Mortalin (GRP75/HSPA9) Promotes Survival and Proliferation of Thyroid Carcinoma Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
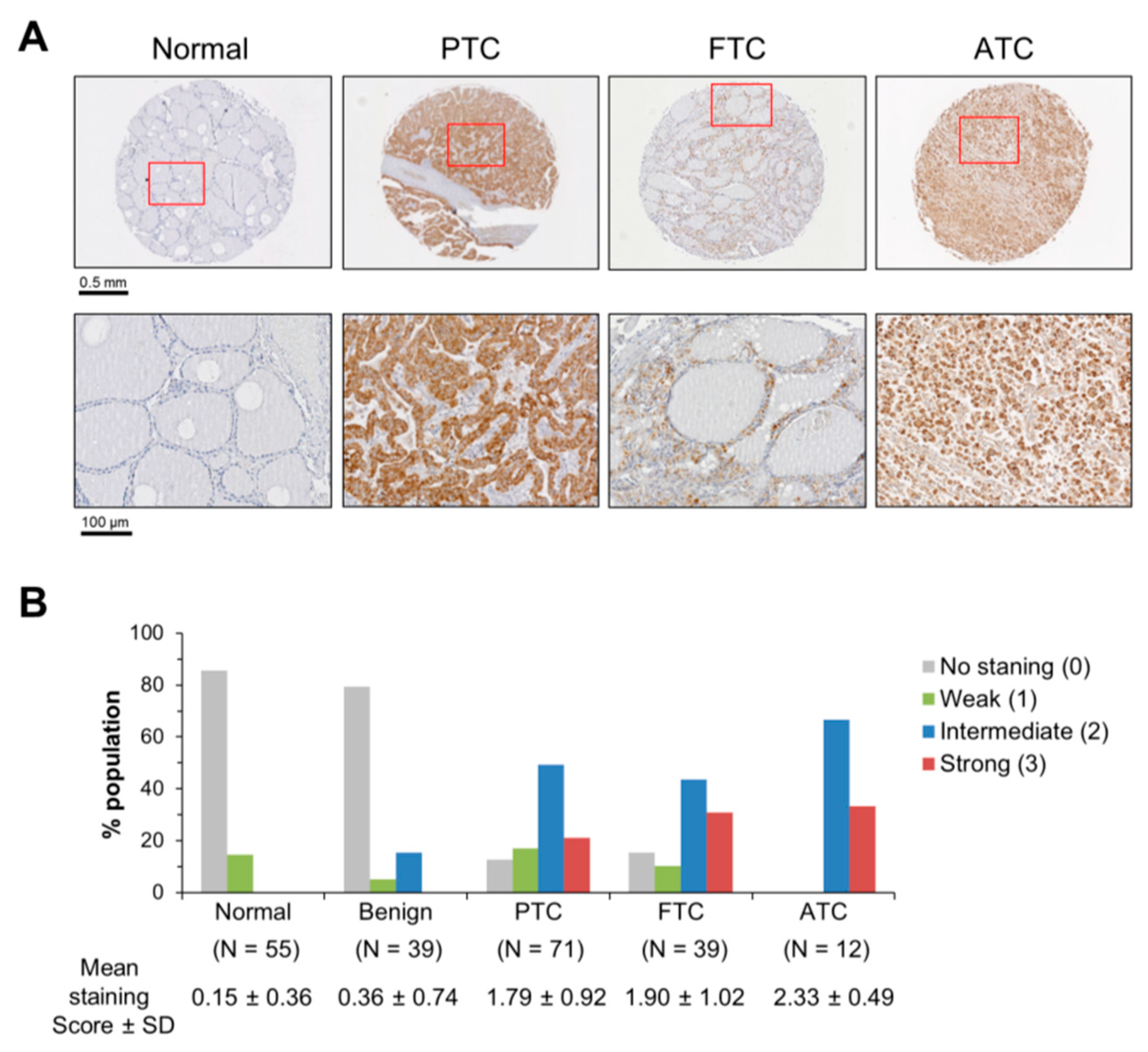
2.1. Mortalin Levels are Upregulated in PTC, FTC and ATC Patient Tissue Biopsies
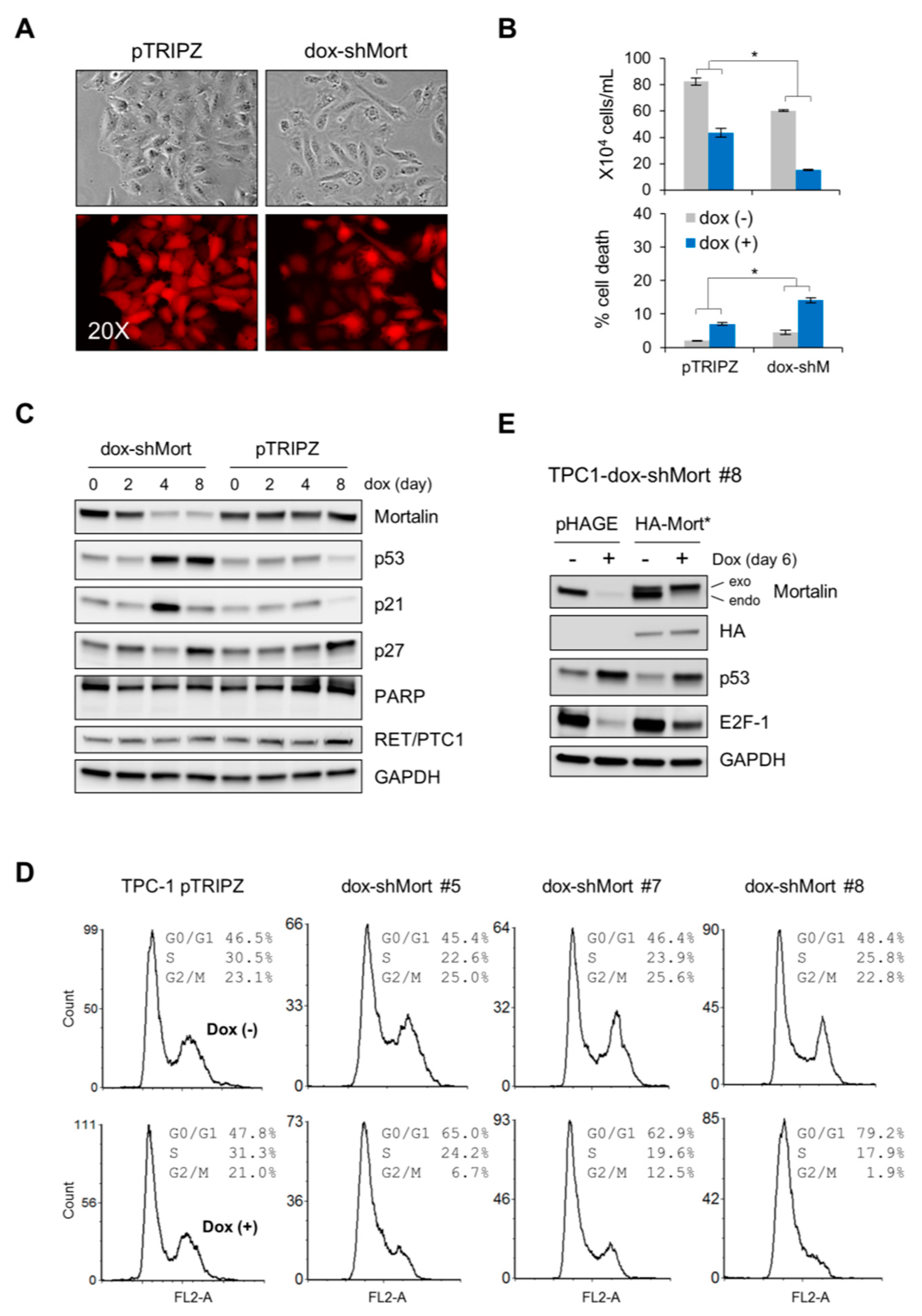
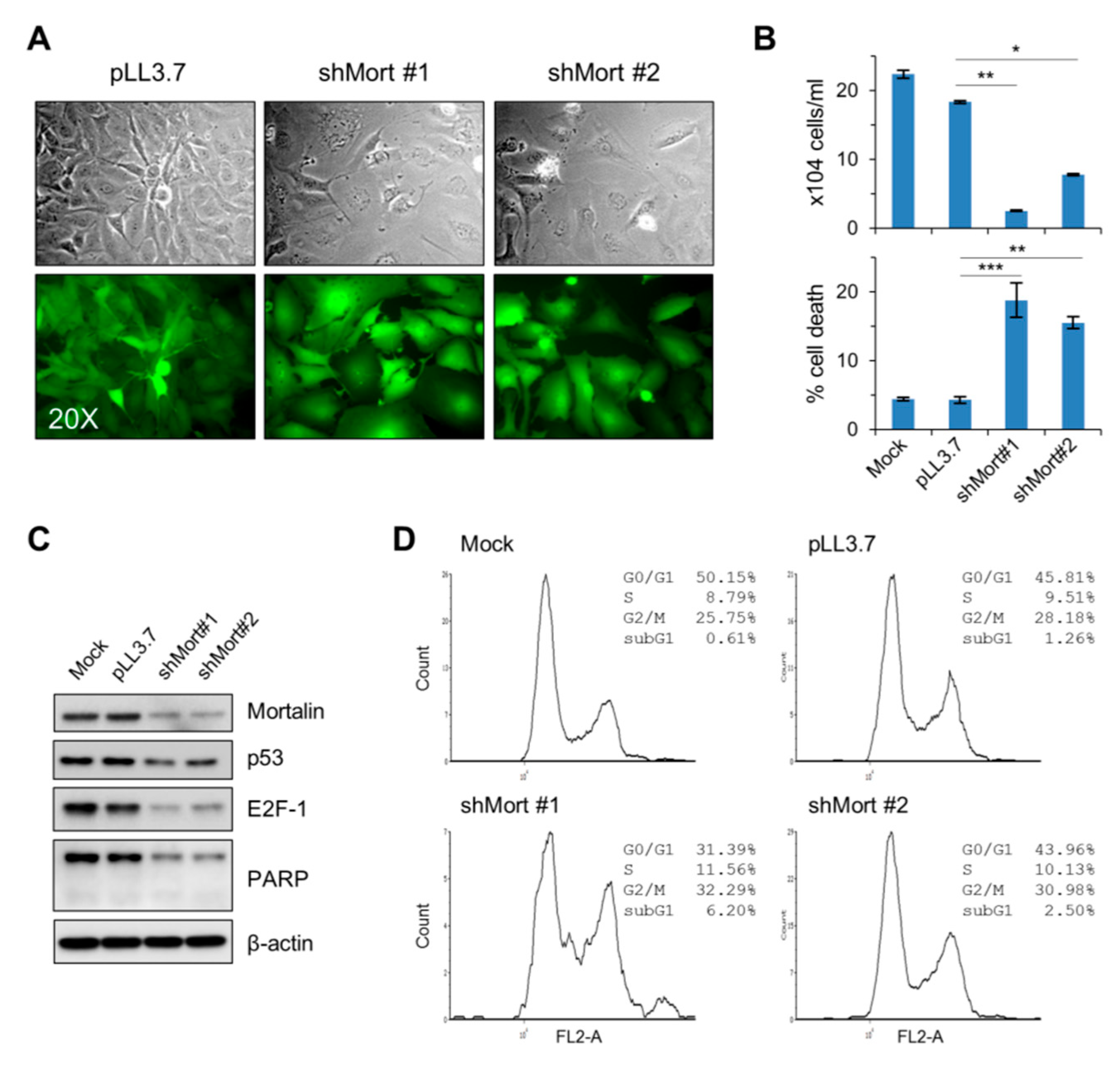
2.2. Mortalin Depletion Induces G0/G1 Phase-Cell Cycle Arrest and TP53 Upregulation in the PTC Cell Line, TPC-1
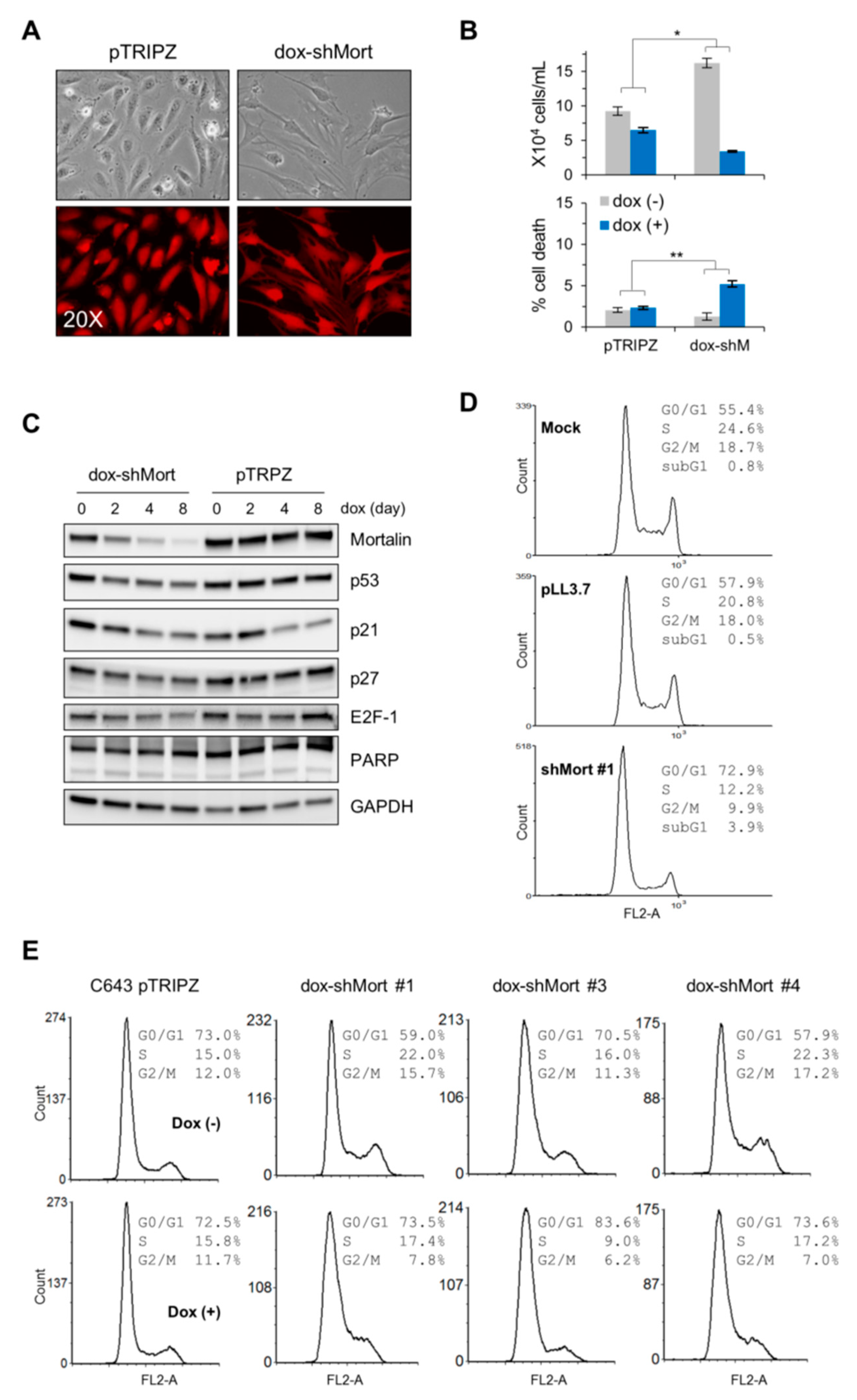
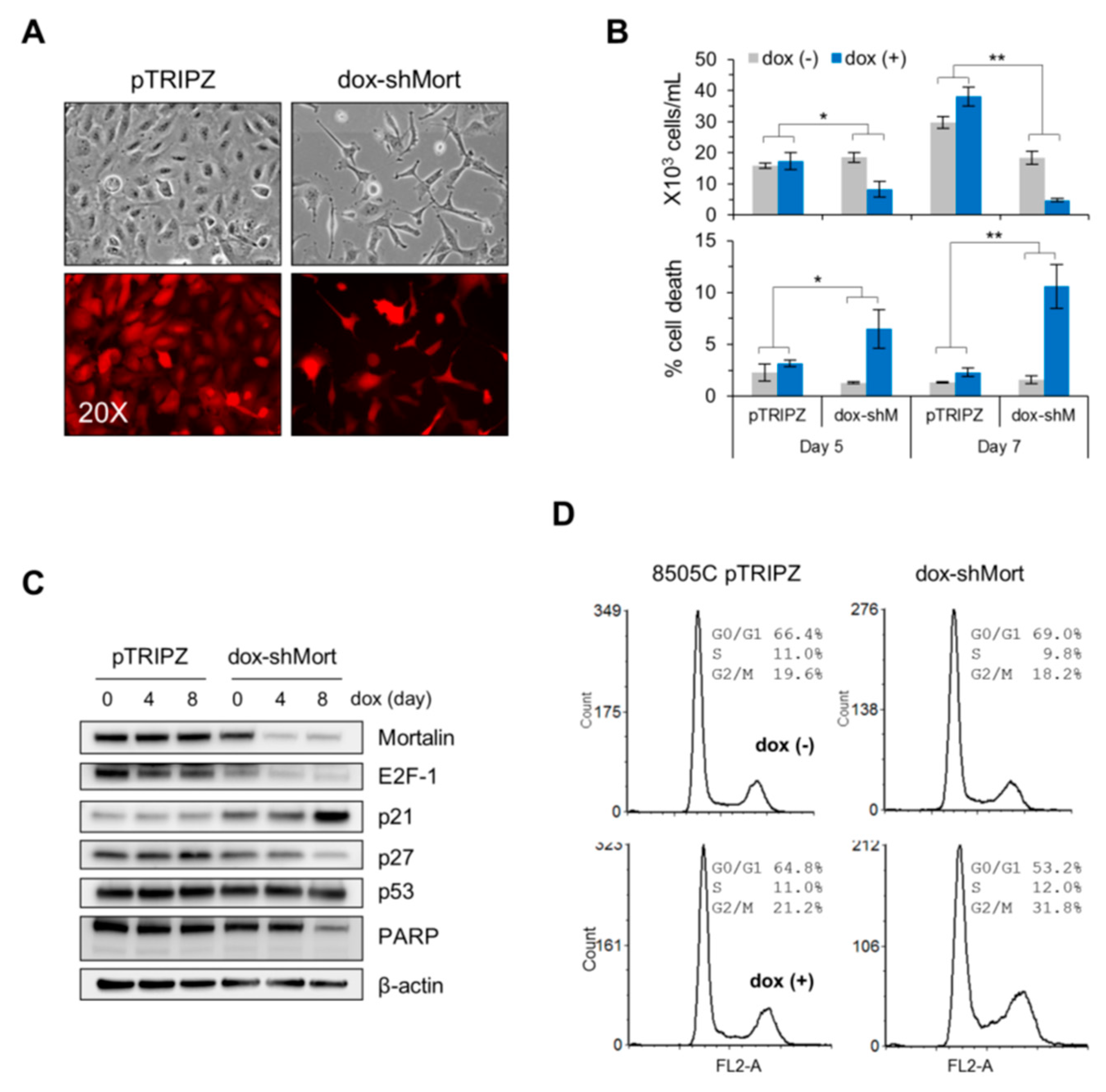
2.3. Mortalin Depletion Suppresses the ATC Cell Lines, C643 and 8505C, and the FTC Cell Line, FTC133, via Cell Cycle Arrest in Different Phases
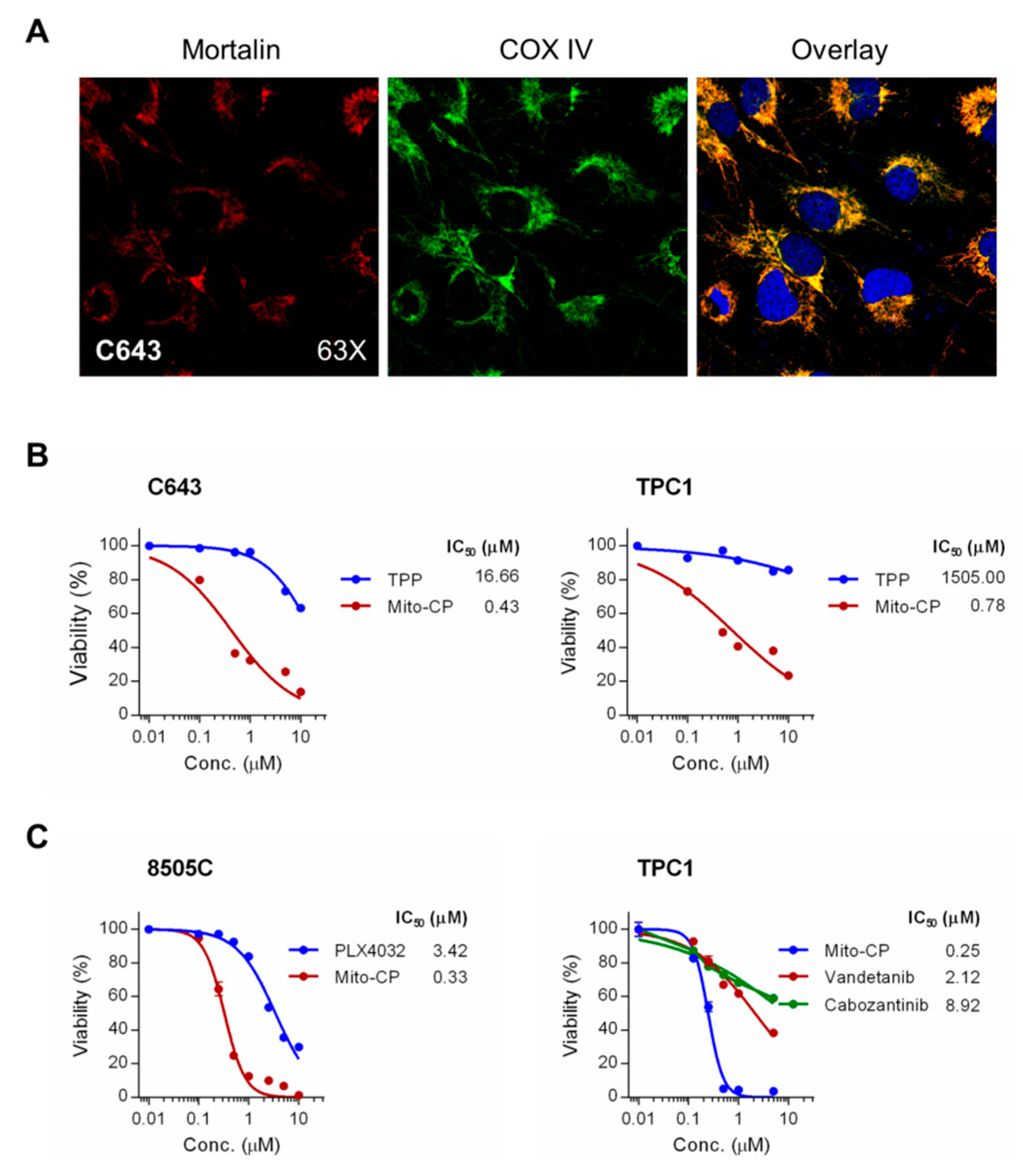
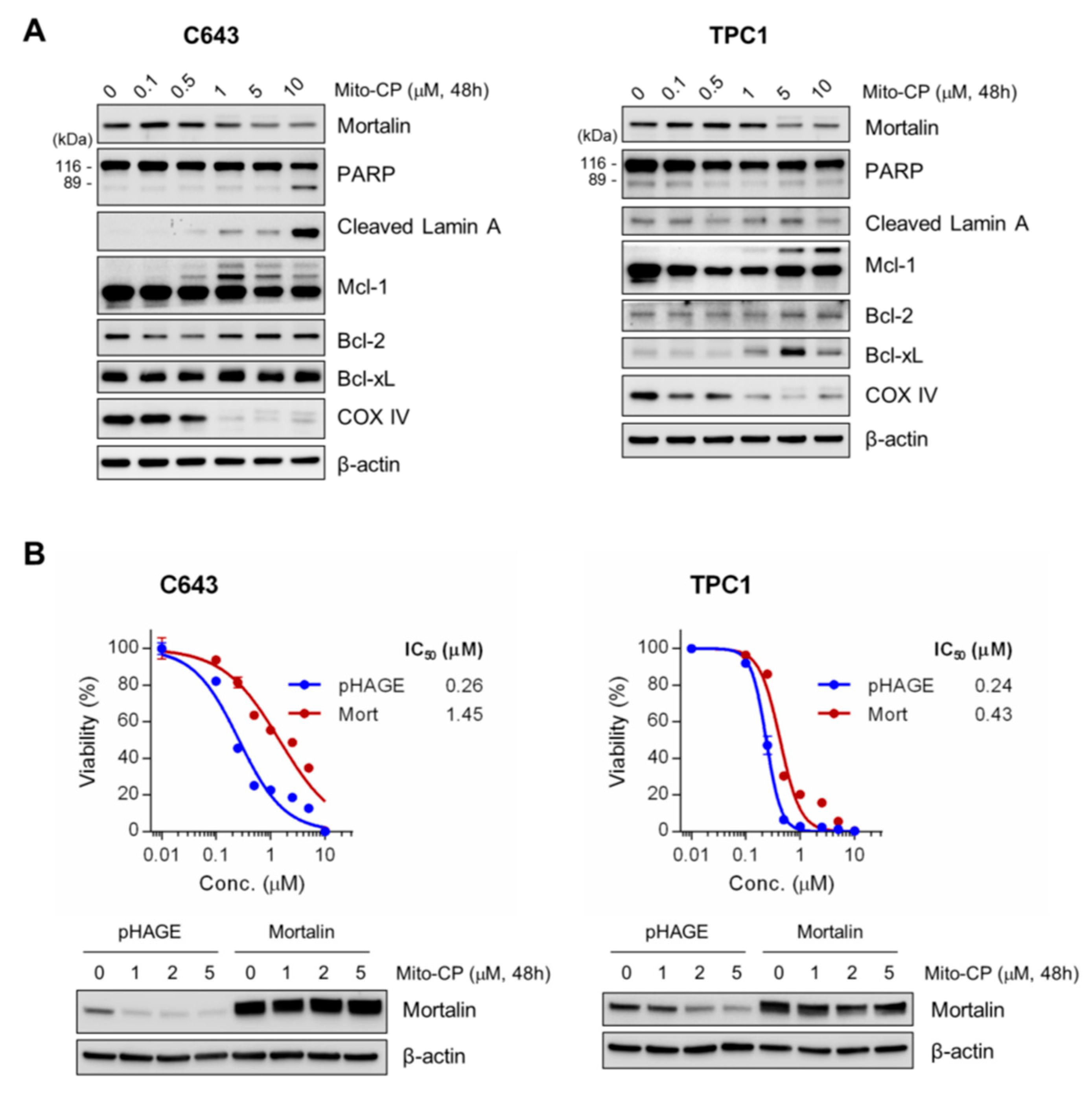
2.4. Mitochondria-Targeted Agent, Mito-CP, can Effectively Suppress Survival of PTC-1, C643, and 8505C Cells In Vitro
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. RNA Interference and Recombinant Lentiviral Constructs
4.3. Cell Proliferation, Death, and Cell Cycle Assays
4.4. Immunoblot Analysis
4.5. Immunofluorescence
4.6. Immunohistochemistry
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Xing, M. Molecular pathogenesis and mechanisms of thyroid cancer. Nat. Rev. Cancer 2013, 13, 184–199. [Google Scholar] [Green Version]
- Lebastchi, A.H.; Callender, G.G. Thyroid cancer. Curr. Probl. Cancer 2014, 38, 48–74. [Google Scholar] [CrossRef] [PubMed]
- Yakushina, V.D.; Lerner, L.V.; Lavrov, A.V. Gene fusions in thyroid cancer. Thyroid 2017. [Google Scholar] [CrossRef]
- Naoum, G.E.; Morkos, M.; Kim, B.; Arafat, W. Novel targeted therapies and immunotherapy for advanced thyroid cancers. Mol. Cancer 2018, 17, 51. [Google Scholar]
- Valerio, L.; Pieruzzi, L.; Giani, C.; Agate, L.; Bottici, V.; Lorusso, L.; Cappagli, V.; Puleo, L.; Matrone, A.; Viola, D.; et al. Targeted therapy in thyroid cancer: State of the art. Clin. Oncol. 2017, 29, 316–324. [Google Scholar] [CrossRef]
- Daugaard, M.; Rohde, M.; Jaattela, M. The heat shock protein 70 family: Highly homologous proteins with overlapping and distinct functions. FEBS Lett. 2007, 581, 3702–3710. [Google Scholar] [PubMed] [Green Version]
- Leustek, T.; Dalie, B.; Amir-Shapira, D.; Brot, N.; Weissbach, H. A member of the Hsp70 family is localized in mitochondria and resembles escherichia coli dnak. Proc. Natl. Acad. Sci. USA 1989, 86, 7805–7808. [Google Scholar] [CrossRef] [PubMed]
- Dundas, S.R.; Lawrie, L.C.; Rooney, P.H.; Murray, G.I. Mortalin is over-expressed by colorectal adenocarcinomas and correlates with poor survival. J. Pathol. 2005, 205, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Wadhwa, R.; Takano, S.; Kaur, K.; Deocaris, C.C.; Pereira-Smith, O.M.; Reddel, R.R.; Kaul, S.C. Upregulation of mortalin/mthsp70/Grp75 contributes to human carcinogenesis. Int. J. Cancer 2006, 118, 2973–2980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaul, S.C.; Deocaris, C.C.; Wadhwa, R. Three faces of mortalin: A housekeeper, guardian and killer. Exp. Gerontol. 2007, 42, 263–274. [Google Scholar] [CrossRef]
- Deocaris, C.C.; Widodo, N.; Ishii, T.; Kaul, S.C.; Wadhwa, R. Functional significance of minor structural and expression changes in stress chaperone mortalin. Ann. NY Acad. Sci. 2007, 1119, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.K.; Hong, S.K.; Veeranki, S.; Karkhanis, M.; Starenki, D.; Plaza, J.A.; Park, J.I. A mortalin/HSPA9-mediated switch in tumor-suppressive signaling of raf/mek/extracellular signal-regulated kinase. Mol. Cell. Biol. 2013, 33, 4051–4067. [Google Scholar] [CrossRef]
- Starenki, D.; Hong, S.K.; Lloyd, R.V.; Park, J.I. Mortalin (GRP75/HSPA9) upregulation promotes survival and proliferation of medullary thyroid carcinoma cells. Oncogene 2015, 34, 4624–4634. [Google Scholar] [CrossRef] [PubMed]
- Starenki, D.; Park, J.I. Selective mitochondrial uptake of MKT-077 can suppress medullary thyroid carcinoma cell survival in vitro and in vivo. Endocrinol. Metab. 2015, 30, 593–603. [Google Scholar] [CrossRef] [PubMed]
- Starenki, D.; Park, J.I. Mitochondria-targeted nitroxide, Mito-CP, suppresses medullary thyroid carcinoma cell survival in vitro and in vivo. J. Clin. Endocrinol. Metab. 2013, 98, 1529–1540. [Google Scholar] [CrossRef]
- Starenki, D.; Hong, S.K.; Wu, P.K.; Park, J.I. Vandetanib and cabozantinib potentiate mitochondria-targeted agents to suppress medullary thyroid carcinoma cells. Cancer Biol. Ther. 2017, 18, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Rosen, A.; Casciola-Rosen, L. Macromolecular substrates for the ice-like proteases during apoptosis. J. Cell. Biochem. 1997, 64, 50–54. [Google Scholar] [CrossRef]
- Hong, S.K.; Starenki, D.; Wu, P.K.; Park, J.I. Suppression of B-RAFV600E melanoma cell survival by targeting mitochondria using triphenyl-phosphonium-conjugated nitroxide or ubiquinone. Cancer Biol. Ther. 2017, 18, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Wadhwa, R.; Takano, S.; Robert, M.; Yoshida, A.; Nomura, H.; Reddel, R.R.; Mitsui, Y.; Kaul, S.C. Inactivation of tumor suppressor p53 by Mot-2, a Hsp70 family member. J. Biol. Chem. 1998, 273, 29586–29591. [Google Scholar] [CrossRef]
- Lu, W.J.; Lee, N.P.; Kaul, S.C.; Lan, F.; Poon, R.T.; Wadhwa, R.; Luk, J.M. Mortalin-p53 interaction in cancer cells is stress dependent and constitutes a selective target for cancer therapy. Cell Death Differ. 2011, 18, 1046–1056. [Google Scholar] [CrossRef]
- Kaul, S.C.; Aida, S.; Yaguchi, T.; Kaur, K.; Wadhwa, R. Activation of wild type p53 function by its mortalin-binding, cytoplasmically localizing carboxyl terminus peptides. J. Biol. Chem. 2005, 280, 39373–39379. [Google Scholar] [CrossRef] [PubMed]
- Iosefson, O.; Azem, A. Reconstitution of the mitochondrial Hsp70 (mortalin)-p53 interaction using purified proteins—identification of additional interacting regions. FEBS Lett. 2010, 584, 1080–1084. [Google Scholar] [CrossRef]
- Lu, W.J.; Lee, N.P.; Kaul, S.C.; Lan, F.; Poon, R.T.; Wadhwa, R.; Luk, J.M. Induction of mutant p53-dependent apoptosis in human hepatocellular carcinoma by targeting stress protein mortalin. Int. J. Cancer 2011, 129, 1806–1814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wadhwa, R.; Yaguchi, T.; Hasan, M.K.; Taira, K.; Kaul, S.C. Mortalin-mpd (mevalonate pyrophosphate decarboxylase) interactions and their role in control of cellular proliferation. Biochem. Biophys. Res. Commun. 2003, 302, 735–742. [Google Scholar] [CrossRef]
- Wu, P.K.; Hong, S.K.; Park, J.I. Steady-state levels of phosphorylated mitogen-activated protein kinase kinase 1/2 determined by mortalin/HSPA9 and protein phosphatase 1 α in KRAS and BRAF tumor cells. Mol. Cell. Biol. 2017, 37. [Google Scholar] [CrossRef] [PubMed]
- Caria, P.; Dettori, T.; Frau, D.V.; Lichtenzstejn, D.; Pani, F.; Vanni, R.; Mai, S. Characterizing the three-dimensional organization of telomeres in papillary thyroid carcinoma cells. J. Cell. Physiol. 2019, 234, 5175–5185. [Google Scholar] [CrossRef]
- Liu, X.; Bishop, J.; Shan, Y.; Pai, S.; Liu, D.; Murugan, A.K.; Sun, H.; El-Naggar, A.K.; Xing, M. Highly prevalent TERT promoter mutations in aggressive thyroid cancers. Endocr. Relat. Cancer 2013, 20, 603–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moon, S.; Song, Y.S.; Kim, Y.A.; Lim, J.A.; Cho, S.W.; Moon, J.H.; Hahn, S.; Park, D.J.; Park, Y.J. Effects of coexistent BRAFV600E and TERT promoter mutations on poor clinical outcomes in papillary thyroid cancer: A meta-analysis. Thyroid 2017, 27, 651–660. [Google Scholar] [CrossRef] [PubMed]
- Shao, H.; Li, X.; Moses, M.A.; Gilbert, L.A.; Kalyanaraman, C.; Young, Z.T.; Chernova, M.; Journey, S.N.; Weissman, J.S.; Hann, B.; et al. Exploration of benzothiazole rhodacyanines as allosteric inhibitors of protein-protein interactions with heat shock protein 70 (Hsp70). J. Med. Chem. 2018, 61, 6163–6177. [Google Scholar] [CrossRef]
- Sarosiek, K.A.; Ni Chonghaile, T.; Letai, A. Mitochondria: Gatekeepers of response to chemotherapy. Trends Cell Biol. 2013, 23, 612–619. [Google Scholar] [CrossRef]
- Modica-Napolitano, J.S.; Aprille, J.R. Delocalized lipophilic cations selectively target the mitochondria of carcinoma cells. Adv. Drug Deliv. Rev. 2001, 49, 63–70. [Google Scholar] [CrossRef]
- Don, A.S.; Hogg, P.J. Mitochondria as cancer drug targets. Trends Mol. Med. 2004, 10, 372–378. [Google Scholar] [CrossRef] [PubMed]
- Dhanasekaran, A.; Kotamraju, S.; Karunakaran, C.; Kalivendi, S.V.; Thomas, S.; Joseph, J.; Kalyanaraman, B. Mitochondria superoxide dismutase mimetic inhibits peroxide-induced oxidative damage and apoptosis: Role of mitochondrial superoxide. Free Radic. Biol. Med. 2005, 39, 567–583. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.K.; Yoon, S.; Moelling, C.; Arthan, D.; Park, J.I. Noncatalytic function of ERK1/2 can promote Raf/MEK/ERK-mediated growth arrest signaling. J. Biol. Chem. 2009, 284, 33006–33018. [Google Scholar] [CrossRef] [PubMed]
- Sosonkina, N.; Nakashima, M.; Ohta, T.; Niikawa, N.; Starenki, D. Down-regulation of ABCC11 protein (MRP8) in human breast cancer. Exp. Oncol. 2011, 33, 42–46. [Google Scholar] [PubMed]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Starenki, D.; Sosonkina, N.; Hong, S.-K.; Lloyd, R.V.; Park, J.-I. Mortalin (GRP75/HSPA9) Promotes Survival and Proliferation of Thyroid Carcinoma Cells. Int. J. Mol. Sci. 2019, 20, 2069. https://doi.org/10.3390/ijms20092069
Starenki D, Sosonkina N, Hong S-K, Lloyd RV, Park J-I. Mortalin (GRP75/HSPA9) Promotes Survival and Proliferation of Thyroid Carcinoma Cells. International Journal of Molecular Sciences. 2019; 20(9):2069. https://doi.org/10.3390/ijms20092069
Chicago/Turabian StyleStarenki, Dmytro, Nadiya Sosonkina, Seung-Keun Hong, Ricardo V. Lloyd, and Jong-In Park. 2019. "Mortalin (GRP75/HSPA9) Promotes Survival and Proliferation of Thyroid Carcinoma Cells" International Journal of Molecular Sciences 20, no. 9: 2069. https://doi.org/10.3390/ijms20092069