Complementary Transcriptomic and Proteomic Analysis Reveals a Complex Network Regulating Pollen Abortion in GMS (msc-1) Pepper (Capsicum annuum L.)

Abstract
:1. Introduction
2. Results
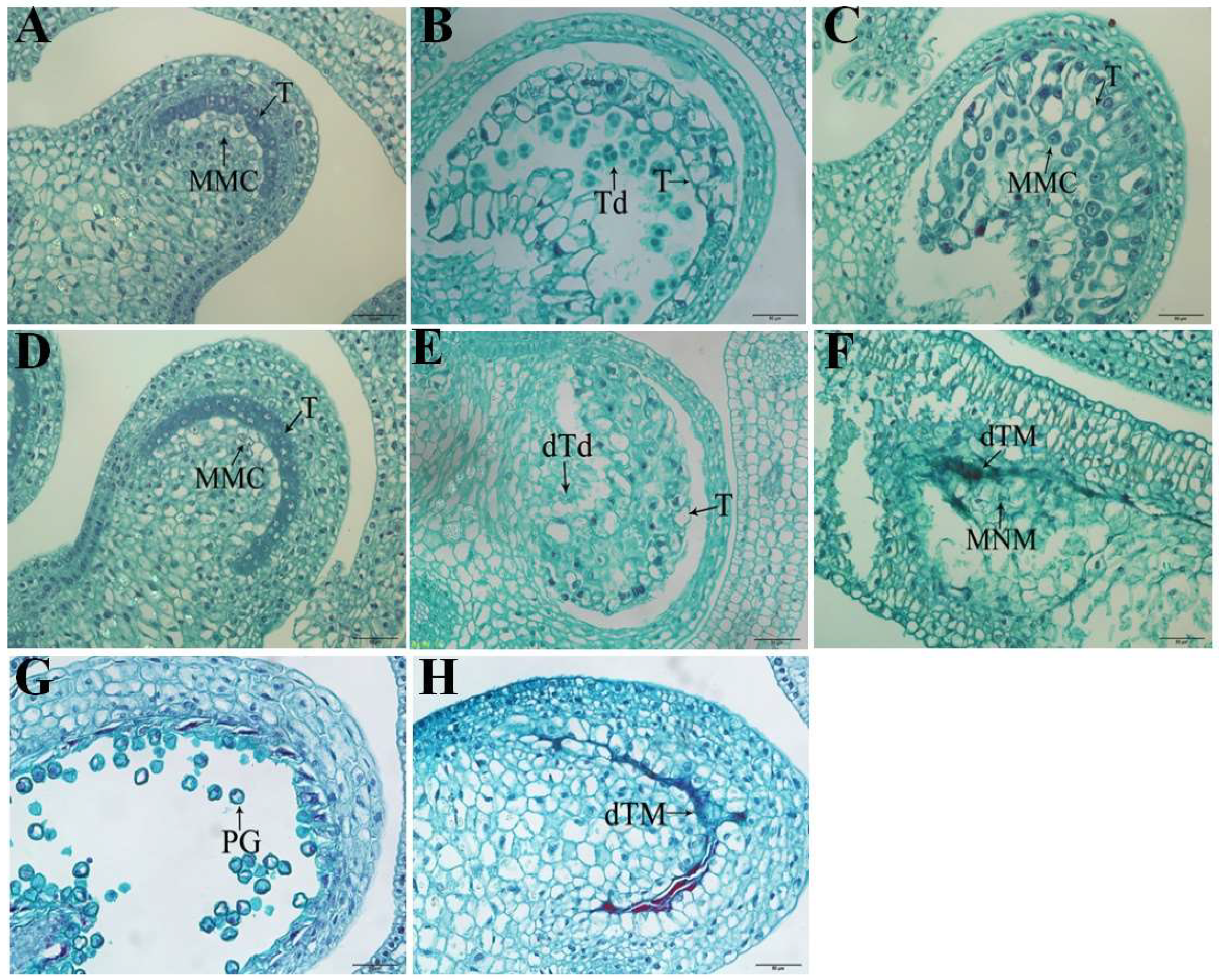
2.1. Cytological Observation of Pepper Anthers at Different Developmental Stages
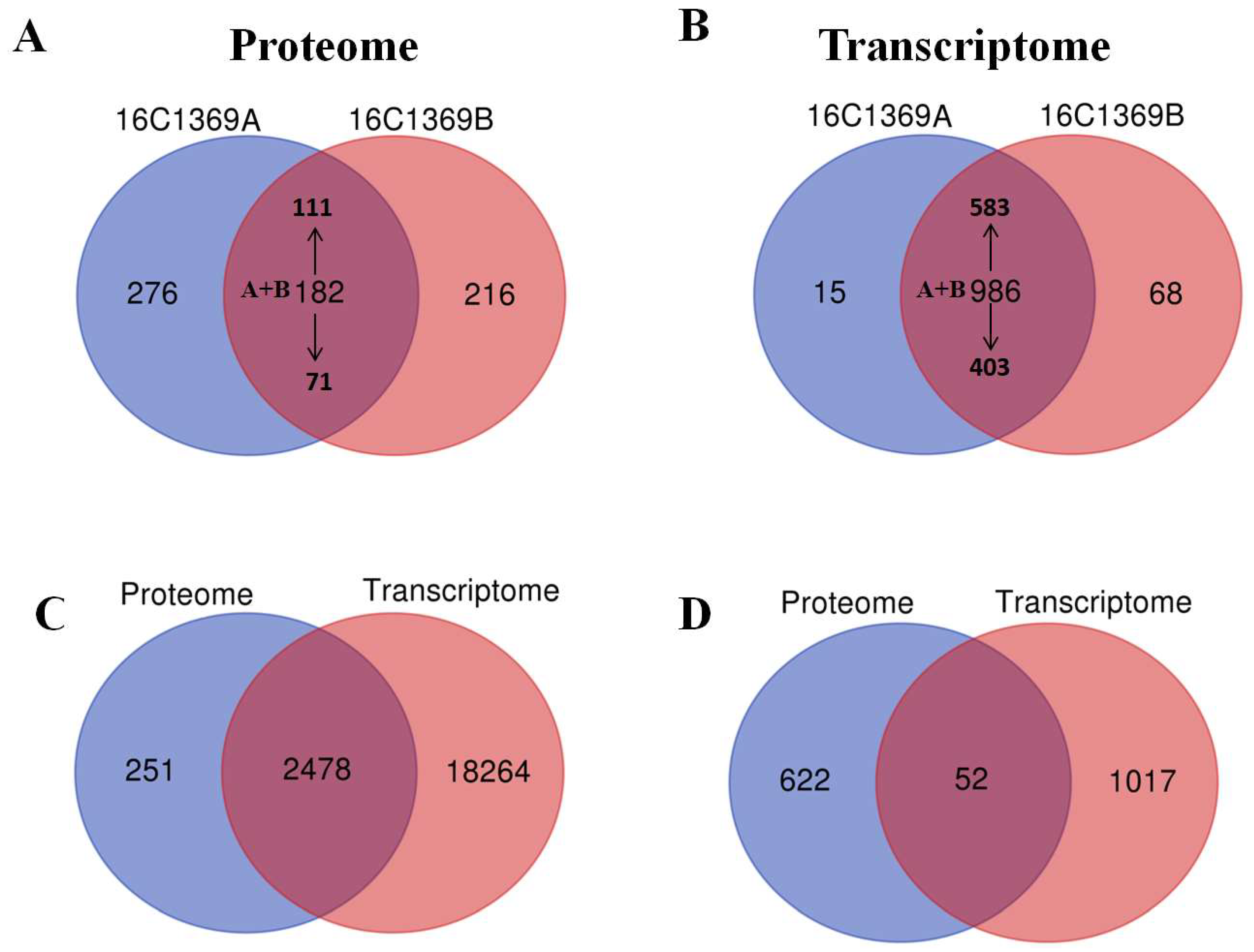
2.2. Overview of Quantitative Proteomics Analysis
2.3. Overview of the Transcriptomic Analysis
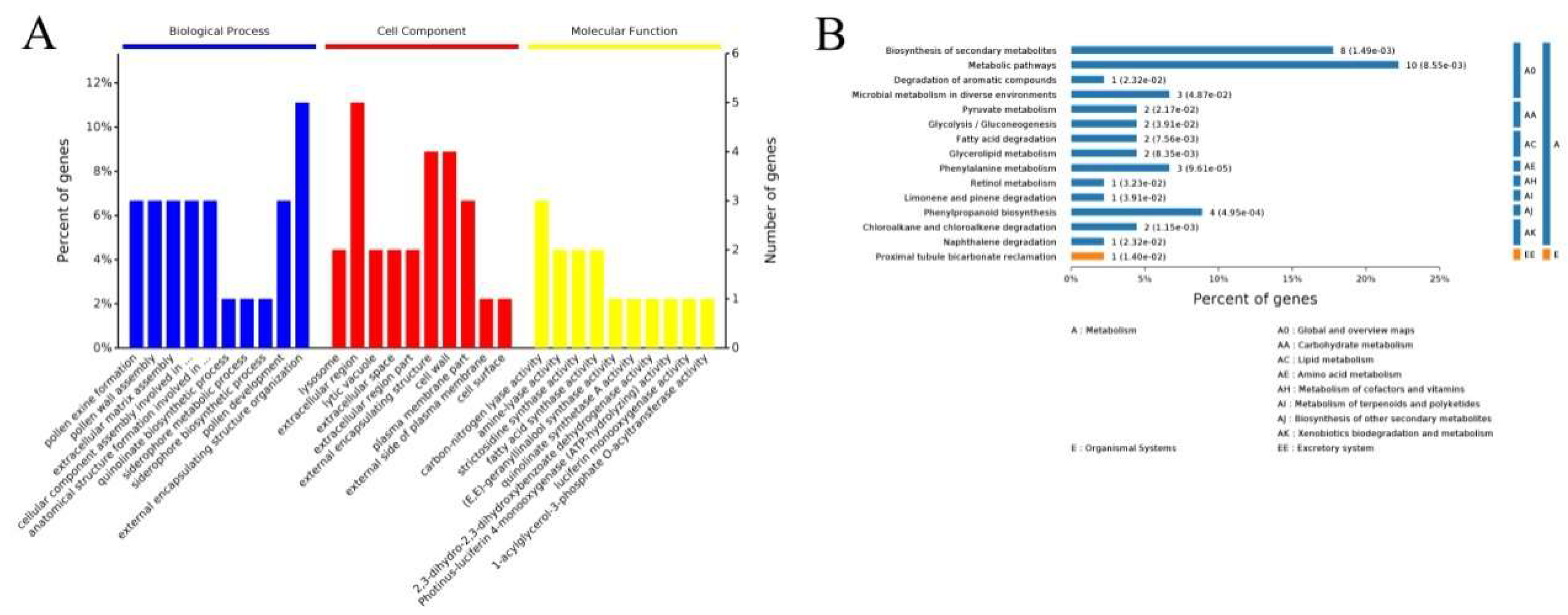
2.4. GO Annotation Function and KEGG Analysis of the DEPs and DEGs
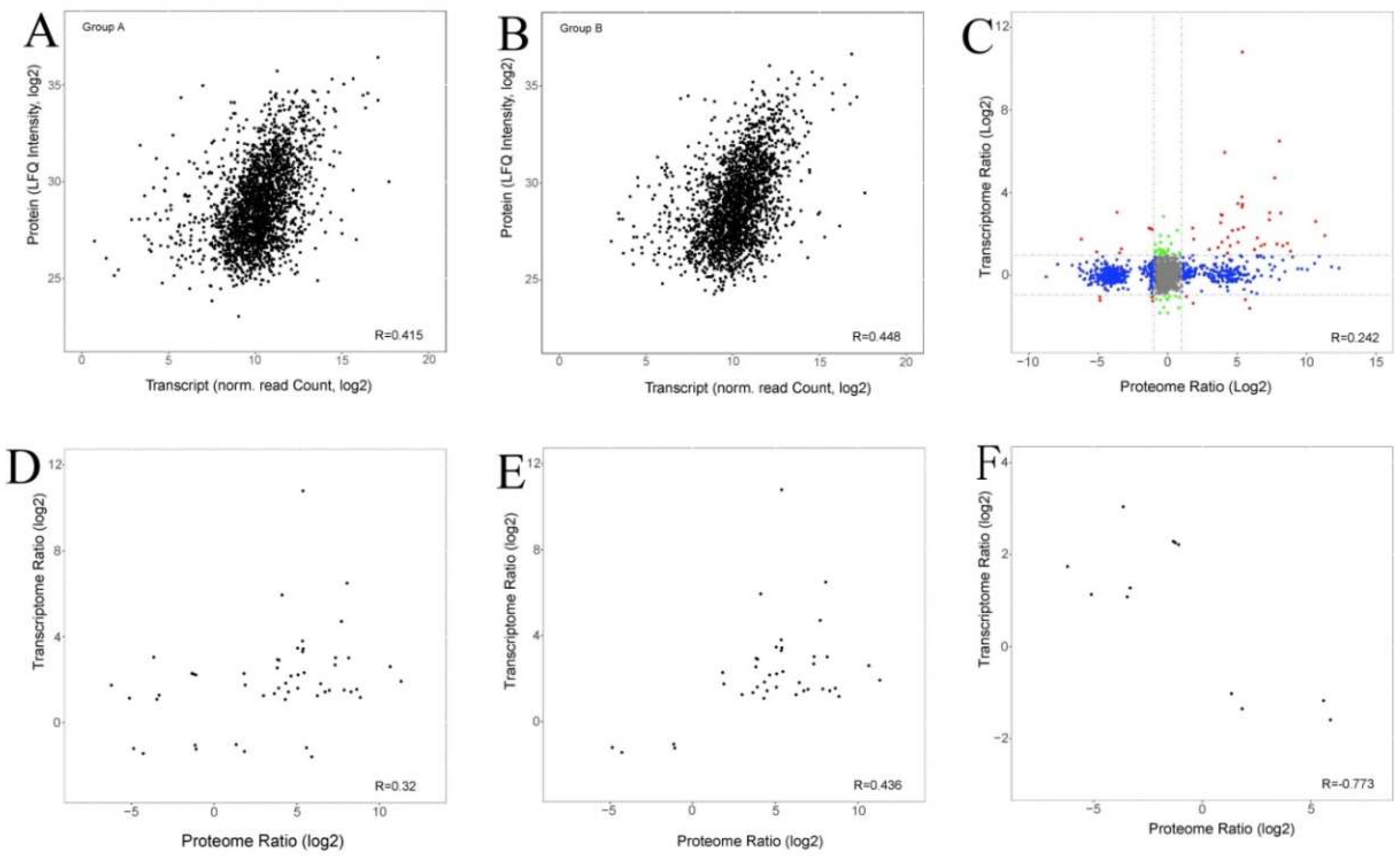
2.5. Comparison of Transcriptome and Proteome Data
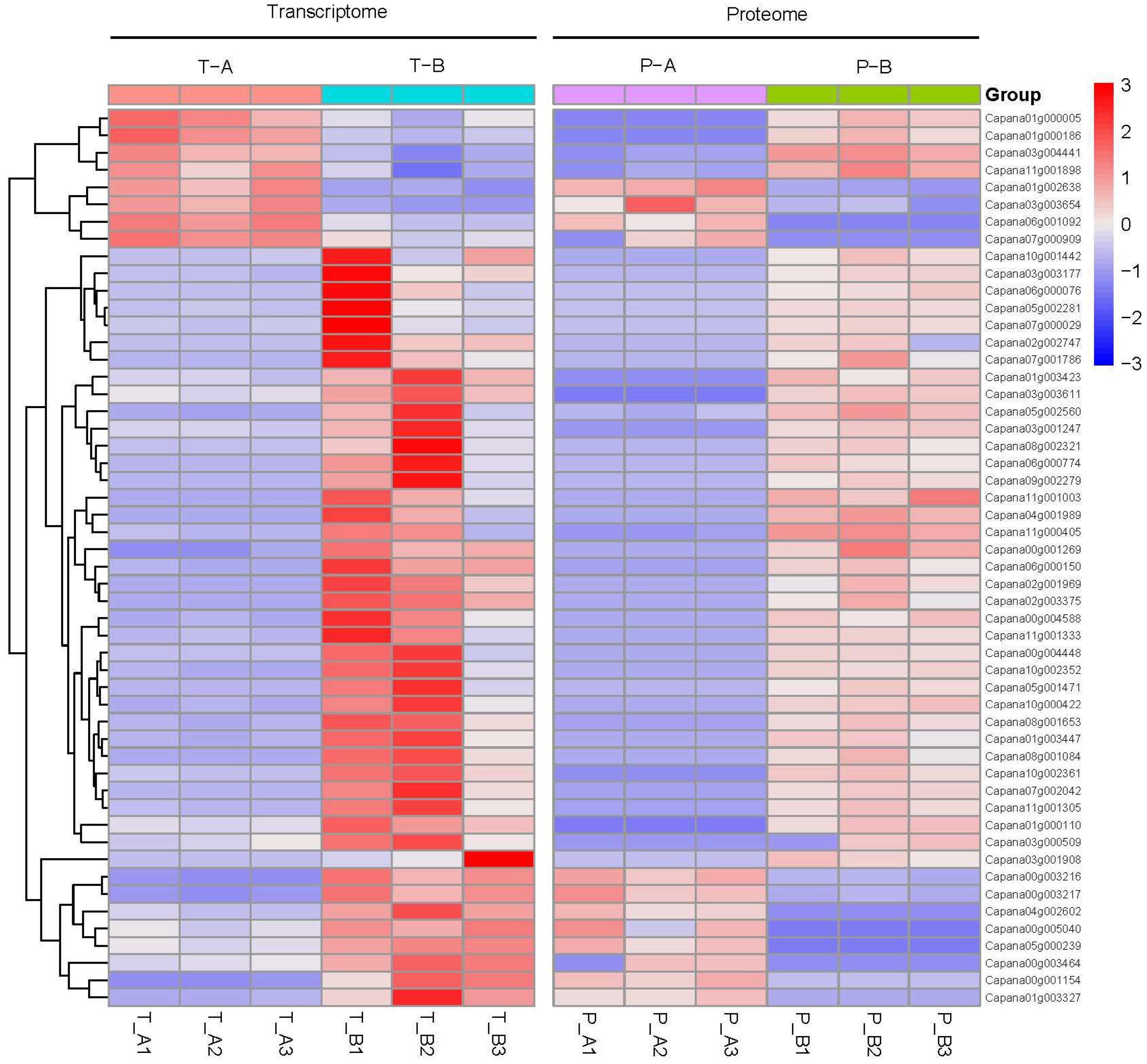
2.6. Cluster Analysis of Expression Patterns in the cor-DEGs-DEPs Genes
2.7. GO and KEGG Analysis of the cor-DEGs-DEPs Genes
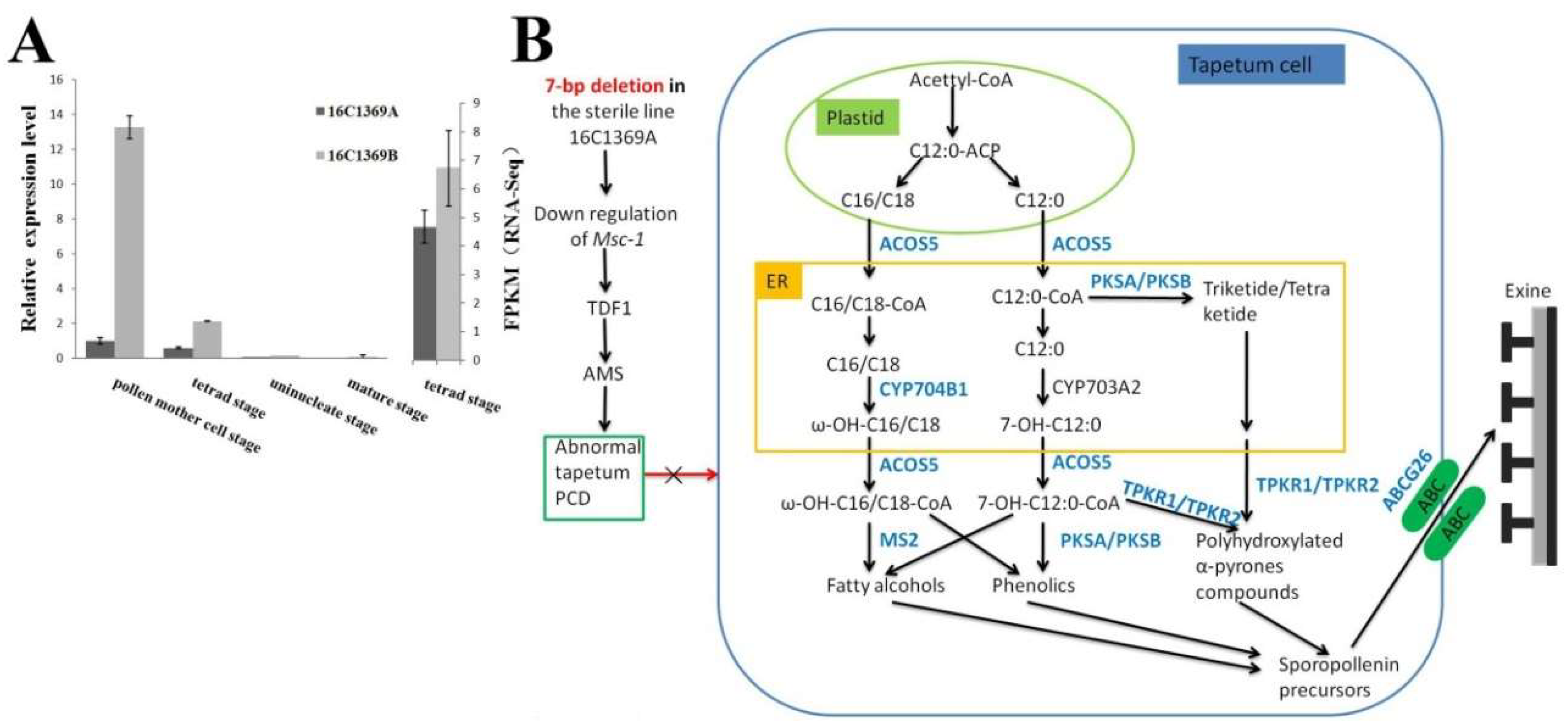
2.8. DEPs and DEGs Related to Anther and Pollen Development
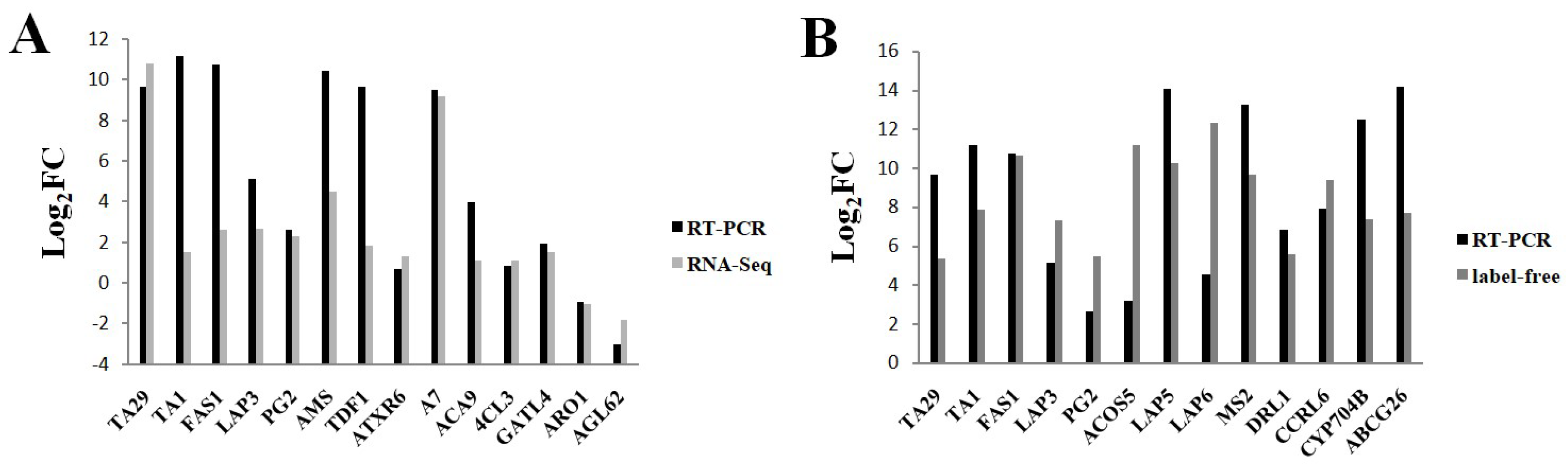
2.9. Validation of Gene Expression Levels
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. Protein Isolation, Enzymolysis, and Label-Free Analysis
4.3. RNA Isolation, Library Preparation, and Sequencing
4.4. Bioinformatics Analysis
4.5. Quantitative Real-Time PCR (qRT-PCR) Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Chen, L.; Liu, Y. Male sterility and fertility restoration in crops. Annu. Rev. Plant Biol. 2014, 65, 579–606. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Zhang, D. Molecular Control of Male Fertility for Crop Hybrid Breeding. Trends Plant Sci. 2018, 23, 53–65. [Google Scholar] [CrossRef]
- Wang, D.; Bosland, P.W. The genes of Capsicum. HortSci 2006, 41, 1169–1187. [Google Scholar] [CrossRef]
- Johri, B.M.; Ambegaoka, K.B.; Srivastava, P.S. Comparative Embryology of Angiosperms; Springer: Berlin, Germany, 1992; Volumes 1–2. [Google Scholar]
- Raghavan, V. Molecular Embryology of Flowering Plants; Cambridge University Press: Cambridge, UK, 1997; pp. 357–393. [Google Scholar]
- Ariizumi, T.; Toriyama, K. Pollen exine pattern formation is dependent on three major developmental processes in Arabidopsis thaliana. Int. J. Plant Dev. Biol. 2007, 1, 106–115. [Google Scholar]
- Heslop-Harrison, J. Pollen wall development. Science 1968, 161, 230–237. [Google Scholar] [CrossRef]
- Blackmore, S.; Wortley, A.H.; Skvarla, J.J.; Rowley, J.R. Pollen wall development in flowering plants. New Phytol. 2007, 174, 483–498. [Google Scholar] [CrossRef] [Green Version]
- Ariizumi, T.; Toriyama, K. Genetic regulation of sporopollenin synthesis and pollen exine development. Annu. Rev. Plant Biol. 2011, 62, 437–460. [Google Scholar] [CrossRef]
- Heslop-Harrison, J. Origin of exine. Nature 1962, 195, 1069–1071. [Google Scholar] [CrossRef]
- Dickinson, H.G.; Heslop-Harrison, J. Common mode of deposition for the sporopollenin of sexine and nexine. Nature 1968, 220, 926–927. [Google Scholar] [CrossRef] [PubMed]
- Mascarenhas, J.P. The biochemistry of angiosperm pollen development. Bot. Rev. 1975, 41, 259–314. [Google Scholar] [CrossRef]
- Stieglitz, H. Role of beta-1,3-glucanase in postmeiotic microspore release. Dev. Biol. 1977, 57, 87–97. [Google Scholar] [CrossRef]
- Pacini, E.; Juniper, B.E. The ultrastructure of pollen grain development in the olive (Olea europaea). II. Secretion by the tapetal cells. New Phytol. 1979, 83, 165–174. [Google Scholar] [CrossRef]
- Hesse, M.; Hess, M.W. Recent trends in tapetum research. A cytological and methodological review. In The Tapetum. Plant Systematics and Evolution (Supplementum 7); Hesse, M., Pacini, E., Willemse, M., Eds.; Springer: Vienna, Austria, 1993; Volume 7, pp. 127–145. [Google Scholar]
- Zhang, Z.B.; Zhu, J.; Gao, J.F.; Wang, C.; Li, H.; Li, H.; Zhang, H.Q.; Zhang, S.; Wang, D.M.; Wang, Q.X.; et al. Transcription factor AtMYB103 is required for anther development by regulating tapetum development, callose dissolution and exine formation in Arabidopsis. Plant J. 2007, 52, 528–538. [Google Scholar] [CrossRef]
- Xu, J.; Yang, C.; Yuan, Z.; Zhang, D.; Gondwe, M.Y.; Ding, Z.; Liang, W.; Zhang, D.; Wilson, Z.A. The ABORTED MICROSPORES regulatory network is required for postmeiotic male reproductive development in Arabidopsis thaliana. Plant Cell 2010, 22, 91–107. [Google Scholar] [CrossRef]
- Goldberg, R.B.; Beals, T.P.; Sanders, P.M. Anther development: Basic principles and partical applications. Plant Cell 1993, 5, 1217–1229. [Google Scholar] [CrossRef]
- McCormick, S. Control of male gametophyte development. Plant Cell 2004, 16, S142–S153. [Google Scholar] [CrossRef]
- Zhang, W.; Sun, Y.; Timofejeva, L.; Chen, C.; Grossniklaus, U.; Ma, H. Regulation of Arabidopsis tapetum development and function by DYSFUNCTIONAL TAPETUM1 (DYT1) encoding a putative bHLH transcription factor. Development 2006, 133, 3085–3095. [Google Scholar] [CrossRef]
- Zhu, J.; Chen, H.; Li, H.; Gao, J.F.; Jiang, H.; Wang, C.; Guan, Y.F.; Yang, Z.N. Defective in Tapetal development and function 1 is essential for anther development and tapetal function for microspore maturation in Arabidopsis. Plant J. 2008, 55, 266–277. [Google Scholar] [CrossRef]
- Wilson, Z.A.; Morroll, S.M.; Dawson, J.; Swarup, R.; Tighe, P.J. The Arabidopsis MALE STERILITY1 (MS1) gene is a transcriptional regulator of male gametogenesis, with homology to the PHD-finger family of transcription factors. Plant J. 2001, 28, 27–39. [Google Scholar] [CrossRef]
- Ito, T.; Nagata, N.; Yoshiba, Y.; Ohme-Takagi, M.; Ma, H.; Shinozaki, K. Arabidopsis MALE STERILITY1 encodes a PHD-type transcription factor and regulates pollen and tapetum development. Plant Cell 2007, 19, 3549–3562. [Google Scholar] [CrossRef]
- Yang, C.; Vizcay-Barrena, G.; Conner, K.; Wilson, Z.A. MALE STERILITY1 is required for tapetal development and pollen wall biosynthesis. Plant Cell 2007, 19, 3530–3548. [Google Scholar] [CrossRef]
- Zhu, J.; Lou, Y.; Xu, X.; Yang, Z.N. A genetic pathway for tapetum development and function in Arabidopsis. J. Integr. Plant Biol. 2011, 53, 892–900. [Google Scholar] [CrossRef]
- Jeong, H.J.; Kang, J.H.; Zhao, M.; Kwon, J.K.; Choi, H.S.; Bae, J.H.; Lee, H.A.; Joung, Y.H.; Choi, D.; Kang, B.C. Tomato Male-sterile 1035 is essential for pollen development and meiosis in anthers. J. Exp. Bot. 2014, 65, 6693. [Google Scholar] [CrossRef]
- Han, Y.; Zhao, F.; Gao, S.; Wang, X.; Wei, A.; Chen, Z.; Liu, N.; Tong, X.; Fu, X.; Wen, C.; et al. Fine mapping of a male sterility gene ms-3 in a novel cucumber (Cucumis sativus L.) mutant. Theor. Appl. Genet. 2017, 131, 449–460. [Google Scholar] [CrossRef]
- Liang, J.; Ma, Y.; Wu, J.; Cheng, F.; Liu, B.; Wang, X. Map-based cloning of the dominant genic male sterile Ms-cd1 gene in cabbage (Brassica oleracea). Theor. Appl. Genet. 2017, 130, 71–79. [Google Scholar] [CrossRef]
- Cheng, Q.; Wang, P.; Liu, J.; Wu, L.; Zhang, Z.; Li, T.; Gao, W.; Yang, W.; Sun, L.; Shen, H. Identification of candidate genes underlying genic male-sterile msc-1 locus via genome resequencing in Capsicum annuum L. Theor. Appl. Genet. 2018, 131, 1861–1872. [Google Scholar] [CrossRef]
- Jeong, K.; Choi, D.; Lee, J. Fine mapping of the genic male-sterile ms1 gene in Capsicum annuum L. Theor. Appl. Genet. 2018, 131, 183–191. [Google Scholar] [CrossRef]
- Guo, J.; Liu, C.; Wang, P.; Cheng, Q.; Sun, L.; Yang, W.; Shen, H. The Aborted Microspores (AMS)-like gene is required for anther and microspore development in pepper (Capsicum annuum L.). Int. J. Mol. Sci. 2018, 19, 1341. [Google Scholar] [CrossRef]
- Voelckel, C.; Gruenheit, N.; Lockhart, P. Evolutionary transcriptomics and proteomics: Insight into plant adaptation. Trends Plant Sci. 2017, 22, 462–471. [Google Scholar] [CrossRef]
- Li, J.; Ren, L.; Gao, Z.; Jiang, M.; Liu, Y.; Zhou, L.; He, Y.; Chen, H. Combined transcriptomic and proteomic analysis constructs a new model for light-induced anthocyanin biosynthesis in eggplant (Solanum melongena L.). Plant Cell Environ. 2017, 40, 3069–3087. [Google Scholar] [CrossRef]
- Luo, X.; Cao, D.; Li, H.; Zhao, D.; Xue, H.; Niu, J.; Chen, L.; Zhang, F.; Cao, S. Complementary iTRAQ-based proteomic and RNA sequencing-based transcriptomic analyses reveal a complex network regulating pomegranate (Punica granatum L.) fruit peel colour. Sci Rep. 2018, 8, 12362. [Google Scholar] [CrossRef]
- Wang, L.; Cao, H.; Chen, C.; Yue, C.; Hao, X.; Yang, Y.; Wang, X. Complementary transcriptomic and proteomic analyses of a chlorophyll-deficient tea plant cultivar reveal multiple metabolic pathway changes. J. Proteom. 2016, 130, 160–169. [Google Scholar] [CrossRef]
- Wang, J.; Islam, F.; Li, L.; Long, M.; Yang, C.; Jin, X.; Ali, B.; Mao, B.; Zhou, W. Complementary RNA-Sequencing based transcriptomics and iTRAQ proteomics reveal the mechanism of the alleviation of quinclorac stress by salicylic acid in Oryza sativa ssp. japonica. Int. J. Mol. Sci. 2017, 18, 1975. [Google Scholar] [CrossRef]
- Wu, Z.; Cheng, J.; Qin, C.; Hu, Z.; Yin, C.; Hu, K. Differential proteomic analysis of anthers between cytoplasmic male sterile and maintainer lines in Capsicum annuum L. Int. J. Mol. Sci. 2013, 14, 22982–22996. [Google Scholar] [CrossRef]
- Fang, X.; Fu, H.F.; Gong, Z.H.; Chai, W.G. Involvement of a universal amino acid synthesis impediment in cytoplasmic male sterility in pepper. Sci. Rep. 2016, 6, 23357. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.J.; Wang, P.; Cheng, Q.; Sun, L.M.; Wang, H.Y.; Wang, Y.T.; Kao, L.N.; Li, Y.N.; Qiu, T.Y.; Yang, W.C.; et al. Proteomic analysis reveals strong mitochondrial involvement in cytoplasmic male sterility of pepper (Capsicum annuum L.). J. Proteom. 2017, 168, 15–27. [Google Scholar] [CrossRef]
- Liu, C.; Ma, N.; Wang, P.Y.; Fu, N.; Shen, H.L. Transcriptome sequencing and de novo analysis of a cytoplasmic male-sterile line and its near-isogenic restorer line in chili pepper (Capsicum annuum L.). PLoS ONE 2013, 8, e65209. [Google Scholar] [CrossRef]
- Qiu, Y.; Liao, L.; Jin, X.; Mao, D.; Liu, R. Analysis of the meiotic transcriptome reveals the genes related to the regulation of pollen abortion in cytoplasmic male-sterile pepper (Capsicum annuum L.). Gene 2018, 641, 8–17. [Google Scholar] [CrossRef]
- Chen, C.; Chen, G.; Cao, B.; Lei, J. Transcriptional profiling analysis of genic male sterile-fertile Capsicum annuum reveal candidate genes for pollen development and maturation by RNA-Seq technology. Plant Cell Tissue Organ Cult. 2015, 122, 465–476. [Google Scholar] [CrossRef]
- Quilichini, T.D.; Grienenberger, E.; Douglas, C.J. The biosynthesis, composition and assembly of the outer pollen wall: A tough case to crack. Phytochemistry 2015, 113, 170–182. [Google Scholar] [CrossRef]
- Piffanelli, P.; Ross, J.H.E.; Murphy, D.J. Biogenesis and function of the lipidic structures of pollen grains. Sex Plant Reprod. 1998, 11, 65–80. [Google Scholar] [CrossRef]
- Hernández-Pinzón, I.; Ross, J.H.; Barnes, K.A.; Damant, A.P.; Murphy, D.J. Composition and role of tapetal lipid bodies in the biogenesis of the pollen coat of Brassica napus. Planta 1999, 208, 588–598. [Google Scholar] [CrossRef]
- Wilson, Z.A.; Zhang, D.B. From Arabidopsis to rice: Pathways in pollen development. J. Exp. Bot. 2009, 60, 1479–1492. [Google Scholar] [CrossRef]
- Vizcay-Barrena, G.; Wilson, Z.A. Altered tapetal PCD and pollen wall development in the Arabidopsisms 1 mutant. J. Exp. Bot. 2006, 57, 2709–2717. [Google Scholar] [CrossRef]
- Li, X.; Gao, X.; Wei, Y.; Deng, L.; Ouyang, Y.; Chen, G.; Li, X.; Zhang, Q.; Wu, C. Rice APOPTOSIS INHIBITOR5 coupled with two DEAD-box adenosine 5′-triphosphate-dependent RNA helicases regulates tapetum degeneration. Plant Cell 2011, 23, 1416–1434. [Google Scholar] [CrossRef]
- Dobritsa, A.A.; Geanconteri, A.; Shrestha, J.; Carlson, A.; Kooyers, N.; Coerper, D.; Urbanczyk-Wochniak, E.; Bench, B.J.; Sumner, L.W.; Swanson, R.; et al. A large-scale genetic screen in Arabidopsis to identify genes involved in pollen exine production. Plant Physiol. 2011, 157, 947–970. [Google Scholar] [CrossRef]
- Dong, X.; Hong, Z.; Sivaramakrishnan, M.; Mahfouz, M.; Verma, D.P. Callose synthase (CalS5) is required for exine formation during microgametogenesis and for pollen viability in Arabidopsis. Plant J. 2005, 42, 315–328. [Google Scholar] [CrossRef]
- Morant, M.; Jorgensen, K.; Schaller, H.; Pinot, F.; Møller, B.L.; Werck-Reichhart, D.; Bak, S. CYP703 is an ancient cytochrome P450 in land plants catalyzing in-chain hydroxylation of lauric acid to provide building blocks for sporopollenin synthesis in pollen. Plant Cell 2007, 19, 1473–1487. [Google Scholar] [CrossRef]
- Dobritsa, A.A.; Lei, Z.; Nishikawa, S.; Urbanczyk-Wochniak, E.; Huhman, D.V.; Preuss, D.; Sumner, L.W. LAP5 and LAP6 encode anther-specific proteins with similarity to chalcone synthase essential for pollen exine development in Arabidopsis. Plant Physiol. 2010, 153, 937–955. [Google Scholar] [CrossRef]
- Lallemand, B.; Erhardt, M.; Heitz, T.; Legrand, M. Sporopollenin biosynthetic enzymes interact and constitute a metabolon localized to the endoplasmic reticulum of tapetum cells. Plant Physiol. 2013, 162, 616–625. [Google Scholar] [CrossRef]
- Wang, K.; Guo, Z.L.; Zhou, W.T.; Zhang, C.; Zhang, Z.Y.; Lou, Y.; Xiong, S.X.; Yao, X.Z.; Fan, J.J.; Zhu, J.; et al. The regulation of sporopollenin biosynthesis genes for rapid pollen wall formation. Plant Physiol. 2018, 178, 283–294. [Google Scholar] [CrossRef]
- Chen, W.; Yu, X.H.; Zhang, K.; Shi, J.; De Oliveira, S.; Schreiber, L.; Shanklin, J.; Zhang, D. Male Sterile 2 encodes a plastid-localized fatty acyl carrier protein reductase required for pollen exine development in Arabidopsis. Plant Physiol. 2011, 157, 842–853. [Google Scholar] [CrossRef]
- Quilichini, T.D.; Friedmann, M.C.; Samuels, A.L.; Douglas, C.J. ATP-binding cassette transporter G26 is required for male fertility and pollen exine formation in Arabidopsis. Plant Physiol. 2010, 154, 678–690. [Google Scholar] [CrossRef]
- Choi, H.; Jin, J.Y.; Choi, S.; Hwang, J.U.; Kim, Y.Y.; Suh, M.C.; Lee, Y. An ABCG/WBC-type ABC transporter is essential for transport of sporopollenin precursors for exine formation in developing pollen. Plant J. 2011, 65, 181–193. [Google Scholar] [CrossRef]
- Dou, X.Y.; Yang, K.Z.; Zhang, Y.; Wang, W.; Liu, X.L.; Chen, L.Q.; Zhang, X.Q.; Ye, D. WBC27, an adenosine tri-phosphate-binding cassette protein, controls pollen wall formation and patterning in Arabidopsis. J. Integr. Plant Biol. 2011, 53, 74–88. [Google Scholar] [CrossRef]
- Xu, J.; Ding, Z.; Vizcaybarrena, G.; Shi, J.; Liang, W.; Yuan, Z.; Werckreichhart, D.; Schreiber, L.; Wilson, Z.A.; Zhang, D. ABORTED MICROSPORES acts as a master regulator of pollen wall formation in Arabidopsis. Plant Cell 2014, 26, 1544–1556. [Google Scholar] [CrossRef]
- Gu, J.N.; Zhu, J.; Yu, Y.; Teng, X.D.; Lou, Y.; Xu, X.F.; Liu, J.L.; Yang, Z.N. DYT1 directly regulates the expression of TDF1 for tapetum development and pollen wall formation in Arabidopsis. Plant J. 2014, 80, 1005–1013. [Google Scholar] [CrossRef]
- Lou, Y.; Xu, X.F.; Zhu, J.; Gu, J.N.; Blackmore, S.; Yang, Z.N. The tapetal AHL family protein TEK determines nexine formation in the pollen wall. Nat. Commun. 2014, 5, 3855. [Google Scholar] [CrossRef]
- Lou, Y.; Zhou, H.S.; Han, Y.; Zeng, Q.Y.; Zhu, J.; Yang, Z.N. Positive regulation of AMS by TDF1 and the formation of a TDF1-AMS complex are required for anther development in Arabidopsis thaliana. New Phytol. 2018, 217, 378–391. [Google Scholar] [CrossRef]
- Xiong, S.X.; Lu, J.Y.; Lou, Y.; Teng, X.D.; Gu, J.N.; Zhang, C.; Shi, Q.S.; Yang, Z.N.; Zhu, J. The transcription factors MS188 and AMS form a complex to activate the expression of CYP703A2 for sporopollenin biosynthesis in Arabidopsis thaliana. Plant J. 2016, 88, 936–946. [Google Scholar] [CrossRef]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef]
- Anders, S.; Huber, W. Differential Expression of RNA-Seq Data at the Gene Level—The DESeqPackage. 2013. Available online: https://bioconductor.org/packages/release/bioc/vignettes/DESeq/inst/doc/DESeq.pdf (accessed on 10 April 2019).
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DEPs | Annotation | Gene ID | Description | Homologous | Regulation (B/A) |
| ACOS5 | Capana02g003302 | ACYL-COA SYNTHETASE 5 | AT1G62940 | UP | |
| PKSA | Capana01g003460 | Chalcone and stilbene synthase family protein | AT1G02050 | UP | |
| PKSB | Capana08g002676 | Chalcone and stilbene synthase family protein | AT4G34850 | UP | |
| MS2 | Capana03g003125 | Jojoba acyl CoA reductase-related male sterility protein | AT3G11980 | UP | |
| TKPR1 | Capana05g000665 | Dihydroflavonol 4-reductase-like 1 | AT4G35420 | UP | |
| TKPR2 | Capana01g002831 | NAD(P)-binding Rossmann-fold superfamily protein | AT1G68540 | UP | |
| CYP704B | Capana01g002203 | Cytochrome P450, family 704, subfamily B | AT1G69500 | UP | |
| ABCG26 | Capana07g002406 | ATP-binding cassette transporter G26 | AT3G13220 | UP | |
| DEGs | Annotation | Gene ID | Description | Homologous | Regulation |
| AMS | Capana08g000254 | ABORTED MICROSPORES | AT2G16910 | UP | |
| TDF1 | Capana04g001901 | DEFECTIVE IN MERISTEM DEVELOPMENT AND FUNCTION 1 | AT3G28470 | UP | |
| ATXR6 | Capana03g001971 | ARABIDOPSIS TRITHORAX-RELATED PROTEIN 6 | AT5G24330 | UP | |
| A7 | Capana07g001721 | ANTHER 7 | AT4G28395 | UP | |
| ACA9 | Capana03g000026 | AUTOINHIBITED CA(2+)-ATPASE 9 | AT3G21180 | UP | |
| 4CL3 | Capana03g001733 | 4-COUMARATE:COA LIGASE 3 | AT1G65060 | UP | |
| GATL4 | Capana01g003063 | GALACTURONOSYLTRANSFERASE-LIKE 4 | AT3G06260 | UP | |
| ARO1 | Capana08g002699 | ARMADILLO REPEAT ONLY 1 | AT4G34940 | DOWN | |
| AGL62 | Trans_newGene_14655 | AGAMOUS-LIKE 62 | AT5G60440 | DOWN | |
| cor-DEGs-DEPs | Annotation | Gene ID | Description | Homologous | Regulation |
| FAS1 | Capana06g000774 | Fasciclin-like arabinogalactan family protein | AT5G16920 | UP | |
| LAP3 | Capana03g003177 | Calcium-dependent phosphotriesterase superfamily protein | AT3G59530 | UP | |
| PG2 | Capana11g001305 | polygalacturonase 2 | AT1G70370 | UP | |
| TA29 | Capana02g001969 | TA29_TOBAC Anther-specific protein TA-29 | NA | UP | |
| TA1 | Capana04g001989 | Arabidopsis homolog of TASSELSEED2. Expressed specifically in tapetal cells | AT3G42960 | UP |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, Q.; Li, T.; Ai, Y.; Lu, Q.; Wang, Y.; Sun, L.; Shen, H. Complementary Transcriptomic and Proteomic Analysis Reveals a Complex Network Regulating Pollen Abortion in GMS (msc-1) Pepper (Capsicum annuum L.). Int. J. Mol. Sci. 2019, 20, 1789. https://doi.org/10.3390/ijms20071789
Cheng Q, Li T, Ai Y, Lu Q, Wang Y, Sun L, Shen H. Complementary Transcriptomic and Proteomic Analysis Reveals a Complex Network Regulating Pollen Abortion in GMS (msc-1) Pepper (Capsicum annuum L.). International Journal of Molecular Sciences. 2019; 20(7):1789. https://doi.org/10.3390/ijms20071789
Chicago/Turabian StyleCheng, Qing, Ting Li, Yixin Ai, Qiaohua Lu, Yihao Wang, Liang Sun, and Huolin Shen. 2019. "Complementary Transcriptomic and Proteomic Analysis Reveals a Complex Network Regulating Pollen Abortion in GMS (msc-1) Pepper (Capsicum annuum L.)" International Journal of Molecular Sciences 20, no. 7: 1789. https://doi.org/10.3390/ijms20071789