mouse hippocampal mossy fibre. J. Physiol. 2000, 523, 653–665. [Google Scholar] [CrossRef]
- Berridge, M.J. Neuronal calcium signalling. Neuron 1998, 21, 13–26. [Google Scholar] [CrossRef]
- Fletcher, E.J.; Lodge, D. New developments in the molecular pharmacology of AMPA and kainate receptors. Pharmacol. Ther. 1996, 70, 65–89. [Google Scholar] [CrossRef]
- Frerking, M.; Schmitz, D.; Zhou, Q.; Johansen, J.; Nicoll, R.A. Kainate receptors depress excitatory synaptic transmission at CA3 CA1 synapses in the hippocampus via a direct presynaptic action. J. Neurosci. 2001, 21, 2958–2966. [Google Scholar] [CrossRef]
- Negrete-Díaz, J.V.; Sihra, T.S.; Delgado-García, J.M.; Rodríguez-Moreno, A. Kainate receptor-mediated presynaptic inhibition converges with presynaptic inhibition mediated by Group II mGluRs and long-term depression at the hippocampal mossy fiber-CA3 synapse. J. Neural Transm. 2007, 114, 1425–1431. [Google Scholar] [CrossRef]
- Grabauskas, G.; Lancaster, B.; O’Connor, V.; Wheal, H.V. Protein kinase signalling requirements for metabotropic action of kainate receptors in rat CA1 pyramidal neurones. J. Physiol. 2007, 579, 363–373. [Google Scholar] [CrossRef]
- Rodríguez-Moreno, A.; Paulsen, O. Spike timing-dependent long-term depression requires presynaptic NMDA receptors. Nat. Neurosci. 2008, 11, 744–745. [Google Scholar] [CrossRef]
- Rodríguez-Moreno, A.; Kohl, M.M.; Reeve, J.E.; Eaton, T.R.; Collins, H.A.; Anderson, H.L.; Paulsen, O. Presynaptic induction and expression of timing-dependent long-term depression demonstrated by compartment-specific photorelease of a use-dependent NMDA receptor antagonist. J. Neurosci. 2011, 31, 8564–8569. [Google Scholar] [CrossRef]
- Reeve, J.E.; Kohl, M.M.; Rodríguez-Moreno, A.; Paulsen, O.; Anderson, H.L. Caged intracellular NMDA receptor blockers for the study of subcellular ion channel function. Commun. Integr. Biol. 2012, 5, 240–242. [Google Scholar] [CrossRef]
- Rodríguez-Moreno, A.; González-Rueda, A.; Banerjee, A.; Upton, A.L.; Craig, M.T.; Paulsen, O. Presynaptic self-depression at developing neocortical synapses. Neuron 2013, 77, 35–42. [Google Scholar] [CrossRef]
- Pérez-Rodríguez, M.; Arroyo-García, L.E.; Prius-Mengual, J.; Andrade-Talavera, Y.; Armengol, J.A.; Pérez-Villegas, E.M.; Duque-Feria, P.; Flores, G.; Rodríguez-Moreno, A. Adenosine receptor-mediated developmental loss of spike timing-dependent depression in the hippocampus. Cereb. Cortex. 2018, 10. [Google Scholar] [CrossRef]
- Perkinton, M.S.; Sihra, T.S. A high-affinity presynaptic kainate-type glutamate receptor facilitates glutamate exocytosis from cerebral cortex nerve terminals (synaptosomes). Neuroscience 1999, 90, 1281–1292. [Google Scholar] [CrossRef]
- Torres-García, M.E.; Solíss, O.; Patricio, A.; Rodríguez-Moreno, A.; Camacho-Abrego, I.; Limón, I.D.; Flores, G. Dendritic morphology changes in neurons from the prefrontal cortex, hippocampus and nucleus accumbens in rats after lesion of the thalamic reticular nucleus. Neuroscience 2012, 223, 429–438. [Google Scholar] [CrossRef]
- Andrade-Talavera, Y.; Duque-Feria, P.; Paulsen, O.; Rodríguez-Moreno, A. Presynaptic spike timing-dependent long-term depression in the mouse hippocampus. Cereb. Cortex. 2016, 26, 3637–3654. [Google Scholar] [CrossRef]
- Hirano, T. Long-term depression and other synaptic plasticity in the cerebellum. Proc. Jpn. Acad. Ser. B Phys. Biol. 1992, 89, 183–195. [Google Scholar] [CrossRef]
- Sihra, T.S.; Flores, G.; Rodríguez-Moreno, A. Kainate receptors: multiple roles in neuronal plasticity. Neuroscientist 2014, 20, 29–43. [Google Scholar] [CrossRef]
- Mateos-Aparicio, P.; Rodríguez-Moreno, A. The impact of studying brain plasticity. Front. Cell. Neurosci. 2019, 13, 66. [Google Scholar] [CrossRef]
- Falcón-Moya, R.; Sihra, T.S.; Rodríguez-Moreno, A. Kainate receptors: Role in epilepsy. Front. Mol. Neurosci. 2018, 11, 217. [Google Scholar] [CrossRef]
- Maiti, A.; Salles, K.S.; Grassi, S.; Abood, L.G. Behavior and receptor changes after kainate lesioning of nodular cerebellum. Pharmacol. Biochem. Behav. 1986, 25, 589–594. [Google Scholar] [CrossRef]
- De Vera, N.; Camón, L.; Martínez, E. Cerebral distribution of polyamines in kainic acid-induced models of status epilepticus and ataxia in rats. Overproduction of putrescine and histological damage. Eur. Neuropsychopharmacol. 2002, 12, 397–405. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Hayashi, K.; Murakami, H.; Maruyama, S.; Yamaguchi, M. Distribution and characterization of the glutamate receptors in the CNS of ataxic mutant mouse. Neurochem. Res. 1984, 9, 497–505. [Google Scholar] [CrossRef]
- Andoh, T.; Kishi, H.; Motoki, K.; Nakanishi, K.; Kuraishi, Y.; Muraguchi, A. Protective Effect of IL-18 on Kainate-and IL-1 β-induced cerebellar ataxia in mice. J. Immunol. 2008, 180, 2322–2328. [Google Scholar] [CrossRef]
- Harrison, P.J.; Barton, A.J.; Najlerahim, A.; Pearson, R.C. Distribution of a kainate/AMPA receptor mRNA in normal and Alzheimer brain. Neuroreport 1990, 1, 149–152. [Google Scholar] [CrossRef]
- Bullock, W.M.; Cardon, K.; Bustillo, J.; Roberts, R.C.; Perrone-Bizzozero, N.I. Altered expression of genes involved in GABAergic transmission and neuromodulation of granule cell activity in the cerebellum of schizophrenia patients. Am. J. Psych. 2008, 165, 1594–1603. [Google Scholar] [CrossRef]
- Korf, J.; Postema, F. Regional calcium accumulation and cation shifts in rat brain by kainate. J. Neurochem. 1984, 43, 1052–1060. [Google Scholar] [CrossRef]
- Savidge, J.R.; Bleakman, D.; Bristow, D.R. Identification of kainate receptor-mediated intracellular calcium increases in cultured rat cerebellar granule cells. J. Neurochem. 1997, 69, 1763–1766. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Falcón-Moya, R.; Losada-Ruiz, P.; Rodríguez-Moreno, A. Kainate Receptor-Mediated Depression of Glutamate Release Involves Protein Kinase A in the Cerebellum. Int. J. Mol. Sci. 2019, 20, 4124. https://doi.org/10.3390/ijms20174124
Falcón-Moya R, Losada-Ruiz P, Rodríguez-Moreno A. Kainate Receptor-Mediated Depression of Glutamate Release Involves Protein Kinase A in the Cerebellum. International Journal of Molecular Sciences. 2019; 20(17):4124. https://doi.org/10.3390/ijms20174124
Chicago/Turabian StyleFalcón-Moya, Rafael, Pilar Losada-Ruiz, and Antonio Rodríguez-Moreno. 2019. "Kainate Receptor-Mediated Depression of Glutamate Release Involves Protein Kinase A in the Cerebellum" International Journal of Molecular Sciences 20, no. 17: 4124. https://doi.org/10.3390/ijms20174124